Article Text

Abstract
Objective We investigated the mutational landscape of mammalian target of rapamycin (mTOR) signalling cascade in hepatocellular carcinomas (HCCs) with chronic HBV background, aiming to evaluate and delineate mutation-dependent mechanism of mTOR hyperactivation in hepatocarcinogenesis.
Design We performed next-generation sequencing on human HCC samples and cell line panel. Systematic mutational screening of mTOR pathway-related genes was undertaken and mutant genes were evaluated based on their recurrence. Protein expressions of tuberous sclerosis complex (TSC)1, TSC2 and pRPS6 were assessed by immunohistochemistry in human HCC samples. Rapamycin sensitivity was estimated by colony-formation assay in HCC cell lines and the treatment was further tested using our patient-derived tumour xenograft (PDTX) models.
Results We identified and confirmed multiple mTOR components as recurrently mutated in HBV-associated HCCs. Of significance, we detected frequent (16.2%, n=18/111) mutations of TSC1 and TSC2 genes in the HCC samples. The spectrum of TSC1/2 mutations likely disrupts the endogenous gene functions in suppressing the downstream mTOR activity through different mechanisms and leads to more aggressive tumour behaviour. Mutational disruption of TSC1 and TSC2 was also observed in HCC cell lines and our PDTX models. TSC-mutant cells exhibited reduced colony-forming ability on rapamycin treatment. With the use of biologically relevant TSC2-mutant PDTXs, we demonstrated the therapeutic benefits of the hypersensitivity towards rapamycin treatment.
Conclusions Taken together, our findings suggest the significance of previously undocumented mutation-dependent mTOR hyperactivation and frequent TSC1/2 mutations in HBV-associated HCCs. They define a molecular subset of HCC having genetic aberrations in mTOR signalling, with potential significance of effective specific drug therapy.
- HEPATOCELLULAR CARCINOMA
- GENE MUTATION
- MUTATION SCREENING
- HEPATITIS B
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hepatocellular carcinoma (HCC) is a common cancer and leading cause of death worldwide.
HCC is heterogeneous with various risk factors including viral infection (HBV and HCV), alcoholism and exposure to aflatoxin B1.
Activation of mammalian target of rapamycin (mTOR) signalling cascade is resulted from ligand-dependent signals from epidermal growth factor and insulin-like growth factor signalling.
What are the new findings?
There are frequent mutational disruptions on multiple key and canonical components of mTOR signalling cascade.
Mutation-dependent mechanism is likely to be another major cause leading to mTOR hyperactivation.
Tuberous sclerosis complex (TSC)1 and TSC2 are the most frequently mutated mTOR pathway-related genes and they collectively define a novel molecular subset of human HCCs with more aggressive tumour behaviours.
Our study signifies personalised therapeutic option for a novel molecular subset of patients with susceptible mutant HCC through the inhibition of mTOR signalling.
How might it impact on clinical practice in the foreseeable future?
TSC1/2 mutations represent one of the most frequent single categories of genetic alterations found in human HCC and mTOR signalling cascade represents one of the most prominent therapeutic targets in HCC.
There is frequent mutation-dependent mTOR hyperactivation, which defines a novel molecular subset of human HCC and is likely to be actionable through inhibition by rapamycin or its derivatives.
Through perfect combination of both high-throughput next-generation sequencing-based mutation screening and patient-derived tumour xenograft model, our study demonstrates, with necessary supportive evidence correlating mutational findings and drug testing, the potential personalised treatment on relevant patients with HCC carrying specific molecular alterations.
Introduction
Liver cancer (hepatocellular carcinoma (HCC)) is a major malignancy worldwide and the second most common fatal cancer in China, including Hong Kong and Southeast Asia. About 55% of all new cases worldwide occur in China.1 ,2 Chronic HBV infection is the major underlying risk factor. HCC is deadly because of high recurrence rate even after surgical resection and frequent metastasis. Unfortunately, the overall response rate of patients with HCC to chemotherapy is unsatisfactory due to the highly chemoresistant nature of the tumour and the toxicity of the chemotherapeutic agents. On the other hand, Sorafenib, a multiple tyrosine-kinase inhibitor and the only Food and Drug Administration-approved molecularly targeted drug for HCC, has demonstrated only a modest survival benefit in patients with HCC, with the median survival only 2–3 months longer than the placebo arm in large-scale trials in both Caucasians and Asians.3 ,4 There is an urgent need to identify strategic molecular targets for developing new and effective molecularly targeted therapy. In this regard, investigation of the mutational landscape of HCC by next-generation sequencing (NGS) for novel, recurrently mutated targets with therapeutic implication would be valuable and would help in rational designing of personalised treatment strategy for patients with HCC.
Recent studies have demonstrated phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB/AKT)/mammalian target of rapamycin (mTOR) signalling pathway to have pivotal role in cancers, including HCC.5 ,6 Components of mTOR signalling pathway are frequently upregulated in human HCCs.7 Activation of mTOR signalling cascade was believed to be the result of ligand-dependent signals from epidermal growth factor and insulin-like growth factor signalling, rather than from a mutation-dependent mechanism.6 In accordance, reported literature has revealed low frequency of mutations in mTOR signalling pathway.7
To systematically exploit the mutational landscape of mTOR signalling cascade in chronic HBV background, we performed deep NGS-based targeted DNA sequencing (targeted-seq) on a panel of 81 selected mTOR pathway-related genes using a large cohort of 95 HBV-associated HCC cases. This initial observation was further augmented by mutational screening using whole-exome sequencing (WES) on 16 additional HBV-associated HCC cases, as well as whole-transcriptome sequencing (WTS) and Sanger sequencing in a panel of HCC cell lines. We found frequent mutations on various components of mTOR signalling cascade. Among the mutant mTOR-related genes identified, tuberous sclerosis complex (TSC) 1 and TSC2 were found to be the most frequently mutated in HCC tumour samples. Such frequent mutational disruption of TSC1/2 complex, which plays negative regulatory on Rheb and in turn inactivates mTORC1 activity, would likely result in hyperactivation of mTOR signalling as suggested by immunohistochemistry (IHC) staining. Of note, we further provide the necessary therapeutic implication of TSC mutations by subjecting relevant TSC-mutant cell lines and in-house established patient-derived tumour xenograft (PDTX) models to rapamycin treatment and confirmed their hypersensitivity. Overall, our study supports personalised therapeutic option for a novel molecular subset of patients with susceptible mutant HCC, through the inhibition of mTOR signalling.
Materials and methods
Patients and samples
A total of 95 human HCCs were selected from our sample collection to perform deep-targeted-seq. Another 16 pairs of human HCCs and their corresponding non-tumourous livers (NTLs) were used in WES. All patients had chronic HBV infection and were positive for hepatitis B surface antigen in their sera (see online supplementary table S1). The samples were obtained immediately after surgical resection from the operation theatre, frozen in liquid nitrogen and kept at −80°C. Frozen sections were cut from tumour and NTL blocks separately and stained for histological examination to ensure a homogenous cell population of tissues. Use of human samples was approved by the institutional review board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (see online supplementary materials and methods for the detailed methodology regarding NGS-based mutation evaluation, subsequent data validation and functional characterisation).
supplementary data
Results
Targeted-seq revealed recurrently mutated genes in mTOR pathway in HCC
To explore the pattern and importance of genetic alterations in mTOR pathway genes, we performed deep-targeted-seq in a large cohort of 95 human HCC tumour samples (targeted-seq cohort) for 81 mTOR pathway-related genes (see online supplementary table S2) selected based on Kyoto Encyclopedia of Genes and Genomes pathway definition and literature search, with mean depth of base coverage of 154.3× (range: 135.9–197.1) (see online supplementary table S3). Normal polymorphic sequence variants were removed by background filtering against reported variants deposited in public repositories. Of the 95 human HCC tumours studied, 69 (73%) carried at least one mutation in our selected mTOR-related gene set. Among the 81 genes, 25 (31%) were found to be recurrently mutated (figure 1A, upper part), while 22 (27%) were singleton (figure 1A, lower part). The remaining 34 genes (42%) showed no mutations in this cohort. All the mutant genes were sorted and tabulated according to their frequency of occurrence (figure 1A) and displayed in a pathway diagram to illustrate their potential roles in the mTOR signalling (figure 1B). In order to identify the potential key signalling components frequently mutated in HCC, we paid particular attention to the top-ranking mutant genes. We found that six genes (and the corresponding proteins), namely TSC2 (TSC2), HIF1A (HIF1α), PABPC3 (PABPC3), RB1CC1 (ATG17), RPS6KA3 (RSK2) and TSC1 (TSC1), were frequently affected in more than 5% of the cases in our cohort (figure 1A, top left and figure 1B). The high mutation frequency of these genes uncovered several key nodes in mTOR pathways which, once genetically altered, may support adaptation benefit and provide survival advantage in HCC. This might possibly involve the dysregulation of mTOR signalling activation (TSC1/2 and RSK2), its activity coupling with downstream biological processes including autophagy and protein translation (ATG17 and PABPC3) and intracellular reprogramming for the tumour microenvironment adaptation (HIF1α). Interestingly, from our findings, mTOR kinase (2%) and other well-known mediators and regulators such as PI3Kβ/γ (3% and 1%), PTEN (1%) and Akt1/2/3 (1%; 1% and 1%) were found to be mutated only in a very limited subset of the HCC samples and no mutation was found for Rheb, which encodes the direct downstream effector of TSC1/2.

Representations of the targeted DNA sequencing (targeted-seq) for mammalian target of rapamycin (mTOR) pathway-related genes in hepatocellular carcinoma (HCC) discovery cohort. (A) A total of 95 HBV-associated human HCC tumour samples were subjected to targeted-seq for mTOR pathway-related genes. Sixty-nine HCC samples were found to carry at least one mTOR-related mutation. Among the 81 mTOR-related genes being screened, 25 were found to be recurrently mutated in more than one sample, while 22 of them were singleton. The mutant genes were ranked and listed accordingly to their frequency. (B) Schematic diagram visualising the mutants listed in (A) to illustrate their implications in mTOR-related signalling. TSC, tuberous sclerosis complex.
Frequent TSC1 and TSC2 mutations defined a novel subgroup of human HCCs
Currently, no reported NGS studies in HCC have identified these high mutation rates of TSC1 and TSC2 in HCC. Our targeted-seq data showed that TSC2 was the top-listing mutant gene, with mutations found in 12 of the 95 HCC tumours (12.6%). The functionally related TSC1 mutation was also found in 5 of the 95 tumours (5.3%) (figure 1A, upper part). Of note, four of the five TSC1 mutants in our cohort were found in the tumours without TSC2 mutation, thus suggesting a mutually exclusive mutation pattern. This mutually exclusive pattern was also observed in the other top-ranking mTOR mutant genes like RB1CC1 and RPS6KA3 (figure 1A, upper part). By further retrieving and analysing the HCC exome sequencing data (n=202) from The Cancer Genome Atlas (TCGA) for the mTOR-related mutants, TSC2 and TSC1 were consistently found to be most frequently mutated, affecting 5.9% and 4.95%, respectively, of the total number of cases (see online supplementary figure S1A). TSC1 and TSC2 mutations among different cancers were also examined using data from TCGA cancer panel. Among the 25 cancer types being examined, 7 of them, were found to carry TSC1 and TSC2 mutations, with HCC ranked second among all the cancer types. High mutation rates of TSC1 and TSC2 in HCC and other cancers were likely suggesting the potential involvement of mTOR signalling dysregulation underlying their development (see online supplementary figure S1B). In addition, we compared the coincidence of TSC mutation with some of the well-reported HCC-related mutant genes, including TP53 (p53), CTNNB1 (β-catenin), AXIN1 (Axin1) and ARID1A (ARID1A) (figure 2A). We found that TSC mutations were present irrespective of the genetic background of TP53 mutation. More importantly, TSC mutations likely stratified a novel subgroup of patients whose tumours were not likely to be affected by CTNNB1, AXIN1 and ARID1A mutations, in which their mutation rates (range: 11.6%–15.8%) were comparable with TSC (figure 2B). We further identified and confirmed two additional TSC mutations from our exome sequencing dataset of 16 HCC cases (see online supplementary table S4 and supplementary figure S2). To investigate the clinical significance of this novel genetic subgroup of HCC, the patient samples were stratified into three groups: (1) with TSC mutation, (2) with known HCC driver mutation in any of TP53, CTNNB1, AXIN1 and ARID1A but not TSC mutation and (3) the others without the aforementioned mutations, followed by clinicopathological correlation analysis. Intriguingly, TSC-mutated HCCs were significantly associated with more aggressive tumour behaviour including larger tumour size (p=0.034) and presence of venous invasion (p=0.041) (table 1). This observation further provided evidence pointing to the potential tumour suppressive role of the TSC complex in human HCC. Loss of function in TSC through genetic alteration likely contributes to HCC progression by supporting tumour growth and enhancing cancer metastasis.
Clinicopathological correlation of patient with HCC samples carrying TSC mutation and other background mutation*

Frequent tuberous sclerosis complex (TSC)1 and TSC2 mutations define a novel subgroup of human hepatocellular carcinomas (HCCs). (A) Diagram listing the key genes found to be mutated in our HCC discovery cohort (n=95). TP53, CTNNB1, AXIN1 and ARID1A are key genes which have been known to be mutated in human HCC. We found that the TSC1 and TSC2, which encodes protein complex as negative regulator of mammalian target of rapamycin signalling, were frequently mutated. (B) Venn diagrams showing the rates of coincidence of TSC2 or TSC1 mutations with TP53, CTNNB1, AXIN1 and ARID1A as highlighted by the black lines.
Mutations in TSC1/2 rendered them functionally inactive
We confirmed all the identified TSC1 and TSC2 mutations in the tumour samples with independent Sanger sequencing (figure 3A, B, left half of the chromatogram, see online supplementary figure S3A,B). The majority of TSC1 and TSC2 mutations were heterozygous, with mutant read frequency at around 50% (figure 3A, B). Thus, noisy and overlapping signals were observed in several HCC cases carrying both copies of the wild type and insertion/deletion (INDEL)-mutant TSC2 (see online supplementary figure S4). On further examination of the results in the correspondingly NTLs (figure 3A, B, right half of the chromatogram, see online supplementary figure S3B, C), intriguingly, among the 14 TSC2 variants in 12 HCC cases, 9 (64.3%) were somatic, while the other 5 (35.7%) were not. Among the five TSC1 variants, two (40%) were confirmed to be somatic, while the remaining three (60%) were not. By combining the mutation data from both the validation and discovery cohorts, we identified that 13 of the 111 total HCC cases (11.7%) carried TSC2 mutations and 6 of the 111 HCC cases (5.4%) carried TSC1 mutations (figure 3C). Overall, TSC1/2 mutations were found in a total of 16.2% (18/111) of the cases and two-third of them (12/18) carried somatic mutations. By considering the somatic and non-somatic variants as a whole, of the six TSC1 variants identified, three (50%) were INDEL mutations, two (33.3%) were non-synonymous ones, resulting in single amino acid change, and the remaining one (16.7%) was stop-gain mutation (figure 3D, right panel). The types of TSC2 mutations identified were more diverse. In addition to non-synonymous mutations (46.7%), INDEL (20%) and two stop-gain mutations (13.3%), splicing-related changes (20%) were also identified in TSC2 (figure 3D, left panel). Collectively, we predicted that the identified INDEL, stop-gain and splicing-related mutations would pose significant deleterious effects on TSC1/2 complex and render them functionally inactive. Similarly, non-synonymous mutations were supported by in silico prediction to confer detrimental outcomes on native protein functions. The TSC inactivation was achieved by limiting the key functional domains (TSC1-binding and GTPase activating protein (GAP) domains in TSC2; TSC2-binding domain in TSC1) and possibly through a range of mechanisms affecting the other structural regions in TSC1 and TSC2 proteins (figure 3E).
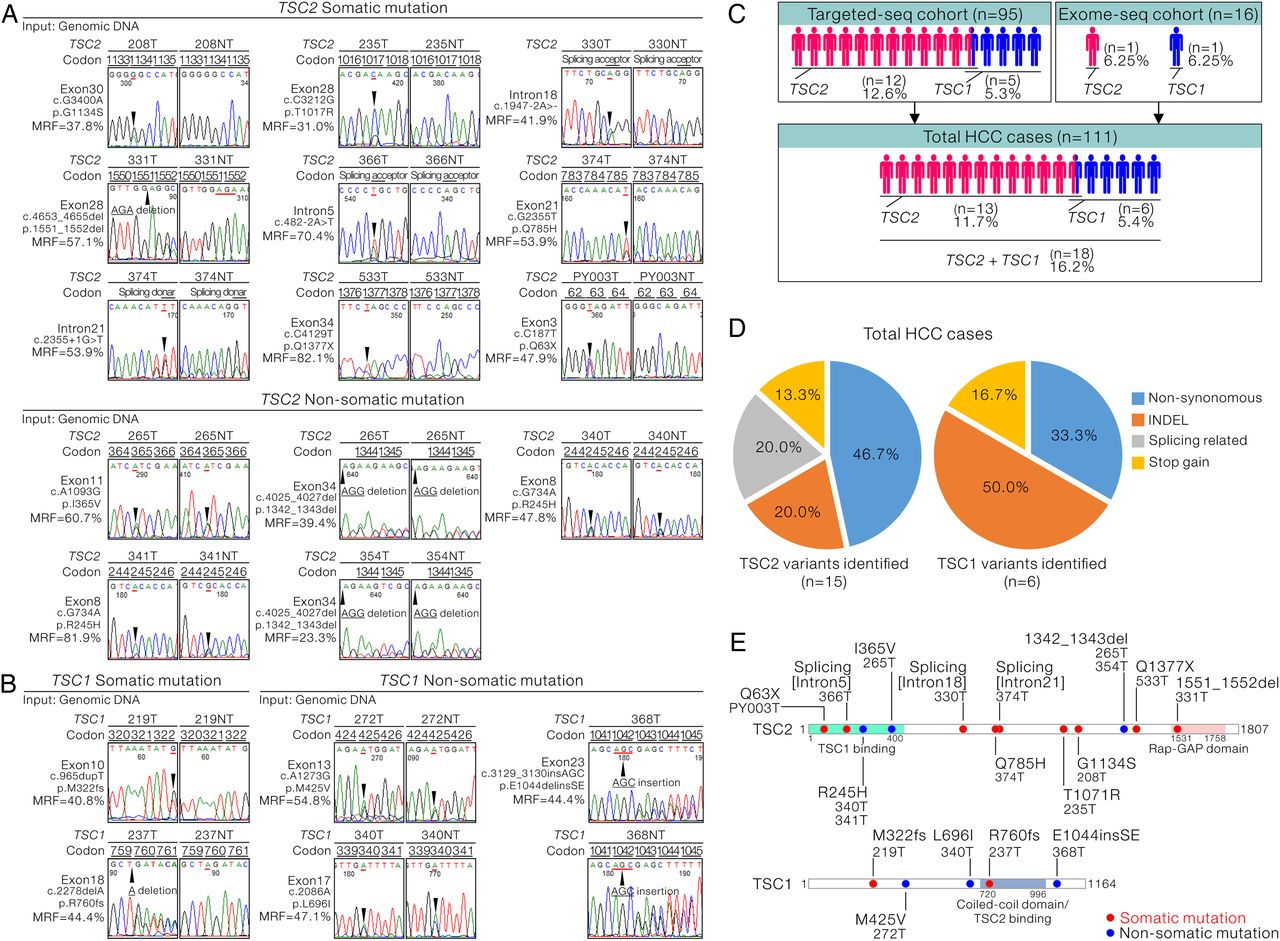
Representations of the confirmation of tuberous sclerosis complex (TSC)1/2 mutations and their possible effects in negatively regulating their tumour suppressing activity. (A) Sanger sequencing confirmation of the TSC2 mutations identified from the targeted DNA sequencing (targeted-seq) in both tumours and corresponding non-tumorous livers is shown. The identified mutations were classified as somatic and non-somatic. (B) Sanger sequencing confirmation of the TSC1 mutations identified from the targeted-seq in both tumours and corresponding non-tumorous livers is shown. The identified mutations were classified as somatic and non-somatic. (C) Flow chart summarising the number of hepatocellular carcinoma (HCC) cases carrying the TSC1 and TSC2 mutations in the discovery cohort, validation cohort or the total combined HCC cases. (D) Diagram summarising and categorising the types of TSC2 (n=15) and TSC1 (n=6) mutations found in the current study. (E) Schematic diagram showing the alterations of the amino acid changes in TSC1 and TSC2 by the identified genetic changes.
TSC1 and TSC2 expressions were reduced in human HCCs carrying TSC mutations
We examined the protein expression levels of TSC1 and TSC2 in human HCCs by IHC in the 12 cases carrying somatic TSC mutations. By scoring the intensity of TSC1 and TSC2 staining, we demonstrated a trend (p=0.05) of downregulation in TSC expression in human HCCs with TSC mutations, when compared with the NTLs (figure 4A). Increase of S6 protein phosphorylation was consistently observed in representative cases carrying somatic TSC2 mutations (figure 4B and see online supplementary figure S5A, B). In contrast, we observed some TSC2 expression in HCC cases carrying wild-type TSC2. More importantly, these wild-type cases showed low phospho-S6 signals in the tumours (figure 4C). Collectively, our results suggest that TSC2 mutations downregulates the TSC protein expression, which in turn upregulates mTOR activity, as shown by the increased phosphorylation of its downstream substrate.
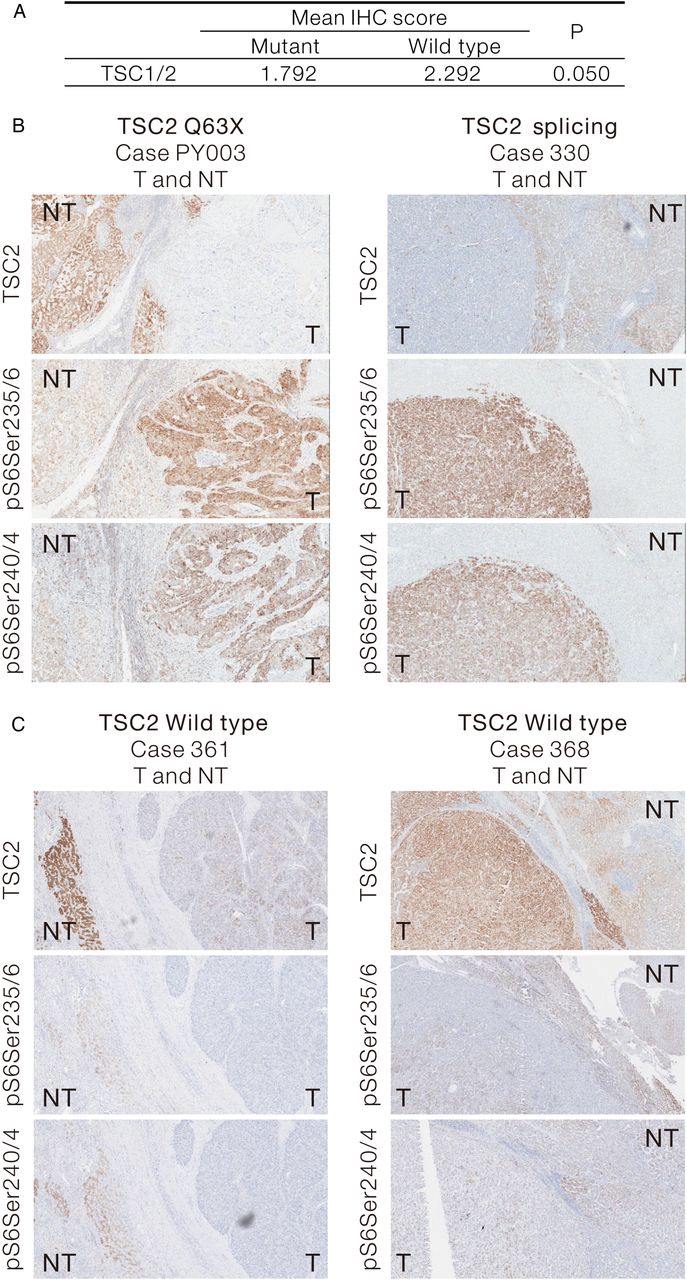
Downregulation of tuberous sclerosis complex (TSC)1/2 expression in human hepatocellular carcinoma (HCC) carrying TSC mutations is shown. (A) Twelve HCC samples carrying somatic TSC1/2 mutations and their corresponding NTL were subjected to IHC staining for TSC1/2. The TSC1/2 staining intensity in the tumour and non-tumour tissues were collectively presented as mean IHC score. Their statistical difference was compared by Wilcoxon signed-rank test (one-sided). (B) Representative IHC images showing the reduced TSC2 expression in HCC tumours carrying TSC2 non-sense mutation (TSC2 Q63X, Case PY003T) and splicing mutation (Case 330T) and their corresponding phospho-S6 staining as downstream markers for mammalian target of rapamycin pathway activation. (C) Representative IHC images showing the TSC2 expression and the downstream phospho-S6 staining in two HCC cases (TSC2 WT, Case 361T and Case 368T) with no TSC2 mutation. IHC, immunohistochemistry; NTL, non-tumorous liver.
Mutational status of TSC1 and TSC2 in HCC cell lines
Next, we examined the TSC1 and TSC2 mutational status in HCC cell lines. On WTS of seven HCC cell lines (BEL7402, HepG2, Hep3B, Huh7, MHCC97L, PLC and SMMC), we found that PLC cells carried TSC2 mutation (figure 5A), which resulted in the N1651S amino acid substitution at the C-terminal Rap-GAP domain. This mutation was confirmed by Sanger sequencing (figure 5B, E). Immunoblotting for TSC1 and TSC2 with corresponding specific antibodies revealed undetectable TSC1 plus a very low level of TSC2 expression in H2P and H2M cells (a pair of HCC cell lines with different metastatic potential established from the same patient) (figure 5C). By Sanger sequencing, we further uncovered that the undetectable TSC1 protein expression in H2P cells were resulted from the c.C1525T substitution which leads to the premature stop gain. In addition, H2P cell was also found to carry a TSC2 c.A2833G substitution which leads to a K945D mutation at the centre region of TSC2 (figure 5E). Taken together, we found that two of the eight HCC cell lines (25%) showed TSC1 and TSC2 mutations.

The mutational status and expression of tuberous sclerosis complex (TSC)1/2 in a panel of human hepatocellular carcinoma (HCC) cell lines and their sensitivity to mammalian target of rapamycin inhibitor treatment are shown. (A) RNA sequencing revealed that PLC HCC cells carried a TSC2 mutation. (B) The TSC2 mutation identified by RNA sequencing in PLC was confirmed by Sanger sequencing at genomic level. (C) Protein expression levels of TSC1 and TSC2 in a panel of HCC cell lines were determined by western blotting with the use TSC1-specific and TSC2-specific antibodies. PLC, H2P and H2M cells showed relatively low TSC1 as well as TSC2 protein expression. H2M was derived from H2P. (D) Sanger sequencing revealed a stop-gain mutation in TSC1 and a missense mutation in TSC2 in H2P cells. The stop-gain mutation may account for the loss of TSC1 protein expression in H2P cells. (E) Schematic diagram showing the TSC1 and TSC2 mutants identified in PLC and H2P cells. (F) Eight HCC cell lines with defined mutational status of TSC1/2 were subjected to rapamycin treatment at the indicated concentrations for 4 days, followed by fixation and crystal violet staining for visualisation.
mTOR Inhibitor effectively suppressed cell proliferation in TSC-mutant HCC cells
Loss-of-function mutation of TSC1/2 relieves Rheb G-protein from inactivation and leads to hyperactivation of mTORC1 activity. We sought to find out the functional implications of TSC mutation in HCC regarding to the tumour sensitivity towards mTOR inhibitor treatment. Eight HCC cell lines with well-defined TSC1/2 mutation status were plated and subjected to rapamycin treatment at three different concentrations ranging between 1, 10 and 100 nM (figure 5F) for 4 days. Among the six HCC carrying wild-type TSC1/2, three of them (Huh7, 97L and HepG2) were sensitive to rapamycin treatment starting from 10 nM, while the remaining three (SMMC, BEL7402 and Hep3B) were insensitive even at the highest concentration. Interesting, for PLC and H2P cells carrying TSC1/2 mutations, their proliferation was marked suppressed by rapamycin treatment at the minimal dosage. The hypersensitivity to the low mTOR inhibitor treatment supports the idea that TSC1/2 mutation may serve as a potential biomarker to assess the reliance of HCC cells on mTOR signalling and predicts their responsiveness to mTOR inhibitor treatment. To further evaluate and mimic the effect of restoration of TSC2 expression in HCC cells, we stably knocked down Rheb in TSC2 wild-type SMMC cells and TSC2-mutant PLC cells by lentiviral-based small hairpin (sh)RNA approach (see online supplementary figure S6A, B). Although stable shRheb SMMC cells could be established (see online supplementary figure S6C), on the contrary, stable shRheb PLC cells could not be established after lentiviral transduction and subsequent puromycin selection. It was likely that TSC2 mutation renders PLC cells to be Rheb-dependent and expression of shRNA against Rheb drastically suppressed its proliferation (see online supplementary figure S6D).
TSC2 mutation-bearing HCCs were hyper-responsive to mTOR inhibitor in PDTX model
To further recapitulate our in-vitro observation, we used two of our HCC PDTX models who carried biologically relevant TSC2 stop-gain mutations (PDTX#5: TSC2 Q1377X and PDTX#9: TSC2 Q63X) and a PDTX with wild-type TSC2 (PDTX#3) as the control. The PDTX models were subjected to rapamycin treatment daily and the body weight and tumour size were monitored. Rapamycin treatment at a concentration of 1.0 and 2.5 mg/kg was both well tolerated, as the body weights of the treatment group were similar to those of the vehicle control (figure 6A). Rapamycin at either 1.0 or 2.5 mg/kg was sufficient to almost completely suppress tumour growth in PDTX#5 (figure 6B, left panel). We then used rapamycin at 1.0 mg/kg as the working concentration. Both PDTX#9 (figure 6B, middle panel) and PDTX#5 (figure 6B, right panel) showed a similar remarkable tumour-suppressive response with rapamycin. In contrast, in PDTX#3 with wild-type TSC2, the rapamycin-treated tumours increased in volume progressively, though moderately (figure 6B, right panel). As an indication of poorer response towards rapamycin treatment, the relative tumour volume (rapamycin: vehicle) of PDTX#3 was significantly higher than those of PDTX#5 and PDTX#9, starting from day 12 onwards (t-test, p<0.001) (figure 6C). A greater difference in tumour size between the vehicle-treated and rapamycin-treated tumours (figure 6D) as well as a higher mean tumour mass ratio (vehicle:rapamycin) was seen in both PDTX#5 and PDTX#9, as compared with PDTX#3 (figure 6E). Interestingly, very small tumours with nearly negligible tumour mass were observed in rapamycin-treated tumours in PDTX#5 and PDTX#9 but not PDTX#3. The hyper-sensitivity towards mTOR inhibitor treatment was further supported by the significantly lower relative tumour mass (rapamycin/vehicle) of PDTX#5 and PDTX#9 when compared with PDTX#3 (t-test, p<0.001) at the end point of experiment (figure 6F). Taken together, our results suggested an overall treatment benefit with the use of mTOR inhibitor in HCC and the treatment response was superior especially in those tumours which bore mutations leading to the activation in this pathway. These observations demonstrated and suggested the therapeutic implication and translational potential of the identified TSC1/2 mutants in human HCCs.

Hepatocellular carcinoma (HCC) patient-derived tumour xenograft (PDTX) models with tuberous sclerosis complex (TSC)2 mutations were more sensitive to mammalian target of rapamycin inhibitor treatment. (A) The graph showing body weight of TSC2-mutant (PDTX#5 and PDTX#9) and TSC2 wild-type (PDTX#3) PDTX tumour-bearing mice throughout the treatment. Body weights were measured every 3 days to assess the general toxicity of the injected vehicle and rapamycin. The relative body weights (rapamycin/vehicle) of PDTX#5, PDTX#9 and PDTX#3 mice are shown. (B) The tumour volumes of the PDTX tumour-bearing mice subjected to vehicle or rapamycin treatment at 1.0 mg/kg/day, unless stated otherwise, are shown. The volumes of the subcutaneous tumours were measured every 3 days throughout the treatment. The numbers in grey represent the mean volume ratio between the vehicle and rapamycin-treated group at the indicated time points. t-test; *p<0.05, **p<0.01; ***p<0.001. (C) The relative tumour volumes (rapamycin/vehicle) of PDTX#5 and PDTX#9 tumours are shown alongside with those of PDTX#3. t-test; ***p<0.001. (D) An overview of the subcutaneous tumours formed and the corresponding dissected tumours at the end point of the experiment is shown. (E) The tumour masses of the dissected tumours at the end point of the treatment are shown. The numbers in grey represent the mean tumour mass ratio between the vehicle and rapamycin-treated groups. t-test; *p<0.05, **p<0.01; ***p<0.001. (F) The relative tumour masses (rapamycin/vehicle) of PDTX#5, PDTX#9 and PDTX#3 are shown. t-test; **p<0.01; ***p<0.001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Continued
Discussion
Despite mutations have been recurrently detected in genes responsible for cell cycle regulation (TP53), cell proliferation (CTNNB1) and switch/sucrose non-fermentable (SWI/SNF) ATP-dependent chromatin remodelling (ARID1A), consensus among mutations or mutant genes detected in recently performed NGS-based studies is suboptimal. High genetic heterogeneity remains the greatest challenge for the delineation of HCC molecular mechanisms. Based on evidence extracted from our NGS-based datasets, extensive components of mTOR signalling cascade are mutated in human HCCs. These data provide novel insight in suggesting significant mutation-dependent regulation mechanism on mTOR signalling, with multiple key and canonical mTOR pathway-related components being shortlisted and confirmed to harbour frequent mutational disruptions. These gene targets are attractive and potentially promising targets for further investigations to further understand the underlying mechanistic processes in driving mTOR hyperactivation.
PI3K/Akt/mTOR pathway has recently drawn great attention as an alternative therapeutic, druggable pathway in HCC to enhance the limited survival advantage delivered by sorafenib treatment. Our group and others have previously demonstrated the overexpression of mTOR in more than 50% of the HCCs.6 ,8 mTOR Signalling is tightly regulated by upstream signalling in response to extracellular growth factor, stresses and nutrients. The signalling axis is dually regulated by both phosphatase and tensin homolog (PTEN) and TSC1/2. We had previously shown that PTEN expression was downregulated in our HCCs from patients and the downregulation was associated with increased invasive behaviour in HCC.9 Intriguingly, our targeted-seq data further revealed that PTEN was rarely mutated in HCC (identified only in a single case among the 95 patient samples). In the current study, we identified a common mechanism in a subset of patients with HCC carrying TSC mutations, which predispose them to have deregulated mTOR activity. Currently, very limited information is known about the expression of TSC1 and TSC2 in human HCCs. A single brief report used IHC to demonstrate the under-expression of TSC2 in human HCCs. The under-expression was also found to be associated with advanced tumour stage, vascular invasion and poor prognosis.10 The inactivation of TSC1/2 complex has been found to be significantly relevant to HCC development, as elegantly demonstrated by the liver-specific TSC1 knockout mouse model.11 ,12 Menon et al have demonstrated that about 80% of the TSC1-null livers developed spontaneous tumours at the age of 9–10 months and 50% of them resembled HCC. HCC formation in TSC1 knockout was found to be a result of chronic mTORC1 activation. The involvement of the chronic mTORC1 kinase activity was further supported by the effective blocking of the TSC1-null tumour formation with the early administration of rapamycin at the age of 5 months before the appearance of macroscopic lesion.12
In the current study, somatic mutations were detected in known HCC genes including TP53, CTNNB1, AXIN1 and ARID1A, revealing the significant involvement of genomic aberrations in the relatively better-characterised genes of HCC. In addition, various key components of the mTOR pathway were identified to be altered by somatic mutations, which highlighted its potential importance and possible translational implication through intervention with the use of mTOR inhibitor in susceptible mutant patients. Based on frequency as well as the clinicopathological correlation, TSC mutation may likely represent a major single driving factor in hepatocarcinogenesis, which confers at least comparable effect to known HCC drivers such as TP53, CTNNB1, AXIN1 and ARID1A.
The study reported by Schulze et al 13 and Totoki et al 14 are currently the latest and largest one, among the reported NGS-based mutation screening studies. Their findings are based on NGS data of 243 cases and 503 cases, respectively. However, HBV infection only accounted for up to a quarter of their cases while our cohorts were entirely HBV-positive. With our study focusing entirely on HBV-associated HCC, homogeneity of the aetiology hopefully could empower us to achieve better intra-study consensus, given our limited sample size. We believe such difference in the underlying aetiological risk factor may likely be responsible for the apparently higher frequency of TSC mutations detected in our cohorts. Such frequent disruption (>15%) on TSC regulation of mTOR activity probably suggests a profound effect of mTOR activation by TSC disruption in HBV-associated hepatocarcinogenesis. Intriguingly, in echo with our current findings, we noted two recent studies highlighting the importance of mTOR signalling in HCC. Janku et al 15 reported a mutation screening on a confined panel of 182 cancer-related genes. More recently, another study concurrently highlighted the hyperactivation of mTOR signalling in hepatocarcinogenesis. Based on a seemingly retrospective approach in explaining their available clinical trial data on mTORC1 inhibitor everolimus, they provided explanatory data pinpointing on TSC2 loss-of-function leading to elevated mTOR signalling.16 More importantly, using HCC cell lines and xenografts, they demonstrated sensitivity to everolimus according to TSC2 status as assayed by IHC. Their sample collection of Asian HBV-positive HCCs (∼20%) frequently identified TSC2 loss and such patients receiving everolimus tended to have longer overall survival than those receiving placebo. In spite of their nice work in highlighting the therapeutic and prognostic value TSC2, the mechanism underlying TSC2 loss in HCC remains unclear. Beyond what they have reported, our current findings systematically delineated the comprehensive mutational landscape for mTOR signalling genes and discovered frequent mutations in TSC1/2 on regulating mTOR signalling. The high frequency of TSC mutations has provided key and novel explanation in addressing the underlying molecular mechanism for the TSC loss in hyperactivation of mTOR signalling. We further demonstrated, through PDTX models carrying TSC2-inactivating mutations, that hyperactivation of mTOR signalling via mutational disruption of TSC regulation is sensitive to rapamycin treatment. PDTXs are established by implanting primary tumours into immunodeficient mice. It provides better conservation and representation of natural tumour heterogeneity and architecture to physiological human tumour. With the use of PDTX model, findings of preclinical testing on drug response will be more relevant and predictive than traditional cancer cell line model.17 Through the perfect combination of both high-throughput NGS-based mutation screening and PDTX model, our study demonstrates, with necessary supportive evidence correlating mutational findings with corresponding drug testing, the potential personalised treatment on relevant patients with HCC carrying specific molecular alterations. These preclinical findings open up the possibility for using rapamycin or its subsequent derivatives as an alternative or in complement with sorafenib in treating specific mTOR hyperactivated patients. We believe TSC mutations represent one of the major and previously undocumented mechanisms in leading to mTOR hyperactivation in HBV-associated HCC. Nevertheless, we strongly agree that mTOR inhibition is likely to be a rational therapeutic option for patients demonstrating mTOR hyperactivation, regardless of its underlying mechanism. Therefore, the treatment efficacy of mTOR inhibition treatment would be greatly improved with further exploration and establishment of the stratification strategy on mTOR hyperactivation status not limited to TSC mutation.
In summary, our study has substantiated the significance of mTOR signalling in HCC through evidence pinpointing its novel mutational aspect with mutant genes recurrently spotted in multiple cases as well as critical mutants in multiple pathway components. Our findings imply the potentially profound involvement of mTOR signalling in HCC and draw further attention on personalised therapeutic option for a novel molecular subset of patients with susceptible mutant HCC, through the inhibition of mTOR signalling.
References
Footnotes
DWHH and LKC contributed equally.
Contributors IOLN and CMW provided study concept and design. DWHH, LKC, MXL and YTC collected and analysed the data. DWHH, LKC, MXL, YTC, IOLN and CMW interpreted the data. LKC, IMJX and YTC performed the experiments. ILOL, DTWY, PWYL, CNT, VWLT, RTPP and TTC collected the patients' samples. LKC, DWHH, IOLN and CMW wrote the manuscript. All authors approved the final version of manuscript.
Funding The study was supported by Hong Kong Research Grants Council General Research Fund (17116414), Research Grants Council Theme-based Research Scheme (T12-704116-R), SK Yee Medical Research Fund 2011, University Development Fund of the University of Hong Kong and Lee Shiu Family Foundation. IOLN is Loke Yew Professor in Pathology.
Competing interests None declared.
Ethics approval Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Commentary