





Abstract
Melatonin is an indolamine synthesized in the pineal gland that has a wide range of physiological functions, and it has been under clinical investigation for expanded applications. Increasing evidence demonstrates that melatonin can ameliorate cadmium-induced hepatotoxicity. However, the potentially protective effects of melatonin against cadmium-induced hepatotoxicity and the underlying mechanisms of this protection remain unclear. This study investigates the protective effects of melatonin pretreatment on cadmium-induced hepatotoxicity and elucidates the potential mechanism of melatonin-mediated protection. We exposed HepG2 cells to different concentrations of cadmium chloride (2.5, 5, and 10μM) for 12 h. We found that Cd stimulated cytotoxicity, disrupted the mitochondrial membrane potential, increased reactive oxygen species production, and decreased mitochondrial mass and mitochondrial DNA content. Consistent with this finding, Cd exposure was associated with decreased Sirtuin 1 (SIRT1) protein expression and activity, thus promoted acetylation of PGC-1 alpha, a key enzyme involved in mitochondrial biogenesis and function, although Cd did not disrupt the interaction between SIRT1 and PGC-1 alpha. However, all cadmium-induced mitochondrial oxidative injuries were efficiently attenuated by melatonin pretreatment. Moreover, Sirtinol and SIRT1 siRNA each blocked the melatonin-mediated elevation in mitochondrial function by inhibiting SIRT1/ PGC-1 alpha signaling. Luzindole, a melatonin receptor antagonist, was found to partially block the ability of melatonin to promote SIRT1/ PGC-1 alpha signaling. In summary, our results indicate that SIRT1 plays an essential role in the ability of moderate melatonin to stimulate PGC-1 alpha and improve mitochondrial biogenesis and function at least partially through melatonin receptors in cadmium-induced hepatotoxicity.
Abbreviations
- AMPK
AMP-activated protein kinase
- CdCl2 (Cd)
cadmium chloride (cadmium)
- Mel
melatonin
- MT1
melatonin receptor 1
- PGC-1 alpha
peroxisome proliferator-activated receptor gamma, coactivator 1 alpha
- ROS
reactive oxygen species
- SIRT1
sirtuin 1
- ΔΨm
mitochondrial membrane potential
Cadmium, a highly cytotoxic metal, is a wide spread environmental and industrial pollutant found in soils, water, and air (Satarug et al., 2003). Humans and animals are exposed to cadmium via a variety of routes, including industrial contamination, food sources, and tobacco smoke (Perlman et al., 2012). The increasing levels of cadmium concentrations in the environment have caused great concern worldwide, especially in China (Larson, 2014). Cadmium is toxic even at low doses, and epidemiological and clinical data have shown that acute exposure to toxic doses of cadmium results primarily in liver damage (Rikans and Yamano, 2000). Among the mechanisms involved in cadmium-mediated hepatotoxicity, oxidative stress and mitochondrial dysfunction have been proposed to play pivotal roles (Cannino et al., 2009). As generally proposed, cadmium causes mitochondrial swelling, decreases expression of genes that control mitochondrial activity, and, finally, decreases mitochondrial oxidative capacity and ATP synthesis (Li et al., 2003).
Melatonin, the chief secretory product of the pineal gland, is best known for its effects on free radical scavenging and antioxidative activity (Galano et al., 2013). Within the past few years, the functional role of melatonin in improving mitochondrial function has been reported. Melatonin is a lipophilic molecule that freely crosses cell membranes and enters cells, where it preserves the cellular redox balance by maintaining mitochondrial homeostasis (Acuna Castroviejo et al., 2011). Moreover, melatonin can also bind to plasma membrane melatonin receptors and initiate signaling to restore mitochondrial function (Dragicevic et al., 2011). Recently, melatonin has been successfully used to reduce oxidative stress in many systems caused by an acute exposure of the liver to cadmium (El-Sokkary et al., 2010). However, the way in which melatonin protects against cadmium-induced oxidative stress and mitochondrial dysfunction in the human liver remains obscure.
SIRT1, a NAD-dependent class III histone deacetylase, plays a pivotal role in various cellular processes, including differentiation, gene silencing, metabolism, and stress resistance by modulating key targets via deacetylation (Imai et al., 2000). A SIRT1 target is PGC-1 alpha, a transcriptional coactivator. Importantly, SIRT1 physically interacts with and deacetylates PGC-1 alpha, consequently increasing PGC-1 alpha activity, promoting mitochondrial biogenesis, and maintaining mitochondrial function. Recently, overexpression of the SIRT1 deacetylase, and subsequent activation of PGC-1 alpha transcriptional activity, produced many health benefits, including protection from metabolic decline, cardiovascular disease, neurodegeneration, and liver injury (Lagouge et al., 2006; Rodgers et al., 2008; Wang et al., 2013; Zhu et al., 2010).
To further extend these findings, we tested whether melatonin, by increasing SIRT1 activity and expression, could modulate PGC-1 alpha function and ultimately influence the regulation of mitochondrial biogenesis and function in cadmium-induced hepatotoxicity. Our results indicate that melatonin efficiently protected HepG2 cells against cadmium-induced mitochondrial damage by promoting SIRT1/ PGC-1 alpha signaling, and that these beneficial effects of melatonin could be partially prevented by luzindole, a melatonin receptor inhibitor.
MATERIALS AND METHODS
Cell culture
The human hepatocellular carcinoma cell line, HepG2, was purchased from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China). The HepG2 cells were cultured in DMEM medium (SH30022.01B, HyClone) that was supplemented with 10% heat-inactivated FCS (SV30087.02, HyClone) and 1% (vol/vol) penicillin/ streptomycin (P4333, Sigma, St Louis, MO) in a 5% CO2 humidified atmosphere at 37°C.
Experimental protocol (Supplementary Data)
Step 1: Evaluation of the effects of cadmium on mitochondrial function and biogenesis in HepG2 cells. At 80% confluence, the cells were treated with CdCl2 (439800, Sigma, St Louis, MO) at different concentrations (0, 2.5, 5, and 10μM) for 12 h.
Step 2: Investigation of whether melatonin pretreatment alleviates cadmium-induced hepatotoxicity. Because 0.1μM melatonin failed to protect against Cd-induced hepatotoxicity in HepG2 cells, we treated the cells with 0.5μM of melatonin for 2 h prior to 5μM Cd treatment (Supplementary Data). Melatonin (M5250, Sigma, St Louis, MO) was freshly dissolved in ethanol to produce a 5mM stock solution and kept at 4°C. Before application, it was further diluted in culture medium. The final ethanol concentration did not exceed 0.1% in the cell culture experiments. The 0.1% ethanol concentration was included in each in vitro assay as vehicle control.
Step 3: Investigation of the role of the SIRT1/ PGC-1 alpha pathway in hepatic cell protection after melatonin pretreatment. (1) The cells were randomly divided into the following treatment groups: 5μM Cd, melatonin (0.5μM) + 5μM Cd, sirtinol (30μM, sc-205976, Santa Cruz, CA) + melatonin (0.5μM) + 5μM Cd, and sirtinol (30μM) + 5μM Cd. Compared with the control group, sirtinol (30μM) had no effect on cell viability or mitochondrial function (data not shown). (2) The cells were randomly divided into the following treatment groups: control siRNA + 5μM Cd, control siRNA + melatonin (0.5μM) +5μM Cd, SIRT1 siRNA + melatonin (0.5μM) + 5μM Cd, and SIRT1 siRNA + 5μM Cd. The cells were transiently transfected with control siRNA or SIRT1 siRNA did not show significant differences in cell viability and mitochondrial function (data not shown).
Step 4: Investigation of whether luzindole treatment abolished melatonin-promoted SIRT1/ PGC-1 alpha signaling pathway. HepG2 cells were treated with 5μM Cd alone or together with melatonin or luzindole (10μM, 0877, Tocris Bioscience) for 12 h.
Cell viability assay
Cell viability was analyzed using a Cell Counting Kit-8 according to the manufacturer's instructions (CK04, Dojindo Molecular Technologies, Kumamoto, Japan). Briefly, 1 × 104 cells were inoculated into 96-well plates. After being treated, 90 μl of medium and 10 μl of CCK-8 solution were added to each well. The cells were then incubated at 37°C for 2 h. After incubation, the absorption at 450 nm was measured using an Infinite M200 Microplate Reader. The results were expressed as a percentage of the values of the control set at 100%.
Oxidative stress determination
Production of intracellular reactive oxygen species (ROS) in HepG2 cells was determined based upon the oxidation of DCFH-DA (S0033, Beyotime Company, Shanghai, China). Fluorescence emission at 525 nm following excitation at 488 nm was measured using a microplate reader. The cellular fluorescence intensity was expressed as the fold change relative to the level observed in the control cells. The cellular fluorescence intensity was expressed as the fold change relative to the level observed in the control cells.
Measurement of mitochondrial membrane potential (ΔΨm)
A mitochondrial membrane potential assay kit containing JC-1 (C2006, Beyotime Company, Shanghai, China) was used to measure the ΔΨm in HepG2 cells. Briefly, 1×104 cells were placed in a 96-well plate and incubated with 1xJC-1 in growth medium at 37°C for 20 min. Monomeric JC-1 green fluorescence emission and aggregate JC-1 red fluorescence emission were measured using an Infinite M200 Microplate Reader (Tecan, Mannedorf, Switzerland). The ΔΨm in each group was calculated as the fluorescence ratio of red to green and expressed as a multiple of the level in the control groups, and a decrease in the fluorescence ratio of red/green indicates an increase of depolarized mitochondria as well as a decrease in the mitochondrial membrane potential.
Real-time PCR analysis to detect mtDNA copy number
Quantitative real-time PCR was used to detect the cellular mtDNA content. The mtDNA primers were designed to detect Complex II (succinate–ubiquinone oxidoreductase). The mtDNA primers were (5’-CAAACCTACGCCAAAATCCA-3’) and (5’-GAAATGAATGAGCCTACAGA-3’); and the GAPDH primers were (5’-TGACAACAGCCTCAAGAT-3’) and (5’-GAGTCCTTCCACGATACC-3’).
Mitochondrial mass
Mitochondrial mass was evaluated using the fluorescent probe N-nonyl acridine orange (NAO) as previously described (Pi et al., 2013). Briefly, HepG2 cells were incubated in DMEM containing 5μM of NAO for 30 min at 37°C in the dark. The NAO fluorescence intensity was determined using an Infinite 200 Microplate Reader (M200, Tecan), and the emitted fluorescence was measured at 530 nm following excitation at 485 nm.
SIRT1 gene silencing
HepG2 cells grown in antibiotic-free DMEM on culture dishes for 24 h were transfected with SIRT1 small-interfering (siRNA; 80pmols; sc-40986, Santa Cruz, CA) and non-targeted control siRNA (sc-37007, Santa Cruz, CA) using Opti-MEM I reduced serum media and Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, CA). Twenty-four hours after transfection, cells were washed and processed for immunoblotting and other studies as described below.
SIRT1 activity
After SIRT1 protein immunoprecipitation, a reaction mixture containing Fluoro-Substrate peptide solution to Protein G agarose beads was added and NAD-dependent deacetylase activity was measured using the SIRT1 Assay Kit (ab156065, abcam, MA) according to the manufacturer's instructions. Fluorescence intensity was read continuously for 30 min at 2 min intervals with excitation at 340 nm and emission at 460 nm using an Infinite M200 Microplate Reader (Tecan, Mannedorf, Switzerland).
Immunoprecipitation
HepG2 cells were treated with Cd and lysed with cell lysis buffer (P0013, Beyotime Company, Shanghai, China). Lysates were clarified by centrifugation at 12,000 g for 15 min and were used for immunoprecipitation. Typically, 2 μg of SIRT1 or PGC-1 alpha antibody was incubated with 500–1000 μg of cell lysate protein overnight at 4°C, then protein A/G beads (P2006 or P2009, Beyotime Company, Shanghai, China) were added and the mixture was incubated overnight at 4°C. Then, the beads were washed three times, solubilized in 40 μl 3xSDS sample buffer (7722, Cell Signaling Technology, MA), and analyzed by immunoblotting with antibodies against PGC-1 alpha or acetylated-lysine, respectively.
Western blot analysis
The HepG2 cell lysates were centrifuged for 15 min at 12,000 g, and the resulting supernatant was transferred to a new tube. The protein concentrations were determined using a Bradford protein assay kit (P0006, Beyotime Company, Shanghai, China). Equal amounts of protein were loaded on to gels and then separated by SDS-PAGE. Following protein transfer to PVDF membranes, the membranes were blocked and then incubated overnight at 4°C with antibodies against SIRT1 (1:1000, ab110304, abcam, MA), PGC-1 alpha (1:200, sc-13067, Santa Cruz, CA), acetylated-lysine (1:1000, 9441, Cell Signaling Technology), Melatonin Receptor 1A (MT1, 1:1000, ab116337, abcam, MA), p-AMPKα1/2 (1:800, sc-33524, Santa Cruz, CA), AMPKα1/2 (1:800, sc-74461, Santa Cruz, CA), Melatonin Receptor 1B (MT2, 1:1000, ab128469, abcam, MA), and β-actin (1:5000, A5441, sigma, St Louis, MO). The membrane was visualized by enhanced chemiluminescence using Super Signal West Pico blotting (34079, Pierce) detection reagents and exposure to Hyper Performance Chemiluminescence film.
Immunofluorescence confocal microscopy
Cells were seeded and cultured on gelatin-coated glass coverslips. HepG2 cells were treated as indicated, incubated with 100nM MitoTracker Red (M7512, Invitrogen, CA), and fixed with 4% (w/v) paraformaldehyde in PBS at room temperature for 30 min. The cells were then permeabilized with 0.5% Triton X-100 in PBS for 15 min at room temperature. After three washes with PBS, the cells were blocked with 10% FBS at 37°C for 30 min and then incubated with a mouse monoclonal to antibody against SIRT1 (ab110304, abcam, MA) at a 1:100 dilution overnight at 4°C. The cells were then incubated with an Alexa Fluor 488 Donkey Anti-Mouse IgG (H+L) antibody (A21202, Invitrogen, CA) at a 1:100 dilution for 1 h at 37°C. The samples were subsequently incubated with a rabbit polyclonal antibody against PGC-1 alpha (ab54481, abcam, MA) at a 1:100 dilution overnight at 4°C. Lastly, the cells were then incubated with an Alexa Fluor 647 goat anti-rabbit IgG (H+L) antibody (A21244, Invitrogen, CA) at a 1:100 dilution for 1 h at 37°C, and were imaged using a Zeiss confocal laser scanning microscope.
Statistical analysis
Data were analyzed using GraphPad Prism-5 software. All of the experimental data are expressed as the mean ± SEM, and each experiment was performed a minimum of three times. One-way ANOVA with a Bonferroni correction was used to determine statistical significance, and p <0.05 was considered to be statistically significant.
RESULTS
Cadmium Exposure Decreases Mitochondrial Function and Impairs Mitochondrial Biogenesis
Mitochondrial dysfunction plays an important role in the hepatotoxicity of cadmium. In our study, we found that treatment of HepG2 cells with 2.5μM, 5μM, or 10μM Cd for 12 h increased the fluorescence intensity of ROS by 1.49- and 1.48-fold that of control cells, respectively (Fig. 1A). In line with the observed ROS production, the ΔΨm level was decreased by 28% and 52% following Cd treatment (Fig. 1B). Moreover, cell viability decreased in a similar pattern (Fig. 1C). Studies over the past few years have implicated impaired mitochondrial biogenesis as a cause of mitochondrial dysfunction in numerous mitochondrial-related diseases (Gomes et al., 2012). To determine if Cd indeed decreases mitochondrial biogenesis, we examined two parameters of mitochondrial biogenesis in HepG2 cells. MtDNA content as well as mitochondrial mass was significantly reduced in HepG2 cells after exposure to Cd (2.5μM, 5μM, or 10μM) for 12 h (Figs. 1D and E).
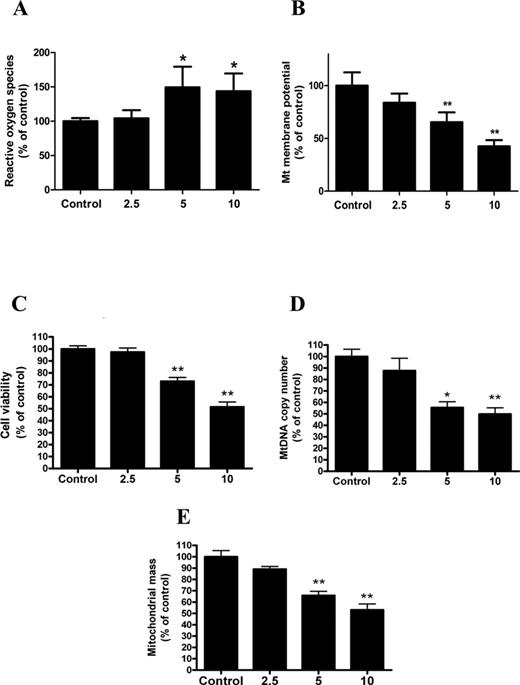
Cd exposure triggers mitochondrial dysfunction and the impairment of mitochondrial biogenesis in a concentration-dependent manner in HepG2 cells. (A) ROS production, (B) ΔΨm level, (C) cell viability, (D) mtDNA copy number, and (E) mitochondrial mass were assayed after HepG2 cells were treated with Cd at different concentrations (0, 2.5, 5, 10μM) for 12 h. The values are presented as the mean ± SEM, *p < 0.05, **p < 0.01 versus the control group (n = 6).
Cadmium Exposure Inhibits Expression and Activity of SIRT1 and Increases Its Downstream Signaling Effect, PGC-1 Alpha Acetylation
PGC-1 alpha, a key transcriptional factor involved in the first step of mitochondrial biogenesis, plays an important role in governing the transcriptional control of mitochondrial biogenesis and function (Scarpulla, 2011). To elucidate the mechanism of cadmium-induced mitochondrial dysfunction, the effects of cadmium on PGC-1 alpha expression were investigated. Interestingly, Cd did not affect the expression of PGC-1 alpha, whereas Cd treatment significantly increased acetylated-PGC-1 alpha in a concentration-dependent manner (Figs. 2A and B).
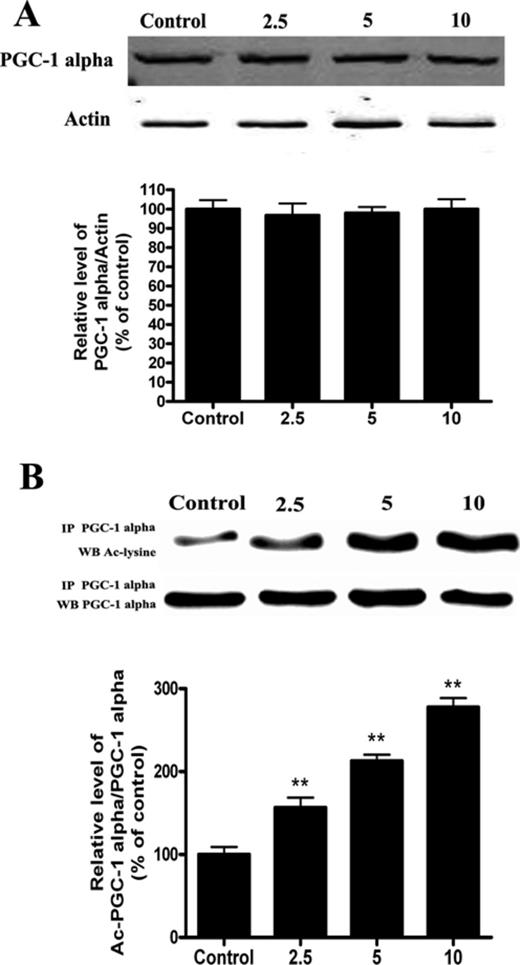
Cd exposure increases the amount of acetylated-PGC-1 alpha expression in a concentration-dependent manner in HepG2 cells. (A) Representative immunoblot of PGC-1 alpha protein levels (90 kDa) in HepG2 cells. (B) Acetylation of PGC-1 alpha after Cd exposure by immunoprecipitation with an anti-PGC-1 alpha antibody, followed by immunoblot analysis of acetylated-lysine. A representative Western blot and the quantification of the ratio of acetylated to total PGC-1 alpha are shown. The results are expressed as a percentage of the control, which is set at 100%. The values are presented as the mean ± SEM, **p < 0.01 versus the control group (n = 4).
Because SIRT1 is key regulator of PGC-1 alpha function through epigenetic regulation of the PGC-1 alpha gene (Gerhart-Hines et al., 2007), we investigated the effect of cadmium on SIRT1 expression and activity. Cd treatment resulted in a significant decrease in SIRT1 protein levels (Fig. 3A). Meanwhile, the SIRT1 enzyme catalyzes NAD+-dependent protein deacetylation (Revollo and Li, 2013). Our experimental data showed that the levels of SIRT1 activity were decreased in the Cd group compared with the control group (Fig. 3B). To obtain additional evidence in support of these findings, we analyzed cultured HepG2 cells stained for endogenous SIRT1 using confocal microscopy. We found that endogenous SIRT1 was decreased in nuclei and that PGC-1 alpha was expressed in both the nuclei and cytoplasm. SIRT1 staining strongly overlapped with PGC-1 alpha staining in the nuclei. These results indicate that SIRT1 protein co-localized with PGC-1 alpha protein (Fig. 3C). In fact, SIRT1 and PGC-1 alpha not only functionally but also physically interact to form a stable complex that regulates the activity and acetylation status of PGC-1 alpha (Nemoto et al., 2005). However, co-immunoprecipitation pull-down assay results indicate that Cd treatment does not disrupt or increase the interaction of SIRT1 and PGC-1 alpha (Fig. 3D).
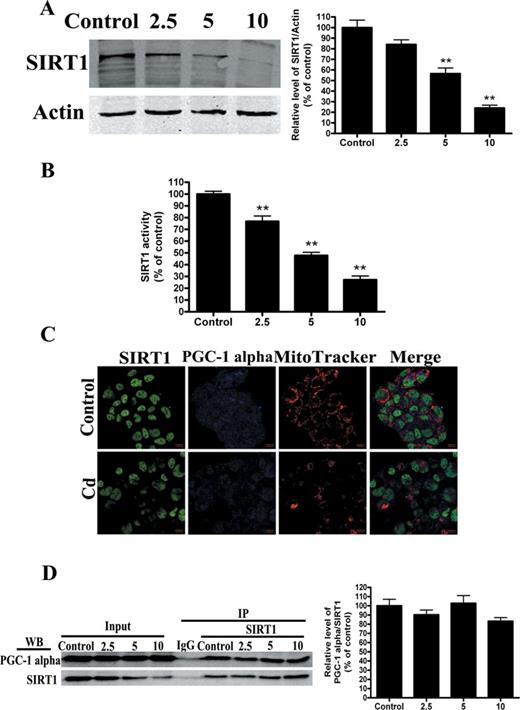
Cd exposure decreases SIRT1 protein expression and activity in a concentration-dependent manner. (A) Representative immunoblot of SIRT1 protein levels (110 kDa) in HepG2 cells. (B) SIRT1 activity was measured based on an enzymatic reaction using a SIRT1 assay kit. (C) After HepG2 cells were treated with 5μM Cd, confocal microscopic images of SIRT1, mitochondria, and PGC-1 alpha in HepG2 cells were obtained. (D) Cell lysates were mixed with various concentrations of Cd for 12 h. Antibodies against SIRT1 were added to immunoprecipitate the SIRT1-containing complexes, and then immunoblotted for PGC-1 alpha or SIRT1. A representative Western blot and the quantification of the ratio of PGC-1 alpha to SIRT1 are shown. The results are expressed as a percentage of the control, which was set at 100%. The values are presented as the mean ± SEM, **p < 0.01 versus the control group (n = 4).
Melatonin Reduces Cadmium-Induced Mitochondrial Dysfunction and Restores Mitochondrial Biogenesis in HepG2 Cells
Because Cd caused cytotoxicity and mitochondrial dysfunction in HepG2 cells, we tested whether melatonin interferes with Cd-induced hepatotoxicity in vitro. To rule out the possibility that 0.1μM melatonin does not inhibit cadmium's effects in the HepG2 cells, we treated the cells with 0.5μM or 1μM melatonin (Supplementary Data). In this study, melatonin markedly decreased ROS production, preserved the mitochondrial membrane potential, inhibited Cd-stimulated cytotoxicity, and maintained mtDNA content and mitochondrial mass in 5μM Cd-treated HepG2 cells (Figs. 4A–E). In addition, melatonin stimulated Cd-suppressed SIRT1 protein expression and activity, and thus inhibited acetylated-PGC-1 alpha expression (Figs. 5A–D).
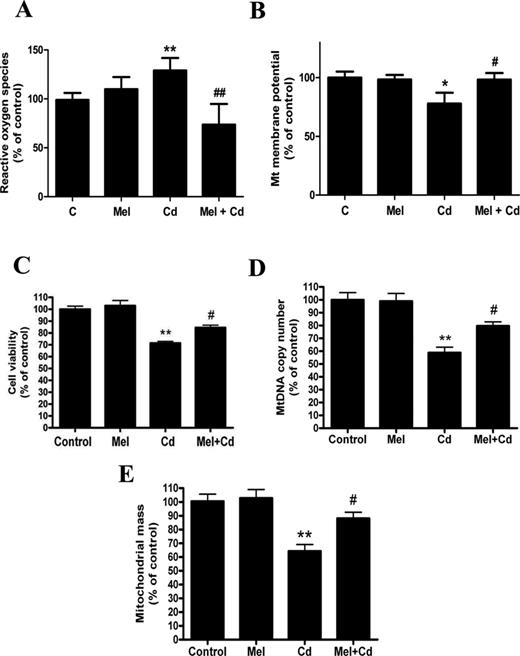
Melatonin protects against mitochondrial dysfunction and the disruption of mitochondrial biogenesis after exposure to Cd in vitro. (A) ROS production, (B) ΔΨm level, (C) cell viability, (D) mtDNA copy number, (E) mitochondrial mass. The results are expressed as a percentage of the control, which was set at 100%. The values are presented as the mean ± SEM, *p <0.05, **p < 0.01 versus control group, #p < 0.05, ##p < 0.01 versus the Cd (5μM) group (n = 6).
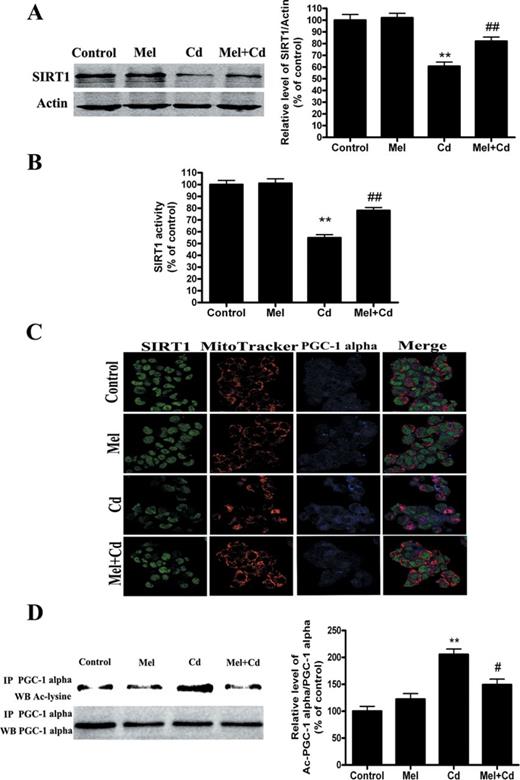
Melatonin increases the expression and activity of SIRT1 and inhibits acetylated- PGC-1 alpha expression after Cd treatment in vitro. Melatonin enhances the expression of SIRT1 (A, C) and SIRT1 activity (B) 12 h after exposure to 5μM Cd. (D) Melatonin induced deacetylation of PGC-1 alpha after Cd treatment. The results are expressed as a percentage of the control, which is set at 100%. The values are presented as the mean ± SEM, **p < 0.01 versus the control group, and #p < 0.05, ##p < 0.01 versus the Cd (5μM) group (n = 4).
Mitochondrial-Protective Action of Melatonin is SIRT1/PGC-1 Alpha Dependent on Cadmium-Induced Hepatotoxicity
To confirm whether melatonin is involved in Cd-induced mitochondrial dysfunction and the disruption of SIRT1-regulated mitochondrial biogenesis, we used SIRT1 inhibitors (sirtinol) and SIRT1 siRNA. Exposure to sirtinol inhibited melatonin-enhanced SIRT1 protein activity but not its expression. However, SIRT1 siRNA successfully inhibited both melatonin-enhanced SIRT1 protein activity and expression (Figs. 6A and 7A, and Supplementary Data). Moreover, sirtinol and SIRT1 siRNA pretreatment reversed the protective effects of melatonin on Cd-induced ROS production, ΔΨm decreases, and cytotoxicity (Figs. 6B–D and 7B–D). Furthermore, mtDNA content and mitochondrial mass were increased in cells with melatonin, but this increase was abolished by sirtinol and SIRT1 siRNA (Figs. 6E–F and 7E–F). Importantly, as shown in Figures 6G and 7G, melatonin-induced decreases in acetylated-PGC-1 alpha expression were significantly attenuated by sirtinol and SIRT1 siRNA in HepG2 cells exposed to Cd. Together, these data suggest a SIRT1-dependent effect of melatonin on acetylated-PGC-1 alpha expression under cadmium exposure.
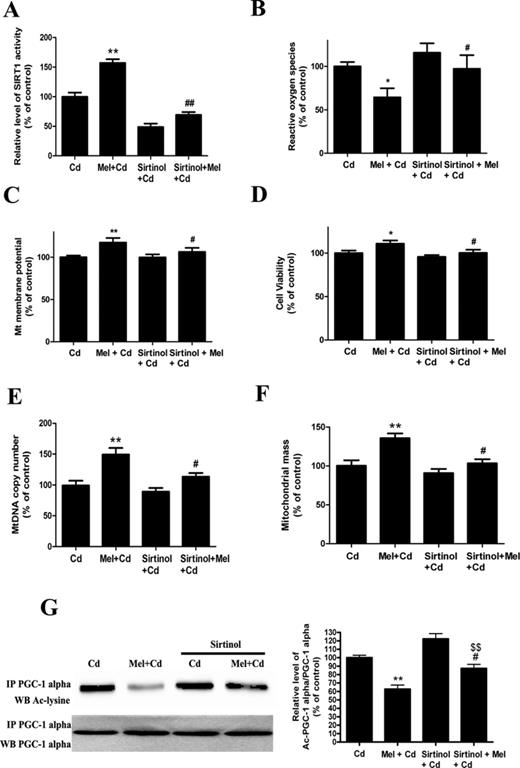
Sirtinol pretreatment abolishes melatonin-enhanced mitochondrial function and biogenesis in Cd-injured HepG2 cells. (A) The effects of melatonin and sirtinol pretreatment on SIRT1 activity, (B) ROS production, (C) ΔΨm level, (D) cell viability, (E) mtDNA copy number, (F) mitochondrial mass, and (G) acetylated-PGC-1 alpha expression. The results are expressed as a percentage of the control (“Cd” group taken as a control), which was set at 100%. The values are presented as the mean ± SEM, **p < 0.01 versus the Cd (5μM) group, and #p < 0.05, ##p < 0.01 versus the Cd (5μM) + melatonin group and $$p < 0.01 versus the sirtinol + Cd (5μM) group (n = 6).
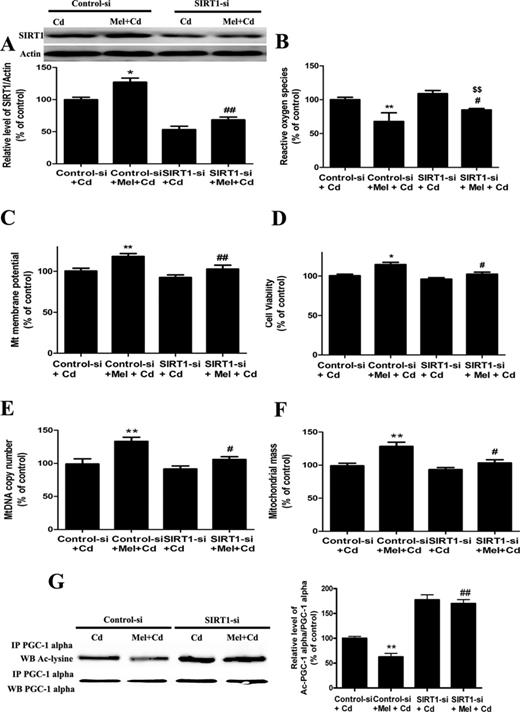
SIRT1 siRNA pretreatment abolishes melatonin-enhanced mitochondrial function and biogenesis in Cd-injured HepG2 cells. (A) The effect of melatonin and SIRT1 siRNA pretreatment on SIRT1 expression, (B) ROS production, (C) ΔΨm level, (D) cell viability, (E) mitochondrial mass, (F) mtDNA copy number, and (G) acetylated-PGC-1 alpha expression. The results are expressed as a percentage of the control (“Cd” group taken as a control), which is set at 100%. The values are presented as the mean ± SEM, *p < 0.05, **p < 0.01 versus the Control siRNA + Cd (5μM) group, #p < 0.05, ##p < 0.01 versus the Control siRNA + Cd (5μM) + melatonin group, and $$p < 0.01 versus the SIRT1 siRNA + Cd (5μM) group (n = 6).
Melatonin Receptor 1 but not AMPK Partially Accounts for the Dual Roles of SIRT1/ PGC-1 Alpha Pathway in the Hepatoprotective Effects of Melatonin
Recently, it was reported that AMPK could act upstream of the SIRT1 pathway (Canto et al., 2009). Therefore, we assessed AMPK signaling using Western blotting for the phosphorylated and total forms of the protein. In our study, Cd treatment resulted in a significant increase in pAMPK protein levels, but melatonin did not alter AMPK signaling (data not shown). Moreover, melatonin exerts many functions through melatonin receptor-dependent mechanism. In the present study, the MT1 but not the MT2 membrane receptor was detected in HepG2 cells (Fig. 8A). To better understand whether melatonin signals to the mitochondria through the MT1 or MT2 receptors, luzindole, a melatonin receptor antagonist, was added alongside melatonin. The melatonin-induced enhancement of SIRT1 activity but not expression level was partially reversed by luzindole (Figs. 8B and C). As expected, the melatonin-induced decrease in the expression of acetylated-PGC-1 alpha was also blocked by luzindole (Fig. 8D).
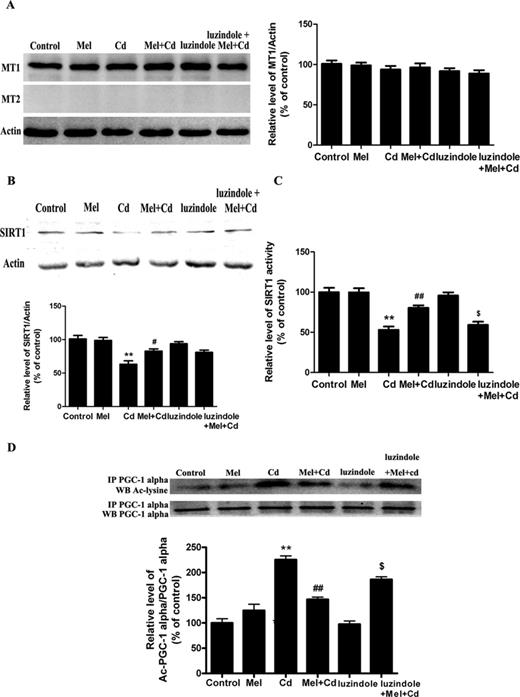
Luzindole treatment abolishes the activity of the melatonin-promoted SIRT1/PGC-1 alpha signaling pathway in Cd-injured HepG2 cells. (A) The effects of melatonin and luzindole treatment on melatonin receptors (MT1 and MT2) expression, (B) SIRT1 expression, (C) SIRT1 activity, and (D) acetylated-PGC-1 alpha expression. The results were expressed as a percentage of the control, which was set at 100%. The values are presented as the mean ± SEM, **p < 0.01 versus control group, #p < 0.05, ##p < 0.01 versus the Cd (5μM) group, and $p < 0.05 versus the Cd (5μM) + melatonin group (n = 4).
DISCUSSION
Cadmium is one of the most toxic metals released into the environment and is one of the important environmental pollutants that can be transferred among various levels of the food chain (Larison et al., 2000). It is well known that cadmium induces hepatotoxicity in humans, as well as in farm and laboratory animals (Ebert-McNeill et al., 2012; Wu et al., 2012). However, its underlying mechanism of toxicity remains poorly understood. In the present study, we found that cadmium exposure significantly impairs mitochondrial biogenesis and function. Notably, SIRT1 coordinates mitochondrial biogenesis with PGC-1 alpha-induced mitochondrial homeostasis in cadmium-induced hepatotoxicity.
Mitochondrial biogenesis requires numerous processes, which involve the expression of nuclear-encoded proteins are essential for the replication of mtDNA and maintenance of mitochondrial mass (Wenz, 2013). In this study, cadmium decreased mtDNA content and mitochondrial mass in HepG2 cells, indicating that cadmium inhibits mitochondrial biogenesis. Much has been learned of the consequences of essential mitochondrial functions by the ablation of mitochondrial biogenesis (Rice et al., 2014). Consistent with these studies, our study confirmed that cadmium exposure decreased the mitochondrial membrane potential and promoted ROS generation in vitro.
PGC-1 alpha is known to be the main regulator of the mitochondrial biogenesis pathway and ultimately regulates mitochondrial function. Ectopic PGC-1 alpha expression increased the levels of respiratory chain proteins, and the steady-state level of mtDNA in cultured cells (Choi et al., 2014). The molecular basis of PGC-1 alpha regulation has been studied extensively and its activity and stability can be regulated on the expressional level (Scarpulla, 2011). However, in the present study, PGC-1 alpha protein expression was unaffected by cadmium treatment. PGC-1 alpha is also regulated at the posttranslational level, as modifications, such as acetylation, significantly impact its activity (Park et al., 2012). Here, we observed that cadmium increased acetylated-PGC-1 alpha expression in a concentration-dependent manner. As a transcriptional co-activator, deacetylated PGC-1 alpha more effectively recruits transcription factors to stimulate the synthesis of its target genes, which are associated with mitochondrial biogenesis, gluconeogenesis, and fatty acid oxidation (Gerhart-Hines et al., 2007). Taken together, these data suggest that cadmium directly affects mitochondrial biogenesis by up-regulating acetylated PGC-1 alpha.
The acetylation/deacetylation of lysine residues on nuclear proteins has been demonstrated to regulate multiple functions, such as transcriptional activity, DNA binding, protein binding, protein stability, and translocation. Recently, SIRT1 acts as a sensor to regulate mitochondrial status by deacetylation of its substrates, including PGC-1 alpha (Higashida et al., 2013). Exercise or nutrient deprivation causes increased SIRT1 deacetylation of PGC-1 alpha, increased transcriptional activity of PGC-1 alpha, and a subsequent increase in mitochondrial gene transcription and mitochondrial biogenesis (Gurd, 2011). Here, we found that SIRT1 localization strongly overlapped with that of PGC-1 alpha in the nucleus, which suggests that SIRT1 may directly interact with PGC-1 alpha, inducing PGC-1 alpha deacetylation in cadmium exposure. Our study also shows that although cadmium decreased SIRT1 protein expression and activity, it did not disrupt the interaction between SIRT1 and PGC-1 alpha, providing evidence for a detailed mechanism by which acetylation modifies the transcription activity of PGC-1 alpha.
Recently, much attention has been paid to exploring different drugs that prevent the adverse effects of cadmium. Melatonin has remarkable antioxidant properties and is a strong candidate for the treatment of cadmium-induced hepatotoxicity (Romero et al., 2014). However, the hepatoprotective effects of melatonin are not simply related to the direct scavenging of free radicals or increasing the activity and expression of antioxidant enzymes but also involve other mechanisms. Recently, the beneficial effects of melatonin in improving cellular function and organism health directly or indirectly depend on its roles in activating SIRT1 (Cuesta et al., 2013). To identify whether the SIRT1/PGC-1 alpha pathway mediates the observed mitochondrial protective effect of melatonin, HepG2 cells were pretreated with sirtinol and SIRT1 siRNA, followed by melatonin treatment and cadmium stimulation for 12 h. We showed that blocking SIRT1 activity through treatment with sirtinol and down-regulating SIRT1 expression through transfection of SIRT1 siRNA reversed the protective effects of melatonin on cadmium-mediated mitochondrial injury. Moreover, melatonin-dependent down-regulated acetylated-PGC-1 alpha expression was strikingly reversed by sirtinol and SIRT1 siRNA. These data suggest that the effects of melatonin in promoting mitochondrial biogenesis and the amelioration of mitochondrial dysfunction are related to up-regulation of the SIRT1 / PGC-1 alpha pathway.
Melatonin exerts this wide range of physiological effects through specific plasma membrane receptors (Wang et al., 2014). In mammals, there are two subtypes of melatonin membrane receptors (MT), MT1 and MT2, both of which are members of the seven transmembrane G protein-coupled receptor family (von Gall et al., 2002). Consistent with initial findings (Carbajo-Pescador et al., 2009), only expression of the MT1 protein was found in HepG2 cells. In addition, luzindole did not completely prevent the effects of melatonin on SIRT1 activity and expression, and the data do not exclude the possibility that melatonin treatment could directly alter SIRT1 protein activity and expression, affecting disease onset, progression, and survival. It is noteworthy that melatonin has been extensively proposed to interact with and freely cross the cell membrane to reach subcellular components and the nucleus, which then generate further signals independently or by secondary interaction with components of receptor-dependent pathways (Park et al., 2013). In fact, we are using 500nM melatonin concentrations, and the kinetic parameter of MT1 receptor is in the pM range. Probability, a portion of the effects of melatonin may be directly as a genomic regulator to control the SIRT1 protein expression in our model, and which is in agreement with the results of a number of studies (Korkmaz et al., 2009). Moreover, SIRT1 is activated downstream, possibly because AMPK activation increases the NAD+/NADH ratio, and SIRT1 is known to be NAD+-dependent (Castano et al., 2014). However, melatonin did not alter AMPK signaling in our study, suggesting that AMPK was not involved in mediating the beneficial effects of this compound in cadmium-induced hepatotoxicity. Further investigation is required to identify other mechanisms of melatonin-mediated effects on the SIRT1/PGC-1 alpha pathway.
In summary, we proposed an intriguing mechanism whereby cadmium induces hepatotoxicity by disrupting mitochondrial biogenesis and function. Most importantly, the protective effect of melatonin is associated with enhanced mitochondrial biogenesis through the SIRT1-dependent PGC-1 alpha pathway, and this effect appears to be mediated partially through the binding of melatonin to the melatonin receptor MT1 to activate signaling to mitochondria (Fig. 9). Taken together, our data illustrate a new molecular mechanism underlying the ability of melatonin to be explored for future clinical treatment of cadmium-induced hepatotoxicity.
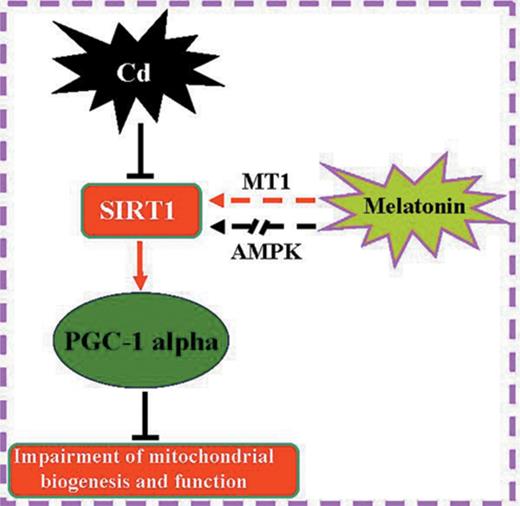
Melatonin improves mitochondrial function by promoting MT1/SIRT1/ PGC-1 alpha -dependent mitochondrial biogenesis in cadmium-induced hepatotoxicity. AMPK is not involved in mediating the beneficial effects of melatonin under these conditions.
FUNDING
The National Natural Science Foundation of China (NO.81422039); the Outstanding Youth Science Fund of Chongqing that provided by Chongqing science and technology commission (CSTC2013JCYJJQ10002).
We appreciate Dr. Guobing Li in our university for his hospitable and professional technical assistance. Conflicts of interest: None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments