





Abstract
Background. Although neutrophils and T cells are important in mediating renal injury following ischaemia/reperfusion, the role of macrophages is still unknown. Using liposomal clodronate (LC), we investigated the effect of systemic monocyte–macrophage depletion on renal damage in ischaemic acute renal failure in rats.
Methods. Male Sprague–Dawley rats were injected by LC or liposomal vehicle and underwent bilateral renal pedicle clamping (40 min) or sham ischaemia. Biochemical and histological renal damage was assessed and gene expression kinetics of tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and monocyte chemoattractant protein-1 (MCP-1) using quantitative real-time reverse transcription–polymerase chain reaction were conducted at 4, 24 and 72 h after reperfusion.
Results. The percentage of peripheral blood monocytes and ectodysplasin-1-positive cells in liver decreased significantly in LC-treated animals at 24 h. Systemic monocyte–macrophage depletion resulted in (a) less severe tubular necrosis, (b) reduced inflammation and (c) reduced apoptosis of renal tubular epithelial cells. Gene expression kinetics showed that IL-6 gene expression peaked early at 4 h after reperfusion, followed by TNF-α, IL-1β and MCP-1 expressions, which peaked at 24 h. Systemic monocyte–macrophage depletion significantly reduced these cytokine and chemokine gene expressions.
Conclusions. These results suggest that macrophages are an important mediator in the initiation period of ischaemia/reperfusion injury and strategies that limit initial macrophage infiltration or activation can be useful in the treatment of acute renal failure.
Introduction
Acute renal failure (ARF) is a significant medical problem and ischaemia/reperfusion (I/R) injury is the leading cause of ARF in native or transplanted kidney [ 1 ]. Despite advances in supportive care, mortality from ARF in hospitalized patients remains high. The pathophysiological mechanism of ischaemic ARF is thought to include a complex interplay between vascular endothelial cell dysfunction, subsequent inflammation and tubular cell damage [ 2 ]. Inflammation is thought to contribute to tissue damage by releasing mediators, such as reactive oxygen species, proteases, cytokines and eicosanoids. Various types of inflammatory cells have been reported to infiltrate into kidney following I/R and the roles of neutrophils and T cells have been demonstrated in many studies using anti-neutrophil serum, anti-ICAM-1 antibody, anti-B7-1 monoclonal antibody or T cell-deficient mice [ 3 , 4 ]. Although macrophages also have been reported to infiltrate in the kidney as early as 24 h after reperfusion, the precise role of macrophages in the pathogenesis of ischaemic ARF is still unknown [ 5 ]. Macrophages might contribute to renal injury by producing a variety of proinflammatory cytokines, like tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interferon-γ, or proteolytic enzymes causing tissue damage, or by promoting the immune response by presenting antigens to lymphocytes. But recent evidence suggests their potential role in the resolution of inflammation or promotion of repair [ 6 ]. The possible mechanisms of their involvement in resolution or recovery are (a) release of cytokine inhibitors or regulatory cytokines, like IL-10 and/or transforming growth factor-β or (b) removal of apoptotic cells that may reduce inflammation. In order to define the precise role of macrophages in ischaemic ARF, depletion of macrophages in vivo is critical.
Liposomal clodronate (LC) is a bisphosphonate encapsulated by liposome and is known to induce macrophage depletion in vivo [ 7 ]. Upon administration, this drug is rapidly taken up by macrophages and by an action of macrophage lysosomal phospholipases, free bisphosphonate is released intracellularly and can induce rapid suicidal apoptosis of macrophages. Several studies have demonstrated the specificity of intravenously administered LC to deplete macrophages, but not neutrophils nor lymphocytes [ 8 ].
Therefore, in the current study, we examined the role of monocytes–macrophages in ischaemic ARF by depleting these cells in vivo through systemic administration of LC. We observed that macrophage depletion afforded a partial renal protection in ischaemic ARF. This beneficial effect was accompanied by decreased expression of proinflammatory cytokine and chemokine mRNA expression, suggesting that macrophages contributed to renal damage through release of these harmful molecules. Inhibition of early macrophage infiltration or activation might be a potential therapeutic strategy in the prevention or treatment of ARF.
Subjects and methods
Animals
Male Sprague–Dawley rats weighing 150–200 g were purchased from Orient (Charles River Korea, Seoul, Korea) and had free access to water and chow before manipulation. Animal care was followed in accordance with the criteria established by the animal care committee of Korea University for the care and use of laboratory animals in research. Rats were anaesthetized with intraperitoneal (i.p.) injection of 100 mg/kg ketamine, 12.5 mg/kg xylazine and subjected to bilateral renal pedicle clamping for 40 min. The animals were kept at a constant temperature (37°C) during the procedure and allowed to recover. Following the procedure rats were given 1 ml of warm normal saline i.p. Sham operation was performed in a similar manner, except for clamping renal pedicles. Following 4, 24 and 72 h of reperfusion, animals were sacrificed, blood was collected by intracardiac puncture and both kidneys were processed for molecular and histological examinations. These time-points were selected because 4 h can represent the initiation phase of renal injury, whereas 24 and 72 h represent the phases when injury peaks or the regeneration process predominates, respectively.
Liposome preparation and in vivo depletion of monocytes–macrophages
Clodronate was purchased from Sigma (Sigma Chemical Co., St. Louis, MO, USA) and Liposomes-encapsulated clodronate was prepared according to methods described previously by Van Roojien et al . [ 8 ]. One millilitre of LC or liposomal vehicle (LV) was injected via a tail vein and I/R injury was induced 24 h after injection.
Peripheral blood examination and biochemical analysis
Blood samples were taken at the time of ischaemia, that is 24 h after LC or LV injection, and were analysed for total and differential white blood cells by an automated cell counter (Cell-Dyn 3700; Abbott Laboratories Abbot Park, IL, USA). Blood urea nitrogen (BUN) and creatinine levels were measured using a Hitachi 747 automatic analyser.
Histological examination
Paraformaldehyde (4%)-fixed and paraffin-embedded kidney tissues were stained with haematoxylin and eosin (H&E) or naphthol AS-D chloracetate esterase (Sigma Aldrich, St Louis, MO, USA). Histological changes in the corticomedullary junction and outer medulla were evaluated semi-quantitatively. Briefly, tubular damage was estimated in 8–10 high-power fields (HPF; ×200) per section by using a scoring system based on the percentage of damaged tubules per field (1: <25%; 2: 25–50%; 3: 50–75%; 4: >75%). The mean score of each animal was compared. Esterase-positive leukocyte infiltration was measured by counting 8–10 HPF (×200) per section and mean numbers of esterase-positive cells were compared.
Immunohistochemical detection of monocytes–macrophages was performed on paraformaldehyde (4%)-fixed and paraffin-embedded kidney and liver tissue sections using the mouse monoclonal antibody against ectodysplasin-1 (ED-1) (Serotec, UK). ED-1 antibody is useful for the detection of a single-chain glycoprotein of 90–100 kDa that is expressed predominantly on the lysosomal membrane of myeloid cells. The antigen is expressed by the majority of tissue macrophages and dendritic cells in blood. Briefly, after deparaffinization, endogenous peroxidase activity was blocked with 3% hydrogen peroxide and then sections were incubated with 10% of normal goat serum, followed by incubation with primary antibody overnight (1:1000). Specific labelling was detected with a biotin-conjugated rabbit anti-mouse immunoglobulin G and avidin–biotin peroxidase complex (Vector Laboratories, Burlingame, CA, USA). ED-1-positive cells in corticomedullary junction and outer medulla were measured by counting 8–10 HPF (×200) per section and mean numbers of ED-1-positive cells were compared.
Detection of apoptosis
Apoptotic cells in paraffin-embedded kidney tissue sections were identified using ApopTag Plus (Intergen, Purchase, NY, USA), according to the manufacturer's protocol. The number of apoptotic cells in outer medulla and cortex was semi-quantitatively measured by counting 8–10 HPF (×200) per section and mean numbers of terminal deoxynucleotidyl transferase nick end labeling TUNEL-positive cells were compared.
Quantitation of TNF-α, IL-1β, IL-6 and MCP-1 mRNA by real-time RT–PCR
Total RNA was isolated by using TRI ZOL reagent (Life Technology, Rockville, MD, USA) according to the manufacturer's protocol. After precipitation by isopropyl alcohol, total RNA was subjected to further purification using RNeasy minikit (Qiagen, Valencia, CA, USA). Then, 1 µg total RNA was reverse transcribed in a reaction volume of 50 μl containing 10× RT buffer, 5.5 mM MgCl 2 , 500 μM of each dNTP, 2.5 μM random hexamer, 0.4 U/μl RNase inhibitor and 3.125 U/μl MultiScribe™ Reverse Transcriptase (Taqman® Reverse Transcription Reagents; Applied Biosystem Inc., Foster City, CA, USA) at 25°C for 10 min, 48°C for 30 min and 95°C for 5 min. Subsequently, real-time polymerase chain reaction (PCR) was run in triplicate using the iCycler system (Bio-Rad, Hercules, CA, USA) under the amplification condition of 40 cycles of 95°C, 15 s and 60°C, 1 min. Gene-specific primer and probe sets provided as a Pre-Developed Taqman Assay Reagents (Applied Biosystem Inc., Foster City, CA, USA) were used according to the manufacturer's protocol (rat TNF-α, IL-1β, IL-6). Primer/probe sets for rat MCP-1 were designed by Beacon Designer software version 2 (Bio-Rad, Hercules, CA, USA) based on the sequences from GenBank and were as follows: sense, 5′-GATCTCTCTTCCTCCACCACTATG-3′; antisense, 5′-GAATGAGTAGCAGCAGGTGAGT-3′; probe: 5′-AGGTCTCTGTCACGCTTCTGGGCC-3′. Taqman® probe was labelled with 6-carboxy-fluorescein as a reporter dye and 6-carboxy-tetramethyl-rhodamine as a quencher dye. 18S ribosomal RNA (Taqman® ribosomal control reagent; Applied Biosystem Inc., Foster City, CA, USA) was used as an internal control to normalize all the data. The dynamic range of each primer/probe set was verified by a serial dilution of cDNA template. The abundance of cytokine and chemokine gene was normalized to that of 18S and was expressed as fold differences relative to sham-operated control animals.
Statistical analysis
For statistical analysis, non-parametric Mann–Whitney rank sum test was done and data were presented as median value. P -values of <0.05 were considered to be statistically significant.
Results
LC-depleted monocytes–macrophages in vivo
We determined the effect of LC injection on the systemic depletion of monocytes–macrophages by examining peripheral blood and liver ED-1 cells. The percentage of blood monocytes was 6.1% and 6.2% in saline- and LV-injected animals, respectively, and was significantly reduced to 0.7% at 24 h after LC injection, whereas total leukocyte or lymphocyte counts remained unchanged ( Figure 1 A). Liver macrophages also decreased markedly 24 h after LC ( Figures 1 C and 1D) and the depletion persisted until 5 days after LC injection (data not shown).
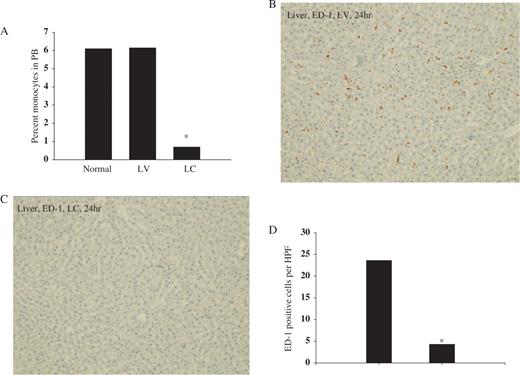
Effect of LC on percentage peripheral blood monocytes and liver macrophages. Rats were injected with either LV or LC via tail vein. At 24 h later, blood samples and liver tissue were obtained to determine blood monocyte–macrophage depletion. ( A ) Percentage blood monocytes, ( B ) LV and ( C ) LC, liver macrophages (ED-1 staining, ×100). ( D ) Number of ED-1-positive cells per HPF (×200) in liver. * P <0.05 compared with LV, n = 5 animals per group.
Systemic monocyte–macrophage depletion reduced I/R injury
To determine the effect of systemic monocyte–macrophage depletion in I/R injury, we first evaluated biochemical and histological renal damage following I/R. Serum BUN and creatinine levels were decreased significantly in monocyte–macrophage-depleted animals compared with LV-treated control animals at 24 h (BUN: 85.8 vs 39.2 mg/dl, P <0.01; creatinine: 2.1 vs 0.7 mg/dl, P <0.01) ( Figures 2 A and 2 B). By 72 h, BUN and creatinine were similar following LC or LV injection. Histological examination showed extensive tubular injury characterized by tubular cell necrosis, dilatation of tubules and cast formation in corticomedullary junction and outer medulla in LV-treated animals ( Figure 2 C). Kidneys from LC-treated animals showed less tubular injury than did kidneys from LV-treated animals ( Figure 2 D) and semi-quantitative assessment of histological damage demonstrating a significant beneficial effect of monocyte–macrophage depletion in I/R-induced renal injury is shown in Figure 2 E.
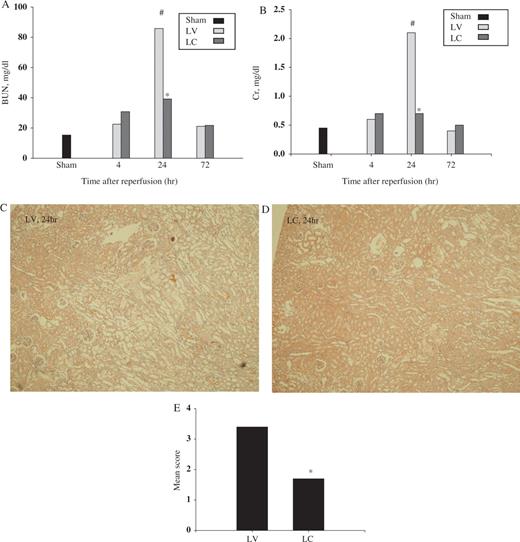
Effect of monocyte–macrophage depletion on renal function and histology in ischaemic ARF in rats. Rats were intravenously injected with either LV or LC. At 24 h later, rat kidneys were subjected to 40 min of bilateral ischaemia or sham operation and sacrificed at 4, 24 or 72 h after reperfusion. ( A ) BUN (mg/dl) and ( B ) creatinine (mg/dl). Data are presented as median value. #P <0.05 compared with sham, * P <0.05 compared with LV, n = 5 animals per group. Representative H&E-stained section of kidney from rats injected with LV ( C ) or LC ( D ) (H&E, ×40). ( E ) Semi-quantitative scoring of histological injury. * P <0.05 compared with LV, n = 5 animals per group.
Systemic monocyte–macrophage depletion reduced not only kidney ED-1 cell infiltration, but also esterase-positive leukocyte infiltration in I/R injury
In the corticomedullary junction and outer medullary area, ED-1 cell infiltration began to increase at 4 h in LV-treated rats ( Figure 3 A) and was noted to be markedly increased at 24 h ( Figures 3 C and 3 E). Systemic monocyte–macrophage depletion with LC resulted in significantly less infiltration at 4 and 24 h following I/R ( Figures 3 D and 3 E). In a similar manner, we also quantified the number of esterase-positive leukocytes. Our results showed the significant increase in esterase-positive leukocytes infiltration at 24 h following I/R in LV-treated animals ( Figures 4 A and 4 C) and macrophage depletion also resulted in reduced infiltration of esterase-positive leukocytes ( Figures 4 B and 4 C).
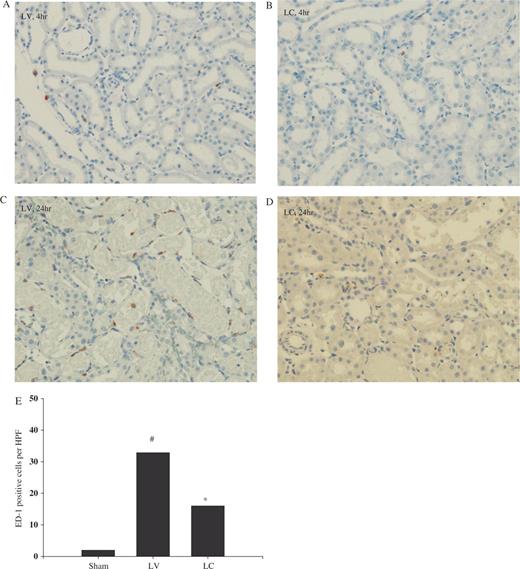
Effect of monocyte–macrophage depletion on ED-1 cell infiltration in ischaemic ARF in rats. Rats were intravenously injected with either LV or LC. At 24 h later, rat kidneys were subjected to 40 min bilateral ischaemia and sacrificed. Immunohistochemical detection of macrophages was performed with anti ED-1 antibody. ( A ) and ( C ) LV, ( B ) and ( D ) LC. ( A ) and ( B ) 4 h, ( C ) and ( D ) 24 h (ED-1 staining, ×200). ( E ) Number of ED-1-positive cells at 24 h. #P <0.05 compared with sham, * P <0.05 compared with LV, n = 5 animals per group.
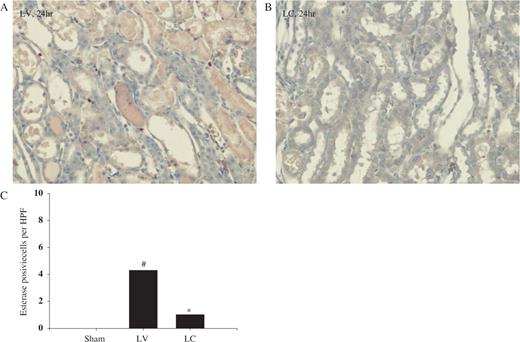
Effect of monocyte–macrophage depletion on esterase-positive leukocyte infiltration in ischaemic ARF in rats. Rats were intravenously injected with either LV or LC. At 24 h later, rat kidneys were subjected to 40 min bilateral ischaemia and sacrificed. ( A ) LV and ( B ) LC (naphthol AS-D chloracetate esterase staining, 24 h, ×200) ( C ) Number of esterase-positive cells at 24 h. #P <0.05 compared with sham, * P <0.05 compared with LV, n = 5 animals per group.
Systemic monocyte–macrophage depletion reduced apoptosis of tubular cells
Apoptosis, demonstrated by TUNEL staining, increased mainly in distal tubules in LV-treated animals at 4 h following I/R ( Figure 5 A) and further increased at 24 h ( Figure 5 C). Systemic depletion of monocytes–macrophages resulted in a significant decrease apoptosis at 24 h ( Figures 5 D and 5 E), when inflammation is evident. However, at 4 h ( Figures 5 B and 5 E) there was no difference in numbers of apoptosis in both groups.
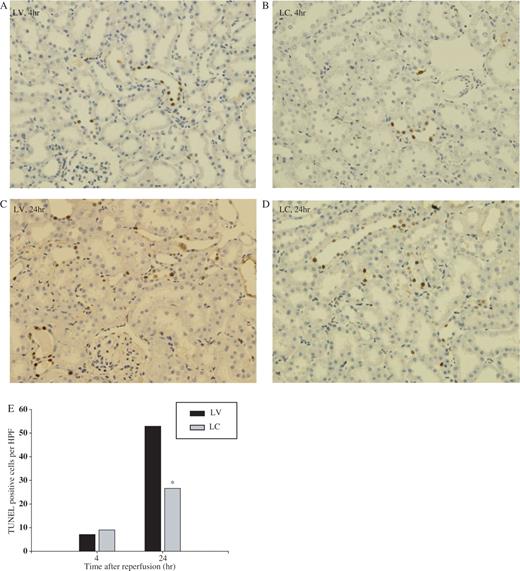
Effect of monocyte–macrophage depletion on apoptosis in ischaemic ARF in rats. Rats were intravenously injected with either LV or LC. At 24 h later, rat kidneys were subjected to 40 min of ischaemia or sham operation and sacrificed. Apoptosis was detected by TUNEL methods at 4 and 24 h after reperfusion. ( A ) and ( C ) LV, ( B ) and ( D ) LC. ( A ) and ( B ) 4 h, ( C ) and ( D ) 24 h, ×200. ( E ) Number of TUNEL-positive cells. * P <0.05 compared with LV, n = 5 animals per group.
Effect of monocyte–macrophage depletion on TNF-α, IL-1β, IL-6 and MCP-1 mRNA expression
We examined the various macrophage-associated factors and also chemokine MCP-1 expression at 4, 24 and 72 h after ischaemia. Renal I/R injury resulted in a 31.3-fold increase of mRNA levels for IL-6 at 4 h after reperfusion compared with sham-operated animals ( Figure 6 C). TNF-α, IL-1β and chemokine MCP-1 gene expressions also increased markedly, peaking at 24 h. Systemic monocyte–macrophage depletion led to a significant decrease in their expressions, suggesting that infiltrating macrophages are partially responsible for the increased gene expressions of these proinflammatory cytokines and chemokines following I/R (IL-6: 31.3/17.8 (fold differences) at 4 h, P <0.05; TNF-α: 13.2/3.2, IL-1β: 2.4/0.8, MCP-1: 14.0/5.6 at 24 h, P <0.05) ( Figure 6 ).
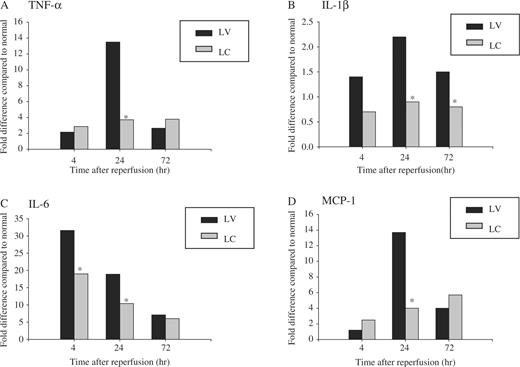
Effect of monocyte–macrophage depletion on cytokine and chemokine gene expression in ischaemic ARF in rats. Rats were intravenously injected with either LV or LC. At 24 h later, rat kidneys were subjected to 40 min of ischaemia or sham operation and sacrificed at 4, 24 or 72 h after reperfusion. TNF-α, IL-1β, IL-6 and MCP-1 mRNA expression was measured by real-time RT–PCR. In each experiment, the expression level was normalized to that of 18S and was expressed as fold differences relative to sham-operated control animals. Data are presented as median value. * P <0.05 compared with LV, n = 5 animals per group.
Discussion
In the present study, we demonstrated that (i) systemic monocyte–macrophage depletion induced by LC afforded a partial renal tissue protection following I/R injury in rats, as assessed from functional and histological studies. (ii) This protection was accompanied by attenuation of tissue macrophages, esterase-positive leukocyte infiltration, as well as decreased tubular cell apoptosis and necrosis with reduced levels of proinflammatory cytokines and chemokines. These results suggest that infiltrating macrophages in response to I/R contribute to maladaptive inflammatory reaction, exacerbating tissue injury by producing a variety of cytokines and chemokines in ischaemic ARF.
I/R-induced vascular endothelial cell dysfunction and consequent inflammation are thought to be important in the initiation and maintenance phases of ARF [ 9 ]. Over the last decade, many studies have highlighted the role of neutrophils and CD-4 T lymphocytes in ischaemic ARF. However, identification of the precise role of macrophages has been difficult, because of a lack of specific macrophage-depleting agents [ 10 , 11 ]. Macrophages infiltrate into post-ischaemic kidney at day 1 and become prominent at days 3–5, suggesting the possibility of their involvement in the regeneration process after I/R. However, there some evidence is that macrophages contribute to the initiation of tissue damage in different models of injury, such as uevitis, endotoxin-induced lung injury and kidney allograft rejection [ 12–14 ]. We, therefore, sought to determine the early role of macrophages in initiating renal injury. Our strategy was to use LC, a compound known to deplete macrophages in the reticuloendothelial system and circulating monocytes and subject rat kidneys to I/R injury.
As in other studies, intravenously administered LC significantly reduced the number of peripheral blood monocytes and liver ED-1-positive macrophages 24 h after injection. Systemic monocyte–macrophage depletion led to decreased ED-1-positive cell infiltration at 4 and 24 h after reperfusion and this was accompanied by functional and histological evidence of renal protection. These findings suggest that infiltrating macrophages in response to an injury signal are important in mediating renal injury. The detrimental effect of macrophages in ischaemic ARF has been suggested previously by Furuichi et al . [ 15 ], who showed that blocking MCP-1/CCR2 signalling using CCR2-deficent mice resulted in decreased infiltration of F4/80-positive macrophages with preserved renal function. Our study, using prior macrophage depletion, extends these observations and provides, more directly, evidence that macrophages play an important role in I/R-induced renal damage.
Next, we quantified the esterase-positive leukocyte infiltration, because previous studies have demonstrated the important role of neutrophils in ischaemic or toxic ARF. In this study, the amount of esterase-positive leukocyte infiltration was also significantly reduced in macrophage-depleted animals. This finding is consistent with a previous report that showed decreased infiltration of granulocytes as well as macrophages in mice whose MCP-1/CCR2 signalling is blocked via CCR2 gene ablation [ 14 ]. How systemic monocyte–macrophage depletion affects neutrophil infiltration following I/R is not certain. Following injury, activated macrophages might have direct cytotoxic effects on other intrinsic renal cells, such as endothelial cells or tubular cells, leading to chemotaxis of inflammatory cells into injured kidney. Interruption of this pathway through macrophage depletion may attenuate the injury.
The beneficial effect of monocyte–macrophage depletion was also accompanied by decreased tubular cell apoptosis at 24 h. Alterations in signalling molecules and various metabolic effectors following I/R, independently or in concert, trigger both apoptotic or necrotic cell death. Generally, the mechanism of cell death induced by a variety of stimuli is determined by the severity of injury, with extremely severe insults leading to necrosis and milder ones to apoptosis [ 16 ]. Intracellular ATP depletion, free radicals and proinflammatory cytokines are thought to be common mediators of cell death in ischaemic ARF. In this study, apoptosis was observed mainly in distal tubules as early as 4 h after reperfusion and peaking at 24 h. Systemic monocyte–macrophage depletion led to a significant decrease in apoptosis at 24 h, when inflammation is prominent, but not at 4 h. The finding that early apoptosis at 4 h after reperfusion was not prevented by macrophage depletion suggests that mechanisms not associated with inflammation, like intracellular ATP depletion or mitochondrial dysfunction, are responsible for early occurrence of apoptosis. However, as inflammation becomes more evident, tissue damage is correlated with ongoing apoptosis. Therefore, the functional, histological protection provided by a variety of anti-inflammatory strategies might be mediated partially by their resultant anti-apoptotic function [ 17 , 18 ]. We did not measure apoptosis at 72 h because delayed apoptosis after I/R might be associated with the regeneration process. Because activated tissue macrophages can release different kinds of effector molecules that can initiate or exacerbate tissue injury, we measured several proinflammatory cytokines and chemokine gene expression at different time-points after reperfusion. We found that IL-6 showed a robust increase at 4 h, followed by IL-1β, TNF-α expression at 24 h and systemic monocyte–macrophage depletion led to partial reduction of their expressions. These results can suggest that these effector molecules released from infiltrating macrophages contribute to tissue damage. The role of IL-6 has been controversial in many types of inflammation, but recent study suggests that IL-6 released by macrophages may be maladaptive and contribute to ischaemic ARF [ 19 ]. Our data are consistent with this hypothesis. Other cytokines, like TNF-α and IL-1β, predominantly products of activated macrophages, and MCP-1 are also thought to contribute to renal tissue injury.
Taken together, our study provides evidence that early infiltrating macrophages play an important pathogenetic role in ischaemic ARF, presumably by releasing proinflammatory cytokine and chemokines. Therefore, strategies that limit early macrophage infiltration or activation may represent a novel approach in the prevention or treatment of ARF. However, macrophage influx into inflamed tissue is a very dynamic process, possibly playing different roles at different time-points, in different kinds of injury. Macrophage infiltration at a relatively later time-point after I/R might participate in repair and regeneration processes. Additional studies which precisely modify organ macrophage content will be needed in order to understand the role of macrophages in injury and repair that might help to provide new avenues in the treatment or prevention of ARF.
We thank Dr Mark D. Okusa (University of Virginia) for careful reading of this manuscript and advice and Mr H.W. Kim, Mrs Y.S. Ko and Mrs K.H. Chang for their excellent technical supports.
Conflict of interest statement . None declared.
References
Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure.
Hellberg PO, Kallskog TO. Neutrophil-mediated post-ischemic tubular leakage in the rat kidney.
Burne MJ, Daniels F, El Ghandour A et al . Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure.
Ysebaert DK, De Greef KE, Vercauteren SR et al . Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury.
Wilson HM, Walbaum D, Rees AJ. Macrophages and the kidney.
Van Rooijen N, Sanders A. Elimination, blocking, and activation of macrophages: three of a kind?
Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications.
Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure.
Hellberg PO, Kallskog TO. Neutrophil-mediated post-ischemic tubular leakage in the rat kidney.
Burne MJ, Daniels F, El Ghandour A et al . Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure.
Baatz H, Puchta J, Reszka R, Pleyer U. Macrophage depletion prevents leukocyte adhesion and disease induction in experimental melanin-protein induced uveitis.
Gaca JG, Palestrant D, Lukes DJ, Olausson M, Parker W, Davis RD, Jr. Prevention of acute lung injury in swine: depletion of pulmonary intravascular macrophages using liposomal clodronate.
Jose MD, Ikezumi Y, van Rooijen N, Atkins RC, Chadban SJ. Macrophages act as effectors of tissue damage in acute renal allograft rejection.
Furuichi K, Wada T, Iwata Y et al . CCR2 signaling contributes to ischemia–reperfusion injury in kidney.
Lieberthal W, Koh JS, Levine JS. Necrosis and apoptosis in acute renal failure.
Jo SK, Yun SY, Chang KH et al . Alpha-MSH decreases apoptosis in ischaemic acute renal failure in rats: possible mechanism of this beneficial effect.
Deng J, Kohda Y, Chiao H et al . Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury.
Author notes
1Division of Nephrology, Department of Internal Medicine, 2Institute of Renal Disease, Korea University Hospital and 3Department of Internal Medicine, Eulji University, Seoul, Korea


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments