Article Text

Abstract
Introduction For resectable soft tissue sarcoma (STS), radical surgery, usually combined with radiotherapy, is the mainstay of treatment and the only potentially curative modality. Since surgery is often complicated by large tumour size and extensive tumour vasculature, preoperative treatment strategies with the aim of devitalising the tumour are being explored. One option is treatment with antiangiogenic drugs. The multikinase inhibitor pazopanib, which possesses pronounced antiangiogenic effects, has shown activity in metastatic and unresectable STS, but has so far not been tested in the preoperative setting.
Methods and analysis This open-label, multicentre phase II window-of-opportunity trial assesses pazopanib as preoperative treatment of resectable STS. Participants receive a 21-day course of pazopanib 800 mg daily during wait time for surgery. Major eligibility criteria are resectable, high-risk adult STS of any location, or metachronous solitary STS metastasis for which resection is planned, and adequate organ function and performance status. The trial uses an exact single-stage design. The primary end point is metabolic response rate (MRR), that is, the proportion of patients with >50% reduction of the mean standardised uptake value (SUVmean) in post-treatment compared to pre-treatment fluorodeoxyglucose positron emission tomography CT. The MRR below which the treatment is considered ineffective is 0.2. The MRR above which the treatment warrants further exploration is 0.4. With a type I error of 5% and a power of 80%, the sample size is 35 evaluable patients, with 12 or more responders as threshold. Main secondary end points are histopathological and MRI response, resectability, toxicity, recurrence-free and overall survival. In a translational substudy, endothelial progenitor cells and vascular epithelial growth factor receptor are analysed as potential prognostic and predictive markers.
Ethics and dissemination Approval by the ethics committee II, University of Heidelberg, Germany (2012-019F-MA), German Federal Institute for Drugs and Medical Devices (61-3910-4038155) and German Federal Institute for Radiation Protection (Z5-22463/2-2012-007).
Trial registration number NCT01543802, EudraCT: 2011-003745-18; Pre-results.
- Sarcoma < ONCOLOGY
- Clinical trials < THERAPEUTICS
- NUCLEAR MEDICINE
- VASCULAR MEDICINE
- Surgical pathology < PATHOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Sarcoma < ONCOLOGY
- Clinical trials < THERAPEUTICS
- NUCLEAR MEDICINE
- VASCULAR MEDICINE
- Surgical pathology < PATHOLOGY
Strengths and limitations of this study
One of the first trials to evaluate pazopanib, a multikinase inhibitor with pronounced antiangiogenic properties, in neoadjuvant treatment of soft tissue sarcoma, using a window-of-opportunity design to minimise potential risks for patients, and yet to provide valid information on efficacy and safety.
Multimodal response assessment: dynamic fluorodeoxyglucose positron emission tomography CT (primary outcome), dedicated MRI protocol, specific protocol for histopathological assessment, has been used.
Translational substudy exploring the role of circulating endothelial progenitor cells and soluble vascular endothelial growth factor receptor as potential biomarkers for antiangiogenic treatment of soft tissue sarcoma.
Heterogeneous study population in terms of histological sarcoma subtype and clinical setting (primary tumours and solitary recurrences/metastases).
Timing of metabolic response assessment is several days after discontinuation of therapy, ‘rebound growth’ is therefore possible; however, metabolic response is assessed close to the time of surgery, which is the clinically most relevant time point; surgery takes place at least 7 days after treatment discontinuation to minimise risk of surgical complications.
Introduction
Soft tissue sarcomas (STS) constitute a heterogeneous group of malignant mesenchymal tumours with varying histological differentiation. High-grade, deeply located, large STS have a poor outcome. For both, primary and recurrent tumours, radical surgery, usually combined with postoperative radiotherapy, is the mainstay of treatment and the only potentially curative modality.1 Likewise, oligometastatic disease with one or few manifestations in one single organ is treated surgically in selected patients with the aim of prolonging survival and reducing symptom burden. Surgery is, however, often hampered by large tumour size with infiltration of adjacent structures, and extensive vasculature of the tumour.
For primary and recurrent tumours, various preoperative treatment strategies have been tested in clinical trials.2 Preoperative doxorubicin/ifosfamide has not been shown to yield any benefit in overall or progression-free survival in a randomised trial when compared with surgery alone.3 The addition of regional hyperthermia to preoperative chemotherapy has improved local progression-free and disease-free survival.4 Regarding preoperative cytotoxic chemotherapy, additional concerns exist because the administration of drugs with a lifetime dose limit, such as anthracyclines, in a non-metastatic setting, might narrow future treatment possibilities in case of recurrence. Preoperative radiation has been shown to have a slight survival benefit compared to postoperative radiation, but the latter is often preferred because of a lower rate of wound complications.5 Isolated limb perfusion is an option for selected patients but requires a high logistic effort and is limited to referral centres.2 In conclusion, there is still no consensus if and which preoperative treatment should be applied in patients with STS,6 ,7 and further modalities are to be tested in clinical trials.
The ideal preoperative treatment for STS would be fast-acting and effective in terms of devitalisation of the tumour and disruption of its hypervasculature, thus facilitating resection. It would have a low incidence of side effects, thus not hampering surgery and postsurgery recovery. In addition, a valid possibility of early response assessment would be highly desirable, since this could spare non-responders from ineffective treatment and a potentially harmful delay of surgery.
The multikinase inhibitor pazopanib has been approved as treatment for metastatic or non-resectable STS based on the results of a phase III trial that compared pazopanib treatment to placebo in patients with metastatic non-adipocytic STS who were angiogenesis inhibitor naïve and had progressed on at least one prior chemotherapy regimen.8 Patients in the pazopanib arm had significantly longer progression-free survival (median: 20 vs 7 weeks; HR=0.31, 95% CI 0.24 to 0.40), which was the primary end point of the trial. There was no significant difference in overall survival (12.5 months with pazopanib vs 10.7 months with placebo; HR=0.86, 95% CI 0.67 to 1.11), but the trial was not powered for this secondary end point. In the heavily pretreated trial population, treatment was sufficiently well tolerated, with a median dose intensity of 96.3% and 14% of toxicity-related treatment interruptions in the pazopanib group. The most frequent adverse events (AEs) were fatigue, hypertension, anorexia and diarrhoea. The trial was restricted to patients with non-adipocytic STS because of the previous phase II trial, which showed no activity in the stratum of adipocytic STS.9 Subsequent central pathology review, however, re-classified two patients with stable disease (internal communication by the trial sponsor). Based on this finding, activity of pazopanib against liposarcoma is probable.
Given this proof of efficacy, its fast and pronounced antiangiogenic effects mediated by kinase inhibition,10 and its favourable safety profile,11 pazopanib might be an ideal candidate for neoadjuvant treatment in STS. Therefore, we decided to conduct a pertinent phase II trial as a ‘window of opportunity’ study.12 ,13 In this design, patients receive an investigational agent in a ‘window period’ before starting the established treatment. The risk that effective treatment is unduly delayed is minimised by keeping the duration of the investigational treatment short. The described window-of-opportunity design is feasible for pazopanib, as shown in a trial for non-small cell lung cancer.14
A crucial precondition for an effective use of preoperative treatment is good and early response monitoring. Dynamic F-18 labelled fluorodeoxyglucose positron emission tomography and CT (dFDG-PET-CT) appears to be ideal for this purpose. It has been prospectively validated as a strong predictor of histopathological response and progression-free survival, for both neoadjuvant and palliative chemotherapy in patients with STS.15–18 Unlike histopathological response assessment, which can only be performed retrospectively after surgery, dFDG-PET-CT allows for early selection of patients who may benefit from preoperative therapy. MRI is another viable option for early response assessment.19 ,20 However, it has not yet been validated as a predictor of progression-free survival. Therefore, up to now, dFDG-PET-CT can be deemed the best available modality for early prospective response assessment of chemotherapy for STS.
Another reason for conducting a neoadjuvant study in patients with STS is that it can provide information on biomarkers for response prediction. Ultimately, such markers would allow distinguishing responders from non-responders, thus facilitating true ‘targeted therapy’. In the context of antiangiogenic therapy, there are several circulating angiogenic factors that might serve this purpose. Circulating endothelial progenitor cells (cEPCs) and soluble vascular epithelial growth factor (sVEGF) correlate with tumour burden in STS,21 and might thus serve as early predictors of clinical and histological response.
We present the trial protocol. The trial was initiated in April 2013, and the first patient recruited in May 2013. At present, the trial is ongoing.
Methods and analysis
Study objectives
The primary objective of the study is to evaluate whether neoadjuvant treatment with pazopanib in patients with STS has therapeutic effects, measured as metabolic response (MR). The secondary study objectives are:
To assess the safety of preoperative pazopanib treatment in patients undergoing resection of STS.
To evaluate the potential correlation between metabolic (dPET-CT), radiological (MRI) and histopathological assessment of tumour response to pazopanib treatment in STS.
To provide exploratory data on blood levels of cEPCs and sVEGF as potential predictive biomarkers during pazopanib treatment of STS.
Study outline
This is a multicentre (three recruiting centres and one centre for PET-CT), single-arm, open-label phase II trial under the auspices of the German Interdisciplinary Sarcoma Group (GISG). Pazopanib is administered for 21 days followed by resection of the tumour after a break of 7–14 days (figure 1).
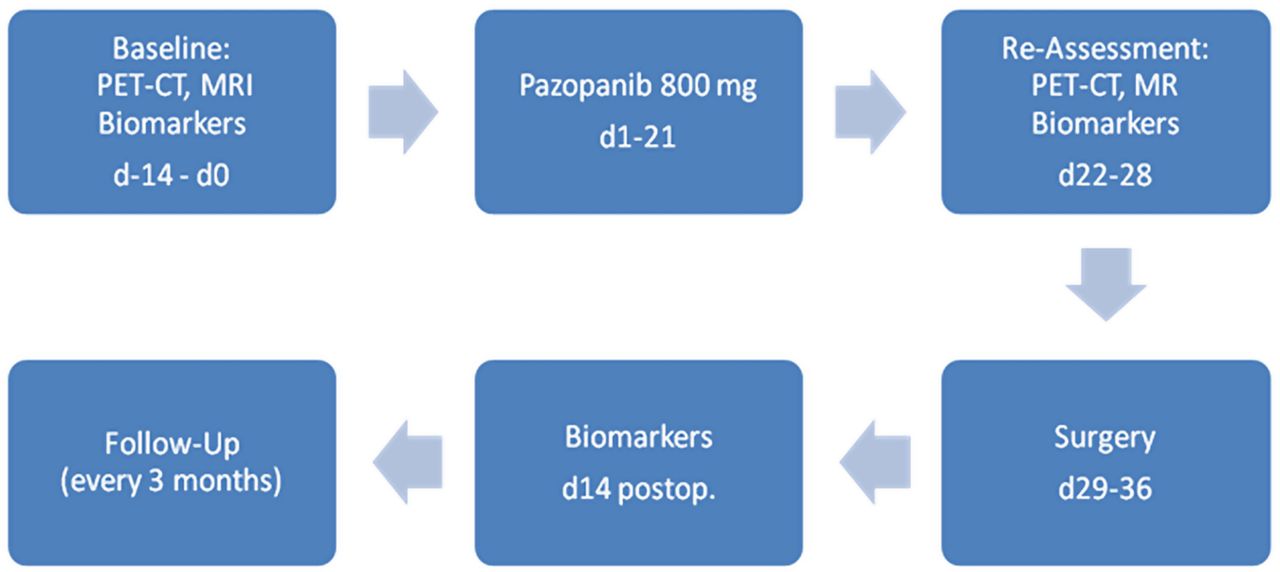
{kind=link}
Study flow chart (d, day; PET, positron emission tomography).
Patient selection
The study population should consist of patients with STS for which efficacy of pazopanib is assumed, and with adequate organ function and performance status to tolerate the treatment. This led to the inclusion and exclusion criteria listed in box 1.
Major inclusion and exclusion criteria of the trial
Inclusion criteria
Non-metastatic primary tumour or locoregional recurrence of histologically confirmed high-risk (G2/3, diameter ≥5 cm) soft tissue sarcoma (STS) of any location; or metachronous solitary metastasis of STS for which surgical resection is planned according to the individual choice of the multidisciplinary treatment team (no grade or size restrictions apply for metastasis)
Resectable and solitary tumour, as assessed by the investigator based on staging examinations (CT scan of the chest, CT or MRI of the abdomen, MRI of the limb in case of extremity STS). Measurable disease according to RECIST 1.1
STS except the following subtypes: embryonal rhabdomyosarcoma, chondrosarcoma, osteosarcoma, Ewing tumour/primitive neuroectodermal tumour (PNET), gastrointestinal stromal tumours, dermofibromatosis sarcoma protuberans, inflammatory myofibroblastic sarcoma
Eastern Cooperative Oncology Group performance status of 0 or 1
Adequate organ system function
Effective contraception
Written informed consent
Age ≥18 years
Exclusion criteria
Prior malignancy other than the STS under study, with active disease within the last 5 years
History or clinical evidence of central nervous system metastases
Prior or concurrent systemic chemotherapy or molecularly targeted therapy or radiotherapy for STS or other malignancies within 5 years before study entry
Clinically significant gastrointestinal abnormalities that may increase the risk for gastrointestinal bleedingvClinically significant gastrointestinal abnormalities that may affect absorption of pazopanib
Corrected QT interval (QTc) >480 ms
Presence of uncontrolled infection
History of severe cardiovascular conditions within the past 6 months
Cardiac angioplasty or stenting
Poorly controlled hypertension (defined as systolic blood pressure of ≥140 mm Hg or diastolic blood pressure of ≥90 mm Hg)
Cerebrovascular accident including transient ischaemic attack, pulmonary embolism or untreated deep venous thrombosis within the past 6 months
Major surgery or trauma within 28 days prior to first dose of pazopanib
Evidence of active bleeding or bleeding diathesis
Known endobronchial lesions and/or lesions infiltrating major pulmonary vessels that increase the risk of pulmonary haemorrhage)
Any serious and/or unstable pre-existing medical, psychiatric or other condition that could interfere with patient's safety, provision of informed consent or compliance to study procedures
Any ongoing toxicity from prior anti-cancer therapy that is >grade 1 and/or that is progressing in severity, except alopecia
Treatment plan
All patients are routinely discussed in an institutional multidisciplinary sarcoma board, and a recommendation for the best available treatment and possible inclusion into the trial is jointly determined.
Pazopanib treatment
The treatment consists of the oral administration of 800 mg (two tablets of 400 mg or four tablets of 200 mg) pazopanib once daily. The treatment is administered for 21 days. Pazopanib should be taken orally without food at least 1 h before or 2 h after a meal. The tablets should be swallowed whole and must not be crushed or broken. The time of day the tablets are taken should be relatively constant. If a dose is missed, the patient should take the dose as soon as possible, but only if there are 12 or more hours remaining before the next dose is due. If the next dose is due in <12 h, the patient should skip the missed dose and take the next dose as scheduled. If vomiting occurs after taking pazopanib, the patient should not take a replacement dose on that day. The patient should resume taking pazopanib at the next scheduled dose on the following day. If vomiting persists, the patient should be instructed to notify the investigator. Pazopanib will be provided by the manufacturer. It should be stored at room temperature up to 25°C. When stored at these temperatures and in unopened bottles, pazopanib tablets will remain stable until the expiration date indicated on the bottle label. For detailed information on the administration of pazopanib, refer to the Investigator's Brochure for pazopanib.22
Given the short duration of 21 days of planned treatment, in case of drug toxicity only two dose reductions are permitted in a stepwise fashion: initially to 600 mg once daily, and subsequently to 400 mg once daily if necessary. Given the short treatment period, no dose re-escalation is foreseen. Recommendations for investigational product dose interruptions/modifications in case of specific treatment-emergent AEs are provided in the online supplementary material.
In each individual patient, pazopanib treatment should be discontinued if one of the following events occurs:
The patient withdraws consent to the study;
The investigator is of the opinion that continuation of treatment would jeopardise the health status of the patient;
A Serious Adverse Drug Reaction (SADR);
A specific AE requiring treatment discontinuation as defined in the online supplementary material;
In female patients: pregnancy;
Severe non-compliance by the patient that jeopardises validity of the data in a relevant way.
Surgery
Surgery should be performed 7–14 days after the end of the study treatment. This interval corresponds to at least five half lives of pazopanib. It was chosen in order to minimise the potential risk of surgical complications (mainly wound or anastomotic complications). Evidence from patients receiving anti-VEGFR treatment with bevacizumab suggests an elevated risk that is inversely correlated to the time interval between treatment discontinuation and surgery.23 Surgery itself is not part of the study protocol and is performed according to the discretion of the treating surgeon. Although not mandatory, for reasons of quality assurance regarding pathological assessment, it is strongly recommended that surgery be performed at one of the study centres.
Postoperative treatment/radiotherapy
A recommendation for possible postoperative treatment is, again, jointly determined in an institutional multidisciplinary sarcoma board. If adjuvant radiotherapy is carried out, it is not part of the study protocol and can be performed according to the choice of the treating physician. However, for patients participating in the translational study, it should not start before blood samples are taken (14 days after surgery) to determine postoperative cEPC/sVEGF levels.
Evaluation, laboratory tests and follow-up
A signed, written informed consent form must be obtained from the patient prior to any study-specific procedures or assessments. Procedures conducted as part of the patient's routine clinical management, and obtained prior to signing of informed consent, may be utsed for screening or baseline purposes provided these procedures are conducted as specified in the protocol. The study assessment schedules and visit windows are summarised in table 1. A certified laboratory is to conduct all clinical laboratory assessments. Assessment at laboratories other than the site laboratory (eg, at a laboratory collaborating with the patient's general practitioner) is possible. Laboratory assessments should be performed as indicated in table 2. All laboratory tests with values that become abnormal and clinically significant while the patient is participating in the study or within 28 days after the last dose of study drug should be repeated until the values return to normal or baseline.
Time and events table
Clinical laboratory assessments
Study evaluation
FDG-dPET-CT
At the time points, days −14–0 and 22–28, patients will undergo dPET-CT with FDG over the tumour area followed by a baseline whole-body PET-CT. All examinations will be performed centrally at the same PET-CT scanner. A topogram and a low-dose CT are used for positioning of the patient, to include the primary volume of interest (VOI) for the dPET-CT. Following the intravenous application of FDG, dynamic data acquisition (4D mode) is performed for 60 min. Then a whole-body PET-CT is acquired using 2 min per bed position. The iteratively reconstructed images are evaluated using dedicated software. Besides the SUV (standardised uptake values), further parameters of the tracer kinetics are assessed using compartment and non-compartment models. The SUVmean 60 min after the FDG injection is used to assess the therapeutic effect. Exploratory analyses of dynamic PET-CT response will be based on a VOI analysis. The following parameters of the FDG kinetics will be calculated: (1) SUV (mean, max) 55–60 min postinjection; (2) influx rates and transport rates based on a support vector machine algorithm and a two-tissue compartment analysis; and (3) calculation of the fractal dimension of the FDG kinetics based on fractal analysis. The percentage change of all parameters of the FDG kinetics as well as discriminant analysis based on the absolute values of the FDG kinetics and their changes will be determined along with CIs. In particular, the SUVmean will be used primarily for the assessment of response. MR is assumed with a change of 50% following pazopanib treatment (21 days). The dynamic parameters are used as secondary parameters for the assessment of treatment.
MRI
MRIs on study entry and on days 22–28 are optional according to local availability and should be performed as follows: 1.5 T scanner with phased-array coil will be used. T1-weighted (T1W) (two-dimensional (2D) or 3D) precontrast and postcontrast sequences with fat suppression will be undertaken. After localiser sequences (T2W HASTE sequences), T1W turbo Spin Echo (TSE) sequences with and T2W TSE without fat suppression precontrast, followed by an Echo planar imaging diffusion-weighted imaging (EPI DWI) sequence with four b-values (0, 50, 400, 800) and quantification of apparent diffusion coefficient (ADC) maps, will be performed. Afterwards, T1W fat-saturated gradient echo sequences in breathhold technique before, and 30, 60 and 90 s after, contrast media application (0.1 mmol/kg body weight (bw) gadolinium chelates), covering at least 20 cm of the tumour size, will be carried out. The matrix size should be at least 256, and the slice thickness should be 3 mm or less.
All quantitative analysis of dynamic and diffusion examinations as well as evaluation according to the RECIST and modified Choi criteria will be performed by an experienced board certified radiologist at the study centre in Mannheim. Data material will be provided for all included patients on CD or by electronic data export.
Evaluation of changes of tumour size during therapy will be performed in accordance with RECIST criteria version 1.124). Complete response (CR) is defined as the disappearance of all lesions. Partial response (PR) is defined as a decrease of at least 30% of the longest diameter of the tumour, taking as reference the longest baseline diameter; progressive disease (PD) is defined as an increase of at least 20% in the longest diameter of the tumour, taking as reference the baseline longest diameter, or the appearance of one or more new lesions; stable disease (SD) is defined as neither sufficient shrinkage to qualify for partial response nor sufficient increase to qualify for progressive disease, taking as reference the baseline longest diameter. For evaluation during therapy, partial response, stable disease and progressive disease will also be defined according to Choi criteria modified for MRI.19 Here, CR is defined as the disappearance of all lesions. PR is defined as ≥10% decrease in the longest maximal diameter or a ≥15% decrease in contrast enhancement; PD is defined as a ≥10% increase in the longest maximal diameter while the criteria for PR by using contrast enhancement are not met, or a ≥15% increase in contrast enhancement while criteria for partial response by using tumour size are not met, or the occurrence of one or more new lesions; SD is defined as all cases that do not meet criteria for CR, PR or PD.
If technically feasible at the local MRI unit, 2D and 3D measurements of the tumour will be performed. The ADC values of two reproducible tumour localisations as well as the plasma flow (PF) and mean transit time (MTT) of the tumour at the same localisations evaluated by Food and Drug Administration (FDA) approved Tissue 4D software or another adequate perfusion quantification tool will be documented and compared between measurements. MRI response will be quantified as the change in per cent between pretherapeutic and post-therapeutic ADC.20
During the follow-up period, all MRIs can be performed according to local clinical practice without adhering to a specific protocol.
Chest CT
For screening, all patients require a multislice chest CT with a slice thickness ≤5 mm (reconstruction interval) covering the whole area from the lung apices to the diaphragm. The use of intravenous contrast medium is recommended unless contraindicated. The images will be assessed by the responsible radiologist, according to local standards.
During the follow-up period (after surgery), all chest CTs can be performed according to local clinical practice without adhering to a specific protocol.
Histopathological procedures
The histopathological report should include the following information:
Tumour size in three dimensions;
Resection status (free margins, margins microscopically infiltrated, margins macroscopically infiltrated);
Smallest distance between resection margin and vital tumour tissue found in the specimen, together with the localisation where it was found;
Histological subtype;
Grading according to the FNCLCC system (G1–3);25
Overall regression grading after slicing of the specimen (in per cent, semiquantitative value);
Most prevalent type of regression: hyalinous necrosis, apoptosis, scar tissue, haemorrhagic necrosis.
Evaluation of efficacy
MR and MR rate
MR is defined as the achievement of at least 50% reduction of the SUVmean over the tumour area in the post-treatment compared to the pretreatment FDG-dPET-CT. The MR rate (MRR) is defined as the proportion of patients achieving an MR.
Other outcome measures
dPET-CT response is defined as the change of FDG influx as well as of transport rates k1–k4, and distribution volume (VB) and fractal dimension. Absolute values of all parameters of FDG kinetics will also be used for discriminant analysis evaluation.
Resection status is determined by the assessing pathologist in the respective report.
Recurrence-free survival is defined as the time from resection to the date of diagnosis of recurrence or date of death, or the last day when the patient was known to be disease free or alive, in case of loss to follow-up (censoring). The date of diagnosis of recurrence is defined as the first day when the below mentioned criteria for recurrence are met. In case of a diagnosis by imaging, the day when the imaging procedure was performed is recorded as the date of recurrence. In case of a clinical diagnosis, the date of the visit is recorded.
Local recurrence-free survival is defined as survival free from local recurrence, which in this context is defined as a newly occurring lesion within a range of 10 cm of the margin of the original tumour. The assessment if a recurrence is considered local or distant should be made by the investigator at the study centre based on the physical examination and the patient's file. Patients with R2 resection are automatically assigned a local recurrence-free survival of zero.
Distant recurrence-free survival is defined as survival free from any recurrence not fulfilling the criteria of local recurrence.
For both local and distant recurrence, prior death will be counted as a competing interest and not included. The date of diagnosis of local/distant recurrence is defined as the first day the criteria for recurrence are met. In case of a diagnosis by imaging, the day the imaging procedure was performed is recorded as the date of recurrence. In case of a clinical diagnosis, the date of the visit is recorded.
Overall survival is computed from the date of resection to the date of death (whatever the cause). Patients not known to be dead at the time of the analysis will be censored at the date of last follow-up.
Toxicity: All AEs will be recorded according to CTCAE, V.4.0. (http://evs.nci.nih.gov/ftp1/CTCAE_4.03_2010-06-14_QuickReference_8.5×11.pdf).
Delay in planned time to resection: Any delay occurring between the actual date of resection and the planned date of resection will be quantified by the treating surgeon, and categorised into ‘treatment related’ and ‘non treatment related’.
Study design, sample size and analysis plan
Study design and sample size
The trial is designed as a non-comparative single-arm phase II trial. Its primary end point is the MRR, defined as the proportion of patients achieving a 50% reduction in the SUVmean in the post-treatment compared to the pretreatment FDG-dPET-CT. The trial uses an exact single-stage design based on the exact binomial distribution.26 The MRR below which the treatment is considered ineffective is set at 0.2 (H0: MRR≤0.2). The MRR above which the treatment warrants further exploration in a subsequent phase III trial is set at 0.4 (H1: MRR≥0.4). This magnitude of response can be expected after preoperative therapy with a supposedly active agent for STS, as shown in several non-controlled trials.15 ,27–29 With a predefined significance level of 5% and a power of 80%, the sample size is calculated to be 35 evaluable patients with an actual significance level of 0.034 and an actual power of 0.805 with a critical value of 12 patients at the upper proportion limit (STPLAN, V.4.5, 2010, The University of Texas, Houston, Texas, USA). If, after treatment of 35 patients, the primary end point MRR cannot be assessed for one or more patients, for example, for technical or medical reasons, up to three additional patients can be enrolled.
Analysis plan
The final analysis of the primary end point MRR will be carried out after all patients have received their post-treatment FDG-PET-CT. All patients registered in the study will be included (intention-to-treat analysis). The number of patients who are not evaluable, or who died or withdrew before treatment began or during treatment, will be specified. According to the exact single-stage design based on the exact binomial distribution, H0 will be rejected if 12 or more patients show MR.26 If, at the end of treatment of all enrolled patients, the number of individuals who can be evaluated for MR is below 35, the border for acceptance of H1 will be modified accordingly with a type I error not exceeding 5%. The MRR will be presented as a percentage with a 95% CI. In addition, MR will be visualised as a waterfall plot with the change in per cent of SUVmean on the y axis.
At the time of analysis of the primary end point, the secondary end points MRI response, dynamic PET-CT response, histopathological response, R0-resectability and toxicity will also be analysed. MRI response will be quantified as the change in per cent between pretherapeutic and post-therapeutic ADC20 as well as the rates of complete response, partial response, stable disease and progressive disease according to Choi criteria modified for MRI and RECIST 1.1, all with 95% CIs. A potential correlation between MRI and MR will be evaluated by calculating Pearson's correlation coefficient for SUVmean change in per cent between the pretherapeutic and post-therapeutic PET-CT and the ADC change in per cent between the pretherapeutic and post-therapeutic MRI. For the categorical variables ‘response according to modified Choi and RECIST 1.1 criteria’, parameters of diagnostic accuracy (sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV)) for complete or partial response will be calculated relating to the primary outcome MR as ‘gold standard’.
Exploratory analyses of dPET-CT response will be based on a VOI analysis. The following parameters of the FDG kinetics will be calculated: (1) SUV (mean, max) 55–60 min postinfusion; (2) influx rates and transport rates based on a support vector machine algorithm and a two-tissue compartment analysis and (3) calculation of the fractal dimension of the FDG kinetics based on fractal analysis. Percentage change of all described parameters of the FDG kinetics as well as discriminant analysis based on the absolute values of the FDG kinetics and their changes will be presented along with CIs.
R0-resectability will be presented as proportion with corresponding 95% CI. Histopathological response will be displayed as the rate of responders with corresponding 95% CI and range as well as a waterfall plot with the percentage of viable tumour tissue on the y axis. Parameters of diagnostic accuracy for histopathological response will be calculated relating to the primary outcome MR as ‘gold standard’. The incidence of AEs will be presented for the safety population, in a descriptive analysis. Delay in planned time to resection due to the treatment will be reported in a descriptive way with appropriate summary measures. The analysis of the secondary end points overall survival, recurrence-free survival, and local and distant recurrence-free survival, will be performed when data are mature. Survival curves will be estimated with the Kaplan-Meier method and displayed graphically.
A first exploratory analysis of biomarker data (see below), that is, cEPC and sVEGF blood levels, will be carried out at the time of analysis of the primary end point. This will assess if the mentioned biomarkers have a predictive value for MR. For this purpose, pretreatment levels of cEPC and sVEGF will be plotted against the change in per cent of SUVmean in order to display a possible predictive relationship. If possible, linear regression will be performed. Likewise, a potential predictive value of cEPC and sVEGF level changes during treatment and MR will be explored by plotting the change in per cent of post-treatment levels (d22–28) compared to pre-treatment levels against the change in per cent of SUVmean. If possible, linear regression will be performed. cEPC and sVEGF levels measured at d14 postsurgery will be compared to levels measured after completion of pazopanib treatment (d22–28). The difference will be presented in a descriptive fashion (mean, median, SD), related to resection status, in an explorative way, and, if meaningful and feasible, formally compared with appropriate parametric or non-parametric tests between patients who received complete and incomplete resection. At the time of analysis of recurrence-free survival, prognostic properties of cEPC and sVEGF levels will be explored. Biomarker levels will be treated as continuous variables. If possible in a meaningful way, recurrence-free survival of patients in the different quartiles of pretreatment and postoperative cEPC and sVEGF levels will be compared by means of Kaplan-Meier curves. In addition, pretreatment and postoperative levels of recurred and recurrence-free patients will be compared with an appropriate parametric test.
Translational research
There is an increasing amount of evidence suggesting that cEPCs, in conjunction with soluble cytokines such as sVEGF, are involved in neoangiogenic processes of solid tumours.30 Studies suggest that the recruitment of cEPC, which is probably mediated through cytokines, is an expression of tumour-induced neoangiogenesis in situations of tumour growth. cEPCs have been shown to contribute to vasculogenesis and neoangiogenesis in several tumour entities.31 ,32 On the contrary, tumour response to chemotherapy or kinase inhibition seems also to be related with a rise in cEPC and sVEGF levels.30 ,33
Based on these preliminary findings, it seems probable that blood levels of cEPCs and sVEGF have a certain prognostic and/or predictive value for the course of disease and treatment of STS. We were able to show a positive association between clinical tumour load and cEPC level in the blood of patients with lung cancer34 and STS (unpublished data). Consequently, pretreatment cEPC levels might reflect tumour load and metabolism, and thus constitute a prognostic marker. The same might hold true for the postoperative cEPC level, which could be an indicator of residual microscopic tumour load and thus of recurrence risk. On the other hand, high cEPC pretreatment levels, indicating an extensive tumour vasculature and thus a high susceptibility to an antiangiogenic treatment, might be a predictive marker for response. The same might be true for a drop in cEPC level during treatment. Similar associations have been observed between sVEGF levels and treatment response.
In order to explore these hypotheses, the study foresees measurement of cEPC and cVEGF levels at various time points. Patients will be eligible for the translational research project if they are eligible for the clinical trial and have given written informed consent to participate in this project. Patients will have the option of accepting or refusing participation in the translational research project, or of accepting participation in only a part of the project, without affecting their participation in the clinical study.
During the specified visits, 20 mL of full blood will be drawn by insertion of a 20-gauge cannula in a peripheral vein and collected in tubes containing sodium citrate (0.105 M) as anticoagulant. In addition, 5 mL of serum will be collected through the same cannula and stored in an appropriate tube. Full blood samples will be processed within 1 h after collection.
Peripheral blood mononuclear cells (PBMCs) will be prepared by gradient centrifugation using Ficoll-Hypaque (Amersham Biosciences, Freiburg, Germany). The expression of cell surface antigens will be determined by four-colour immunofluorescence staining. From each sample, 100 μL of PBMC (containing 1×106 cells) will be incubated with 10 μL of FcR-blocking reagent (Miltenyi Biotec, Bergisch-Gladbach, Germany) for 10 min, to inhibit non-specific bindings. Thereafter, the cells will be incubated at 4°C for 30 min with 10 μL PE-conjugated antihuman CD133 mAb (Miltenyi Biotec, Bergisch-Gladbach, Germany), 10 μL PerCP-conjugated antihuman CD34 mAb (BD Biosciences, Heidelberg, Germany), 10 μL allophycocyanin (APC)-conjugated VEGF R2 mAb (R&D Systems, Wiesbaden-Nordenstadt, Germany) and 10 μL fluorescin isothiocyanate (FITC)-conjugated Annexin V mAb (BD Biosciences, Heidelberg, Germany). PE-conjugated, PerCP-conjugated, APC-conjugated and FITC-conjugated isotype-matched immunoglobulin (Ig)-G1 and IgG2a antibodies (DakoCytomation, Hamburg Germany) will be used for each patient and as measurement for negative controls. The cells will be washed three times to remove unbound antibodies and finally re-suspended in 400 μL of fluorescence-activated cell sorting (FACS) solution (BD Biosciences, Heidelberg, Germany). FACS analysis will be performed on a FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany) and the data will be analysed using WinMDI V.2.8 software. A minimum of 500 000 events is to be collected. FACS analysis of each probe will be performed in triplicate. The frequency of cEPCs in peripheral blood is determined by a two-dimensional side scatter/fluorescence dot plot analysis of the samples, after exclusion of Annexin V-positive cells and appropriate gating. The exclusion of Annexin V-positive cells is performed to rule out contamination with apoptotic cells. cEPC counts will be expressed as percentage of total PBMC in each patient.
sVEGF serum concentrations will be assessed using an ELISA kit (R&D Systems, Wiesbaden-Nordenstadt, Germany) in triplicate samples obtained from 5 mL serum. ELISA will be performed according to the manufacturer’s instructions. The cVEGF concentration will be measured in pg/mL. Further details will be described in a laboratory manual.
Ethics and dissemination
After due consideration, all participating investigators are convinced that the trial has a positive risk–benefit ratio. Based on results from a phase III trial, which showed efficacy for pazopanib treatment in STS,8 patients who are treated according to this protocol can be expected to have a higher chance of complete tumour resection and thus a lower risk of tumour recurrence, which should positively affect progression-free and overall survival. Moreover, the preoperative treatment has the potential to result in a less radical, tissue-sparing resection with fewer side effects. In contrast, the expected magnitude of potential risks and harmful side effects of the study treatment are small. Pazopanib is a registered drug. Overall, it is well tolerated, and most side effects are manageable and quickly subside once the dosage is reduced or the therapy discontinued. Therefore, the risk that surgery is delayed or perioperative morbidity is increased due to pazopanib side effects is expected to be small. Given the short preoperative treatment period in the window-of-opportunity design and the demonstrated efficacy of pazopanib in STS, the risk of relevant tumour progression during treatment is judged to be very low.
Study-related diagnostic procedures performed in addition to standard of care include two dPET-CTs. These expose the patient to radiation. However, the overall radiation dose of these two procedures does not exceed the annual dose threshold stipulated by relevant German laws for individuals who are professionally exposed to radiation. Thus, the risk of radiation-induced secondary malignancy is judged to be very low and outweighed by the scientific benefit of the study as well as the individual clinical benefit of each participating individual. dPET-CT, although not yet part of standard clinical algorithms for primary STS, does yield additional relevant information. It can detect occult metastases leading to a change in treatment strategy. Moreover, the planning of resection is facilitated for the surgeon, as PET-CT provides additional information on tumour extension and depicts the metabolism in different tumour areas, which potentially changes the surgical approach.
Before the start of the study, patients are informed, in writing and verbally, about the nature and implications of the proposed study, and especially about the possible benefits for their health, and possible risks are explained. Patients document their consent by signing the informed consent form.
Medical confidentiality and the provisions of the German Federal Data Protection Act are complied with. Moreover, the German Medicines Act (AMG) and the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use—Good Clinical Practice (ICH-GCP) regulations are complied with. The principal investigator can decide to discontinue the entire study if he concludes that continuation would pose a relevant threat to the health or well-being of the individual participants, or if he concludes that the risk-benefit-ratio of the study is unfavourable, that is, if the risks clearly outweigh the potential benefits.
Prior to initiation, the trial was registered at clinicaltrials.gov (NCT01543802) and the European Clinical Trials database (EudraCT: 2011-003745-18). The University of Heidelberg is the legal sponsor of this trial.
The originals of all central study documents will be archived at the principal study site for at least 10 years after preparation of the final report. The principal investigator will retain the generated administrative documents (correspondence with Ethics Committee, supervisory authority, etc), patient identification list, signed informed consent forms and copies of the general study documentation (protocol, amendments) for the time period stated above. Original data of study patients (medical source records) are to be retained for the applicable archiving period of the study centre but for not <15 years, starting from study completion.
We aim to publish results from this study in the form of one or several manuscripts in international medical journals. The principal investigator will review all manuscripts to prevent forfeiture of patent rights to data not in the public domain. Publication of the first manuscript reporting study results is planned to take place as soon as possible after analysis of the primary end point.
At the time of manuscript submission (September 2015), 18 patients have been recruited into the trial.
Discussion
We present the protocol of a trial that uses pazopanib for preoperative treatment of STS in a window-of-opportunity study. While pazopanib has shown efficacy in metastatic and unresectable STS8 and therefore been approved for this indication, the drug has not yet been extensively evaluated in the preoperative setting in resectable STS. Results of a study on preoperative pazopanib combined with radiation therapy were recently published.35 The study assessed the safety of this combination but, being a phase I trial, it was not designed to provide sufficient information on efficacy. An ongoing pilot trial (NCT01446809) evaluates pazopanib induction therapy prior to preoperative chemotherapy. Results, to appear in 2016, are awaited. Another ongoing phase II/III trial (NCT02180867) compares the combination of pazopanib and chemoradiotherapy or radiotherapy with chemoradiotherapy or radiotherapy without pazopanib. Results, to appear in 2018, are awaited.
Our trial is unique in that it tests preoperative pazopanib monotherapy in a window-of-opportunity design. The relatively short duration of pazopanib therapy was chosen to minimise the risk of tumour progression and consequent unresectability in non-responders. Notwithstanding, given the drug's mechanism of action, 21 days of treatment are considered sufficient to detect efficacy by assessing dPET-CT response, which is the trial's primary outcome.36 The wide range of secondary outcomes allows validating other potential parameters for response assessment. In particular, the trial offers the unique possibility to prospectively compare different modalities of response assessment, that is, MRI and histopathological features, with dFDG-PET-CT, which is considered ‘gold standard’.
The timing of MR assessment with dFDG-PET-CT is important for the validity of the results and their interpretation. For several tumour entities, the phenomenon of ‘rebound growth’, that is, an increase in tumour proliferation and possibly neoangiogenesis shortly after discontinuation of antiangiogenic therapy, has been described.37–39 Therefore, to assess the strongest achievable response to an antiangiogenic drug such as pazopanib, it would probably be best to perform MR assessment at the last day of therapy. Following this rationale, one would also perform surgery without previous discontinuation of therapy. However, surgical complications, mainly wound and anastomotic morbidity, have been described in patients with prior antiangiogenic treatment. Evidence suggests an inverse correlation of their incidence with the time interval between treatment discontinuation and surgery. To minimise this complication risk, we established a wash-out period of at least 7 days between end of drug treatment and surgery, corresponding to five half lives of the drug. The same 7-day period was used in a recent trial on neoadjuvant pazopanib treatment for renal cell carcinoma.40 We believe that this would also be a rational approach if pazopanib were to be used as neoadjuvant treatment outside of trials. Consequently, we aimed at assessing MR after treatment discontinuation and close to the date of surgery. This mirrors the metabolic state of the tumour at the time of surgery, thus providing the best estimate of the clinically meaningful therapeutic benefit of pazopanib.
Another important element of the trial is its translational substudy. The trial design offers the opportunity to measure two potential biomarkers, cEPCs and sVEGF, during antiangiogenic treatment and in the perioperative setting, and to correlate them with several parameters of treatment response.
In summary, this trial will provide initial evidence regarding preoperative pazopanib treatment of STS, and, if efficacy can be demonstrated, lead to a pertinent phase III trial. Moreover, cEPCs and sVEGF are further explored regarding their use as biomarkers for antiangiogenic treatment of STS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors UR and PH conceived of and planned the trial. UR drafted the trial protocol and edited its final version. AD-S wrote the parts of the protocol relating to dPET-CT. JJ, MS and PH provided advice on the parts of the protocol relating to surgical treatment. BK, GE, SF and H-GD provided advice on the parts of the protocol relating to medical treatment. KN wrote the parts of the protocol relating to the translational study. LRP designed the final trial, devised the analysis plan and wrote the corresponding parts of the trial protocol. UA provided advice on the parts of the protocol relating to MRI diagnostics. TG provided advice on the parts of the protocol relating to pathology. All the authors read and approved the final version of the protocol and of the manuscript.
Funding This trial was initially funded by GlaxoSmithKline Oncology. Funding is continued by Novartis, who acquired pazopanib from GlaxoSmithKline Oncology in 2015.
Competing interests UR, BK, JJ and PH have received travel reimbursements from GlaxoSmithKline Oncology/Novartis.
Ethics approval The trial was approved by the ethics committee II of the University of Heidelberg, Germany (reference number 2012-019F-MA). Furthermore, it was approved by the German Federal Institute for Drugs and Medical Devices (Reference number 61-3910-4038155) and the German Federal Institute for Radiation Protection (Z5-22463/2-2012-007).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.