Efficacy and safety of betahistine treatment in patients with Meniere’s disease: primary results of a long term, multicentre, double blind, randomised, placebo controlled, dose defining trial (BEMED trial)
BMJ 2016; 352 doi: https://doi.org/10.1136/bmj.h6816 (Published 21 January 2016) Cite this as: BMJ 2016;352:h6816
- Christine Adrion, biostatistician1 2,
- Carolin Simone Fischer, otorhinolaryngologist1,
- Judith Wagner, neurologist3,
- Robert Gürkov, otorhinolaryngologist4,
- Ulrich Mansmann, director and chair of biostatistics and bioinformatics2,
- Michael Strupp, professor of neurology1 3
- On behalf of the BEMED study group
- 1German Center for Vertigo and Balance Disorders, University Hospital Munich, Campus Grosshadern, Munich, Germany
- 2Institute for Medical Informatics, Biometry, and Epidemiology, University of Munich, Campus Grosshadern
- 3Department of Neurology, University Hospital Munich, 81377 Munich
- 4Department of Otorhinolaryngology, Head and Neck Surgery, University Hospital Munich
- Correspondence to: M Strupp Michael.Strupp{at}med.uni-muenchen.de
- Accepted 2 November 2015
Abstract
Study question What is the long term efficacy of betahistine dihydrochloride on the incidence of vertigo attacks in patients with Meniere’s disease, compared with placebo?
Methods The BEMED trial is a multicentre, double blind, randomised, placebo controlled, three arm, parallel group, phase III, dose defining superiority trial conducted in 14 German tertiary referral centres (for neurology or ear, nose, and throat). Adults aged 21-80 years (mean age 56 years) with definite unilateral or bilateral Meniere’s disease were recruited from March 2008 to November 2012. Participants received placebo (n=74), low dose betahistine (2×24 mg daily, (n=73)), or high dose betahistine (3×48 mg daily, (n=74)) over nine months. The primary outcome was the number of attacks per 30 days, based on patients’ diaries during a three month assessment period at months seven to nine. An internet based randomisation schedule performed a concealed 1:1:1 allocation, stratified by study site. Secondary outcomes included the duration and severity of attacks, change in quality of life scores, and several observer-reported parameters to assess changes in audiological and vestibular function.
Study answer and limitations Incidence of attacks related to Meniere’s disease did not differ between the three treatment groups (P=0.759). Compared with placebo, attack rate ratios were 1.036 (95% confidence interval 0.942 to 1.140) and 1.012 (0.919 to 1.114) for low dose and high dose betahistine, respectively. The overall monthly attack rate fell significantly by the factor 0.758 (0.705 to 0.816; P<0.001). The population based, mean monthly incidence averaged over the assessment period was 2.722 (1.304 to 6.309), 3.204 (1.345 to 7.929), and 3.258 (1.685 to 7.266) for the placebo, low dose betahistine, and high dose betahistine groups, respectively. Results were consistent for all secondary outcomes. Treatment was well tolerated with no unexpected safety findings. Without a control group of patients who did not receive any intervention to follow the natural course of the disease, the placebo effect could not be accurately assessed and differentiated from spontaneous remission and fluctuation of symptoms.
What this study adds Current evidence is limited as to whether betahistine prevents vertigo attacks caused by Meniere’s disease, compared with placebo. The trial provides information on symptom relief on placebo intervention which is relevant for the design of future studies on potential disease modifying treatments in patients with Meniere’s disease.
Funding, competing interests, data sharing Support from the German Federal Ministry of Education and Research (BMBF support code 01KG0708). Potential competing interests have been reported in full at the end of the paper on thebmj.com. Data are available from the corresponding author (Michael.Strupp@med.uni-muenchen.de) or biostatistician (mansmann@ibe.med.uni-muenchen.de).
Study registration EudraCT no 2005-000752-32; ISRCTN no ISRCTN44359668.
Introduction
Meniere’s disease is characterised by recurrent attacks of vertigo, fluctuating sensorineural hearing loss, aural fullness, and tinnitus.1 Its histopathological hallmark is endolymphatic hydrops.2 3 Lifetime prevalence of the disease in the United States is reported as 190 per 100 000 people, with a ratio of 1.89 women to every man. 4 5 Annual incidence of the disease in the USA was 15.3 per 100 000 people (age adjusted rate).6 The peak age of onset is during the fifth and sixth decade.7
For patients with Meniere’s disease, unpredictable vertigo attacks are the most important and unpleasant symptom. Although the disease is clinically problematic and the target of several treatments, there are so far no validated instruments related to vertigo that are based on patient reported outcomes (PRO) for comprehensively evaluating disease severity in a clinical trial. Treatment should aim to stop or reduce the number and severity of acute attacks of vertigo, reduce or eliminate tinnitus, and prevent impaired vestibular function and hearing loss. Given the chronic nature of the disease and the fluctuating and episodic pattern of symptoms, the long term effectiveness of any prophylactic drug should be investigated.
Many therapeutic approaches to Meniere’s disease have been studied. These include a low salt diet and diuretics,8 intratympanic steroid application,9 10 or minimal invasive interventions (such as insertion of a ventilation tube into the tympanic membrane,11 12 endolymphatic sac surgery,13 or pulsed low pressure delivery (using Meniett devices)).14 15 16 17 For patients who do not respond to these treatments, more aggressive procedures can be considered, such as intratympanic application of gentamycin,18 19 plugging of the semicircular canal, labyrinthectomy, or neurectomy.20 21 22 23 However, these interventions are irreversible and could damage the cochlear and vestibular organ; furthermore, a recent Cochrane review could not show any evidence of benefit in a surgical approach.24 25
Betahistine is a licensed drug for Meniere’s disease-like symptom complexes, which contains the active ingredient betahistine dihydrochloride (maximum daily dose 48 mg) or betahistine dimesylate (maximum daily dose 36 mg). Betahistine is a strong H3 antagonist and a weak H1 agonist26 with three sites of action. Firstly, it increases dose-dependent cochlear blood flow,27 mainly via the H3 receptor as an inverse agonist.28 Because betahistine has a strong first pass effect and is metabolised in the liver into three metabolites, not only betahistine but also its major metabolite aminoethylpyridine increases cochlear blood flow.29 Secondly, betahistine increases histamine turnover in the central nervous and vestibular system, also mainly via the H3 receptor. Thirdly, it decreases vestibular input in the peripheral vestibular system, with possible involvement with the H3 and H4 receptors.
How betahistine might have an effect in the prophylactic treatment of Meniere’s disease is so far unknown. It could lead to an improvement of labyrinthine microcirculation, thereby rebalancing the production and resorption of endolymph. The drug was first registered in Europe in the 1970s and has been administered to more than 100 million patients so far. In Germany, betahistine is the first line treatment for Meniere’s disease in clinical practice, before consideration of endolymphatic sac surgery or ablative gentamicin treatment.30 The drug is inexpensive and well tolerated, and is one of the most frequently prescribed drugs for Meniere’s disease in Europe.31 32 In the USA, betahistine is not approved by the Food and Drug Administration but can be easily obtained through US compounding pharmacies with a prescription.
Several clinical studies assessing the effect of betahistine on the vestibular system and, to a lesser degree, audiological symptoms suggested that the drug improved these symptoms.33 34 According to a Cochrane systematic review of betahistine for Meniere’s disease or syndrome, there is, however, insufficient evidence to indicate whether betahistine has any effect.33 So far, randomised controlled trials that meet high quality standards are lacking, either due to inadequate diagnostic criteria or methods,35 or because the effect of betahistine treatment on vertigo was assessed inadequately. To summarise, the limitations of the evidence base for preventive treatment strategies for Meniere’s disease include:
• Predominance of trials investigating short term effects (treatment periods of six months or less)
• Inclusion criteria of enrolled patients (for instance, no differentiation between patients with the disease and patients with other causes of vertigo)
• High dropout rates35 with potential for considerable attrition bias
• Small trials or few placebo controlled trials36
• Varying quality of outcome measures for assessing efficacy (including quality of life scores, functional impairment, disability, and the number and severity of acute attacks of vertigo).33
The dose of betahistine in these studies varied between 16 and 72 mg per day, which might explain the differences in symptom relief observed. Even higher doses of up to 480 mg per day have shown benefit for severe cases in a small case series, suggesting a possible effect of high dose regimens in the treatment of Meniere’s disease.37 The drug seems to retain a good tolerability profile. On the basis of many years’ clinical experience, the dose was successively increased to 48 mg three times a day, pointing towards the role of long term treatment (up to 12 months). This dose increase was supported by an open, uncontrolled, non-masked study without a placebo arm that compared a high dose regimen of 48 mg three times daily with the recommended standard dose of 16 or 24 mg three times daily.36 This non-interventional study showed that the higher dose was superior to the lower dose, and that the treatment effect of betahistine on the incidence of attacks of vertigo became more prominent over time.
Owing to variable methodological rigour and shortcomings in previous trials including the potential risk of bias, the medical treatment of Meniere’s disease with betahistine (BEMED) trial was designed. This investigator initiated, prospective, longitudinal, multicentre, double blind, randomised, placebo controlled, three arm, parallel group, phase III superiority trial aimed to assess the long term prophylactic effects of betahistine dihydrochloride in two different doses and placebo. The doses and placebo were administered continuously for nine months, and investigators observed their effect on the frequency, duration, and severity of acute attacks caused by Meniere’s disease, vertigo related impairment of quality of life, and vestibular and audiological function.
The trial also aimed to ascertain the speed of effect—that is, whether the two active doses can be distinguished from each other or from placebo by how quickly reduction in attack frequency is achieved.38 Additionally, the tolerance and adverse events were examined. We report the prespecified efficacy and safety analyses at nine months for the BEMED trial.
Methods
Study population and protocol
Study participants were recruited by the outpatient dizziness services in the neurology department or the ear, nose, and throat department of 14 German university hospitals. Patients were enrolled in the study from 31 March 2008 (first patient, first visit) to 5 November 2013 (last patient, last visit), including a three month follow-up. Patients aged 18-80 years were eligible for enrolment if they presented with two or more definitive spontaneous episodes of vertigo of at least 20 minutes’ duration, had audiometrically documented hearing loss on at least one occasion, and tinnitus or aural fullness in the treated ear, excluding other possible causes of vertigo. These factors made up a diagnosis of definite unilateral or bilateral Meniere’s disease, fulfilling the criteria of the 1995 American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) guideline.39 Furthermore, patients had to be in an active phase of the disease, with at least two vertigo attacks per month in at least three consecutive months before enrolment. Female patients of childbearing potential were only included if they had a negative serum pregnancy test within seven days before initiation of treatment and were willing to practice acceptable methods of birth control during treatment and for three months after treatment.
Exclusion criteria were diagnosis of other central or peripheral vestibular disorders such as vestibular migraine, benign paroxysmal positioning vertigo, paroxysmal brainstem attacks, as well as phobic postural vertigo. Patients were excluded if they had known contraindications or sensitivity to betahistine, such as bronchial asthma, pheochromocytoma, treatment with other antihistaminic drugs, ulcer of the stomach or duodendum, or severe dysfunction of liver or kidney. Safety related exclusion criteria were severe coronary heart disease or heart failure, persistent uncontrolled hypertension with systolic blood pressure higher than 180 mm Hg or diastolic blood pressure higher than 110 mm Hg, life expectancy less than 12 months, other serious illness, or a complex disease that might confound treatment assessment. General exclusion criteria were participation in another trial with an investigational drug or device within the past 30 days, previous participation in the present study, or planned participation in another trial. We excluded pregnant and breastfeeding women and women contemplating pregnancy during the trial from enrolment.
Written informed consent was obtained from all patients before initiation of the first study specific procedure. The protocol was approved by local independent ethics committees and was performed in accordance with the Declaration of Helsinki and other applicable guidelines, laws, and regulations. The study was a longitudinal, multicentre, double blind, randomised, placebo controlled, three arm, parallel group, dose defining phase III trial conducted at 14 academic sites throughout Germany.
The individual study duration was 12 months: nine months of treatment and three months of follow-up. Both the examinations and the study, treatment were performed in an outpatient setting. At the baseline visit, patients received their study drug treatment together with a paper based vertigo diary, and returned to the study centre at months one, four, and six; and at the end of the treatment period at month nine. In addition to these four clinic visits, five standardised telephone interviews were performed after postbaseline months two, three, five, seven, and eight, to verify compliance and increase protocol adherence. In particular, these interviews reminded patients to complete their vertigo diary every day and to record any treatment discontinuation, change in relevant concomitant drug treatment, or adverse events they might have experienced in the meantime (which were also documented regularly on the case report forms during each telephone and clinic visit).
All patients underwent a standardised physical, neurological, and neuro-orthoptic examination; peripheral vestibulocochlear testing; assessment of medical history (for up to five years before enrolment); laboratory examination; and measurement of blood pressure and heart rate. We also performed electronystagmography, including bithermal caloric irrigation to measure caloric nystagmus response, and pure tone audiometry. At each clinic visit, patients had to complete three self-administered questionnaires on quality of life aspects, and their diaries were checked for data completeness and quality of documentation to ensure patient comprehension of the diary items.
Randomisation, concealment, and blinding
A total of 221 eligible patients at 14 study sites were randomly assigned in a 1:1:1 ratio to receive either high dose or low dose betahistine, or placebo for nine months (fig 1⇓). Each site received a pool of study medication kits including the treatment assignment in a sealed opaque emergency envelope. If a patient dropped out before receiving the kit, he or she was replaced by the next eligible patient enrolled at the same centre. The concealed allocation was performed by an internet based randomisation schedule (https://wwwapp.ibe.med.uni-muenchen.de/randoulette), stratified by study site. The fixed block size was three (starting with six), which was not disclosed during the trial. The random number list was generated by an investigator with no clinical involvement in the trial. Patients, clinicians, core laboratories, and trial staff (data analysts, statisticians) were blind to treatment allocation.

Fig 1 Study flowchart, according to the consolidated standards of reporting trials (CONSORT). The diagram shows enrolment and primary efficacy endpoints based on patient diaries, from prescreening to data collection; and the extent of exclusions, loss to follow-up, and completeness of diary documentation available across months one to nine. Stages following randomisation are allocation, follow-up, and analysis. FAS=full analysis set; PP=per protocol
{kind=link}
Study treatments
Betahistine dihydrochloride tablets were over-encapsulated with mannitol and aerosil as filling material. Capsules containing the active ingredient were refilled from original pharmacy packaging into vials under sterile conditions and relabelled by the pharmacy of the university hospital of the University of Heidelberg. In the control group, an identically appearing capsule filled with mannitol and aerosil but not containing any active ingredient was administered as placebo.
Patients were instructed to take six capsules per day (two capsules in the morning, two at noon, and two in the evening). The first drug intake started as soon as possible after receipt of the study medication kits containing the vials during the baseline visit. Patients assigned to the experimental arms were given low doses or high doses of betahistine dihydrochloride (Vasomotal, manufactured by Abbott Pharma, Hannover, Germany). A dose of 24 mg, which is the highest clinically admitted dose, was administered orally two times each day to the low dose group, and 2×24 mg three times each day were given to the high dose group; both groups received treatment for nine months. In the low dose group, patients took one betahistine capsule and one placebo capsule in the morning; two placebo capsules at noon; and one betahistine capsule together with one placebo capsule in the evening.
The nine month treatment duration was deemed necessary and adequate to reliably assess the long term prophylactic effect of continuous treatment on the incidence and severity of vertigo symptoms caused by Meniere’s disease. There were no disallowed concomitant drugs used during the study except for antihistaminic drugs, because we aimed to assess the efficacy of the assigned prophylactic treatment irrespective of rescue medication use by measuring efficacy conditional on real life adherence. Hence, rescue medication for managing of acute vertigo related symptoms such as vomiting or nausea could also be prescribed, because a possible effect on the occurrence of vertigo attacks is unknown.
Study outcomes and data collection
Blinded diary assessment
Participants were instructed to record acute attacks of vertigo related to Meniere’s disease, coexisting symptoms (such as aural fullness, changes in tinnitus, changes in hearing) and other characteristics of their vertigo attack. Other characteristics included time of onset, type of vertigo (rotatory or postural vertigo, gait unsteadiness, or lightheadedness), duration, and severity in a paper based diary for the full 12 month study duration. We also monitored additional symptoms that could occur simultaneously with attacks caused by Meniere’s disease and symptoms of other diseases with vertigo symptoms, to catching real attacks related to Meniere’s disease. Web appendix 1 shows a template of the vertigo diary.
Typically, attack data were recorded by the patients whenever they experienced vertigo related symptoms. However, owing to the complexity of vertigo symptoms, erroneously documented perseverative or persistent episodes of vertigo, and differing individual perceptibility, counting of vertigo attacks caused by Meniere’s disease is challenging.40 Therefore, all raw patient ratings (this is, the patient’s opinion of the occurrence of vertigo episodes) were evaluated in a blinded manner by trained professionals (CSF; CA) at the site of the principal investigator. The decision process was performed according to a consensus document (unpublished standard operating procedure) before unblinding in order to define conclusive primary efficacy data from a clinical perspective on the basis of the whole attack information documented in the patient’s diary. In particular, since multiple classifications concerning the type of vertigo episode were documented in the original patient diaries, the hierarchy displayed above was used to derive type specific efficacy outcomes, with rotatory vertigo being the most severe of four different types used to characterise an attack.
The primary efficacy outcome was the individual attack rate standardised on a 30 day interval (starting from timepoint 1—defined as the date of first intake, with the day of first study drug intake being day 1). The number of evaluated days was defined as the number of days with non-missing information about the patient’s vertigo status, as provided by the daily diary recordings. For example, a patient with 12 attacks during 75 documented days (that is, 75÷30=2.5 intervals) has the rate 12÷2.5=4.8. The 15 undocumented days out of the prespecified 90 day assessment period (starting day 181, ending day 270) were considered as missing at random.41
Secondary efficacy outcomes
Diary based secondary efficacy endpoints were the median duration and median severity of evaluated Meniere’s attacks during months seven to nine within the nine month treatment period.
We measured handicap and impairment of quality of life due to vertigo or tinnitus with the following three well established self-administered questionnaires: the dizziness handicap inventory score based on 25 items,42 the vestibular disorders activities of daily living (VDADL) score,43 and the mini-tinnitus impairment questionnaire score based on 12 items (MiniTF12).44 45 The total VDADL score is defined as the median value of answers across all 28 questions and is thus not affected significantly by missing values. To deal with missing items for both the dizziness handicap inventory score and the MiniTF12 questionnaire, we derived the mean total scores for the dizziness handicap inventory and the MiniTF12 as secondary outcome variables, averaging for the number of available answers. For all three scores, higher values reflect greater perceived disability and impairment of QoL. Web appendix 2 shows the definition of the total scores for the dizziness and self-assessment scales.
A key secondary endpoint measured during clinic visits was peripheral vestibular function determined by electronystagmography under caloric irrigation (two test conditions for the right and left ear: 30°C for the cool irrigation, 44°C for the warm irrigation). The parameter of interest was the peak slow phase velocity (recorded in °/sec) of the caloric nystagmus response of the selected ear. Web appendix 2 provides the definition of the “selected ear”; web appendix 3 shows the trial protocol. Furthermore, we used pure tone audiometry to determine hearing loss (recorded in dB) during bone conduction for test conditions 250 Hz, 500 Hz, 1000 Hz, and 2000 Hz, and to determine the tinnitus intensity (in dB). Likewise, these secondary outcomes were defined for the selected ear.
The three quality of life scores as well as the observer reported secondary efficacy outcomes measured during clinic visits were assessed at baseline and at the nine month visit. Explorative, not preplanned efficacy analyses were performed on specific types of vertigo spells: rotatory or postural attacks, and rotatory only attacks.
Safety
We assessed safety from reports of adverse events as well as laboratory parameters, vital signs (blood pressure, pulse, height, weight, and body mass index), and physical or neurological examinations. Safety was assessed over the entire treatment period at months one, four, six, and nine (including adverse events occurring in the first three weeks after cessation of treatment).
Hypotheses and statistical methodology
Principal analysis
The BEMED trial aimed to determine whether treatment with high or low dose betahistine or placebo differed in effectiveness. We assumed that the maximum effect would probably occur during the prespecified 90 day assessment period (months seven to nine). Our first idea of how to analyse the primary endpoint was to perform a robust comparison of the number of attacks observed during the last three months of the nine month treatment period by non-parametric tests. During the trial, it became apparent that dropouts and incomplete diary documentation created missing data that could not be adequately handled by the intended robust comparison. To deal with the missing data structure in the longitudinal individual observations, we used a negative binomial, generalised linear mixed effects model (NB GLMM) that not only yields unbiased parameter estimates when missing observations are missing at random (MAR),46 but also provides reasonably stable results even when the assumption of MAR is violated.47 48
The NB GLMM has fixed effects for treatment group, time (numerical variable for months one to nine), and treatment by time interaction. The model contains normally distributed random intercepts and random slopes associated with time, as well as an offset term for the log-transformed number of evaluated days within each 30 day interval.49 The offset term quantifies the observed information time per 30 days and delivers the denominator for the attack rate.
Patients who did not provide any diary data (leading to zero evaluable days) were excluded from the MAR based primary efficacy analysis, according to an “all observed data approach” as proposed by White and colleagues.50 This approach is statistically efficient without using multiple imputation techniques.51 Data retrieved after withdrawal of randomised study treatment were also included in the analysis.
The target estimates consist of the attack decay rate for the placebo group (fixed effect for time) as well as rate ratios for both betahistine dose groups (treatment by time interaction) to assess whether the magnitude of the difference between treatment groups varies over time (speed of effect). This model also allows individual attack rates over time to be calculated. To approximate the robust comparison as mentioned at the beginning of this section, we averaged the individual attack rates over the prespecified assessment period (months seven to nine) to derive a (marginal) population based, mean attack rate per 30 days for each treatment arm.
The principal model was established by use of data from a previous open non-interventional study36 together with statistical methodology that has been published elsewhere.49
Analyses of secondary outcomes
Secondary outcomes assessed during clinic visits—that is, both the observer-reported outcome and the quality of life scores, were analysed in a descriptive manner. The absolute change from baseline to the nine month visit was prespecified as the parameter of interest. For differences between treatment groups, we used an analysis of covariance (ANCOVA) for absolute change scores, with factor for treatment group and the baseline value as covariates. We used a closed testing approach to avoid the adjustment of the significance level because of multiple testing. In case of a high proportion of missing values at baseline or at the nine month visit, multiple imputation techniques based on chained equations (the MICE method52 53) assuming MAR were applied within the ANCOVA.
Both diary based endpoints (attack duration and severity) were reported on an ordinal rating scale by use of predetermined codes (codes for attack duration: 2 (1-20 min), 3 (20-60 min), 4 (60-180 min), 5 (>180 min); codes for attack severity: 1 (mild), 2 (moderate), 3 (severe), 4 (very severe)). For each patient, we calculated the median duration and severity of attacks within months seven, eight, and nine (time period of primary interest). Hence, only patients with a total number of evaluated days larger than zero across the assessment period were considered for analysis. To quantitatively describe treatment effects together with 95% confidence intervals, we applied a cumulative logit model based on the threshold approach (proportional odds model). According to the consensus document, the variable duration was necessary and sufficient for a Meniere’s attack to be defined on the basis of the raw diary recordings. Hence, there were no missing values concerning the duration of an evaluated attack.
Analysis sets
Analyses were based on the intention to treat principle; safety analyses were done on all patients who received at least one dose of study drug. The full analysis set (FAS) population included all patients randomised (irrespective of whether they were treated or not), and who did not fail to satisfy a major entry criterion. We excluded patients who provided neither primary nor secondary efficacy data from efficacy analyses, assuming MAR.
The per protocol set consisted of all patients from the FAS population who did not substantially deviate from the protocol; they had three characteristics. Firstly, this group included patients for whom no major protocol violations were detected (for example, poor compliance, errors in treatment assignment). Secondly, they were on treatment for at least eight months—that is, more than 240 days, counting from day of first intake (completion of a certain prespecified minimal exposure to the treatment regimen). Thirdly, they provided diary information within the 90 day assessment period (that is, there was availability of measurements of the primary variable within the period of interest). Hence, patients who prematurely discontinued the study or treatment before month 7 were excluded from the per protocol sample.
Determination and recalculation of sample size
We planned to use non-parametric tests to assess differences in the number of attacks during the last three months of the nine month treatment period. The closed testing procedure was intended to handle multiple comparison issues between the three arms. A general difference between the three arms would be tested by a Kruskal-Wallis test, and if the global test was significant (5% level), three two-group comparisons using the Mann-Whitney U test would be performed (5% level). Therefore, determination of sample size focused on a two group comparison based on the U test.
The basic effect quantity of a U test is the probability to achieve a better result in the experimental arm than in the control arm. Pilot data from an observational study by Strupp and colleagues36 supported the assumption that the probability to achieve a better result on betahistine than placebo is 0.75. Hence, a sample size of 21 patients in each group would have 80% power to detect the difference between two groups using a two sided Mann-Whitney U test on a 5% significance level. Initially, a dropout rate of about 25% was assumed. Thus, a total of 84 patients (28 in each treatment group) had to be enrolled.
This optimistic sample size approach was questioned. A simulation study was performed to calculate a model based proposal for the probability to achieve a better result on betahistine than on placebo. We applied the asinh-transformed linear mixed model of Adrion and Mansmann49 to simulate potential study results under more conservative clinical scenarios. The decay rate of the daily attack rate (fixed effect for time) on placebo was set to −0.06, and a treatment effect of −0.08 (treatment by time interaction) was chosen. A random intercept with a standard deviation of 0.8 and a measurement error of 0.5 was assumed. These planning figures reflect the data of Strupp and colleagues.36 Based on 1000 simulated samples, the probability to achieve a better result under betahistine than under placebo was estimated as 0.67. Based on this target parameter, the recalculated sample size of 46 participants per group (138 in total) will have 80% power to detect the difference between two independent groups using a Mann-Whitney U test with a two sided 5% significance level (nQuery Advisor 7.0). We assumed a dropout rate of about 37%. Hence, a total of 220 patients had to be enrolled in the trial. Web appendix 2 describes preplanned sensitivity analyses and additional efficacy analyses as well as the statistical strategies applied for multiple testing.
The study database was stored in SAS (Unix Version 9.2, SAS Institute). Statistical analyses were performed using the statistical software package R version 3.1.1.54 We used the R packages lme4 (version lme4_1.1-7) to fit frequentist generalised linear mixed effects models,55 56 ordinal to fit cumulative logit models,57 and mice for multiple imputation techniques applied for key secondary efficacy outcomes.52 53 All statistical tests were two sided, and P<0.05 was considered significant.
Previous and concomitant drug treatments were coded using the World Health Organization Drug Dictionary (version 1 March 2014). Medical history and adverse events were coded using the Medical Dictionary for Regulatory Activities (version 17.0).
Patient involvement
No patients were involved in setting the research question or the outcome measures, nor were they involved in developing plans for participant recruitment, or the design and implementation of the study. There are no plans to explicitly involve patients in dissemination. Final results will be sent to all participating sites.
Results
Enrolment and subject attrition
A total of 1450 patients were screened for eligibility at 17 outpatient sites; 221 patients were randomised at 14 sites. The largest site was the sponsor’s site located at the Department of Neurology, University Hospital and German Center of Vertigo and Balance Disorders (DSGZ) in Munich, Germany. At this site, 410 patients were screened, and 86 of 221 patients randomised (that is, about 40% of all study participants). Overall, 74 patients were assigned to the placebo group, 73 to the low dose betahistine group, and 74 to the high dose betahistine group. Figure 1⇑ shows the flow of participants through the trial together with the completeness of diary information over the entire nine month treatment period.
Of 1450 patients, 1229 (85%) did not pass the screening stage. The most common reason was that patients did not meet the inclusion criteria regarding attack frequency (n=255), followed by general refusal to participate for no specific reasons (n=204), and concerns about the protocol, especially fear of placebo (n=100). Some did not meet the inclusion criteria of definite Meniere’s disease (n=123), fulfilled exclusion criteria (n=173), or could not tolerate or were allergic to betahistine (n=31). Other patients were being treated with betahistine and did not want to change or stop treatment (n=93). In some patients, the cause of vertigo was not clear (n=137). Other reasons (for example, a desire for another treatment option such as an operation; or moving abroad) were named in 158 patients. In total, 45 patients were judged ineligible because they fulfilled two of these criteria.
No patient prematurely terminated study participation before allocation to treatment. One patient in the low dose group did not receive the allocated intervention because of fear of placebo. Figure 1⇑ follows the CONSORT PRO reporting guideline58 and reveals that within the three month assessment period, 79% (174 of 221 patients) provided attack data for the primary endpoint. In each group, a few patients did not submit any diaries, giving no specific reason for this. Completeness of the patient diaries did not differ between the three treatment groups.
Participants’ baseline characteristics
Table 1⇓ shows the demographic and clinical characteristics as well as three total scores on quality of life assessed at the baseline visit of all 221 patients randomised. Overall, about half of the randomised patients were female; the total age range was 21-80 years. The treatment groups were well balanced for demographics; clinical factors; and tinnitus related, dizziness, and self-assessment scores. Prerandomisation attack frequency was not documented, although considered as an inclusion criterion. Initial evaluation of the post-treatment frequency of attacks caused by Meniere’s disease within the first 30 days of treatment (pseudobaseline) showed the three groups to be comparable at the outset (table 2⇓). Moreover, groups were well balanced with regard to disease duration and age at onset of vertiginous symptoms (data not shown).
Baseline characteristics of intention to treat sample according to study treatment
Postrandomisation data regarding initial attack frequency and treatment compliance (FAS population)
Dosing and protocol adherence
Treatment compliance based on drug accountability was not calculated owing to insufficient data quality and a high proportion of missing data. Instead, we used the treatment duration (defined as the difference between the end of treatment and the first study drug intake) as a measure of treatment adherence. In the FAS population, mean treatment duration was 222.5 days (95% confidence interval 201.99 to 243.10), 225.8 (204.55 to 246.99), and 215.8 (192.63 to 239.04) in the placebo, low dose betahistine, and high dose betahistine groups, respectively (table 2⇑). We saw no significant difference between the three groups concerning treatment duration (Kruskal-Wallis test used as global testing procedure; FAS P=0.824; per protocol P=0.600).
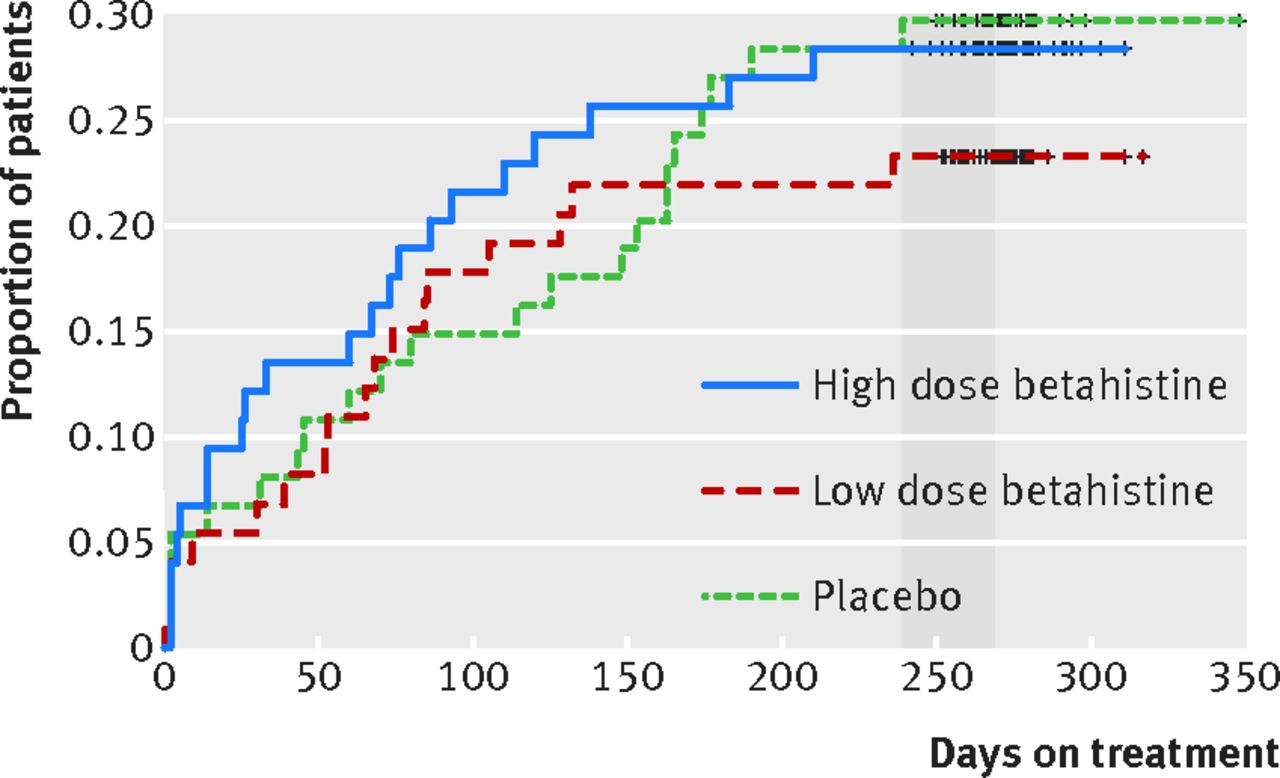
Figure 2⇓ shows for each treatment group the time to withdrawal and the proportion of patients who stopped treatment before the day indicated on the x axis. In this figure, an event indicating treatment dropout was defined as end of treatment before day 241 (according to the definition for per protocol). For example, about 10% of the placebo patients discontinued the assigned treatment before day 50, compared with about 14% of patients on high dose treatment. The figure also indicates that about 77% of low dose patients (versus 72% of high dose patients and 70% of placebo patients) were on treatment for at least eight months (241 days). We found no evidence for a differential dropout (attrition bias) from the administered treatment between the three groups (log rank test P=0.703).

Fig 2 Proportion and timing of patient withdrawal for all 221 patients randomised to each treatment group. According to the protocol, 270 days was the preplanned treatment duration. An event was defined as end of treatment before day 241 (start of grey region), according to the prespecified minimum exposure to the treatment regimen defined as per protocol and the corresponding definition of a major protocol deviation
{kind=link}
Primary efficacy outcome measures
An NB GLMM (negative binomial mixed effects model) assessed a general decline in the incidence of attacks caused by Meniere’s disease over the nine 30 day intervals. For the FAS population, the mean attack rate for placebo patients was significantly lowered by the factor 0.758 per additional 30 day interval on treatment (95% confidence interval 0.705 to 0.816; P<0.001). We hypothesised that the assigned experimental treatment (low or high dose betahistine) would make this overall decay rate even smaller. The corresponding estimated factors, representing rate ratios compared with placebo, were 1.036 (0.942 to 1.140) and 1.012 (0.919 to 1.114) for the low and high dose groups, respectively (table 3⇓). We saw no evidence for a treatment by time interaction (global testing, likelihood ratio test P=0.759 for FAS population; P=0.493 for per protocol sample), indicating no significant differences in attack rates across the treatment groups.
Primary efficacy analysis on full analysis set, plus use of two varying definitions of Meniere’s attacks as supportive efficacy analyses
During the 90 day assessment period, the population based, mean attack rate per 30 days was on average 2.722 attacks (95% confidence interval 1.304 to 6.309), 3.204 (1.345 to 7.929), and 3.258 (1.685 to 7.266) for the placebo, low dose betahistine, and high dose betahistine groups, respectively (table 4⇓).
Marginal mean attack rates per month over study assessment periods for each treatment group (FAS population)
In figure 3⇓, the upper panels show the observed individual profiles of monthly incidences of attacks caused by Meniere’s disease during the nine month treatment period, stratified by treatment group. The lower panels show the estimated individual incidences per month over time.

Fig 3 Profile plots of observed and estimated incidence of vertigo attacks caused by Meniere’s disease. Upper panels: Observed individual trajectories of monthly incidence of attacks over nine month treatment period (divided into nine 30 day intervals). Lower panels: Estimated individual trajectories of incidence of attacks per month depending on fixed and random effects after fitting an NB GLMM (that is, conditional estimates resulting from the longitudinal model used for the primary efficacy analysis). Thick solid lines in lower panels (indicated by arrows)=smoothing lines with standard error bounds. Ten patients (n=5 placebo; n=2 low dose betahistine; n=3 high dose betahistine) submitted no diary for the entire study period for various reasons (no specific reasons (n=1), loss to follow-up (n=3), informed consent withdrawn (n=4), analysis dropout due to adverse events (n=2))
{kind=link}
For all 221 patients randomised, a total of 5003 episodes of vertigo were evaluated according to the consensus document on the basis of the raw diary entries. Of these evaluated vertigo episodes, 1833 (37%) could be classified at least as an attack of postural vertigo (P attack); 2633 (53%) were classified as an attack of rotatory vertigo (R attack) and were interpreted as the most severe type of vertigo attack. Furthermore, 4229 (85%) evaluated episodes of vertigo were documented as rotatory, postural, or both (RP attack). Only 237 (5%) episodes of vertigo were characterised as both an R and P attack.
Table 3⇑ displays the estimated rate ratios if two alternative definitions of a Meniere’s disease attack were considered for statistical analysis. Notably, these supportive efficacy analyses demonstrated the robustness of the key results with respect to the definition of the primary endpoint. If either RP attacks or R attacks of vertigo were considered for model based primary analysis (by leaving out episodes of vertigo which were classified as gait unsteadiness or lightheadedness), these retrospective analyses reflected no betahistine effect in a consistent way.
The primary analysis considered time courses for each patient in a longitudinal manner, taking into account patients who did not provide attack information for the three month assessment period at the end of the treatment period. To check whether early analysis dropouts affected the main efficacy results, we did a preplanned sensitivity analysis to calculate attack rates across months seven, eight, and nine by taking into account only patients who provided attack information within the 90 day assessment period.
The results of the generalised linear model approach confirmed the robustness of the longitudinal model applied for primary efficacy analysis (table 3⇑). For the FAS population, the estimated mean attack rate per 30 days was 2.360 (95% confidence interval 1.581 to 3.712), 1.996 (1.321 to 3.016), and 2.094 (1.370 to 3.200) for the placebo, low dose betahistine, and high dose betahistine groups, respectively. We saw no evidence for a difference in attack rates between the three treatment groups for the FAS population as well as for the per protocol set (global likelihood ratio test, FAS P=0.850; per protocol P=0.808).
Exploratory adjusted efficacy analyses
A preplanned additional analysis to investigate centre effects also yielded no significant treatment by time interaction (P>0.100) for the number of attacks per 30 days. Pooling of sites within the catchment area of the DSGZ in Munich, which recruited about 40% of all randomised patients, showed no evidence of a centre effect (P=0.542, global likelihood ratio test comparing the main model with the adjusted one (pooled pseudosite Munich yes v no)). The overall decline of attacks over time in the three treatment groups was not significantly affected by whether a patient was recruited in a study centre outside of Munich or not. This finding was confirmed when pooling small investigator sites with fewer than 15 randomised patients (P=0.080, global likelihood ratio test).
Adjustment for sex effect did not significantly improve the model used for the primary efficacy analysis (P=0.202, global likelihood ratio test). Hence, sex did not affect the time course of Meniere’s attacks, nor did it affect the decline in attack rates. The main result of no treatment induced changes in attack rates was therefore confirmed.
A second prespecified adjusted analysis explored whether estimated treatment effects varied significantly between age subcategories of trial participants. However, adjustment for age (using age categories as defined in web appendix 2) did not significantly improve the model used for primary efficacy analysis (P=0.771, global likelihood ratio test). This finding indicated that age did not affect the decline in attack rates as was seen in the model used for the primary efficacy analysis.
Attack duration and severity
The duration and severity of an attack for those patients with at least one evaluated attack of Meniere’s disease within the assessment period was analysed by a cumulative logit modelling approach to compare ordinal duration and severity data across the study treatment groups. We wanted to examine whether the percentages of patients with attacks of a longer duration and a higher severity, respectively, were reduced by the assigned treatment. For the FAS population as well as the per protocol sample, the percentages of patients with longlasting attacks or more severe attacks did not significantly differ across treatment groups (duration: P=0.348 (FAS), P=0.515 (per protocol); severity: P=0.390 (FAS), P=0.438 (per protocol)). The data showed that the experimental treatment with low dose or high dose betahistine did not lead to higher probabilities of attacks in the low categories of duration and severity, respectively, as compared with placebo (table 5⇓).
Cumulative logit model for ordinal secondary efficacy outcomes (attack duration and severity), assessed over months seven to nine (FAS population)
Patient questionnaires, and vestibular and audiological parameters
The three tinnitus related or vertigo specific quality of life scores remained fairly stable at the end of the treatment period compared with the baseline scores. Table 6⇓ displays the results of the ANCOVA applying multiple imputation techniques to account for missing outcome data. The analysis found no evidence for between treatment differences in mean change scores. Web appendix 2 provides results of the complete case analyses. With regards to changes in vestibular and audiological function, no therapeutic gain of drug treatment was found. The efficacy of placebo treatment in tinnitus intensity, peak slow phase velocity during caloric irrigation with water at 30°C and 44°C, and pure tone audiometrically assessed hearing level was not significantly better than the efficacy of either betahistine dose. P values of more than 0.05 were generated by a global F test for the FAS population as well as the per protocol sample, with ANCOVA for absolute change values applied.
Analysis of absolute change from baseline at month nine for secondary efficacy outcomes, by ANCOVA (FAS population)
Clinical safety events
Table 7⇓ summarises adverse events deemed clinically important. The majority of patients (>85%) in the safety set reported one or more treatment emergent adverse events (TEAEs) within the nine month treatment period, with no clinically relevant difference between the three treatment groups. The most commonly reported TEAEs were headache, balance disorder, nausea, nasopharyngitis, feeling hot, eye irritation, and palpitations. Balance disorder and nausea were more commonly reported in the betahistine groups than in placebo. Eye irritation and palpitations were more commonly reported with high dose betahistine than with low dose betahistine and placebo. Differences were, however, small and probably not clinically relevant.
Safety assessment (safety sample), by study treatment group. Frequency of clinically important adverse events occurring in nine month treatment period (plus post-treatment adverse events occurring within a three week gap period)
Between 54% and 64% of patients in each treatment group had one or more TEAEs that were considered to be treatment related by the investigator: most of these were reported with low dose betahistine treatment (table 7⇑). Most TEAEs were of mild or moderate intensity. TEAEs of severe intensity were reported for 20 (27%), 20 (28%), and 19 (26%) patients in the placebo, low dose betahistine, and high dose betahistine groups, respectively. The only adverse event of severe intensity reported by more than 5% of patients in any treatment group was headache. There were no deaths during the study.
Forty treatment emergent serious adverse events (TESAEs) were reported for 15%, 14%, and 14% of patients in the placebo, low dose betahistine, and high dose betahistine groups, respectively. TESAEs reported by more than one patient during the study were vertigo (4.1% of patients in the placebo and high dose betahistine groups) and inguinal hernia and intervertebral disc protrusion (both 2.8% of patients in the low dose betahistine group). These events were all considered to be unrelated to study treatment. We saw a higher incidence of drug discontinuations due to adverse events in the high dose betahistine group (15% v 7% (placebo) and 6% (low dose betahistine)). The most commonly reported adverse events leading to drug discontinuation were tinnitus, vertigo, ear discomfort, and nervous system disorders, which were all more commonly reported with high dose betahistine than with low-dose betahistine treatment and placebo.
Discussion
Principal findings
For patients with Meniere’s disease, unpredictable vertigo attacks are the most unpleasant symptom, leading to not just physical but also psychological strain. Clinical experience and several studies have supported a potential beneficial effect of prophylactic drug treatment with betahistine on the attacks of vertigo as well as on vestibular and, to a lesser degree, audiological symptoms.34 However, according to a Cochrane review of betahistine for Meniere’s disease or Meniere’s syndrome,33 there is insufficient evidence to say whether betahistine has any effect.
The key findings of the BEMED trial are as follows:
• A significant decline of attack rates in each treatment arm was observed over the nine month treatment period
• The effects of two different doses of betahistine could not be distinguished from a patient reported effect caused by placebo intervention in terms of the incidence of attacks as well as vestibular and audiological function and quality of life. Therefore, the results do not give clear evidence that patients have a relevant clinical reduction in the number of attacks after nine months of treatment with betahistine at a daily dose of 48 mg or 144 mg, compared with a placebo (sham) intervention
• There were no safety concerns, and betahistine was well tolerated even in the high dose group of 144 mg betahistine per day.
Results in context
Meniere’s disease is a disorder with interindividual differences in a complex mixture of specific symptoms represented by vertigo attacks, hearing loss, tinnitus, pressure on the affected ear, and accompanying symptoms such as nausea or vomiting. The clinical course of the disease is cyclical and unpredictable.59 Furthermore, knowledge about the natural history and the underlying progression of episodes of vertigo in the long term is limited so far. The spectrum of symptoms tends to reflect the stage of the disorder. Some patients develop bilateral disease and non-relapsing symptoms. Variability also exists in the length of time required before symptoms improve. Perez-Garrigues and colleagues60 provide data that even without therapeutic intervention, the vertigo spells subside with time as vestibular function burns out. It might be the case that for some BEMED trial participants, a degree of compensation had already occurred.
Separation of the effect of treatment from the cyclical natural history of the disorder poses difficulties for all studies of Meniere’s disease. Because the natural history is one of remission and recurrence, and because participants must have active vertigo to enrol in a study, spontaneous improvement through regression to the mean in terms of symptom frequency and severity is expected, creating the illusion of a therapeutic efficacy.61 62 Thus, a control group is vital to contrast the long term treatment effect against spontaneous improvement.
The possibility of a patient experiencing an episode free year increases as the disease progresses.60 Therefore, assessment of the efficacy of treatments for Meniere’s disease needs a randomised approach, including a placebo or no treatment (wait and see) control group. Following the concept of Perez-Garrigues and colleagues,60 the BEMED population consisted of patients at different stages of Meniere’s disease, which might be reflected by individual baseline rates as well as individual time slopes for decay rate of attacks (as displayed in the upper panels of fig 3⇑). This consideration also influenced the choice of our statistical model.
The BEMED trial is, to our knowledge, the first pragmatic randomised controlled trial that specifically focuses on how betahistine prevents attacks caused by Meniere’s disease, taking into account different types of vertigo. It was designed as an investigator initiated, prospective, longitudinal, multicentre, double blind, randomised, placebo controlled, three arm, parallel group, phase III superiority trial. It specifically assessed the frequency, duration, and severity of acute attacks caused by Meniere’s disease during a nine month treatment period. As secondary endpoints, the trial also studied the treatment effect on vertigo related impairment in quality of life as well as on vestibular and audiological function. The BEMED trial also ascertains the speed of effect—that is, whether the two active doses can be distinguished from each other or from placebo by how quickly reduction in attack frequency is achieved.38 A series of sensitivity analyses supported the consistency and robustness of the BEMED efficacy results.
Studies that support a beneficial effect of betahistine on Meniere’s disease have been mostly observational. On the one hand, non-randomised studies tend to show larger treatment effects compared to randomised controlled trials, and tend to overestimate the magnitude of a potential treatment effect.63 On the other hand, there is the question of whether bias alone can explain the large effect differences between observational and experimental studies. There could also be a problem of external validity for the trial under consideration. Below, we reconsider these aspects using the PICO approach (patient, intervention, comparison, outcome) to discuss the strengths and limitations of the BEMED trial.
Strengths and limitations
Patient population
The BEMED trial population (n=221) was selected from 1450 screened patients. Patients were diagnosed with definite Meniere’s disease according to the criteria of the 1995 AAO-HNS guideline39 and the new schema for diagnosis of Meniere’s disease previously ratified by the Bárány Society,64 which are widely accepted and provide sufficient diagnostic accuracy.33 The mean monthly attack rate during the first month of treatment was about 5.7, which is considered as representative for patients with Meniere’s disease and being treated with betahistine. The population could have also included patients with vestibular migraine, benign paroxysmal positional vertigo, and secondary functional dizziness, which is typical for many Meniere’s disease studies.65
Intervention
The duration of exposure to study treatment was similar across the three treatment groups and ranged between a mean of 214 and 224 days for the intention to treat population. About 75% of patients completed the nine month treatment period.
Control group
The BEMED trial decided to implement a placebo arm for ethical and compliance reasons. Our placebo results may not fully reflect Meniere’s disease’s natural history.
Outcome
Electing the “(number of) Meniere’s attacks in a given time period” as the efficacy endpoint, documented by diaries in the patients’ natural environment (PRO), runs the risk of having missing or inaccurate information compared with objective measurements such as audiogram or questionnaires. In previous trials, the frequency of vertigo spells was mainly documented by a symptom report card using a Likert scale of 0 (“no vertigo”) to 4 (“worst vertigo attack ever”) to characterise a vertigo symptom and to perform a vertigo control categorisation as a simple and convenient summary statistic of a patient’s vertigo experience.15
The BEMED trial used a more complex vertigo symptom diary as an instrument to enable the patient to differentiate between several types of vertigo feelings to consider the multifaceted nature of symptoms in Meniere’s disease. To establish efficacy or effectiveness from a patient’s perspective, there are no reasonable alternatives to patient diaries that might be superior to alternative PROs, such as self-assessment scales or disease specific and validated quality of life scores that reflect fluctuations of disease severity over time. Derivation of definite or probable Meniere’s attacks based on the original daily patient recordings obtained by paper diaries is methodologically challenging.
Other studies have used quality of life scores, functional impairment, and disability instruments. In the BEMED trial, these PROs were implemented as secondary endpoints. A wide spectrum of efficacy endpoints is needed to measure any treatment related effect, because it is not known how the complex symptom clusters are modified by the treatment.
Generalisability to other populations
These results are valid for patients with definite unilateral or bilateral Meniere’s disease who had at least two monthly vertigo attacks in the three months before enrolment and who would receive betahistine as first line treatment, irrespective of whether they had received betahistine before. The results might not hold for patients with other vestibular disorders or taking higher doses of betahistine.
Preliminary implications and recommendations for clinical practice, and future research
We presented the primary results of the BEMED trial and articulated open questions that might guide future studies on treatment options in Meniere’s disease (for example, planning figures for sample size calculation). Several aspects of our design and experiences during the trial might also be relevant for clinical trials of other vertigo diseases that cause recurring attacks of spontaneous vertigo, such as vestibular migraine or vestibular paroxysmia, as well as for treatment and prevention of acute episodes of vertigo.
Further long term randomised, placebo controlled trials with higher betahistine doses than examined in this trial could be considered to confirm or disprove our findings and explore the potential prophylactic capacities of betahistine for Meniere’s disease. Clinical research should also focus more specifically on identifying predictors for betahistine treatment success, to broaden knowledge of this challenging field, and ultimately improve patients’ quality of life. Additionally, reliable and valid instruments should be developed to assess self-reported vertigo symptoms (in particular, vertigo attacks associated with Meniere’s disease). The definition of definite or probable Meniere’s attacks based on the raw patient recordings documented by paper diaries requires the development of prespecified rules for outcome derivation, and specific approaches to handle such complex PRO data.
What is already known on this topic
Acute vertigo attacks caused by Meniere’s disease greatly affect patients’ quality of life and perceived wellbeing
The disease’s natural history is one of remission and recurrence; because participants must first have active vertigo to enrol in a study, spontaneous improvement through regression to the mean is expected
Observational studies or low quality randomised controlled trials of low and moderate betahistine doses have produced contradictory results on treatment efficacy, and have not investigated the effect of an experimental intervention from the patient’s perspective with respect to vertigo attack prophylaxis
What this study adds
Long term prophylactic treatment with betahistine dihydrochloride (at daily doses 2×24 mg or 3×48 mg) does not change the time course of vertigo episodes related to Meniere’s disease compared with placebo
Placebo intervention as well as betahistine treatment showed the same reduction of attack rates over the study’s nine month treatment period
Reliable and valid instruments that measure subjective vertigo symptoms (in particular, vertigo attacks caused by Meniere’s disease) are lacking; derivation of definite or probable attacks caused by Meniere’s disease, on the basis of raw patient recordings in vertigo diaries, is methodologically challenging and requires prespecified rules
Footnotes
We thank Sheila Knoche (Department of Neurology and DSGZ, University Hospital Munich, Campus Grosshadern, Ludwig-Maximilians-University) for her assistance in carrying out the trial, and Katie Ogston for copyediting the manuscript; we also thank all patients included in the present study and their families, the study centres, and investigators who took part in this trial. Descriptive safety analyses were supported by Abbott Healthcare Products BV, Established Pharmaceuticals Division, The Netherlands, which was involved neither in the design and conduct of the study, collection, management, statistical efficacy analyses, and interpretation of the data, nor in the preparation and approval of the manuscript. This manuscript is part of the doctoral thesis of Christine Adrion.
This article is published on behalf of the BEMED investigators, who are listed in full with their affiliations in web appendix 2. Collaborators on behalf of the of BEMED (medical treatment of Meniere’s disease with betahistine) study group: Michael Strupp, Carolin Simone Fischer, Eike Krause, Robert Gürkov, Sabrina Holzapfel, Martin Westhofen, Thomas Lempert, Hubert Löwenheim, Michael v Brevern, Thomas Lenarz, Hans-Christoph Diener, Hermann Hilber, Ines Repik, Türker Basel, Daniel Weiß, Dirk Beutner, and Holger A Rambold.
Contributors: CA and CSF have shared first authorship. MS, JW, CSF, CA, and UM contributed to the design of the trial, wrote the study protocol and subsequent amendments, and interpreted the work. As coordinating investigator, MS initiated the collaborative clinical trial project and is the guarantor. MS, JW, CSF, RG, and all investigators of the 14 study sites acquired the data. RG was responsible for the assessment of the audiometry and tinnitus intensity of the patients recruited at the site of the coordinating investigator and contributed to the interpretation of the audiological data. CA and UM wrote the statistical analysis plan, performed the statistical analyses, interpreted the results, wrote the first draft, and substantially contributed to the writing of all subsequent versions of the manuscript. CSF and CA evaluated the original patient ratings provided by paper based vertigo diaries. All authors revised the work critically for important intellectual content, approved submission of the final report for publication, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding: This study was not industry sponsored. The study was supported by grants from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung (BMBF), support code 01KG0708; sponsor’s protocol code no 04T-617). This work was supported by the German Centre for Vertigo and Balance Disorders (DSGZ), University Hospital Munich, Campus Grosshadern, Munich, Germany. The funder had no role in the design, management, data collection, analyses, or interpretation of the data or in the writing of the manuscript or the decision to submit for publication.
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: support from the German Federal Ministry of Education and Research and the German Centre for Vertigo and Balance Disorders for the submitted work; MS is joint chief editor of the Journal of Neurology, editor in chief of Frontiers of Neuro-otology, and section editor of F1000; MS has received speaker honorariums from Abbott, Actelion, UCB SA Belgium, GlaxoSmithKline, TEVA GmbH, Biogen, Pierre Fabre, Eisai GmbH, MSD Sharp & Dohme, and Hennig Pharma; MS has worked as a consultant for Abbott; CSF has received travel grants from Abbott and Pierre Fabre; RG reports grants from the German Federal Ministry of Education and Research and the Volkswagen Foundation, and non-financial support from Interacoustics, outside the submitted work; the other authors declare no interests. According to a contract approved by the German Federal Ministry of Education and Research (BMBF/DLR), University of Munich, and University Hospital of Munich, Abbott had access to the data after the study and statistical analyses were completed in order to use the data for approval of betahistine for the treatment of Meniere’s disease. Appropriate financial compensation was paid for this service, which was approved by the DLR, University of Munich, and University Hospital of Munich. Abbott did not have any influence on the analyses or interpretation of the data or on the content or form of the manuscript.
Ethical approval: All participants provided written informed consent before initiation of the first study specific procedure, according to the procedures approved by the ethics committee of the University of Hospital Munich. The protocol was approved by local independent ethics committees and was performed in accordance with the Declaration of Helsinki and other applicable guidelines, laws, and regulations.
Data sharing: Data to reproduce the intention to treat analyses of the primary and secondary efficacy endpoints, together with the used R code are available from the corresponding author (Michael.Strupp@med.uni-muenchen.de) or biostatistician (mansmann@ibe.med.uni-muenchen.de). Informed consent for data sharing was not obtained from participants, but the data presented are anonymised and the risk of identification is very low.
The guarantor affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned and registered have been explained.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/.