





Abstract
Objective. Genome scans for rheumatoid arthritis (RA) have yielded inconsistent results. The absence of replication of linkage might be due to lack of power of individual studies. We performed a genome scan meta-analysis of published data to increase statistical power and to assess evidence for linkage of RA across genome scan studies.
Methods. Four RA whole-genome scans containing 767 families with 964 sibling pairs were included for the genome scan meta-analysis (GSMA). The GSMA method was applied to pool the results obtained from four genome scans. For each study, 120 genomic bins of ∼30 centimorgans were defined and ranked according to maximum evidence for linkage within each bin. Bin ranks were weighted and summed across all studies. The summed rank for each bin was assessed empirically for significance using permutation methods.
Results. A total of nine bins lay above the 95% confidence level (P=0.05) and four bins were above the 99% confidence level (P=0.01) in the RA GSMA, suggesting that these bins contain RA-linked loci: bins 6.2, 6.4, 8.1, 18.3, 12.3, 12.2, 1.5, 6.3 and 16.2. The strongest evidence for linkage occurred on chromosome 6p22.3-6p21.1 (bin 6.2), containing the HLA region (Psumrnk=0.0000008).
Conclusion. This RA GSMA confirmed the evidence for HLA loci as the greatest susceptibility factor to RA and showed evidence for linkage at non-HLA loci, such as chromosomes 1p, 6, 8p, 12, 16 and 18q, across studies. These data may provide a basis to carry out targeted linkage and candidate gene studies, particularly in the regions.
Rheumatoid arthritis (RA) is a chronic inflammatory disease predominantly involving synovial joints and affecting up to 1% of the population worldwide [1]. Although the aetiology of RA remains unsolved, a genetic component of RA susceptibility has been established by data from twin and family studies. Twin studies estimated the heritability of RA liability as up to 60% [2]. Family-based studies have also suggested an increased risk for siblings of RA probands compared with that for the general population (λs = 5.0–7.2) [3]. A consistent association between RA and human leucocyte antigen (HLA) loci has been observed in many populations [4]. The HLA class II molecules constitute the most powerful recognized genetic factor for RA. However, results of family studies suggest that this association accounts for only one-third of the genetic susceptibility and non-HLA genes are also involved in disease susceptibility [5].
Several whole-genome scans for RA have been performed to identify the HLA and non-HLA loci involved in RA susceptibility [6–11]. They identified several linkage loci for RA, but the findings of most of the studies have not been replicated. It is not surprising that linkage studies have shown inconsistent results, because they have been limited by small sample size, low statistical power and clinical or genetic heterogeneity [12].
Meta-analysis combines the linkage results from several studies, providing greater statistical power. Meta-analysis may also identify regions where the genetic effect is too small to be detected in an individual study. Recently, the genome search meta-analysis (GSMA) method has been developed to combine genome scan results in an attempt to enhance evidence of linkage [13]. The GSMA is a non-parametric ranking method to identify genomic regions that show consistent linkage evidence based on the linkage scores obtained in each scan. We applied the GSMA method to genome scans of RA to assess evidence for linkage across the studies.
Materials and methods
Selection of genome scans
Genome scans for RA were identified via literature databases. We included RA genome scans using more than 300 microsatellite markers covering the whole genome. When there were genome scans using duplicated data, we chose the study that used larger numbers of markers because genotyping more markers increases the likelihood of detecting true susceptibility loci. The GSMA method assumes a uniform map in each scan, so we did not consider the second stages of genome scans where candidate regions were more densely mapped or samples were modified. For consistency of this meta-analysis, genome scans that did not show all linkage results with P<0.05 were excluded from this study. All loci with a P-value <0.05 were obtained from each study for the rank-ordering procedure. The linkage scores were input as P-values in the bins and linkage scores with P>0.05 were substituted as P-values of 0.5 and were considered ties.
Genome search meta-analysis
The GSMA was performed as described [13]. In brief, the autosomes were divided into 120 30-centimorgan (cM) bins defined by Genethon markers (CEPH-Genethon Integrated Map web site http://www.cephb.fr/ceph-genethon-map.htm). On the Marshfield map (available at http://www.marshfieldclinic.org/research/genetics), the average bin width was 29.1 cM. Each marker was placed within one of these bins on the basis of its location on the Genethon or Marshfield map. For each study, each bin was assigned a within-study rank (Rstudy) based on the maximum linkage score within the bin. Bins were ranked in descending order (120 = most significant result). The summed rank (Rsum) across studies was computed for each bin. A weighted GSMA was carried out to allow results to reflect the relative contribution of each study. For the weighted analysis, each Rstudy value was multiplied by its study's weight (square root of the number of sibling pairs divided by the mean of this value over all studies). Two pointwise P-values were determined, Psumrnk and Pord, as described and determined by 10 000 permutations of the weighted data set. Psumrnk is the probability of observing a bin's summed rank by chance, and Pord is the probability of observing the jth place bin's summed rank in jth place bins in randomly permuted data. The empirical criterion for bins likely to contain linked loci was Psumrnk<0.05 and the criterion for genome-wide significance was Psumrnk<0.000417 (0.05 corrected for 120 bins).
Results
Individual genome scans
Four RA genome scans were used for this GSMA based on the inclusion criteria [6–9]. Two genome scan studies of RA were excluded due to duplicated data [11] or absence of all linkage data with P<0.05 [10]. This meta-analysis included 767 families with 964 sibling pairs. The main characteristics of genome scans included in the meta-analysis are summarized in Table 1.
Summary of genome searches included in the genome search meta-analysis
| Study characteristics | European study | US study | US study | UK study |
|---|---|---|---|---|
| Author (year) | Cornelis (1998) | Jawaheer (2001) | Jawaheer (2003) | John (2004) |
| Study population | France, Italy Spain, Belgium | USA (90.1% Caucasian) | USA (92.9% Caucasian) | UK (Caucasian) |
| No. of families | 97 | 257 | 256 | 157 |
| No. of siblings pairs | 114 | 301 | 332 | 217 |
| Relative weighting factor | 0.70 | 1.13 | 1.20 | 0.97 |
| No. of autosomal markers | 313 | 379 | 379 | 11 245 |
| Analysis program | SIBPALNA | SIBPAL | SIBPAL | MERLIN |
| Test statistic output | P-value | P-value | P-value | P-value |
| Study characteristics | European study | US study | US study | UK study |
|---|---|---|---|---|
| Author (year) | Cornelis (1998) | Jawaheer (2001) | Jawaheer (2003) | John (2004) |
| Study population | France, Italy Spain, Belgium | USA (90.1% Caucasian) | USA (92.9% Caucasian) | UK (Caucasian) |
| No. of families | 97 | 257 | 256 | 157 |
| No. of siblings pairs | 114 | 301 | 332 | 217 |
| Relative weighting factor | 0.70 | 1.13 | 1.20 | 0.97 |
| No. of autosomal markers | 313 | 379 | 379 | 11 245 |
| Analysis program | SIBPALNA | SIBPAL | SIBPAL | MERLIN |
| Test statistic output | P-value | P-value | P-value | P-value |
Summary of genome searches included in the genome search meta-analysis
| Study characteristics | European study | US study | US study | UK study |
|---|---|---|---|---|
| Author (year) | Cornelis (1998) | Jawaheer (2001) | Jawaheer (2003) | John (2004) |
| Study population | France, Italy Spain, Belgium | USA (90.1% Caucasian) | USA (92.9% Caucasian) | UK (Caucasian) |
| No. of families | 97 | 257 | 256 | 157 |
| No. of siblings pairs | 114 | 301 | 332 | 217 |
| Relative weighting factor | 0.70 | 1.13 | 1.20 | 0.97 |
| No. of autosomal markers | 313 | 379 | 379 | 11 245 |
| Analysis program | SIBPALNA | SIBPAL | SIBPAL | MERLIN |
| Test statistic output | P-value | P-value | P-value | P-value |
| Study characteristics | European study | US study | US study | UK study |
|---|---|---|---|---|
| Author (year) | Cornelis (1998) | Jawaheer (2001) | Jawaheer (2003) | John (2004) |
| Study population | France, Italy Spain, Belgium | USA (90.1% Caucasian) | USA (92.9% Caucasian) | UK (Caucasian) |
| No. of families | 97 | 257 | 256 | 157 |
| No. of siblings pairs | 114 | 301 | 332 | 217 |
| Relative weighting factor | 0.70 | 1.13 | 1.20 | 0.97 |
| No. of autosomal markers | 313 | 379 | 379 | 11 245 |
| Analysis program | SIBPALNA | SIBPAL | SIBPAL | MERLIN |
| Test statistic output | P-value | P-value | P-value | P-value |
Two genome scans were performed in Europe [6, 9] and the other scans were conducted in the USA [7, 8]. The number of sibling pairs ranged from 114 to 332 and individual weighting factors were 0.70, 1.13, 1.20 and 0.97, respectively. The results of four genome scans were inconsistent except for HLA loci. The strongest linkage findings were shown at HLA (6p21.3) in all of four RA genome scans. Major linkage findings except for chromosome 6 in the individual studies were 13q32-qter (D13S1315), 17q25.1 (D17S1301), 10q21.1 (D10S1221) and 1q (152 cM), respectively [6–9].
Meta-analysis of RA genome scans
Figure 1 shows the summed ranks for each bin giving Psumrnk. The summed ranks (vertical axis) are plotted against the bin location, by a single point plotted for the summed rank for each bin with chromosome numbers (horizontal axis). A total of nine bins lay above the 95% confidence level (Psumrnk = 0.05) and four bins were above the 99% confidence level (Psumrnk = 0.01): bins 6.2 (Psumrnk = 0.0000008), 6.4 (Psumrnk = 0.0007950), 8.1 (Psumrnk = 0.00075842), 18.3 (Psumrnk = 0.0077142), 12.3 (Psumrnk = 0.0224458), 12.2 (Psumrnk = 0.0227133), 1.5 (Psumrnk = 0.0254791), 6.3 (Psumrnk = 0.00289908) and 16.2 (Psumrnk = 0.0495275), suggesting that these bins contain RA-linked loci. Table 2 summarizes the highest 10% of bins when ordered by summed rank. Nine bins achieved a significant Psumrnk. The strongest evidence for linkage occurred on chromosome 6p22.3-6p21.1 (bin 6.2), containing the HLA region. The RA GSMA produced genome-wide evidence for linkage on this chromosome 6p22.3-6p21.1 (Psumrnk = 0.0000008). Two clusters of adjacent bins occurred (6.2–6.4, 12.2–12.3), which would be expected if a susceptibility locus were present in this region, since, for a complex disease, true linkage may extend up to 30 cM from the true locus position.
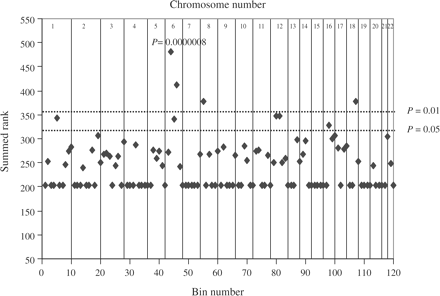
GSMA results of RA whole-genome linkage scans. Individual chromosomes were sub-divided into ∼30 cM bins (represented by a dot), and bins were ranked by the significance after summing weighted data across the studies. Significance levels corresponding to 99% (Psumrnk<0.01) and 95% (Psumrnk<0.05) were shown by the horizontal lines.
Genome search meta-analysis results, showing chromosomal bins with a summed rank within the highest 10% of all observed summed rank values in weighted analyses
| Marshfield location (cM) | Weighted analyses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bin | Begin | End | Cytogenetic location | Rsum | Psumrnk | Pord | |||
| 6.2 | 32.62 | 65.14 | 6p22.3-p21.1 | 480.0 | 0.0000008 | 0.0001000 | |||
| 6.4 | 99.01 | 131.07 | 6q15-q23.2 | 411.1 | 0.0007950 | 0.0037996 | |||
| 8.1 | 0 | 27.4 | 8pter-p22 | 378.4 | 0.0075842 | 0.0538946 | |||
| 18.3 | 62.84 | 96.48 | 18q12.3-q22.1 | 378.0 | 0.0077142 | 0.0105989 | |||
| 12.3 | 53.28 | 82.12 | 12p11.21-q15 | 347.0 | 0.0224458 | 0.1039900 | |||
| 12.2 | 24.45 | 53.28 | 12p12.1-p11.21 | 346.9 | 0.0227133 | 0.0337966 | |||
| 1.5 | 113.69 | 142.24 | 1p31.1-p13.3 | 344.1 | 0.0254791 | 0.0143986 | |||
| 6.3 | 65.14 | 99.01 | 6p21.1-q15 | 341.3 | 0.0289908 | 0.0092991 | |||
| 16.2 | 32.07 | 67.62 | 16p13-q12.2 | 328.3 | 0.0495275 | 0.0914909 | |||
| 2.9 | 206.74 | 233.62 | 2q34-q35 | 307.5 | 0.0998799 | 0.8566140 | |||
| Marshfield location (cM) | Weighted analyses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bin | Begin | End | Cytogenetic location | Rsum | Psumrnk | Pord | |||
| 6.2 | 32.62 | 65.14 | 6p22.3-p21.1 | 480.0 | 0.0000008 | 0.0001000 | |||
| 6.4 | 99.01 | 131.07 | 6q15-q23.2 | 411.1 | 0.0007950 | 0.0037996 | |||
| 8.1 | 0 | 27.4 | 8pter-p22 | 378.4 | 0.0075842 | 0.0538946 | |||
| 18.3 | 62.84 | 96.48 | 18q12.3-q22.1 | 378.0 | 0.0077142 | 0.0105989 | |||
| 12.3 | 53.28 | 82.12 | 12p11.21-q15 | 347.0 | 0.0224458 | 0.1039900 | |||
| 12.2 | 24.45 | 53.28 | 12p12.1-p11.21 | 346.9 | 0.0227133 | 0.0337966 | |||
| 1.5 | 113.69 | 142.24 | 1p31.1-p13.3 | 344.1 | 0.0254791 | 0.0143986 | |||
| 6.3 | 65.14 | 99.01 | 6p21.1-q15 | 341.3 | 0.0289908 | 0.0092991 | |||
| 16.2 | 32.07 | 67.62 | 16p13-q12.2 | 328.3 | 0.0495275 | 0.0914909 | |||
| 2.9 | 206.74 | 233.62 | 2q34-q35 | 307.5 | 0.0998799 | 0.8566140 | |||
Genome search meta-analysis results, showing chromosomal bins with a summed rank within the highest 10% of all observed summed rank values in weighted analyses
| Marshfield location (cM) | Weighted analyses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bin | Begin | End | Cytogenetic location | Rsum | Psumrnk | Pord | |||
| 6.2 | 32.62 | 65.14 | 6p22.3-p21.1 | 480.0 | 0.0000008 | 0.0001000 | |||
| 6.4 | 99.01 | 131.07 | 6q15-q23.2 | 411.1 | 0.0007950 | 0.0037996 | |||
| 8.1 | 0 | 27.4 | 8pter-p22 | 378.4 | 0.0075842 | 0.0538946 | |||
| 18.3 | 62.84 | 96.48 | 18q12.3-q22.1 | 378.0 | 0.0077142 | 0.0105989 | |||
| 12.3 | 53.28 | 82.12 | 12p11.21-q15 | 347.0 | 0.0224458 | 0.1039900 | |||
| 12.2 | 24.45 | 53.28 | 12p12.1-p11.21 | 346.9 | 0.0227133 | 0.0337966 | |||
| 1.5 | 113.69 | 142.24 | 1p31.1-p13.3 | 344.1 | 0.0254791 | 0.0143986 | |||
| 6.3 | 65.14 | 99.01 | 6p21.1-q15 | 341.3 | 0.0289908 | 0.0092991 | |||
| 16.2 | 32.07 | 67.62 | 16p13-q12.2 | 328.3 | 0.0495275 | 0.0914909 | |||
| 2.9 | 206.74 | 233.62 | 2q34-q35 | 307.5 | 0.0998799 | 0.8566140 | |||
| Marshfield location (cM) | Weighted analyses | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bin | Begin | End | Cytogenetic location | Rsum | Psumrnk | Pord | |||
| 6.2 | 32.62 | 65.14 | 6p22.3-p21.1 | 480.0 | 0.0000008 | 0.0001000 | |||
| 6.4 | 99.01 | 131.07 | 6q15-q23.2 | 411.1 | 0.0007950 | 0.0037996 | |||
| 8.1 | 0 | 27.4 | 8pter-p22 | 378.4 | 0.0075842 | 0.0538946 | |||
| 18.3 | 62.84 | 96.48 | 18q12.3-q22.1 | 378.0 | 0.0077142 | 0.0105989 | |||
| 12.3 | 53.28 | 82.12 | 12p11.21-q15 | 347.0 | 0.0224458 | 0.1039900 | |||
| 12.2 | 24.45 | 53.28 | 12p12.1-p11.21 | 346.9 | 0.0227133 | 0.0337966 | |||
| 1.5 | 113.69 | 142.24 | 1p31.1-p13.3 | 344.1 | 0.0254791 | 0.0143986 | |||
| 6.3 | 65.14 | 99.01 | 6p21.1-q15 | 341.3 | 0.0289908 | 0.0092991 | |||
| 16.2 | 32.07 | 67.62 | 16p13-q12.2 | 328.3 | 0.0495275 | 0.0914909 | |||
| 2.9 | 206.74 | 233.62 | 2q34-q35 | 307.5 | 0.0998799 | 0.8566140 | |||
Discussion
To date, whole-genome linkage studies for RA have revealed a number of regions of the genome that are likely to harbour genes predisposing to RA [6–11]. Four linkage studies for RA included in the meta-analysis have shown the HLA region as containing the most significant linkage loci, but they have revealed inconsistent non-HLA linkage loci [6–9]. Susceptibility to RA is likely to involve several genes of weak effect, and consequently individual studies may have insufficient power to detect linkage. Here we have conducted a meta-analysis of RA genome scans using the GSMA method in an attempt to increase statistical power and to enhance evidence of linkage.
The highest evidence for linkage was observed in bin 6.2, which includes the HLA loci, with genome-wide significance (Psumrnk = 0.0000008). The nominally significant Psumrnk and Pord of the adjacent bins, 6.3 and 6.4, provided additional evidence for linkage. This might have been because, in simulated data, bins adjacent to those containing disease loci often also achieve nominal significance [14], or it may suggest the possibility that genes on non-HLA chromosome 6 regions may also play an important role in susceptibility to RA. Previous analyses of HLA associations have shown that the majority of RA-associated HLA-DRB1 alleles share a common structural feature at positions 70–74 of the DRB1 chain. This finding has given rise to the shared epitope hypothesis [15], and there is a hypothesis suggesting a role for DQ alleles or protective effects of DRB1 alleles [16, 17]. Another candidate gene, tumour necrosis factor (TNF), located in the HLA class III region, has been reported to play a role in susceptibility to RA [18].
Besides confirming linkage of the HLA region, the GSMA found evidence for linkage at non-HLA loci, such as chromosome 1p, 6, 8p, 12, 16 and 18q, across studies. Although all genome scans of RA included in this analysis have shown the HLA region to contain the strongest linkage loci, one or two studies did not reveal significant linkage in the non-HLA loci defined by this meta-analysis. This suggests that a gene with low frequency or weak penetrance may be present in this region, and an extensive collection of RA families will be required to provide sufficient power to detect linkage. Notably, the major linkage findings at non-HLA loci in individual studies were not replicated in this meta-analysis, and several other loci, although ranked high in individual studies, did not achieve a high summed rank. This suggests that some linkage loci observed in individual genome scans may be false-positive results. Also, there was no new locus identified by this meta-analysis that was not previously identified individually in any of the four genome-wide scans.
The linkage loci shown by this meta-analysis may provide a basis for the location of RA susceptibility genes. It is interesting to examine these regions for candidate genes. A possible candidate gene on chromosome 18q21 is the TNFRSF11A gene, which encodes receptor activator of nuclear factor κB (RANK). RANK is critically involved in the differentiation of osteoclasts in inflamed synovium and is important for the development of bone resorption in inflammatory arthritis [19]. A risk allele of a haematopoietic-specific protein tyrosine phosphatase, PTPN22, located at 1p13.3-1p13.1, changes the function of the protein, which functions as a negative regulator of T-cell activation, leading to T cells with a lower threshold for T-cell activation. The risk allele was significantly increased in RA patients compared with controls in Caucasians (28 vs 17%) [20] and the minor allele of this SNP recently was implicated in type 1 diabetes [21], suggesting that the variant of PTPN22 may increase susceptibility to autoimmune diseases. CD94 and NKG2 are members of the NK cell receptor families, and are encoded in the natural killer gene complex (NKC) on human chromosome 12p12-13. Although one study did not find a significant association between variations in CD94 and NKG2 genes and RA and systemic lupus erythematosus (SLE), chromosome 12p has been considered to be one of the potential candidate chromosomal regions for RA [22].
Many candidate genes in other chromosome regions have been reported to be associated with susceptibility to RA, such as TNFR2 on 1p36.3-p36.2 [23], PADI4 on 1p36.13 [24], FcGR3A on 1q23 [25], IL10 on 1q32.1 [26], PARP on 1q41-1q42 [27], CTLA4 on 2q33 [28], SLC22A4 on 5q31.1 [29], IFNG on 12q21.1 [30] and MIF on 22q11.23 [31]. This might reflect the limited power of linkage studies compared with association analyses.
There were limitations to our meta-analysis. First, we looked at published genome scans for meta-analysis. Our analysis was a little different from the RA GSMA method performed by Fisher et al. [32]. They analysed genome data obtained from linkage graphs, whereas we used linkage scores with P<0.05. Fisher et al. found significant results on chromosomes 6, 16, 12, 1q, 14q, 8p, 9q, 4q and 3q based on weighted P-values <0.05. There was consistency in the four bins 6.2, 16.2, 6.4 and 8.1 between the previous study and the present meta-analysis. The differences in results between the two studies could also have been due primarily to the use of different sets of scan analyses. Fisher et al. included two data sets that were used in this analysis [7, 8], a different data set for one scan [11] and data from one scan that we excluded because of absence of all linkage data with P<0.05 [10]. Although there might be a bias in our analysis, we believe that our meta-analysis using published data is not invalid for the assessment of evidence for linkage of RA across genome scan studies. For example, a previous meta-analysis using published data instead of whole-genome data has shown that the analysis is more powerful than one of several genome scans in detecting significant linkage without increasing false positives and also robust to a considerable amount of heterogeneity [33]. Secondly, although meta-analysis significantly increases the sample size, it also may introduce heterogeneity arising from phenotypic differences between different populations and ethnic and geographic differences. Although most populations included in the meta-analysis were Caucasian, it was possible that the Caucasian population in the USA was admixed with other populations, such as populations with African or Hispanic ancestry, and there was a difference in admixture of Caucasians between Europe and the USA. These might confound the analysis. There is a need for the judicious application of phenotypic stratification, including ethnicity, as a means of reducing heterogeneity in gene mapping studies. Thirdly, this RA GSMA did not consider the X or Y chromosome data, so no conclusions can be reached on possible linkage on these chromosomes.
In conclusion, this RA GSMA confirmed the evidence that the HLA loci is the greatest susceptibility factor for RA and revealed evidence for chromosomes 1p, 6, 8p, 12, 16 and 18q being non-HLA susceptibility loci across studies. We believe that these data may provide a basis for carrying out targeted linkage and candidate gene studies in these chromosomal regions.

The authors have declared no conflicts of interest.
References
Harris ED Jr. Rheumatoid arthritis. Pathophysiology and implications for therapy.
MacGregor AJ, Snieder H, Rigby AS et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins.
Wordsworth P, Bell J. Polygenic susceptibility in rheumatoid arthritis.
Ollier W, Thomson W. Population genetics of rheumatoid arthritis.
Deighton CM, Walker DJ, Griffiths ID, Roberts DF. The contribution of HLA to rheumatoid arthritis.
Cornelis F, Faure S, Martinez M et al. New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study.
Jawaheer D, Seldin MF, Amos CI et al. A genomewide screen in multiplex rheumatoid arthritis families suggests genetic overlap with other autoimmune diseases.
Jawaheer D, Seldin MF, Amos CI et al. Screening the genome for rheumatoid arthritis susceptibility genes: a replication study and combined analysis of 512 multicase families.
John S, Shephard N, Liu G, Zeggini E et al. Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites.
Shiozawa S, Hayashi S, Tsukamoto Y et al. Identification of the gene loci that predispose to rheumatoid arthritis.
MacKay K, Eyre S, Myerscough A et al. Whole-genome linkage analysis of rheumatoid arthritis susceptibility loci in 252 affected sibling pairs in the United Kingdom.
Altmuller J, Palmer LJ, Fischer G, Scherb H, Wjst M. Genomewide scans of complex human diseases: true linkage is hard to find.
Levinson DF, Levinson MD, Segurado R, Lewis CM. Genome scan meta-analysis of schizophrenia and bipolar disorder, part I: methods and power analysis.
Cordell HJ. Sample size requirements to control for stochastic variation in magnitude and location of allele-sharing linkage statistics in affected sibling pairs.
Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis.
Zanelli E, Gonzalez-Gay MA, David CS. Could HLA-DRB1 be the protective locus in rheumatoid arthritis?
Zanelli E, Krco CJ, Baisch JM, Cheng S, David CS. Immune response of HLA-DQ8 transgenic mice to peptides from the third hypervariable region of HLA-DRB1 correlates with predisposition to rheumatoid arthritis.
Mulcahy B, Waldron-Lynch F, McDermott MF et al. Genetic variability in the tumor necrosis factor-lymphotoxin region influences susceptibility to rheumatoid arthritis.
Li J, Sarosi I, Yan XQ et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism.
Begovich AB, Carlton VE, Honigberg LA et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis.
Bottini N, Musumeci L, Alonso A et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes.
Hikami K, Tsuchiya N, Yabe T, Tokunaga K. Variations of human killer cell lectin-like receptors: common occurrence of NKG2-C deletion in the general population.
Dieude P, Petit E, Cailleau-Moindrault S et al. Association between tumor necrosis factor receptor II and familial, but not sporadic, rheumatoid arthritis: evidence for genetic heterogeneity.
Suzuki A, Yamada R, Chang X et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis.
Kyogoku C, Tsuchiya N, Matsuta K, Tokunaga K. Studies on the association of Fc gamma receptor IIA, IIB, IIIA and IIIB polymorphisms with rheumatoid arthritis in the Japanese: evidence for a genetic interaction between HLA-DRB1 and FCGR3A.
Crawley E, Kay R, Sillibourne J, Patel P, Hutchinson I, Woo P. Polymorphic haplotypes of the interleukin-10 5′ flanking region determine variable interleukin-10 transcription and are associated with particular phenotypes of juvenile rheumatoid arthritis.
Pascual M, Lopez-Nevot MA, Caliz R et al. A poly(ADP-ribose) polymerase haplotype spanning the promoter region confers susceptibility to rheumatoid arthritis.
Vaidya B, Pearce SH, Charlton S et al. An association between the CTLA4 exon 1 polymorphism and early rheumatoid arthritis with autoimmune endocrinopathies.
Tokuhiro S, Yamada R, Chang X et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis.
Khani-Hanjani A, Lacaille D, Hoar D et al. Association between dinucleotide repeat in non-coding region of interferon-gamma gene and susceptibility to, and severity of, rheumatoid arthritis.
Baugh JA, Chitnis S, Donnelly SC et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis.
Fisher SA, Lanchbury JS, Lewis CM. Meta-analysis of four rheumatoid arthritis genome-wide linkage studies: confirmation of a susceptibility locus on chromosome 16.

{kind=link}
Comments