





Abstract
Background. Coronary artery calcification (CAC) is an extensive and common complication in patients with end-stage renal disease (ESRD). The aim of this study was to assess prospectively the change in CAC over a 2-year period and to identify the factors that may be associated with CAC progression in ESRD patients.
Methods. The final analysis was performed on 40 of 43 stable haemodialysis patients who initially entered into the study. The study population underwent multirow spiral computed tomography to derive CAC scores at baseline and after a minimum of 12 months (24 months in 30 patients, 18 months in four, and 12 months in the remaining six patients). To provide a stable estimate that was unbiased with respect to the baseline CAC, square root-transformed CAC scores were used for the analyses of the changes in CAC.
Results. The median CAC score was 191 (range, 0–2403) mm3 at baseline and increased to 253 (range, 0–2745) mm3 at follow-up (P<0.001) and the median annualized change in square root-transformed CAC score was 1.48 (range, −0.95–8.64) mm3/year. The annualized change of the square root-transformed CAC score positively correlated with the time-integrated levels of C-reactive protein (R = 0.521, P = 0.001), phosphorus (R = 0.433, P = 0.005) and calcium × phosphorus product (R = 0.394, P = 0.012), but did not correlate with the levels of fetuin-A or lipid parameters. Even after adjusting for age, gender and baseline CAC score, C-reactive protein levels were independently associated with CAC progression.
Conclusion. These data suggest that chronic inflammation as well as altered mineral metabolism contributes to a rapid progression of CAC in ESRD patients. Additional, larger scale studies are required to confirm our findings.
Introduction
Cardiovascular disease is the leading cause of death in patients with end-stage renal disease (ESRD). The risk of dying from a cardiovascular event is 10–20 times higher than in the general population, and the prevalence of coronary artery disease in ESRD patients is approximately 40%. Although patients develop cardiovascular abnormalities early in the disease process and have substantial traditional risk factors such as diabetes, hypertension and dyslipidaemia, a feature of many patients in the years preceding a cardiovascular event is the presence of extensive vascular calcification.
New imaging techniques have recently been used for reliably detecting and objectively measuring the extent of vascular calcification. These techniques include the use of electron-beam computed tomography (EBCT) and multirow spiral computed tomography (MSCT) to quantify coronary artery calcification (CAC). The degree of calcification within the coronary arteries is measured to obtain a CAC score.
The CAC is an extensive and common complication in patients with ESRD [1]. In addition to cross-sectional studies, several longitudinal studies have documented rapid progression of CAC among these patients and have attempted to define the risk factors associated with this rapid progression [2–4]. However, only limited and conflicting data exist concerning possible factors that may lead to increased risk of CAC progression. A major limitation in assessing change in CAC using currently available technologies, including EBCT and MSCT, is interscan variability, which is not constant but depends on the baseline CAC score [5]. Thus, assessing the progression of CAC without accounting for interscan variability may lead to biased estimates of the rates and risk factors associated with CAC progression.
The aim of this study was to assess prospectively the change in CAC over a 2-year period after accounting for interscan variability and to identify factors that may be associated with CAC progression in ESRD patients.
Subjects and methods
Subjects
This study was carried out in a single haemodialysis unit in South Korea. Forty-three patients were entered into the study from April 2003 to October 2004. All patients were older than 18 years of age, with no metallic objects (e.g. stents and clips) in their chest, and were clinically stable (i.e. no symptoms of acute coronary syndrome, congestive heart failure or infectious disease) for at least 2 months prior to the baseline assessment. For the study, 40 out of 43 patients remained on haemodialysis and underwent follow-up MSCT after at least 12 months (30 had a final MSCT scan at month 24, four at month 18 and six at month 12). Only the results for these 40 patients were analysed. The three excluded patients included one patient with incomplete results secondary to a transfer and two patients who died of malignancy before 12 months. Ten of the remaining 40 patients could not undergo MSCT at 24 months because six patients were transferred or died between 12 and 24 months, and four had only recently entered the study. The study protocol was approved by our institutional Review Board and written informed consent was obtained from each enrolled patient.
The initial clinical evaluation included a medical history questionnaire, a physical examination and a review of the patient's medical records. The mean age of the analysed group was 56±12 years. There were 26 men and 14 women. The median duration of haemodialysis was 27 (range, 1–111) months. The causes of ESRD included chronic glomerulopathy in 11 patients, diabetic nephropathy in 17 patients, hypertensive nephropathy in three patients and polycystic kidney disease in one patient. The cause of renal failure was unknown in eight patients. Thirteen of the patients had a previous history of cardiovascular disease, including acute myocardial infarction, congestive heart failure, stroke and peripheral vascular disease.
During the study period, all of the patients were treated with conventional bicarbonate haemodialysis using Polyflux 6L (Gambro Dialysatoren GmbH, Hechingen, Germany) or F6 HPS (Fresenius Medical Care AG, Bad Homburg, Germany) synthetic membranes. The duration of each dialysis session was 4 h. The calcium concentration of dialysate was either 3.0 or 2.5 mEq/l. The patients received calcium carbonate or calcium acetate as a phosphate binder throughout the study period. The dose of phosphate binder and the dialysate calcium concentration could be titrated every month. Patients could use aluminium as a rescue binder if the calcium × phosphorus (Ca × P) product exceeded 5.64 mmol2/l2. Active vitamin D was used if the serum level of intact parathyroid hormone (PTH) exceeded 300 pmol/l.
Biochemical analysis
The results of the monthly serum biochemical determinations were collected for the study period. Routine serum chemistry variables, which included calcium, phosphorus and albumin, were analysed using standard methods. Serum calcium was adjusted for the serum albumin concentration and the Ca × P product was calculated, using these results.
The total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol and triglycerides were determined every 3 months for the lipid profile. Intact PTH and C-reactive protein (CRP) were also measured every 3 months during the study period. A high-sensitivity assay (detection limit, 0.1 mg/l) for CRP was performed using the latex agglutination method (Denka, Tokyo, Japan). If a patient was affected by an acute episode (e.g. infection, bleeding or cardiovascular event), laboratory results for at least 1 month after complete resolution of the episode were excluded from the data collection.
Predialysis blood samples were collected with a 6-month interval for fetuin-A determinations. Blood samples collected without anticoagulant were allowed to coagulate for at least 30 min and were then centrifuged at 520 g for 10 min. The resulting serum was divided into aliquots, frozen and stored at −70°C. Serum fetuin-A was determined using a human fetuin-A ELISA kit (BioVendor GmbH, Heidelberg, Germany). The assay is a sandwich enzyme immunoassay with a detection limit of 0.35 ng/ml. Intra-assay precision was 3.3–4.8% and inter-assay precision was 2.5–7.4%.
Imaging procedure
The MSCT (LightSpeed Plus; GE Medical Systems, Milwaukee, WI, USA) scans were repeated under the same standardized conditions at 6- or 12-month intervals for all patients to obtain the CAC scores. Details of the methods of MSCT imaging have been published elsewhere [6]. Briefly, the data were acquired with a rotation time of 500 ms and a table feed of 4 × 2.5 mm/rotation, and the tube current was 370 mA at 120 kVp. Data were obtained during the diastolic phase of the heart cycle and stored on an optical disk for later analysis. The acquired images were reviewed on an Advantage Workstation (Smart Score; GE Medical Systems). To ensure the continuity and consistency of the calcium score interpretation, a single investigator reviewed both the baseline and follow-up scans. Calcification scores were calculated using the volumetric method, which is more reproducible than the traditional area-based Agaston method [7].
In this study, we defined the progression as outlined by Hokanson et al. [5], who found that the square root transformation of the coronary artery calcium volume score provides a stable estimate of interscan variability. They suggested using the difference between the baseline and follow-up square root-transformed coronary artery calcium volume score of ≥2.5 mm3 to signify a significant change in CAC because a change of this magnitude has a <1% likelihood of being the result of interscan variability. We computed an annualized rate of change in the square root-transformed CAC score [(final square root CAC − baseline square root CAC)/follow-up interval in years] to provide a stable estimate of progression in CAC that was unbiased with respect to baseline CAC scores or follow-up intervals.
Statistical analysis
The results of the biochemical determinations were averaged to obtain geometric (for CRP and PTH) or simple (for the other measurements) means for each measurement. Continuous clinical variables are presented as medians and ranges or as means and SDs. Paired comparisons of baseline and follow-up CAC scores were performed using Wilcoxon's signed-ranks test. The patients were classified into three groups according to the annualized change in the square root-transformed CAC score: <1.25 mm3/year (i.e. <2.50 mm3/2 years) as non-progressors, 1.25–2.50 mm3/year as slow progressors and >2.50 mm3/year as rapid progressors. These three groups were compared using the Jonckheere–Terpstra test to explore the existence of trends [8]. Comparisons of proportions were made using a chi-square test. The associations between the annualized changes of the square root-transformed CAC score and the clinical and biochemical parameters were analysed using Pearson's correlation coefficient. Multivariate linear regression analysis was used to assess the relative importance of the different risk factors associated with a change in CAC score. All statistical analyses were performed with the SPSS software package (version 12.0; SPSS, Chicago, IL, USA). Two-sided P-values<0.05 were considered to be statistically significant.
Results
On baseline MSCT examination, nine patients had no evidence of CAC, whereas 31 patients had CAC of differing severities. The median CAC score was 191 (range, 0–2403) mm3 at baseline and increased to 253 (range, 0–2745) mm3 at follow-up (P<0.001). The median annualized change in square root-transformed CAC score was 1.48 (range, −0.95–8.64) mm3/year.
Based on an annualized change in square root-transformed CAC score, the patients were classified into three groups: 17 patients were non-progressors with a change of <1.25 mm3/year, 14 patients were slow progressors with a change of 1.25–2.50 mm3/year and nine patients were rapid progressors with a change of >2.50 mm3/year. The characteristics of the patients are presented in Table 1. The degree of CAC progression was associated with the time-integrated level of CRP; median CRP levels gradually increased (P = 0.001) from non-progressors through slow progressors to rapid progressors. Additionally, the sex ratio, phosphorus and Ca × P product were associated with the degree of CAC progression; the patients were more frequently male (P<0.001) and had higher phosphorus (P = 0.018) and Ca × P product values (P = 0.026) with increasing CAC progression. Non-progressors tended to receive a lower dose of oral calcium than slow or rapid progressors, but the difference was not statistically significant. The serum levels of fetuin-A, the lipid parameters, the parameters of diabetes history and the haemodialysis duration did not differ significantly across the three groups.
Comparison between non-progressors, slow progressors and rapid progressors of CAC.
| Non-progressors (n = 17) | Slow progressors (n = 14) | Rapid progressors (n = 9) | P | |
|---|---|---|---|---|
| Age (years) | 56±15 | 52±11 | 60±8 | 0.458 |
| Male (n) | 5 (29%) | 13 (93%) | 8 (89%) | <0.001 |
| Diabetic (n) | 7 (41%) | 6 (43%) | 4 (44%) | 0.987 |
| CVD history (n) | 4 (24%) | 4 (29%) | 5 (56%) | 0.234 |
| Dialysis duration (months) | 27 (1–99) | 38 (1–111) | 20 (11–97) | 0.970 |
| Baseline CAC score | 45 (0–1660) | 704 (0–2403) | 356 (0–1338) | 0.060 |
| CRP (mg/l) | 0.9 (0.2–7.4) | 2.1 (0.5–10.3) | 3.9 (1.8–18.6) | 0.001 |
| Fetuin-A (mg/l) | 300±45 | 279±31 | 285±34 | 0.555 |
| Total cholesterol (mmol/l) | 4.22±0.85 | 4.19±0.80 | 3.75±0.62 | 0.360 |
| HDL cholesterol (mmol/l) | 1.19±0.31 | 1.09±0.28 | 1.01±0.21 | 0.132 |
| LDL cholesterol (mmol/l) | 2.33±0.54 | 2.30±0.49 | 2.12±0.47 | 0.415 |
| Triglycerides (mmol/l) | 1.95±1.20 | 2.03±1.92 | 1.64±1.06 | 0.429 |
| Albumin (g/l) | 38±2 | 38±2 | 39±1 | 0.366 |
| Haemoglobin (g/l) | 94±4 | 94±7 | 93±5 | 0.960 |
| Calcium adjusted (mmol/l) | 2.37±0.17 | 2.40±0.15 | 2.45±0.17 | 0.228 |
| Phosphorus (mmol/l) | 1.52±0.16 | 1.71±0.32 | 1.74±0.26 | 0.018 |
| Ca × P product (mmol2/l2) | 3.55±0.49 | 4.11±0.97 | 4.30±0.97 | 0.026 |
| Intact PTH (ng/l) | 41 (6–238) | 49 (10–347) | 24 (7–115) | 0.309 |
| Alkaline phosphatase (U/l) | 187±96 | 180±100 | 138±30 | 0.387 |
| Calcium dose (mg/day) | 960 (260–2040) | 1530 (170–3000) | 1480 (400–2390) | 0.075 |
| Non-progressors (n = 17) | Slow progressors (n = 14) | Rapid progressors (n = 9) | P | |
|---|---|---|---|---|
| Age (years) | 56±15 | 52±11 | 60±8 | 0.458 |
| Male (n) | 5 (29%) | 13 (93%) | 8 (89%) | <0.001 |
| Diabetic (n) | 7 (41%) | 6 (43%) | 4 (44%) | 0.987 |
| CVD history (n) | 4 (24%) | 4 (29%) | 5 (56%) | 0.234 |
| Dialysis duration (months) | 27 (1–99) | 38 (1–111) | 20 (11–97) | 0.970 |
| Baseline CAC score | 45 (0–1660) | 704 (0–2403) | 356 (0–1338) | 0.060 |
| CRP (mg/l) | 0.9 (0.2–7.4) | 2.1 (0.5–10.3) | 3.9 (1.8–18.6) | 0.001 |
| Fetuin-A (mg/l) | 300±45 | 279±31 | 285±34 | 0.555 |
| Total cholesterol (mmol/l) | 4.22±0.85 | 4.19±0.80 | 3.75±0.62 | 0.360 |
| HDL cholesterol (mmol/l) | 1.19±0.31 | 1.09±0.28 | 1.01±0.21 | 0.132 |
| LDL cholesterol (mmol/l) | 2.33±0.54 | 2.30±0.49 | 2.12±0.47 | 0.415 |
| Triglycerides (mmol/l) | 1.95±1.20 | 2.03±1.92 | 1.64±1.06 | 0.429 |
| Albumin (g/l) | 38±2 | 38±2 | 39±1 | 0.366 |
| Haemoglobin (g/l) | 94±4 | 94±7 | 93±5 | 0.960 |
| Calcium adjusted (mmol/l) | 2.37±0.17 | 2.40±0.15 | 2.45±0.17 | 0.228 |
| Phosphorus (mmol/l) | 1.52±0.16 | 1.71±0.32 | 1.74±0.26 | 0.018 |
| Ca × P product (mmol2/l2) | 3.55±0.49 | 4.11±0.97 | 4.30±0.97 | 0.026 |
| Intact PTH (ng/l) | 41 (6–238) | 49 (10–347) | 24 (7–115) | 0.309 |
| Alkaline phosphatase (U/l) | 187±96 | 180±100 | 138±30 | 0.387 |
| Calcium dose (mg/day) | 960 (260–2040) | 1530 (170–3000) | 1480 (400–2390) | 0.075 |
Values expressed as mean±SD, median (range) or number (%) of patients.
Comparison between non-progressors, slow progressors and rapid progressors of CAC.
| Non-progressors (n = 17) | Slow progressors (n = 14) | Rapid progressors (n = 9) | P | |
|---|---|---|---|---|
| Age (years) | 56±15 | 52±11 | 60±8 | 0.458 |
| Male (n) | 5 (29%) | 13 (93%) | 8 (89%) | <0.001 |
| Diabetic (n) | 7 (41%) | 6 (43%) | 4 (44%) | 0.987 |
| CVD history (n) | 4 (24%) | 4 (29%) | 5 (56%) | 0.234 |
| Dialysis duration (months) | 27 (1–99) | 38 (1–111) | 20 (11–97) | 0.970 |
| Baseline CAC score | 45 (0–1660) | 704 (0–2403) | 356 (0–1338) | 0.060 |
| CRP (mg/l) | 0.9 (0.2–7.4) | 2.1 (0.5–10.3) | 3.9 (1.8–18.6) | 0.001 |
| Fetuin-A (mg/l) | 300±45 | 279±31 | 285±34 | 0.555 |
| Total cholesterol (mmol/l) | 4.22±0.85 | 4.19±0.80 | 3.75±0.62 | 0.360 |
| HDL cholesterol (mmol/l) | 1.19±0.31 | 1.09±0.28 | 1.01±0.21 | 0.132 |
| LDL cholesterol (mmol/l) | 2.33±0.54 | 2.30±0.49 | 2.12±0.47 | 0.415 |
| Triglycerides (mmol/l) | 1.95±1.20 | 2.03±1.92 | 1.64±1.06 | 0.429 |
| Albumin (g/l) | 38±2 | 38±2 | 39±1 | 0.366 |
| Haemoglobin (g/l) | 94±4 | 94±7 | 93±5 | 0.960 |
| Calcium adjusted (mmol/l) | 2.37±0.17 | 2.40±0.15 | 2.45±0.17 | 0.228 |
| Phosphorus (mmol/l) | 1.52±0.16 | 1.71±0.32 | 1.74±0.26 | 0.018 |
| Ca × P product (mmol2/l2) | 3.55±0.49 | 4.11±0.97 | 4.30±0.97 | 0.026 |
| Intact PTH (ng/l) | 41 (6–238) | 49 (10–347) | 24 (7–115) | 0.309 |
| Alkaline phosphatase (U/l) | 187±96 | 180±100 | 138±30 | 0.387 |
| Calcium dose (mg/day) | 960 (260–2040) | 1530 (170–3000) | 1480 (400–2390) | 0.075 |
| Non-progressors (n = 17) | Slow progressors (n = 14) | Rapid progressors (n = 9) | P | |
|---|---|---|---|---|
| Age (years) | 56±15 | 52±11 | 60±8 | 0.458 |
| Male (n) | 5 (29%) | 13 (93%) | 8 (89%) | <0.001 |
| Diabetic (n) | 7 (41%) | 6 (43%) | 4 (44%) | 0.987 |
| CVD history (n) | 4 (24%) | 4 (29%) | 5 (56%) | 0.234 |
| Dialysis duration (months) | 27 (1–99) | 38 (1–111) | 20 (11–97) | 0.970 |
| Baseline CAC score | 45 (0–1660) | 704 (0–2403) | 356 (0–1338) | 0.060 |
| CRP (mg/l) | 0.9 (0.2–7.4) | 2.1 (0.5–10.3) | 3.9 (1.8–18.6) | 0.001 |
| Fetuin-A (mg/l) | 300±45 | 279±31 | 285±34 | 0.555 |
| Total cholesterol (mmol/l) | 4.22±0.85 | 4.19±0.80 | 3.75±0.62 | 0.360 |
| HDL cholesterol (mmol/l) | 1.19±0.31 | 1.09±0.28 | 1.01±0.21 | 0.132 |
| LDL cholesterol (mmol/l) | 2.33±0.54 | 2.30±0.49 | 2.12±0.47 | 0.415 |
| Triglycerides (mmol/l) | 1.95±1.20 | 2.03±1.92 | 1.64±1.06 | 0.429 |
| Albumin (g/l) | 38±2 | 38±2 | 39±1 | 0.366 |
| Haemoglobin (g/l) | 94±4 | 94±7 | 93±5 | 0.960 |
| Calcium adjusted (mmol/l) | 2.37±0.17 | 2.40±0.15 | 2.45±0.17 | 0.228 |
| Phosphorus (mmol/l) | 1.52±0.16 | 1.71±0.32 | 1.74±0.26 | 0.018 |
| Ca × P product (mmol2/l2) | 3.55±0.49 | 4.11±0.97 | 4.30±0.97 | 0.026 |
| Intact PTH (ng/l) | 41 (6–238) | 49 (10–347) | 24 (7–115) | 0.309 |
| Alkaline phosphatase (U/l) | 187±96 | 180±100 | 138±30 | 0.387 |
| Calcium dose (mg/day) | 960 (260–2040) | 1530 (170–3000) | 1480 (400–2390) | 0.075 |
Values expressed as mean±SD, median (range) or number (%) of patients.
The significant correlates with the annualized change of square root-transformed CAC score in Pearson's correlation coefficient were log-transformed CRP (R = 0.521, P = 0.001) and phosphorus (R = 0.433, P = 0.005) (Figure 1). The annualized change also correlated with the Ca × P product (R = 0.394, P = 0.012) and log-transformed baseline CAC score (R = 0.336, P = 0.034), but did not correlate with the serum levels of intact PTH, fetuin-A or lipid parameters. Patient age positively correlated with the annualized change of square root-transformed CAC score in male patients (R = 0.500, P = 0.009) but not in female patients. Further analyses in 30 patients, excluding 10 who could not undergo final scans at month 24, were performed to determine if the difference in follow-up intervals altered the results. These analyses demonstrated no changes in the above-stated relationships.
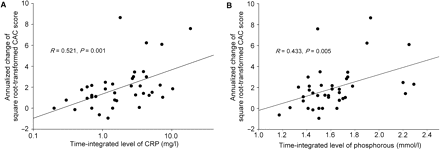
Scatter plots of the association between CRP levels and the change of square root-transformed CAC scores (A) and of the association between phosphorus levels and the change of square root-transformed CAC scores (B).
Multiple linear regression analysis revealed that CRP levels were independently associated with CAC progression even after adjusting for age, gender and baseline CAC score (Table 2).
Multiple linear regression analysis predicting the annualized change of the square root-transformed CAC score
| β-coefficient | Standard error | Standardized-β | P | |
|---|---|---|---|---|
| Age (years) | 0.029 | 0.025 | 0.176 | 0.248 |
| Male | 1.365 | 0.639 | 0.317 | 0.040 |
| Baseline log (CAC score + 1) | 0.257 | 0.231 | 0.150 | 0.273 |
| Log CRP (mg/l) | 2.725 | 1.079 | 0.335 | 0.016 |
| Phosphorus (mmol/l) | 0.693 | 0.355 | 0.271 | 0.059 |
| β-coefficient | Standard error | Standardized-β | P | |
|---|---|---|---|---|
| Age (years) | 0.029 | 0.025 | 0.176 | 0.248 |
| Male | 1.365 | 0.639 | 0.317 | 0.040 |
| Baseline log (CAC score + 1) | 0.257 | 0.231 | 0.150 | 0.273 |
| Log CRP (mg/l) | 2.725 | 1.079 | 0.335 | 0.016 |
| Phosphorus (mmol/l) | 0.693 | 0.355 | 0.271 | 0.059 |
The model F is 6.958 (P<0.001), R2 = 0.506.
Multiple linear regression analysis predicting the annualized change of the square root-transformed CAC score
| β-coefficient | Standard error | Standardized-β | P | |
|---|---|---|---|---|
| Age (years) | 0.029 | 0.025 | 0.176 | 0.248 |
| Male | 1.365 | 0.639 | 0.317 | 0.040 |
| Baseline log (CAC score + 1) | 0.257 | 0.231 | 0.150 | 0.273 |
| Log CRP (mg/l) | 2.725 | 1.079 | 0.335 | 0.016 |
| Phosphorus (mmol/l) | 0.693 | 0.355 | 0.271 | 0.059 |
| β-coefficient | Standard error | Standardized-β | P | |
|---|---|---|---|---|
| Age (years) | 0.029 | 0.025 | 0.176 | 0.248 |
| Male | 1.365 | 0.639 | 0.317 | 0.040 |
| Baseline log (CAC score + 1) | 0.257 | 0.231 | 0.150 | 0.273 |
| Log CRP (mg/l) | 2.725 | 1.079 | 0.335 | 0.016 |
| Phosphorus (mmol/l) | 0.693 | 0.355 | 0.271 | 0.059 |
The model F is 6.958 (P<0.001), R2 = 0.506.
Discussion
In this cohort, elevated CRP was one of the strongest and the most consistent predictor of the progression of CAC. This provides prospective evidence that high CRP levels are associated with progressive CAC in ESRD patients, adding substantially to the previously limited evidence from cross-sectional data for a relationship between CRP levels and vascular calcification [9]. In addition, this study showed the association between altered mineral metabolism, such as elevated serum phosphate and elevated serum Ca × P products, and the progression of CAC. This is in agreement with the results obtained by previous cross-sectional and longitudinal studies [1,4].
There are several distinctions between this and previous observational studies investigating changes of CAC in ESRD patients (Table 3). Most studies estimated progression by calculating the absolute change in the CAC score. However, the absolute difference in the untransformed CAC score may overestimate the degree of progression as the baseline CAC score increases because the interscan variability in the CAC score increases with the level of increase in CAC [5]. Unlike other studies, ours defined progression in the same manner as Hokanson et al. [5] to provide a stable estimate that was unbiased with respect to baseline CAC, and our study had a relatively small number of withdrawal patients and a longer follow-up period. In addition, CRP was frequently measured at regular intervals and averaged to obtain time-integrated levels in our study. These CRP levels may represent the degree of chronic inflammation during the follow-up period. Using these methods, we found that elevated concentrations of CRP were associated with the progression of CAC independently of confounding factors. However, it is not yet clear that our methods including the square root transformation of the CAC score are more reliable than others, and more studies regarding the progression of CAC are required.
Observational studies assessing the progression of CAC in dialysis patients
| Authors | n, final/ baseline | Interval (months) | Estimates of progression | CRP measurements | Predictors of progression |
|---|---|---|---|---|---|
| Current study | 40/43 | 24 in 30, 18 in 4, 12 in 6 | Change of square root-transformed CAC score | ×5–9, 3-month interval | Baseline CAC, male, CRP, P, CPP in U, CRP in M |
| Goodman et al. [1] | 22/39 | 22±7 (12–41) | Absolute change | Not measured | Baseline CAC, P, CPP in U |
| Tamashiro et al. [2] | 24/35 | 17±3 (12–19) | Absolute change | Not measured | Baseline CAC, high TG, low HDL-C in U |
| Moe et al. [3] | 17/33 | ≤15 | Absolute change | ×2 baseline | Baseline CAC, age in U |
| Stompor et al. [4] | 47/61 | 12 | Absolute change | ×3, 6-month interval | P and CPP in U, no in M |
| Authors | n, final/ baseline | Interval (months) | Estimates of progression | CRP measurements | Predictors of progression |
|---|---|---|---|---|---|
| Current study | 40/43 | 24 in 30, 18 in 4, 12 in 6 | Change of square root-transformed CAC score | ×5–9, 3-month interval | Baseline CAC, male, CRP, P, CPP in U, CRP in M |
| Goodman et al. [1] | 22/39 | 22±7 (12–41) | Absolute change | Not measured | Baseline CAC, P, CPP in U |
| Tamashiro et al. [2] | 24/35 | 17±3 (12–19) | Absolute change | Not measured | Baseline CAC, high TG, low HDL-C in U |
| Moe et al. [3] | 17/33 | ≤15 | Absolute change | ×2 baseline | Baseline CAC, age in U |
| Stompor et al. [4] | 47/61 | 12 | Absolute change | ×3, 6-month interval | P and CPP in U, no in M |
C = calcium; CAC = coronary artery calcification; CPP = calcium × phosphorus product; CRP = C-reactive protein; HDL-C = high-density lipoprotein cholesterol; M = multivariate analysis; P = phosphorus; TG = triglycerides; U = univariate analysis.
Observational studies assessing the progression of CAC in dialysis patients
| Authors | n, final/ baseline | Interval (months) | Estimates of progression | CRP measurements | Predictors of progression |
|---|---|---|---|---|---|
| Current study | 40/43 | 24 in 30, 18 in 4, 12 in 6 | Change of square root-transformed CAC score | ×5–9, 3-month interval | Baseline CAC, male, CRP, P, CPP in U, CRP in M |
| Goodman et al. [1] | 22/39 | 22±7 (12–41) | Absolute change | Not measured | Baseline CAC, P, CPP in U |
| Tamashiro et al. [2] | 24/35 | 17±3 (12–19) | Absolute change | Not measured | Baseline CAC, high TG, low HDL-C in U |
| Moe et al. [3] | 17/33 | ≤15 | Absolute change | ×2 baseline | Baseline CAC, age in U |
| Stompor et al. [4] | 47/61 | 12 | Absolute change | ×3, 6-month interval | P and CPP in U, no in M |
| Authors | n, final/ baseline | Interval (months) | Estimates of progression | CRP measurements | Predictors of progression |
|---|---|---|---|---|---|
| Current study | 40/43 | 24 in 30, 18 in 4, 12 in 6 | Change of square root-transformed CAC score | ×5–9, 3-month interval | Baseline CAC, male, CRP, P, CPP in U, CRP in M |
| Goodman et al. [1] | 22/39 | 22±7 (12–41) | Absolute change | Not measured | Baseline CAC, P, CPP in U |
| Tamashiro et al. [2] | 24/35 | 17±3 (12–19) | Absolute change | Not measured | Baseline CAC, high TG, low HDL-C in U |
| Moe et al. [3] | 17/33 | ≤15 | Absolute change | ×2 baseline | Baseline CAC, age in U |
| Stompor et al. [4] | 47/61 | 12 | Absolute change | ×3, 6-month interval | P and CPP in U, no in M |
C = calcium; CAC = coronary artery calcification; CPP = calcium × phosphorus product; CRP = C-reactive protein; HDL-C = high-density lipoprotein cholesterol; M = multivariate analysis; P = phosphorus; TG = triglycerides; U = univariate analysis.
In agreement with our findings, a recent study demonstrated that sevelamer, a non-calcium-based phosphate binder, inhibited progressive CAC and reduced inflammatory markers in ESRD patients [10]. This is indirectly suggestive of an association between progressive CAC and the inflammatory process. In addition, several studies have shown that inflammatory biomarkers are associated with the progression of carotid intima-media thickness in ESRD patients [11]. However, the clinical relevance of CAC may differ from that of carotid intima-media thickness, which may indicate the severity of atherosclerosis. The CAC might represent arteriosclerosis or arterial stiffness [12], which is an indicator of cardiovascular mortality, distinct from atherosclerosis. We believe that our study provides additional essential knowledge for the understanding of progressive CAC in ESRD patients.
Although the mechanism by which vascular calcification develops in ESRD patients is complex and is not well known, a possible explanation of our finding is that inflammation may participate in arterial calcification in the presence of an increased serum phosphorus and calcium load [13]. The CRP may be a trigger for calcium deposition in the arteries of dialysis patients; at the end of each dialysis session, when back-filtration can occur, plasma calcium concentrations reach their maximum levels and patients become alkalotic. In this context, it should also be considered that CRP, a member of the pentraxin family, binds to damaged tissue in a calcium-dependent manner, associating with membranes in the presence of multiple calcium ions [14]. A recent study showed that the calcification rate of an arterial wall increases as the concentration of CRP increases when the arterial wall is exposed to an excess amount of CRP in an in vitro simulation model [15]. Moreover, recent evidence from different cell types suggests that CRP is not only a risk marker, but might also be a participant in atherogenesis [16].
Based on studies that have implicated low levels of serum fetuin-A, a calcification inhibitor, as a risk factor for CAC and poor cardiovascular outcome [17,18], we evaluated the associations between progressive CAC and serum levels of fetuin-A. In agreement with a previous cross-sectional study on ESRD patients [17], we also found that fetuin-A levels negatively correlated with baseline and final CAC scores (R = −0.399 and −0.355, P = 0.011 and 0.025, respectively; cross-sectional data not presented in the results). However, we were unable to identify the time-integrated level of fetuin-A as a major predictor for change in CAC over the follow-up period. Furthermore, a positive relationship between serum fetuin-A levels and CAC scores in non-dialysed patients with diabetic nephropathy has been demonstrated [19]. Thus, the role of fetuin-A in vascular calcification may be far more complex than previously thought. Serum fetuin-A may possibly represent the anti-calcification profile of patients with a certain degree of calcification or renal dysfunction, but it appears to have no direct impact on the progression of CAC.
Our results should be interpreted with caution because of several limitations of our study. First, subjects were on long-term haemodialysis at a single centre, which can make it difficult to draw general conclusions and can lead to survivor or other selection biases. Second, the sample size was small, and the power to detect associations between potential risk or protective factors and progressive CAC was limited. Third, we did not make any assessments of the functional activity of fetuin-A. Only the phosphorylated form of fetuin-A appears to be functionally active, and it is unclear whether the presence of renal disease modifies the proportion of circulating fetuin-A that is phosphorylated [20]. Finally, this study could not provide follow-up for specific cardiovascular outcomes in relation to progressive CAC, because the number of cardiovascular events was too small for valid analyses.
In conclusion, this study provides the prospective evidence that chronic inflammation, in addition to altered mineral metabolism, contributes to the rapid progression of CAC in ESRD patients. The small number of patients who participated in this study necessitates the confirmation of our findings in additional, larger-scale studies.
Conflict of interest statement. None declared.
References
Goodman WG, Goldin J, Kuizon BD et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis.
Tamashiro M, Iseki K, Sunagawa O et al. Significant association between the progression of coronary artery calcification and dyslipidemia in patients on chronic hemodialysis.
Moe SM, O’Neill KD, Reslerova M, Fineberg N, Persohn S, Meyer CA. Natural history of vascular calcification in dialysis and transplant patients.
Stompor TP, Pasowicz M, Sulowicz W et al. Trends and dynamics of changes in calcification score over the 1-year observation period in patients on peritoneal dialysis.
Hokanson JE, MacKenzie T, Kinney G et al. Evaluating changes in coronary artery calcium: an analytic method that accounts for interscan variability.
Jung HH, Ma KR, Han H. Elevated concentrations of cardiac troponins are associated with severe coronary artery calcification in asymptomatic haemodialysis patients.
Ohnesorge B, Flohr T, Fischbach R et al. Reproducibility of coronary calcium quantification in repeat examinations with retrospectively ECG-gated multisection spiral CT.
Stompor T, Pasowicz M, Sullowicz W et al. An association between coronary artery calcification score, lipid profile, and selected markers of chronic inflammation in ESRD patients treated with peritoneal dialysis.
Ferramosca E, Burke S, Chasan-Taber S, Ratti C, Chertow GM, Raggi P. Potential antiatherogenic and anti-inflammatory properties of sevelamer in maintenance hemodialysis patients.
Stompor T, Krasniak A, Sulowicz W et al. Changes in common carotid artery intima-media thickness over 1 year in patients on peritoneal dialysis.
Haydar AA, Covic A, Colhoun H, Rubens M, Goldsmith DJ. Coronary artery calcification and aortic pulse wave velocity in chronic kidney disease patients.
Brancaccio D, Tetta C, Gallieni M, Panichi V. Inflammation, CRP, calcium overload and a high calcium-phosphate product: a “liaison dangereuse”.
Nelsestuen GL, Ostrowski BG. Membrane association with multiple calcium ions: vitamin-K-dependent proteins, annexins and pentraxins.
Warrier B, Mallipeddi R, Karla PK, Lee CH. The functional role of c-reactive protein in aortic wall calcification.
Venugopal SK, Devaraj S, Jialal I. Effect of C-reactive protein on vascular cells: evidence for a proinflammatory, proatherogenic role.
Moe SM, Reslerova M, Ketteler M et al. Role of calcification inhibitors in the pathogenesis of vascular calcification in chronic kidney disease (CKD).
Ketteler M, Bongartz P, Westenfeld R et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study.
Mehrotra R, Westenfeld R, Christenson P et al. Serum fetuin-A in nondialyzed patients with diabetic nephropathy: relationship with coronary artery calcification.
Author notes
1Department of Internal Medicine and 2Department of Radiology, Kangwon National University Hospital, Kangwon National University School of Medicine, Chunchon, Kangwon-do, Republic of Korea


{kind=link}
Comments