Article Text

Abstract
Objective To determine the efficacy of acotiamide, an acetylcholinesterase inhibitor, in patients with functional dyspepsia (FD) in a 4-week trial
Methods A multicentre, randomised, placebo-controlled, parallel-group, phase III trial was carried out, in which patients with FD received 100 mg of acotiamide or placebo three times a day for 4 weeks, with 4 weeks post-treatment follow-up. The primary efficacy end points were global assessment of overall treatment efficacy (OTE) and elimination rate of all three meal-related symptoms (postprandial fullness, upper abdominal bloating and early satiation), as derived from daily diaries. Secondary efficacy end points were individual symptom scores and quality of life. Adverse events were monitored.
Results 52.2% of those receiving acotiamide and 34.8% in the placebo group (p<0.001) were classified as responders according to a global assessment of OTE. Over 4 weeks, the elimination rate for all three meal-related symptoms was 15.3% among patients receiving acotiamide compared with 9.0% in the placebo group (p=0.004). The significant benefit of acotiamide over placebo in OTE and elimination rate was maintained during the 4 week post-treatment follow-up. All other secondary efficacy end points, including quality of life, were significantly improved with 100 mg of acotiamide as compared with placebo. The number needed to treat was 6 for OTE and 16 for symptom elimination rate. The incidence of adverse events was similar between the acotiamide group and placebo group and no significant cardiovascular effects due to treatment were seen.
Conclusions Over 4 weeks, acotiamide significantly improved symptom severity and eliminated meal-related symptoms in patients with FD.
Trial registration number http://ClinicalTrials.gov number, NCT00761358.
- Gastro-oesophageal reflux disease
- enteric nervous system
- irritable bowel syndrome
- appetite
- gastrointestinal motility
- visceral sensitivity
- gastric emptying
- dyspepsia
- gastroduodenal motility
- functional dyspepsia
- functional bowel disorder
- achalasia
- pancreatic cancer
- neurogastroenterology
- hepatobiliary cancer
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Gastro-oesophageal reflux disease
- enteric nervous system
- irritable bowel syndrome
- appetite
- gastrointestinal motility
- visceral sensitivity
- gastric emptying
- dyspepsia
- gastroduodenal motility
- functional dyspepsia
- functional bowel disorder
- achalasia
- pancreatic cancer
- neurogastroenterology
- hepatobiliary cancer
Significance of this study
What is already known on this subject?
In phase II trials in Europe, the USA and Japan, acotiamide has been shown to have beneficial effects in functional dyspepsia (FD), particularly for meal-related FD symptoms such as postprandial fullness, upper abdominal bloating and/or early satiation, without major adverse events.
What are the new findings?
Acotiamide was better than placebo in improving overall treatment efficacy and meal-related symptoms and this benefit started during the second week of treatment.
Acotiamide achieved a higher rate of elimination of meal-related FD symptoms, which was associated with significant improvement in all subdomains of the disease-specific Short Form-Nepean Dyspepsia Index (SF-NDI) quality-of-life assessment.
How might it impact on clinical practice in the foreseeable future?
FD is one of the most common disorders seen in clinical practice, and there are no treatments of established efficacy.
The findings will assist many clinicians seeing dyspeptic patients in general and specialised practice.
Introduction
Dyspeptic symptoms, defined as the presence of symptoms considered to originate from the gastroduodenal region, occur commonly in the general population.1 2 Epidemiological surveys suggest that 20–30% of the general population has dyspepsia over the course of a year and this percentage is reasonably consistent around the world.2 In Japan, about 25.6% of the population report dyspeptic symptoms.2 3 In most of these subjects, additional examinations fail to identify a cause for the symptoms and this is referred to as functional dyspepsia (FD).4 5 According to the Rome III consensus, FD is defined as the presence of symptoms thought to originate in the gastroduodenal region (postprandial fullness, early satiation, epigastric pain or burning), in the absence of any organic, systemic or metabolic disease that might explain the symptoms. FD has considerable impact on quality of life (QoL), healthcare use and loss of productivity.6
The underlying pathophysiology in FD is incompletely understood, possibly because of the heterogeneous nature of the disorder.1 7 Based on a presumed relationship between symptom pattern and underlying pathophysiological mechanisms, the Rome III consensus subdivided FD into two subcategories: postprandial distress syndrome (PDS) and epigastric pain syndrome.1 PDS; characterised by postprandial fullness and early satiation, is also referred to as meal-related FD, and is thought to be caused by disturbed gastric motor function.1 7 8 It has been suggested that this group of patients may respond to drugs that alter gastric motility, such as prokinetic and fundus-relaxing drugs, but to date no drug has been approved for the treatment of FD or PDS.1 7 8
Acotiamide (Z-338) is a first-in-class drug which exerts gastroprokinetic activity by enhancement of acetylcholine release.8 9 It does so by acting as an antagonist on muscarinic autoreceptors in the enteric nervous system and by inhibiting cholinesterase activity.9–11 Acotiamide may act directly on the gut and also indirectly through the brain–gut axis via actions in the central nervous system.12 In animal models and in man, acotiamide enhances gastric emptying and gastric accommodation, two factors which have been implicated in the pathogenesis of PDS symptoms.8–11 In a rat model, acotiamide reversed stress-induced inhibition of gastric emptying and food intake.12 In phase II trials in Europe, the USA and Japan, acotiamide has been shown to have beneficial effects in FD, particularly for meal-related FD symptoms such as postprandial fullness, upper abdominal bloating and/or early satiation, without major adverse events.13–16 Based on the Japanese and European studies, 100 mg three times a day was identified as the most effective dose.13 14
In the present phase III, placebo-controlled trial, we aimed to determine the efficacy, safety and effect on QoL of oral acotiamide at a dose of 100 mg three times a day for 4 weeks, in patients with FD as defined by the Rome III classification.1
Methods
Study design
This multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase III trial was conducted at 67 centres in Japan over a period of 21 months, from October 2008 to June 2010. For all evaluations, patients were asked to attend the centres after having fasted in the morning. The study design is summarised in figure 1.

Trial design. Daily dose: three times a day before meal.
The trial was conducted in accordance with the Good Clinical Practice Guidelines of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, the Declaration of Helsinki and local laws and regulations. The protocol was reviewed and approved by the ethics committees of participating centres. Written informed consent was obtained from every patient.
Eligibility of patients
We recruited Japanese patients with FD aged between 20 and 64 years, who had FD-PDS as defined by the Rome III classification.1
In agreement with the Rome III PDS definition, patients were included if they had had postprandial fullness or early satiation for at least 6 months before inclusion. Patients were also eligible if they had had two or more of the following symptoms at a moderate or severe level within the previous 3 months: upper abdominal pain, upper abdominal discomfort, postprandial fullness, upper abdominal bloating, early satiation, nausea, vomiting or excessive belching. Patients were not necessarily excluded if they had coexisting epigastric pain syndrome symptoms (epigastric pain, epigastric burning), but the symptom causing the most distress at the time of obtaining informed consent had to be one of the following meal-related symptoms: postprandial fullness, upper abdominal bloating or early satiation. All patients underwent upper abdominal endoscopy in the screening period. Those with any changes in the oesophagus, duodenum or in the stomach were excluded. Patients who had experienced heartburn within 12 weeks before the baseline period were excluded. Patients with the comorbidity irritable bowel syndrome were excluded. Helicobacter pylori status was not assessed as part of the study.
The following exclusion criteria were also applied: presence of any symptom indicating serious or malignant disease, drug or alcohol abuse and severe abnormality in the electrocardiogram at rest or the clinical or laboratory examination during the baseline period. Anti-secretory drugs, antacids, prokinetics, non-steroidal anti-inflammatory drugs and antidepressant drugs were not allowed after the baseline period. Pregnant or breastfeeding women and those of childbearing age who were not using an approved method of contraception were also excluded.
Randomisation
The 4-week treatment period was preceded by an 8-day baseline period after the screening visit and followed by a 4-week post-treatment follow-up period. Before undergoing randomisation at the end of the baseline period, patients had symptoms evaluated each week during the baseline period. Randomisation was performed with a computer-generated program. At the end of the baseline period, eligible patients were assigned a randomisation number according to a predetermined list at each centre. These numbers were allocated in sequential order and registered in the patient enrolment list and the allocation was concealed. Emergency envelopes containing the randomisation code were provided to the investigators and were examined at the end of the trial to ensure that the trial blinding had been maintained.
Assessments
No universally accepted end point has been established for therapeutic trials in FD and the Food and Drug Administration (FDA) has provided no specific guidelines.17 A well-recognised approach is the assessment of global outcome by the ‘overall treatment effect’ (OTE) approach, where the patients is asked at intervals during, or at completion of the treatment, whether symptoms have remained the same, improved or deteriorated compared with pretreatment phase.17 18 The advantage of this end point is that it closely resembles the way in which doctors evaluate treatment benefit in clinical practice, but it does imply recall of pretreatment symptom severity. OTE has been used as a secondary outcome measure in several therapeutic trials in FD.19–23 OTE was used most recently in the tegaserod FD studies, where a weekly global assessment of change was rated on a seven-point Likert scale and this end point generated the most consistent improvements with tegaserod over placebo.24 For our phase III study, OTE was evaluated weekly and severity ratings of individual symptoms were obtained in daily diaries using Likert scales. QoL was assessed using a validated scale.25 26
Symptoms and global assessment
During the 8-day baseline period, the 4-week treatment period and the 4-week post-treatment follow-up period, patients rated each of nine symptoms (upper abdominal pain, upper abdominal discomfort, postprandial fullness, upper abdominal bloating, early satiation, excessive belching, nausea, vomiting and heartburn) on a severity scale of 0–3 (none, mild, moderate and severe) using paper diaries. Each week, patients completed a global assessment of the OTE questionnaire. The question asked was ‘How were your gastric symptoms during the past week in comparison with the baseline period?’. This was scored in the paper diaries on a seven-point Likert scale, ranging from ‘extremely improved compared with the baseline period’, ‘improved compared with the baseline period’, ‘slightly improved compared with the baseline period’, ‘not changed’, ‘slightly aggravated compared with the baseline period’, ‘aggravated compared with the baseline period’ and ‘extremely aggravated compared with the baseline period’.
Primary efficacy end points
The two primary efficacy end points were OTE and elimination rate of all three cardinal symptoms: postprandial fullness, upper abdominal bloating and early satiation (no symptoms) at the last survey point.
Disease-specific QoL
To assess the effects of treatment on validated disease-specific QoL, the Japanese version of the Short Form-Nepean Dyspepsia Index questionnaire (SF-NDI)25 26 was used at baseline, at week 4 of treatment and week 4 of the post-treatment follow-up period. Data for 10 items were obtained and consisted of five subscales: tension, interference with daily activities, eating/drinking, knowledge/control and work/study. The scores ranged from 1 to 5, with lower scores indicating a better QoL.
Safety assessments
Data from patients who took at least one dose of the trial drug were included in the full analysis set (FAS) and safety population. Adverse events were reported at weeks 2 and 4 of treatment and week 4 of the post-treatment follow-up period. Vital signs were evaluated at the baseline visit, at week 4 of treatment and week 4 of the post-treatment follow-up period. Results of electrocardiography, clinical laboratory tests and physical examinations were evaluated at the baseline visit, at week 4 of treatment and at week 4 of the post-treatment follow-up period.
Statistical analysis
Based on our previous phase IIb trials, the proportions of the acotiamide and placebo responders for OTE were expected to be 50% and 40%. For elimination of all the three symptoms, proportions were expected to be 20% and 10%, respectively. The two-sided significance level was 0.05. A sample size of 820 (410 patients in each arm) was estimated to have at least 80% power to detect the differences. The power was calculated as the product of the power of each end point, 81.7%, 98.0%.
The efficacy analysis was based on both the FAS and per protocol (PP) populations. Adverse effects were evaluated in the safety population.
For evaluation of the primary end point, patients who were “extremely improved” or “improved” on the OTE scale were considered responders. The elimination rate was the proportion of patients who achieved elimination of all three symptoms—that is, postprandial fullness, upper abdominal bloating and early satiation. Primary efficacy variables were evaluated over the entire treatment period using Fisher's exact test method. The level of significance was set at 0.05 (two sided).
A frequency distribution table was prepared for the OTE at the last survey point and improvement rates were determined from this. Treatment effects and 95% CIs were obtained for each variable.
Recently and well after the design and start of this phase III study, the FDA issued a statement on the use of patient-reported outcome measures in the evaluation of therapeutic efficacy of drugs for medical conditions, in general, and irritable bowel syndrome, in particular.27 28 In these documents, the FDA no longer recommends overall symptom assessment in binary questions, or in comparison with a previous time points, but advocates assessment of the severity of individual clinically important symptoms and defining responders on the basis of a clinically meaningful improvement of these symptoms.28 In the absence of an existing validated instrument for FD and in order to allow better estimation of the magnitude of the therapeutic response to acotiamide, we performed a post hoc responder analysis on changes in severity of individual symptoms. For each symptom, a responder was defined as a patient with a more than 50% reduction in symptom severity during the last observation week in comparison with baseline severity (weekly average), as derived from the daily diaries.
Results
Enrolment and baseline characteristics of the patients
We screened 1394 outpatients with a suspected diagnosis of patients with FD (figure 2) and of these, 497 were excluded during the baseline period. The remaining 897 patients were randomly assigned to receive one of two trial drugs: 100 mg of acotiamide (452 patients) or placebo (445 patients) three times a day. Five patients withdrew from the trial, thus, 892 patients were included in the FAS. The baseline characteristics of the trial population were similar between the groups (table 1).

Summary of patient flow. FAS, full analysis set.
Baseline characteristics of the patients receiving at least one dose of study medication (full analysis set)
Primary efficacy end point
The responder rate based on the OTE at the last survey point on FAS was 52.2% for patients receiving acotiamide and 34.8% with placebo (p<0.001, table 2).
Efficacy end points (full analysis set)
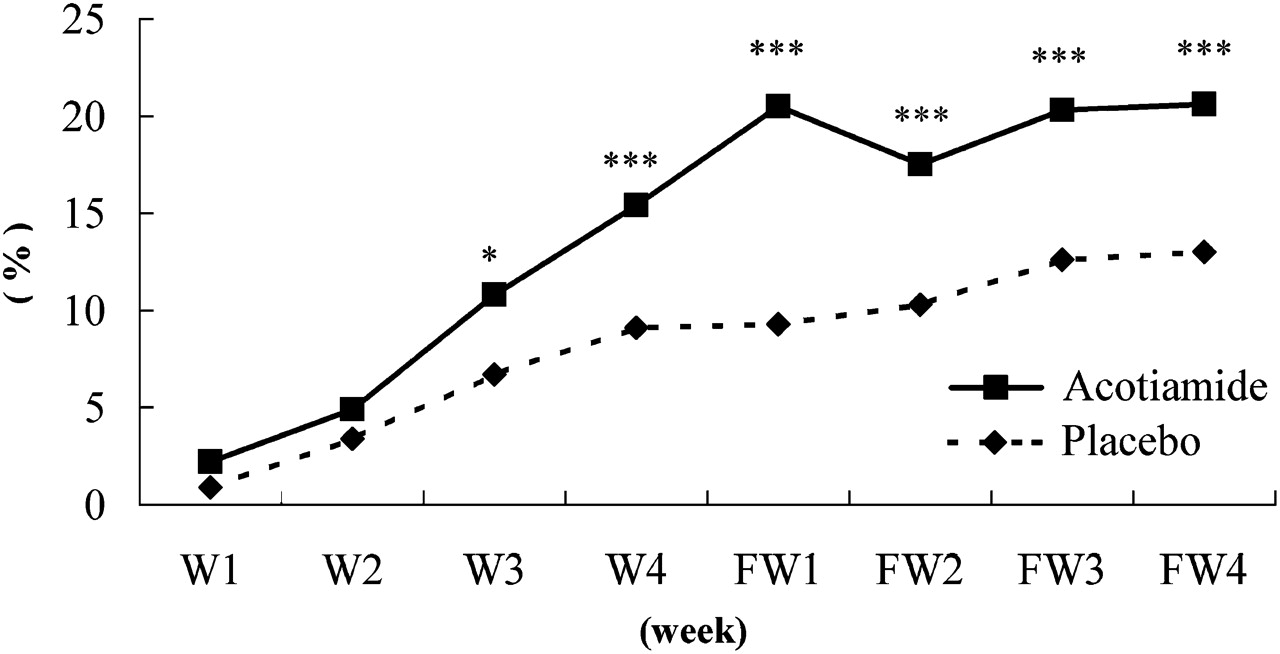
The elimination rate of all three meal-related symptoms (postprandial fullness, upper abdominal bloating and early satiation) at the last survey point on FAS was 15.3% with acotiamide and 9.0% with placebo (p=0.004, table 2).
Secondary efficacy end points
PP analysis
The PP analysis of OTE data at the last survey point showed similar response rates of 53.3% with acotiamide group and 34.7% with placebo group (p<0.001). Moreover, the elimination rate of all of the three symptoms at the last survey point on PP was 15.2% with acotiamide and 9.0% with placebo (p=0.006).
Elimination rate of individual symptoms
Significantly higher elimination rates were obtained with acotiamide than with placebo for early satiation and for postprandial fullness (table 2). The highest elimination rate was seen for early satiation with acotiamide (37.8% vs 25.4% with placebo, p<0.001) (table 2).
Weekly evaluations on FAS
For OTE, a significant difference became apparent from week 2 (figure 3). For the elimination rate of all of the three symptoms, there was a significant difference from week 3 (figure 4). After discontinuing acotiamide or placebo, neither group returned to baseline within the 4-week post-treatment follow-up period. A significantly higher response rate persisted in the acotiamide group compared with the placebo group for all 4-week post-treatment follow-up periods (all p<0.005).
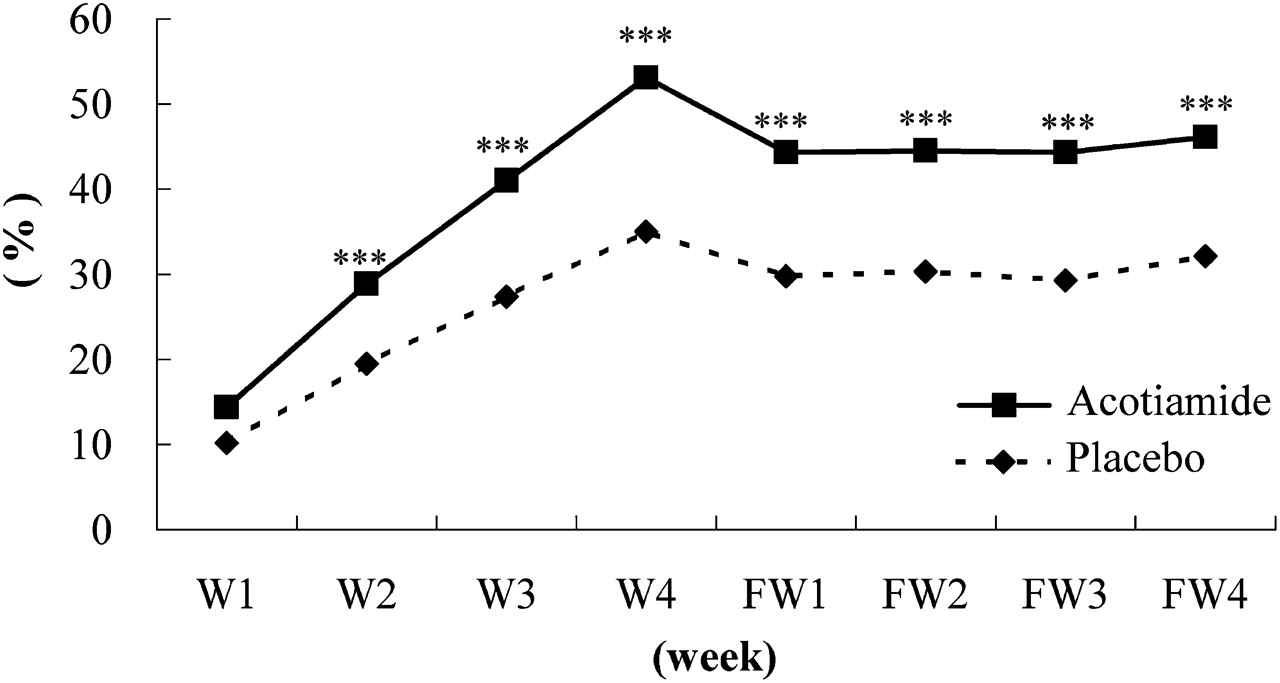
Weekly improvement rate in overall treatment efficacy. ***p<0.005 compared with placebo. FW, follow-up week after treatment; W, week.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Elimination rate for all three symptoms. *p<0.05 compared with placebo. ***p<0.005 compared with placebo. FW, follow-up week after treatment; W, week.
Individual symptom scores for postprandial fullness, upper abdominal bloating and early satiation were significantly improved in the acotiamide group compared with the placebo group. The improvement of symptom scores for upper abdominal pain, upper abdominal discomfort, nausea, vomiting and excessive belching did not differ significantly between the groups at the last survey point.
The number needed to treat (NNT)
For the interpretation of treatment efficacy, the NNT was an important measure to compare the different therapeutic options for an FD. Based on the results, the NNT as indicated by OTE was calculated as 6 and the NNT for the elimination rate of all of the three symptoms at the end of the treatment period were calculated as 16.
Post hoc analysis of individual symptom severities
When a decrease in symptom severity of >50% was considered, the responder rates for postprandial fullness, early satiation, upper abdominal pain and upper abdominal bloating were significantly higher during acotiamide treatment than with placebo (table 3). For the meal-related symptoms of postprandial fullness, early satiation and upper abdominal bloating, the responder difference over placebo ranged between 11.8% and 19.2%.
Responder rates for individual symptoms: (average symptom score at the last observation period)/(average symptom score at baseline) ≦0.5
Quality-of-life scores
Overall SF-NDI scores at the last observation in the FAS showed a significantly greater improvement from baseline among the patients with acotiamide (−3.66) than among those with placebo (−2.84) (p<0.001). Similarly, all five SF-NDI subscale scores at the last observation showed improvements from baseline in the acotiamide group compared with the placebo group (table 4).
Summary of overall and subscale symptom scores on the Short Form-Nepean Dyspepsia Index (SF-NDI) questionnaire
Safety
Adverse events were reported by 252 of the 450 patients (56.0%) receiving acotiamide and 267 of the 442 patients (60.4%) with placebo. The most commonly reported adverse events were increase of serum triglycerides, serum prolactin, or serum γ-glutamyltransferase and nasopharyngitis (table 5). Most adverse effects were mild or moderate in severity. No deaths occurred during the trial. Only one serious adverse event was reported: an intervertebral disc herniation occurred in one of the 450 patients (0.2%) treated with acotiamide, judged as ‘not related’. No serious adverse events were reported in the 442 patients receiving placebo.
Incidence of adverse events (>4% in any group)
Discussion
Despite its high prevalence and impact, treatment options for FD are limited. Systematic reviews of the available literature indicate that antisecretory drugs and prokinetic agents may be better than placebo in relieving symptoms of FD,19 20 but the studies were hampered by flaws in patient selection and trial design.18 21–23 29–31 No treatment has been approved for FD. Proton pump inhibitors are mainly effective in patients with symptoms of heartburn and FD, but they have limited efficacy compared with placebo when the patients' symptoms are not acid-related.29 30 32 33 Attempts to establish efficacy for prokinetic drugs in FD have been disappointing. Mosapride, a 5-HT4 receptor agonist, was no better than placebo in a dose-finding trial in FD.34 Positive results were obtained in a phase IIb trial with itopride, a dopamine-2 receptor antagonist with cholinesterase inhibitory properties, but in two phase III trials the drug was no better than placebo.35 36 Tegaserod, another 5-HT4 agonist, showed some promise after phase II studies,37 but the result of two phase III trials was disappointing.24 Therefore, a true unmet medical need for FD exists and many patients turn to alternative treatments of unproven value.38
Acotiamide is a first-in-class drug that was developed for the treatment of FD and other upper gastrointestinal symptomatic conditions. Placebo-controlled phase II studies in Europe and in Japan showed beneficial effects of acotiamide for FD symptoms of postprandial fullness, upper abdominal bloating and early satiation.13 14 The analysis of the Japanese phase II studies showed that acotiamide had significant benefit in comparison with placebo using OTE as a primary end point in a post hoc analysis of patients who fulfilled Rome III criteria for PDS. In the Japanese studies, a higher elimination rate of meal-related symptoms was also seen with acotiamide, and 100 mg three times a day was established as the optimal dose.13 In a phase II trial in the USA, the randomised treatment phase was preceded by an open-label proton pump inhibitor (PPI) trial to eliminate PPI responders. In the 12-week double-blind, placebo-controlled treatment phase, using OTE, acotiamide showed significant efficacy over placebo in the PDS group, with the highest benefit occurring during the first 4 weeks. In addition, the beneficial effects were confirmed using adequate relief for >50% of the time as the end point. Furthermore, acotiamide showed significant improvement in several domains of the generic Short Form-36 QoL scale and in three of five domains in the SF-NDI.25 26
In this phase III randomised, 4-week placebo-controlled trial, patients who fulfilled the Rome III criteria for FD-PDS were enrolled and both primary end points were met. A significantly higher rate of patients achieved the first primary end point—namely, improved or extremely improved on the OTE, in the acotiamide group than in the placebo group. OTE evaluation allows the individual to integrate all aspects of his condition into a single treatment outcome. OTE has previously been used as an outcome measure in studies evaluating H. pylori eradication or acid suppression in FD.18 21–23 29–31 More recently, OTE was used in the tegaserod FD studies, where this end point generated the most consistent improvements with tegaserod over placebo.24 37 The advantage of this end point is that it closely resembles the way in which doctors evaluate treatment benefit in clinical practice. The bidirectional construct makes it particularly suitable for evaluating symptom improvement and also deterioration. Psychometric analysis supports the concept that a rating of ‘improved’ or ‘extremely improved’ on the OTE indicates substantial and clinically meaningful improvement.31 Moreover, with a margin of more than 17% over placebo in reaching this end point and a NNT of 6, the benefit of acotiamide is well above the minimal range of efficacy that was proposed to be clinically relevant by a group of experts.39 On the other hand, because of the inherent recall, which may lead to bias, the FDA has recently argued against the use of OTE in future drug development trials.28 However, the clinical relevance of the improvement with acotiamide is further supported by the significant improvement of several prespecified secondary efficacy end points, including disease-related QoL. For the latter, the changes seen using the SF-NDI were of the order of 1 SD or 10% of the total scale range on all five domains and are above the minimal clinically relevant difference.40 The post hoc analysis of responder rates for individual symptoms, using a 50% decrease during the last evaluation week compared with baseline, further confirms substantial benefit of acotiamide over placebo, especially for meal-related symptoms.
The other primary end point, elimination of all of three meal-related symptoms, was also reached by significantly more patients in the acotiamide group than in the placebo group. Acotiamide was better than placebo in eliminating symptoms of postprandial fullness and upper abdominal bloating, but the margin over placebo at the end of the 4-week trial was modest, with an estimated NNT of 16. More robust efficacy in symptom resolution was seen for early satiation (37.8% vs 25.4% with acotiamide and placebo, p<0.001). The elimination rate of all three symptoms was also significantly higher for acotiamide than with placebo at the last survey point and further increased during the 4-week post-treatment week. Gradual symptom improvement over the 4-week treatment period was seen in both arms, but acotiamide showed a higher rate of improvement, and significantly better responses on OTE were already reached in week 2. Moreover, the efficacy over placebo persisted during the 4 weeks after the end of the treatment period and there were no signs of immediate relapse or rebound symptom aggravation. Both the rapid onset of efficacy and the lack of rapid relapse upon interruption of treatment are attractive aspects when considering the use of acotiamide in clinical practice.
Similar to previous studies,13–16 acotiamide was well tolerated and the incidences of adverse events were similar between the acotiamide group and placebo group. Furthermore, no clinically relevant changes in vital signs or electrocardiographic variables were obtained. A number of drugs that have been used or were under development for the treatment of FD, such as cisapride or tegaserod, have been associated with major cardiovascular side effects.41 42 Affinities of these drugs for the HERG channel or for 5-HT receptors have been implicated in the occurrence of major cardiovascular events.43 Acotiamide has little or no affinity for these receptors, which contributes to its favourable benefit–risk profile, and thorough QTc studies in the USA with acotiamide up to 900 mg showed no arrhythmogenic potential.
Strengths of the study are the large number of patients studied and the multicentre double-blind, placebo-controlled design. Limitations are the short treatment duration of only 4 weeks for a condition which is often chronic relapsing, but this study duration was chosen in agreement with the regulatory authorities in Japan. The study does not examine efficacy upon prolonged treatment or re-treatment. Reassuringly, however, the post-treatment follow-up period does not show rebound tachyphyllaxis but, on the contrary, shows sustained symptomatic benefit. Future studies will need to examine the relapse rate over longer post-treatment periods and the response to re-treatment.
The mechanism underlying the clinical benefit seen with acotiamide in FD-PDS is incompletely elucidated and may well be multifactorial. Animal studies have shown that acotiamide can enhance the gastric emptying rate, and meal-induced gastric accommodation and impaired accommodation as well as delayed gastric emptying have been established as key pathophysiological mechanisms in FD.12 Mechanistic studies in patients with FD have confirmed the ability of acotiamide to enhance gastric accommodation and to enhance gastric emptying, and these may be particularly relevant in the improvement of symptoms of early satiation and postprandial fullness.44 In addition, many patients with FD have increased anxiety levels, which are associated with decreased gastric compliance and enhanced gastric sensitivity in patients with FD and in healthy controls.45 46 In an animal model, acotiamide pretreatment inhibited the expression of central mediators of induced stress (neuromedin U and BDNF) and the associated delayed gastric emptying and feeding inhibition.12
In conclusion, in this 4-week randomised placebo-controlled phase III trial, acotiamide was effective, safe and well tolerated in the treatment of patients with FD-PDS according to the Rome III diagnostic criteria. Acotiamide 100 mg three times a day was better than placebo in improving OTE and meal-related symptoms and this benefit became apparent during the second week of treatment. Acotiamide achieved a higher rate of elimination of meal-related FD symptoms and this was associated with significant improvement on all subdomains of the disease-specific SF-NDI QoL assessment. When treatment was stopped, no rapid symptom relapse or rebound occurred. Taking into account the favourable tolerability and safety profile in this and previous studies, acotiamide is a drug for the treatment of FD, a highly prevalent condition for which there is no treatment with established or approved efficacy.
Acknowledgments
We thank the site investigators for their participation in the trial.
References
Footnotes
See Commentary, p 789
Funding Financial support for this study was provided by Zeria Pharmaceutical Co, Ltd and Astellas Pharma Inc.
Competing interests KM has provided scientific advice to Astellas, Zeria, Ajinomoto, Abbott. MH has provided scientific advice to Astellas, Zeria, Takeda, Eisai, AstraZeneca, Sucampo. JT has provided scientific advice to Addex Pharma, Almirall, Aryx, AstraZeneca, Danone, Given, Ipsen, Menarini, Movetis, Norgine, Novartis, Nycomed, Ocera, Rose Pharma, SK Life Sciences, Smartpill, Sucampo, Theravance, Tranzyme, Xenoport and Zeria. YS and HK are employees of Zeria.
Ethics approval This study was conducted with the approval of the local institutional review boards and medical ethics committees in Japan.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Commentary