





The human Duffy blood group (FY) antigens are transmembrane glycoproteins that function as receptors for chemokines and for the malarial parasite Plasmodium vivax (reviewed in Hadley and Peiper 1997 ). Most of the FY antigenic variation is determined by three common alleles (FY*A, FY*B, and FY*O) in a gene located on chromosome 1q21-q22. DNA sequence characterization of these alleles and interspecies comparisons with the orthologous genes from nonhuman primates have shown that FY*A and FY*O are derived variants, each resulting from a single mutation in an ancestral FY*B background (Chaudhuri et al. 1995 ; Tourmamille et al. 1995 ). Whereas the FY*A gene product is a functional protein with a Gly44Asp substitution, FY*O has a T-46C promoter mutation that disrupts a binding site for the GATA1 erythroid transcription factor leading to a tissue-specific loss of expression of FY antigens in red blood cells (Tourmamille et al. 1995 ). In contrast to most human autosomal polymorphisms where common alleles tend to be shared by different, geographically distant populations, the distribution of FY alleles is peculiar: FY*O has reached near fixation over a vast area of sub-Saharan Africa, whereas FY*A and FY*B are the only alleles present across Eurasia and the Americas. This peculiarity, together with the observation that homozygous individuals for the FY*O allele are completely resistant to P. vivax malaria (Miller et al. 1976 ), has led to the concept that the observed pattern of allele frequencies has been driven by positive selection. According to this model, selection by vivax malaria led to the replacement of FY*A and FY*B by the advantageous FY*O allele in west and central Africa and to the extinction of P. vivax by lack of susceptible hosts. Alternatively, because no significant mortality is associated with P. vivax and an Asian origin of the parasite is conceivable, it is possible that it was the earlier fixation of FY*O that has prevented vivax malaria from becoming endemic in Africa (Livingstone 1984 ). If this hypothesis is correct, different scenarios may account for the present distribution of FY alleles, including selection by an unknown agent other than P. vivax or the possibility of an entirely fortuitous event linked to the dynamics of population movements within and out of Africa. To search for a signature of natural selection at the FY locus, the patterns of DNA sequence variation linked to the three FY common alleles have been recently characterized (Hamblin and Di Rienzo 2000 ; Hamblin, Thompson, and Di Rienzo 2002 ). Consistent with the expectations of models of directional selection, the level of DNA sequence variation associated with FY*O was found to be significantly reduced. But the observation that the FY*O mutation occurs in two divergent haplotypes with intermediate frequencies in most samples from sub-Saharan Africa indicated that the signature of selection may be more complex than that predicted by a simple selective sweep.
We have approached the evolutionary history of the FY polymorphism by studying the distribution of the faster-mutating D1S2635 microsatellite polymorphism within more stable lineages carried by FY*A, FY*B, and FY*O alleles.
The FY*A and FY*B alleles were sampled from a total of 123 Portuguese individuals (FY*A = 0.35; FY*B = 0.62; FY*O = 0.03). The FY*O alleles were sampled from 141 individuals from the island of São Tomé (FY*A = 0.03; FY*B = 0.07; FY*O = 0.90). This previously uninhabited island, located 300 km off the coast of Gabon, started to be peopled by the end of the 15th century with slaves imported by Portuguese colonists from the adjacent coasts of the Gulf of Guinea and the Congo-Angola area. As a consequence of this settlement pattern, the population of São Tomé has retained the high levels of genetic diversity that are generally observed in the African mainland and has an estimated European admixture of 11% (Tomás et al. 2002 ). Identification of FY alleles was done by using previously described polymerase chain reaction (PCR)-restriction length fragment polymorphism methods (Tourmamille et al. 1995 ). The D1S2635 microsatellite (GenBank accession number Z52215; table 1 ) was typed by PCR amplification with fluorescently labeled primers (GDB: 603410; http://www.gdb.org/) followed by separation of amplification products in an ABI 310 DNA sequencer. Two flanking polymorphic sites described by Hamblin and Di Rienzo (2000) in the region around the FY locus were additionally characterized (nucleotide positions as in BAC bk134P22; GenBank accession number AL35403; table 1 ): (1) a C→T transition in position 70628, which has previously been found to be always associated with a 69596 T→C transition in a C-T haplotype shared by both FY*O and non-FY*O alleles; (2) a CT deletion at nucleotides 75336 and 75337 that defines one of the two major common lineages associated with FY*O. Both polymorphisms were typed after PCR amplification of DNA fragments containing each of the corresponding positions. The 70628 C-T variation was detected by StyI restriction enzyme digestion. Length variation at nucleotides 75336 and 75337 was scored by electrophoretic separation of amplification products in 12% polyacrylamide gels. Microsatellite alleles were sequenced in both directions from PCR products cloned into a pCR4 plasmid vector with the TOPO TA cloning kit (Invitrogen) using the ABI Prism Big Dye Dydeoxy Terminator Cycle sequencing kit. Sequencing products were analyzed in an ABI 377 automatic DNA sequencer. Human sequences were compared with homologous regions from one chimpanzee (Pan troglodytes) and two gorilla (Gorilla gorilla) specimens. Allele frequencies at the individual loci were calculated by direct gene counting. Maximum-likelihood haplotype frequencies were estimated using the expectation-maximization algorithm implemented in the ARLEQUIN package (Schneider et al. 1997 ). Unbiased estimates of heterozygosity were calculated according to Nei (1987, p. 178) . Significant differences among heterozygosity estimates were tested by comparing the corresponding 95% confidence intervals established by 10,000 bootstrap simulations with the GENETIX software (Belkhir et al. 1998 ).
The D1S2635 microsatellite allele frequency distribution within the FY alleles is depicted in figure 1A. Although the FY*O distribution presents a decreased variance in allele length and a significantly lower heterozygosity than does FY*B, there is no drastic reduction in diversity levels, suggesting that the signature of directional selection in microsatellite variation is not as evident as it is at the DNA sequence level (Hamblin and Di Rienzo 2000 ; Hamblin, Thompson, and Di Rienzo 2002 ). But the three microsatellite distributions still have noticeable differences in shape, with FY*O presenting a more smoothly peaked pattern than did both non-FY*O alleles. Most interestingly it was found that the D1S2635 allele size changes within FY*A and FY*B were not always in increments of 2 bp. To examine the cause of this heterogeneity, 17 D1S2635 alleles from Portugal and 24 alleles from São Tomé were sequenced. Sequence information has revealed four types of D1S2635 lineages that were defined by different flanking polymorphisms (table 1 ). The lack of regularity in D1S2635 allele size increments was found to be caused by the deletion of a single C within a run of five T's located in the 3′-flanking region of the (CA)n repeat. In addition, two further D1S2635 lineages were defined by the presence of one or three CG dinucleotides immediately before the variable (CA)n repeat. The presence of a single CG dinucleotide could have resulted from a CA→CG misincorporation promoted by slipped mispairing in the (CA)n repeated motif. Reiteration of this process has probably led to the occurrence of a (CG)3 motif, although no intermediate sequences bearing a (CG)2 were found. All interspecies differences in the microsatellite sequence structure between humans, chimpanzee, and gorillas were found to be the likely result of mutation accumulation in an ancestral CpG dinucleotide stretch lying between an invariable (CA)5 array and a polymorphic (CA)n repeated block (table 1 ). The joint distribution of the four types of D1S2635-flanking sequences and the polymorphisms at nucleotides 70628, 75336, and 75337 was further analyzed in an extended survey, which led to the definition of seven distinct haplotypes (table 1 ). In this survey, the D1S2635 indel could be directly typed by the observation of even (insertion) or odd (deletion) numbers of base pairs in the different microsatellite alleles. The presence and number of CG dinucleotides adjacent to the variable (CA)n repeat could be assessed through the digestion of D1S2635 alleles of known length with the Hin6I restriction enzyme that recognizes the G!CGC sequence. For example, if two alleles have the same size but …AAGCG(CA)10… and …AAGCGCGCG(CA)8… sequences, Hin6I digestion will produce in both cases a fragment of constant size, corresponding to the 5′ portion of the D1S2635 amplimer, and additional CG (CA)10… and CG (CA)8… fragments, which can be easily discriminated upon polyacrylamide gel electrophoresis. In the absence of CG dinucleotides, microsatellite alleles will remain uncut.
The seven haplotypes had different distributions among the FY alleles (table 1 ). Haplotype H1, which shows the highest similarity with nonhuman primates and is likely to be the ancestral lineage, was found to be shared by all three alleles and is most frequent among FY*A and FY*O. Haplotypes H4 and H5 were found exclusively in non-FY*O alleles, whereas H6 and H7 were detected only among FY*O. Within FY*O exclusive lineages, haplotype H6, defined by the 75336-75337 CT deletion, has a 19% frequency and corresponds to one of the two major FY*O haplotype branches previously found in 23% of FY*O alleles from five sub-Saharan African populations (Hamblin and Di Rienzo 2000 ). Haplotype H7 adds an additional common FY*O sublineage and strengths the disagreement between the observed haplotype structure and the skew of the frequency spectrum toward rare alleles that should be expected under a simple selective sweep. These patterns of lineage sorting along with either FY*O or non-FY*O alleles are consistent with the present geographic distribution of the FY variants and agree with previous observations on sequence variation linked to the FY locus (Li et al. 1997 ; Hamblin and Di Rienzo 2000 ; Hamblin, Thompson, and Di Rienzo 2002 ). But two derived lineages were found to be shared by all FY alleles. Haplotype H2, is defined by the 70628C→T transition, that was previously found in 6% of FY*O alleles from other African populations and in 50% of Italian FY*B alleles (Hamblin and Di Rienzo 2000 ). Haplotype H3 adds further evidence for gene conversion or recombination-driven sequence transfer between FY*O and non-FY*O alleles. This sharing of derived haplotypes between alleles that currently occupy distinct geographical areas shows that both FY*O and non-FY*O lineages were indeed present in the same ancestral population before the fixation of FY*O in Africa.
Figure 1B presents the distributions of the faster-mutating D1S2635 alleles within the haplotypes carried by FY*A, FY*B, and FY*O. Derived haplotypes that are shared by at least two FY variants (H2, H3, H4, and H5) have modal (CA)n alleles with the same, or a very similar, number of repeats in each FY allele, thus providing additional evidence for lineage spread through recombination or gene conversion. Analysis of the (CA)n repeat variation within the lineages defined by sequence polymorphisms allows the comparison of diversity levels accumulated since the origin of each haplotype and provides information on the relative antiquity of different lineages that cannot be directly inferred from sequence data alone. Haplotype H6, which corresponds to one of the two major FY*O haplotypes previously described, has been found to be characterized by the joint occurrence in absolute linkage disequilibrium of the 75336-75337 CT deletion together with two additional mutations: 75082A→G and 75872T→C (Hamblin and Di Rienzo 2000 ). In spite of its derived sequence structure, this haplotype has the highest (CA)n repeat heterozygosity and is likely to be the oldest lineage linked to FY*O. On the contrary, the FY*O-linked haplotype H1, which would be included in the other major FY*O haplotype branch defined by Hamblin and Di Rienzo (2000) , is associated with lower levels of (CA)n diversity although it bears a more primitive sequence structure. Taking this evidence into account, it is probable that FY*O has arisen in Africa by two independent mutational events. According to this hypothesis, a first FY*O mutation is likely to have occurred long after the origin of FY*B in a derived background carrying haplotype H6 that has been lost from currently sampled populations. More recently, a second FY*O mutation occurring in a less-derived FY*B chromosome, represented here by haplotype H1, would have given rise to a second FY*O major branch to which haplotypes H2, H3, and H7 are connected. Alternatively, gene conversion could have occurred between a FY*O-linked haplotype H6 and a FY*B-linked haplotype H1, but the recurrence of the FY*O mutation is further supported by the finding of a recent independent GATA1 T-46C transition in a FY*A allelic background with a 2% frequency in P. vivax endemic region of Papua New Guinea (Zimmerman et al. 1999 ). In any case, the high levels of (CA)n variation within haplotype H6 and the sharing of the H1 haplotype both by FY*O and non-FY*O haplotypes, indicate that the two major FY*O branches had arisen before the action of positive selection, as previously noted (Hamblin and Di Rienzo 2000 ). Because, under the recurrent mutation scenario, the two FY*O mutations could have had different geographical origins, it is conceivable that they could provide replication evidence for selection-driven independent increase in FY*O allele frequencies. Further studies of FY haplotypes and (CA)n variation in an extended panel of African populations, including those with remnant FY*A and FY*B alleles, will be necessary to identify the major paths of spread of FY*O and to confirm this hypothesis.
We have also attempted to estimate an upper limit to the date of fixation of FY*O by using the (CA)n variation to infer the age of FY*O-linked H2- and H3-derived haplotypes, which were found to be shared with non-FY*O alleles (fig. 1B ). The age of the most recent common ancestor of each haplotype was approximated by simulating the overtime decay in the frequency of the microsatellite allele originally associated with each lineage under the stepwise mutation model in a population of infinite size (Seixas et al. 2001 ). Assuming a 0.001 mutation rate at the microsatellite locus (Weber and Wong 1993 ), a rough calculation of the time necessary for the ancestral (CA)14 allele to reach its current 65% frequency within the 22 FY*O-linked H3 haplotypes was estimated at 490 generations. A minimum 170–1,060 generations support interval was calculated as ±2 × the SD of the binomial distribution with parameters n = 22 and P = 0.65 (Goldstein et al. 1999 ). If a generation time of 30 years is assumed (Tremblay and Vézina 2000 ), our calculations would imply that non-FY*O alleles were still not replaced by FY*O as late as 14,700 (5,100–31,800) years ago in West Africa. Similar calculations using the (CA)n variation within the FY*O-linked haplotype H2 led to a 11,100 years estimate, but because only seven FY*O chromosomes were found to have this lineage the estimation is associated with a very wide uninformative interval (0–44,000 years). A more recent coalescent time of 9,300 years (4,350–15,750) was calculated for the less diverse haplotype H7, which is exclusive to FY*O and might have arisen after the fixation of this allele. Under our set of assumptions, these estimates point to a more recent date for replacement of FY*A and FY*B alleles in Africa than a previous 33,000 years (6,500–97,200) calculation based on single nucleotide polymorphisms (Hamblin and Di Rienzo 2000 ). Although absolute age estimates are strongly dominated by uncertainties about relevant parameters such as mutation rates, we note that our calculations place the fixation date of FY*O closer to the origins of agriculture and to the concomitant spreading of malaria as a generalized selective pressure (Livingstone 1984 ). This would imply that the FY polymorphism may have become subject to malarial selection only shortly before known P. falciparum protective mutations (Tishkoff et al. 2001 ; Currat et al. 2002 ) and that P. vivax might have been indeed the selective agent that promoted FY*O fixation.
Supplementary Material
The GenBank accession numbers of the D1S2635 microsatellite sequences referred to are as follows: AF515840 (included in haplotypes H1, H2, and H6); AF515841 (included in haplotype H3); AF515842 (included in haplotypes H4 and H5); AF515843 (included in haplotype H7); AF515844 (Chimpanzee); AF515845 (Gorilla).
Naruya Saitou, Reviewing Editor
Keywords: human Duffy blood group microsatellite variation malarial selection
Address for correspondence and reprints: Jorge Rocha, Instituto de Patologia e Imunologia Molecular, Universidade do Porto (IPATIMUP), R. Dr. Roberto Frias s/n, 4200-465 Porto, Portugal. jrocha@ipatimup.pt .
Table 1 Distribution of Haplotypes Defined by Sequence Variation at the D1S2635 Microsatellite and Flanking Polymorphisms Among FY alleles. Location of the Different Polymorphic Regions is Shown in Inset
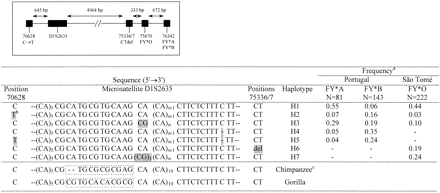
Table 1 Distribution of Haplotypes Defined by Sequence Variation at the D1S2635 Microsatellite and Flanking Polymorphisms Among FY alleles. Location of the Different Polymorphic Regions is Shown in Inset
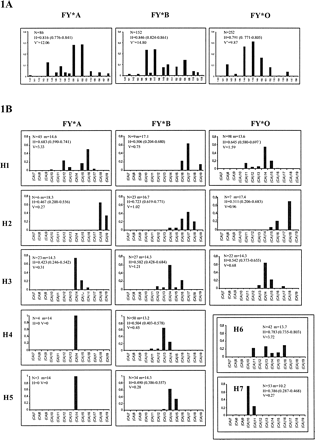
Fig. 1.—A D1S2635 microsatellite allele frequency distributions within FY alleles. B. Distribution of D1S2635 (CA)n variation within the seven haplotypes (H1-H7) carried by FY*A, FY*B, and FY*O alleles. N = number of chromosomes, V′ = variance in allele length, V = variance in repeat number, m = average repeat number
We thank the encouragement and suggestions of Drs. Sarah Tishkoff and James Harris. This work was partially supported by POCTI. Field work in São Tomé was supported by Instituto de Cooperação Científica e Tecnológica Internacional (ICCTI). S.S. is supported by grant BD/13885/97 from Praxis XXI.
References
Belkhir K., P. Borsa, J. Goudet, L. Chikhi, F. Bonhomme,
Chaudhuri A., J. Polyakova, V. Zbrzezna, A. O. Pogo,
Currat M., G. Trabuchet, D. Rees, P. Perrin, R. M. Harding, J. B. Clegg, A. Langaney, L. Excoffier,
Goldstein D. B., D. E. Reich, N. Breidman, S. Usher, U. Seligsohn, H. Peretz,
Hadley T. J., S. C. Peiper,
Hamblin M. T., A. Di Rienzo,
Hamblin M. T., E. E. Thompson, A. Di Rienzo,
Li J., S. S. Iwamoto, N. Sugimoto, H. Okuda, E. Kajii,
Livingstone F. B.,
Miller L. H., S. J. Mason, D. F. Clyde, M. H. McGiniss,
Schneider S., J.-M. Kueffer, D. Roessli, L. Excoffier,
Seixas S., O. Garcia, M. J. Trovoada, M. T. Santos, A. Amorim, J. Rocha,
Tishkoff S., R. Varkonyi, N. Cahinhinan, et al. (17 co-authors)
Tomás G., L. Seco, S. Seixas, P. Faustino, J. Lavinha, J. Rocha,
Tourmamille C., Y. Colin, J. P. Cartron, C. Le Van Kim,
Tremblay M., H. Vézina,

{kind=link}