Article Text

Abstract
Objectives Oesophageal squamous cell carcinoma (OSCC) and adenocarcinoma (OAC) are distinct cancers in terms of a number of clinical and epidemiological characteristics, complicating the design of clinical trials and biomarker developments. We analysed 1048 oesophageal tumour-germline pairs from both subtypes, to characterise their genomic features, and biological and clinical significance.
Design Previously exome-sequenced samples were re-analysed to identify significantly mutated genes (SMGs) and mutational signatures. The biological functions of novel SMGs were investigated using cell line and xenograft models. We further performed whole-genome bisulfite sequencing and chromatin immunoprecipitation (ChIP)-seq to characterise epigenetic alterations.
Results OSCC and OAC displayed nearly mutually exclusive sets of driver genes, indicating that they follow independent developmental paths. The combined sample size allowed the statistical identification of a number of novel subtype-specific SMGs, mutational signatures and prognostic biomarkers. Particularly, we identified a novel mutational signature similar to Catalogue Of Somatic Mutations In Cancer (COSMIC)signature 16, which has prognostic value in OSCC. Two newly discovered SMGs, CUL3 and ZFP36L2, were validated as important tumour-suppressors specific to the OSCC subtype. We further identified their additional loss-of-function mechanisms. CUL3 was homozygously deleted specifically in OSCC and other squamous cell cancers (SCCs). Notably, ZFP36L2 is associated with super-enhancer in healthy oesophageal mucosa; DNA hypermethylation in its super-enhancer reduced active histone markers in squamous cancer cells, suggesting an epigenetic inactivation of a super-enhancer-associated SCC suppressor.
Conclusions These data comprehensively contrast differences between OSCC and OAC at both genomic and epigenomic levels, and reveal novel molecular features for further delineating the pathophysiological mechanisms and treatment strategies for these cancers.
- oesophageal cancer
- gene mutation
- methylation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Oesophageal squamous cell carcinoma (OSCC) and adenocarcinoma (OAC) exhibit a number of different clinical and epidemiological characteristics.
The genomic landscapes of both OSCC and OAC have been established; however, molecular features at both genomic and epigenomic levels have not been systematically compared between these two subtypes.
Significantly mutated genes (SMGs) have been discovered in OSCC and OAC; however, due to limited number of samples in individual studies, many additional SMGs await to be identified.
What are the new findings?
OSCC and OAC display strikinglyfig distinct sets of driver genes, mutational signatures and prognostic biomarkers.
A novel mutational signature is discovered as OSCC-specific; it is also correlated with the survival time of patients with OSCC.
A number of novel subtype-specific SMGs are identified, two of which (CUL3 and ZFP36L2) are functionally validated as tumour-suppressors.
SCC-specific hypermethylation of the super-enhancer of ZFP36L2 silences this tumour suppressor.
How might it impact on clinical practice in the foreseeable future?
This work reveals many novel molecular features that will help develop novel treatment strategies for both patients with OAC and OSCC. Specifically, the molecular similarity between OSCC and other SCCs (likewise between OAC and stomach adenocarcinoma) suggests a unified perspective of these malignancies which has important implications in the design of future clinical trials.
Several gene mutations and mutational signatures are correlated with the overall survival of patients with oesophageal cancer, which might serve potential prognostic biomarkers.
Introduction
Ranking sixth in cancer mortality and eighth in incidence worldwide,1 oesophageal cancer is histologically classified as either squamous cell carcinoma (OSCC) or adenocarcinoma (OAC). These two subtypes have unique clinical and epidemiological characteristics. For instance, geographically, OSCC predominantly occurs in Eastern Asia and some regions of Africa, while OAC is more common in Western countries and its incidence is rising rapidly.2 The risk factors for OSCC include smoking and alcohol consumption; OAC is associated with Barrett’s oesophagus and reflux. Effective targeted agents are unavailable in either subtype, partly due to pervasive intertumour and intratumour heterogeneity.3–5
Recent sequencing efforts characterising OSCC and OAC genomes have revealed many significantly mutated genes (SMGs) using computational frameworks that model the genomic features of potential driver and passenger mutations.6–12 However, our understanding of driver genes in oesophageal cancer is far from complete, as these individual studies typically profiled 100–150 individuals. Recent saturation analysis has estimated that, in order to confidently identify SMGs present in 2%–3% of the population, an analysis of 1000–2000 tumour-normal pairs are required based on the background mutational rate of oesophageal cancer.13 We, therefore, reasoned that combining samples across data sets and performing an SMG re-analysis will allow us to identify additional driver genes, by increasing the statistical power to distinguish infrequent driver mutations from passengers. This re-analysis will also permit a comprehensive comparison of the SMG landscape between OSCC and OAC, providing the molecular basis underpinning the distinct clinical and pathological presentations of these two subtypes beyond those previously identified in individual studies.6 14 In addition, an unbiased and systematic comparison of the mutational patterns (eg, mutational signatures, clonal composition) of these two subtypes might shed light into the different pathophysiologies of OSCC and OAC.
Material and Methods are provided in online supplementary information.
Supplementary file 1
Results
Subtype-specific driver genes in oesophageal cancer
To compare comprehensively the mutational landscapes between OSCC and OAC, mutational profiles from large-scale genomic studies were aggregated,7–12 including those from The Cancer Genome Atlas (TCGA)6 and International Cancer Genome Consortium (ICGC).15 In total, we compiled 602 and 446 tumour-germline pairs for OSCC and OAC, respectively, representing the largest collection of each subtype to date (see online supplementary table 1). Associated clinical-pathological parameters were retrieved, and prior to analysis, we confirmed that important parameters were generally comparable between different cohorts, including age at diagnosis, gender, primary tumour size, as well as distant metastasis. The status of lymph node involvement was also largely consistent among different cohorts, except for the one reported by Sawada et al 10 which contained more cases of lymph node metastasis (see online supplementary figure 1). To minimise multicentre bias and batch effects due to differences in either sequencing platforms or analytical pipelines, extensive curation, reannotation and filtering steps were performed (see Material and Methods). With the awareness that these steps could not entirely eliminate intrinsic biases, results of individual analyses were examined for batch effects; for instance, mutation spectra across different studies were highly similar, and differences between OSCC and OAC subtypes were reproducible within all cohorts (see online supplementary figure 2). Additional analysis of SMGs also strongly suggested that cohort bias or batch bias had a minimal impact in the downstream analysis (see below).
Supplementary file 2
Supplementary file 3
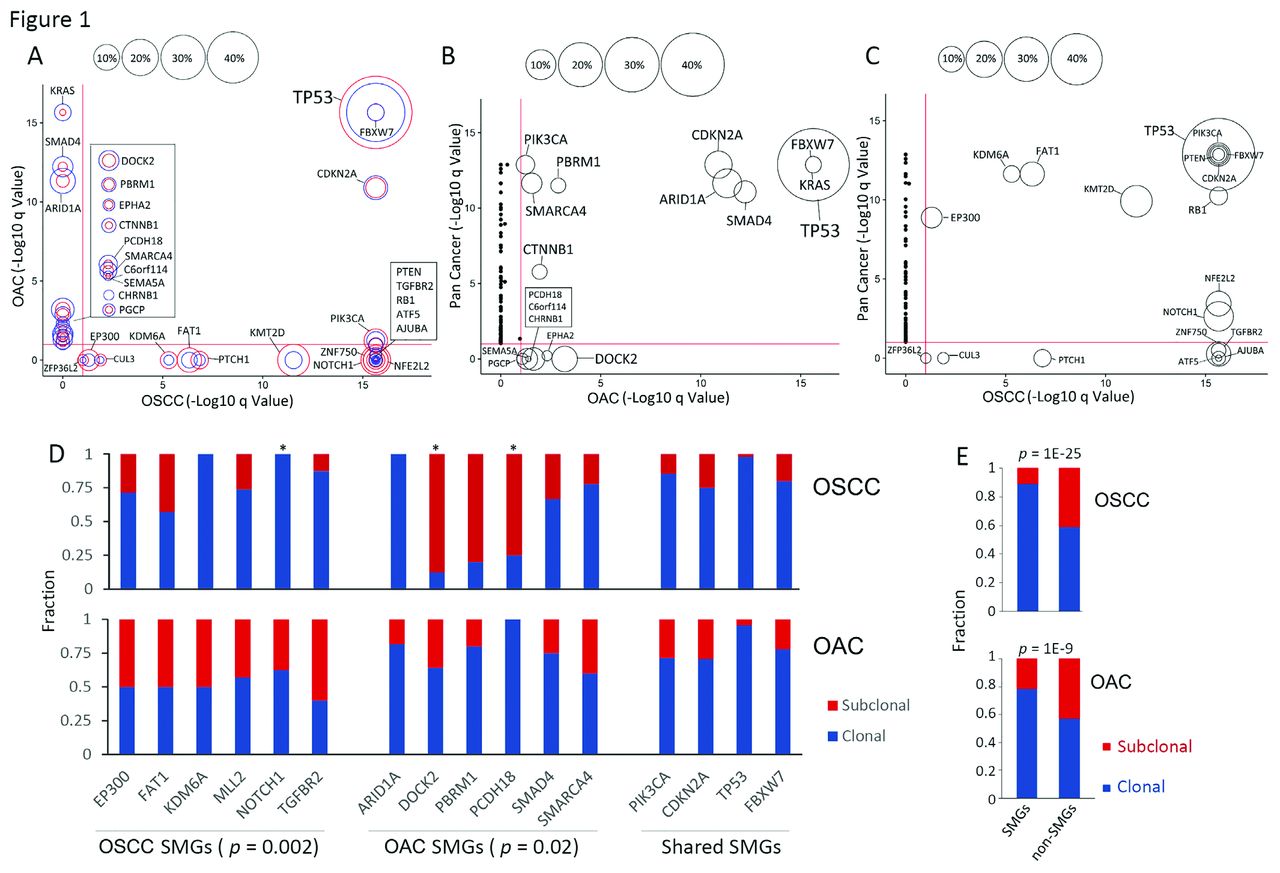
SMGs in OSCC, OAC and other cancer types. SMGs identified by MutSig2CV are shown on the basis of their q values and mutational fractions. Comparisons were made between OSCC against OAC (A), OAC against pan-cancer (B), OSCC against pan-cancer (C). In (A), the red circle denotes the fraction in OSCC, and the blue circle denotes OAC. q values in 20 other tumour types were retrieved from the TCGA pan-cancer study.13 (D) CCF analysis of SMGs. Only SMGs with at least three mutations in both cancer types are shown. p Values were derived by Fisher’s exact tests. *, p<0.05; (E) The fraction of clonal and subclonal mutations in both SMGs and non-SMGs. CCF, cancer cell fraction; OAC, oesophageal adenocarcinoma; OSCC, oesophageal squamous cell carcinoma; SMG, significantly mutated gene; TCGA, The Cancer Genome Atlas.
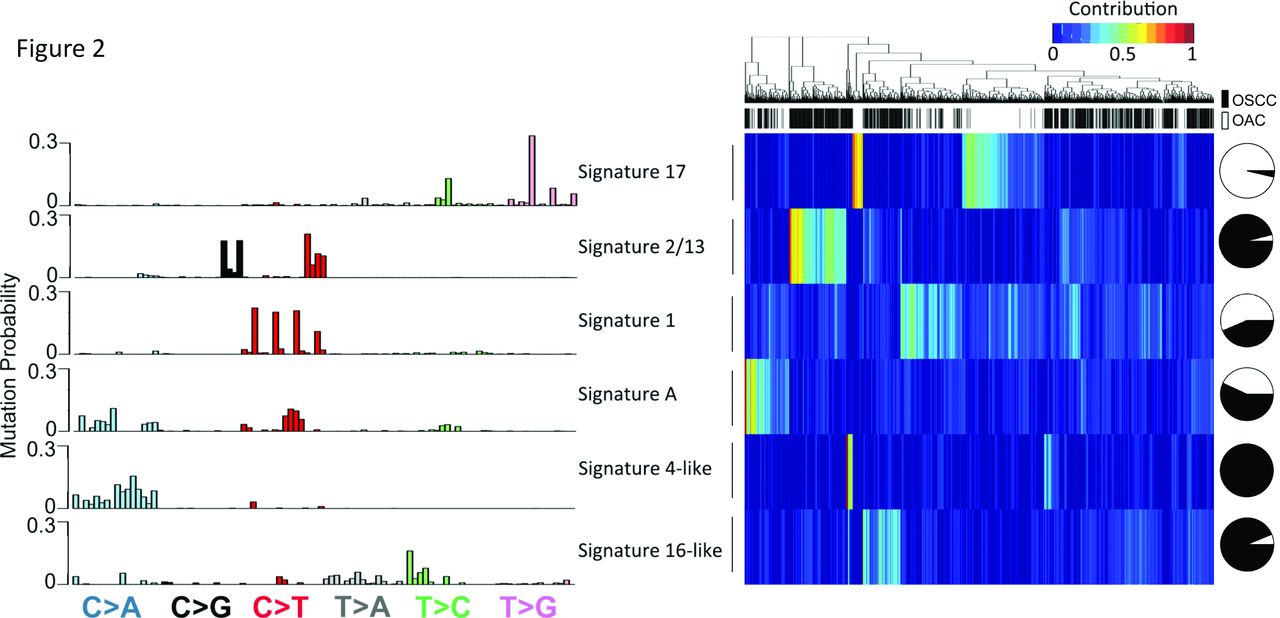
De novo mutational signature analysis of both types of oesophageal cancers. (Left panel) 96 trinucleotide substitutions of 6 mutational signatures identified by the NMF method in oesophageal cancers. (Right panel) Heatmap showing unsupervised hierarchical clustering of both OSCC and OAC tumours based on the weight of each signature in each sample. Black, OSCC; white, OAC. Pie chart showing the fraction of samples from either subtypes having the indicated signature. OAC, oesophageal adenocarcinoma; OSCC, oesophageal squamous cell carcinoma; NMF, negative matrix factorisation.
We first identified SMGs using MutSigCV,13 after excluding genes with undetectable expression in TCGA RNA-seq data, similarly as we and others have performed previously9 16 17 (see Methods). With q value=0.1 as a cut-off, 19 and 17 genes were significantly mutated in OSCC and OAC, respectively (figure 1A). Confirming previous findings, the majority of SMGs were well established oesophageal cancer genes, with highly similar mutational frequencies across individual cohorts and the combined cohort (see online supplementary figure 3). Notably, of these 32 genes, only 4 (TP53, CDKN2A, FBXW7 and PIK3CA) were significantly mutated in both subtypes. In fact, SMGs from these two tumour types had a greater overlap with other cancer types than they had with each other (figure 1B,C). OSCC shared more SMGs with squamous cell cancer (SCC) of the lung (LUSC), head and neck (HNSC) as well as the bladder (BLCA, a significant proportion of which are molecularly squamous-like18) than any other tumour types (see online supplementary figure 4). This similarity suggests that some of these SMGs are uniquely required to promote the transformation of the squamous cell lineage, in keeping with a recent molecular classification.18 On the other hand, SMGs of OAC had more in common with those from stomach adenocarcinoma (STAD), supporting a recent finding of the molecular similarity between these two cancer types.6 Importantly, the large number of samples empowered us to identify many novel SMGs which were mutated at moderate or low frequencies, including ATF5, PTCH1, CUL3, ZFP36L2 in OSCC and EPHA2, PCDH18, C6orf114, CHRNB1, PGCP in OAC. Some of these, like ATF5 and ZFP36L2, had mutations that occurred at low frequency but were associated with very strong features of driver mutations, leading to significant q values. We investigated some of these novel SMGs in more detail, as shown below. Several lines of evidence (see online supplementary figure 3; Methods) verified that these novel SMGs were consistently mutated across cohorts; that is, they were the result of increased statistical power associated with sample pooling, rather than cohort-specific biases.

Survival-associated mutational signatures and SMGs. (A) Kaplan Meier survival analysis was performed for both OSCC and OAC; p value was determined using log-rank test. (B) Cox regression analyses was performed to identify independent prognostic factors. See also online supplementary figure 6. OAC, oesophageal adenocarcinoma; OSCC, oesophageal squamous cell carcinoma; SMGs, significantly mutated genes.
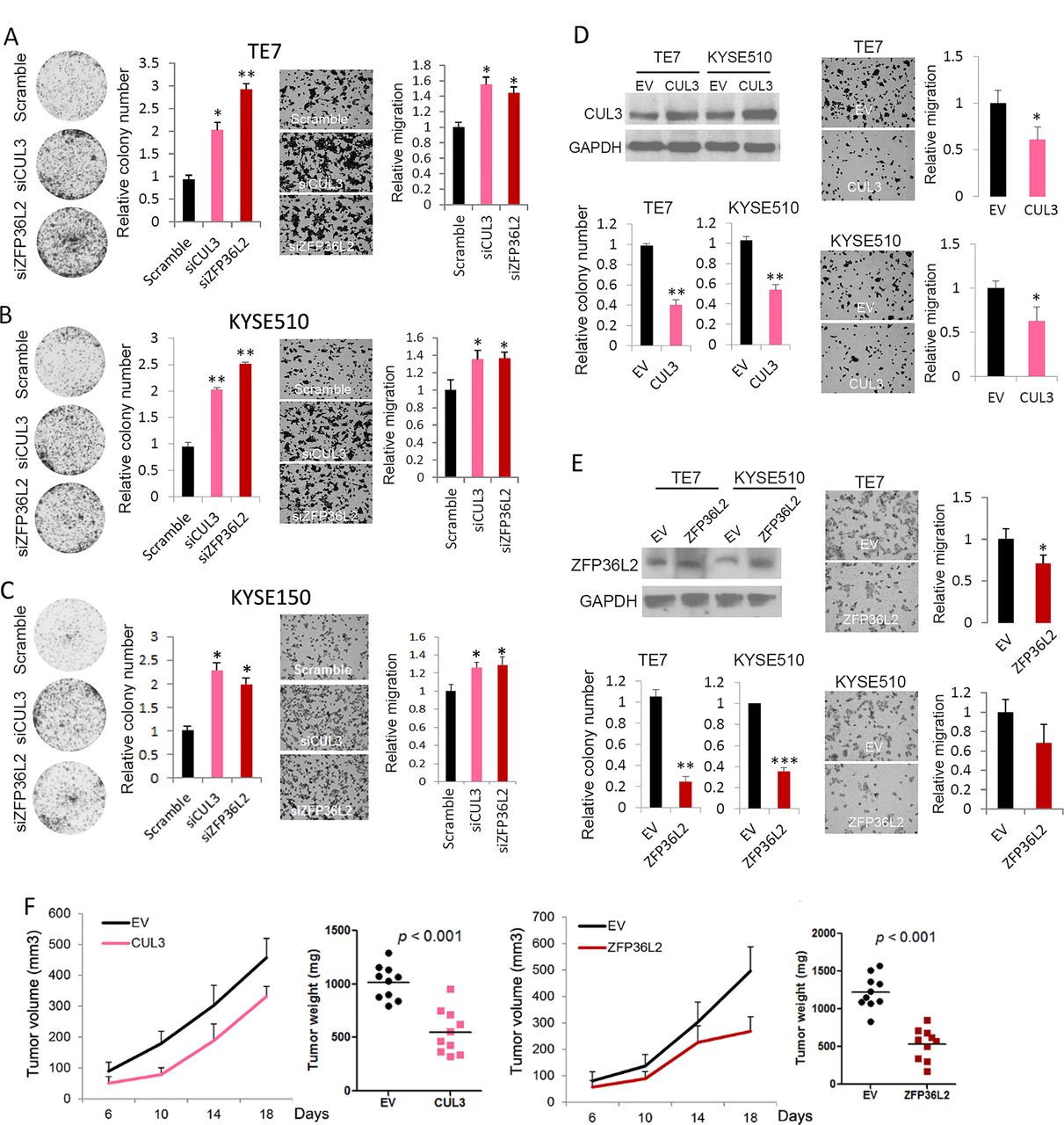
Tumour-suppressive property of CUL3 and ZFP36L2 in oesophageal squamous cell carcinoma (OSCC) cells. CUL3 and ZFP36L2 expression were depleted via siRNAs in TE7 (A), KYSE510 (B) and KYSE150 (C) cell lines and subjected to colony formation and cell migration assay. (D,E) TE7 and KYSE510 cells ectopically expressing either CUL3 (D) or ZFP36L2 (E) were subjected to western blotting (upper left), colony formation (lower left), as well as cell migration assays (right). Data represent mean±SD. Values were determined using t-test; *, p<0.05; **, p<0.01; ***, p<0.001. (F) TE7 cells ectopically expressing either CUL3 or ZFP36L2 were grown as xenograft; tumour volumes and weight are shown. Horizontal bar denotes mean value.
We next determined the clonal status of these SMGs by assessing their cancer cell fractions through integrative analysis of tumour cellularity (online supplementary table 2), variant allele frequency as well as variant copy number, as described previously.3 19 20 Notably, subtype-specific SMGs tended to be significantly more clonal in the corresponding subtype (figure 1D). Specifically, OSCC-specific SMGs had more clonal mutations in OSCC relative to OAC (p=0.002), and vice versa (p=0.02). This tendency was observed in the majority of subtype-specific SMGs, although only three genes (NOTCH1, DOCK2 and PCDH18) reached statistical significance because the number of mutations having matched copy number value was small. In contrast, shared SMGs exhibited similar clonality between the two subtypes (figure 1D, right). This result demonstrates that mutations within these SMGs in the non-corresponding subtype (eg, MLL2 mutations in OAC, or DOCK2 mutations in OSCC) arise late during tumorigenesis and may reflect random passenger events rather than drivers. Not surprisingly, SMGs generally had significantly more clonal mutations compared with non-SMGs in both malignancies, in line with the findings in most cancer types reported by us and others19 20 (figure 1E).
Mutational signatures in oesophageal cancer
To compare the mutational processes operative between OSCC and OAC, we performed de novo signature analysis based on negative matrix factorisation framework.21 22 Mutational signature patterns of these two subtypes were conspicuously distinct from each other (figure 2). For example, signature 17 (associated with gastric acid reflux) stood out as the most prominent OAC-specific signature, in agreement with the findings from an ICGC Study which was based on whole-genome sequencing data.11 OAC cases dominated by signature 17 were found to have more mutations in DOCK2 and PCDH18 (online supplementary table 3). Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) signatures (signatures 2 and 13, associated with APOBEC deaminases activity) were exclusively associated with OSCC tumours, validating recent observations from our group3 and others.6 23 Notably, APOBEC signature-positive OSCC samples had significantly more mutations targeting driver genes including ZNF750, PIK3CA, MLL2, MLL3 and RB1 (online supplementary table 3), with some of these associations having been reported previously.11 Not surprisingly, signature 1, associated with age of cancer diagnosis, was detected in both subtypes (figure 2).
Importantly, the large sample size enabled the discovery of novel mutational signatures which have not been established in these cancers. Signature 16-like, which was very similar to the established signature 16, was detected specifically in OSCC cases (figure 2). Signature 16 is characterised by T > C transitions at ApT dinucleotides with a strong transcriptional strand bias (figure 2). With unknown aetiology, signature 16 has been observed only in liver cancer.22 24 We noted a signature highly similar with signature 4 (signature 4-like) exclusively in OSCC. Signature 4 features frequent C > A mutations resembling a mutational profile induced in vitro by exposing cells to benzo[a]pyrene (a tobacco carcinogen). Indeed, a recent statistical study linked signature 4 directly to tobacco exposure in many cancer types,25 which has been confirmed by the TCGA consortium.6 Due to the lack of smoking history, we could not test the association between signature 4-like and tobacco exposure in the combined cohort. In addition, we found a novel signature, signature-A, which exhibited both C > A transversion and C > T transitions (with a sharp increase in frequency in GCN context) in both OSCC and OAC cohorts. Interestingly, signature-A-positive cases had significantly more ATM mutations in both oesophageal cancer subtypes (online supplementary table 3), indicating that a link between DNA damage and this signature might exist. Supporting this hypothesis, enriched C > T mutations in the GCN context are also a feature of the established signature 15, which is associated with defective DNA mismatch repair in STAD and small cell lung cancers.22 Since signature-A has not been observed in any cancer types, its aetiology remains to be explored.
To validate the stability and robustness of these mutational signature results, a different statistical framework, pmsignature, was used for comparison. As these two methods are based on different statistical models, we expected to see some level of disagreement. Despite this, highly consistent patterns were observed in five out of six signatures between these two different analytical methods (online supplementary figure 5A), validating our present results. Even the only signature that was not in strong agreement (signature 4-like) did show a highly statistically significant correlation. Moreover, we confirmed highly consistent contributions of each mutational signature in each individual cohort, thus ruling out cohort-specific biases (online supplementary figure 5B).

Genomic abnormalities of CUL3 enhance both NRF2 and wingless in Drosophila (WNT)/β-catenin pathways. (A) Integrative Genomics Viewer (IGV) snapshot of CUL3 homozygous deletions in TCGA pan-SCC samples. Relative copy number value was denoted by the colour bar. (B) Frequency of homozygous deletions and somatic mutations in CUL3 across cancer types. Squamous lineage tumours are highlighted in pink squares. BLCA is also highlighted as a significant proportion of them are molecularly squamous-like. Insert panel showing the distribution of CUL3 somatic mutations in both OSCC and pan-SCC (including LUSC, HNSC and cervical squamous cell carcinoma (CESC), summarised from TCGA results). Black and purple dots, truncating mutations; green dots, missense mutations. pRCC, papillary renal cell carcinoma; LUAD, lung adenocarcinoma; ccRCC, clear cell renal cell carcinoma; ACC, adenoid cystic carcinoma; BRCA, breast invasive carcinoma; HCC, hepatocellular carcinoma; chRCC, chromophobe renal cell carcinoma; DLBC, diffuse large B cell lymphoma; GBM, glioblastoma multiforme; PCPG, pheochromocytoma and paraganglioma. (C) mRNA levels of CUL3 in TCGA pan-SCC cohorts. Samples are grouped based on their gene dosage. Hem Loss, heterozygous deletion; Hom Del, homozygous deletion. (D) CUL3 was either silenced by siRNA (left) or ectopically expressed (right) in OSCC cells, followed by western blotting analysis or (E) qRT-PCR assay. Scr, scramble siRNA; EV, empty vector. p Values were determined using t-test; ***, p<0.001. (F) Comparison of β-catenin or NRF2 protein levels between CUL3 intact (wildtype (WT)) against mutant or deleted (altered (ALT)) TCGA SCC samples. (G) Colony formation assay demonstrating the synergistic effect of ICG001 and Trig. (H) A model of CUL3 genomic inactivation in SCC cells and its implication. BLCA, bladder cancer; LUSC, squamous cell cancer of the lung; HNSC, squamous cell cancer of the head and neck; OSCC, oesophageal squamous cell carcinoma; SCC, squamous cell carcinoma; STAD, stomach adenocarcinoma; TCGA, The Cancer Genome Atlas;
Identification of novel genomic prognostic biomarkers
Available follow-up data in this large group of patients with oesophageal cancer also allowed us to explore the prognostic value of driver genes, as well as mutational signatures. As a validation, all existing clinical prognostic parameters, including primary tumour size, lymph node involvement, as well as distant metastasis were strongly associated with patients’ overall survival (online supplementary figure 6), confirming that the combined cohort was valid for assessing prognostic biomarkers. We found that mutations of several driver genes (all SMGs except for BRCA1, which was tested because it has been associated with treatment response and survival in oesophageal cancer26–28) were associated with patients’ outcome in univariate log-rank analysis, including EP300, AJUBA and BRCA1 in OSCC, and FBXW7 and PTEN in OAC (figure 3A, online supplementary figure 6). EP300 mutation and PTEN protein loss were observed to have significant detrimental effects on survival in independent OSCC and OAC cohorts,8 29 respectively, supporting the present finding. When controlling for all covariates in a Cox regression model, KDM6A, EP300 and AJUBA mutations were either significant or borderline significant for OSCC, while FBXW7 remained significant for OAC (figure 3B).

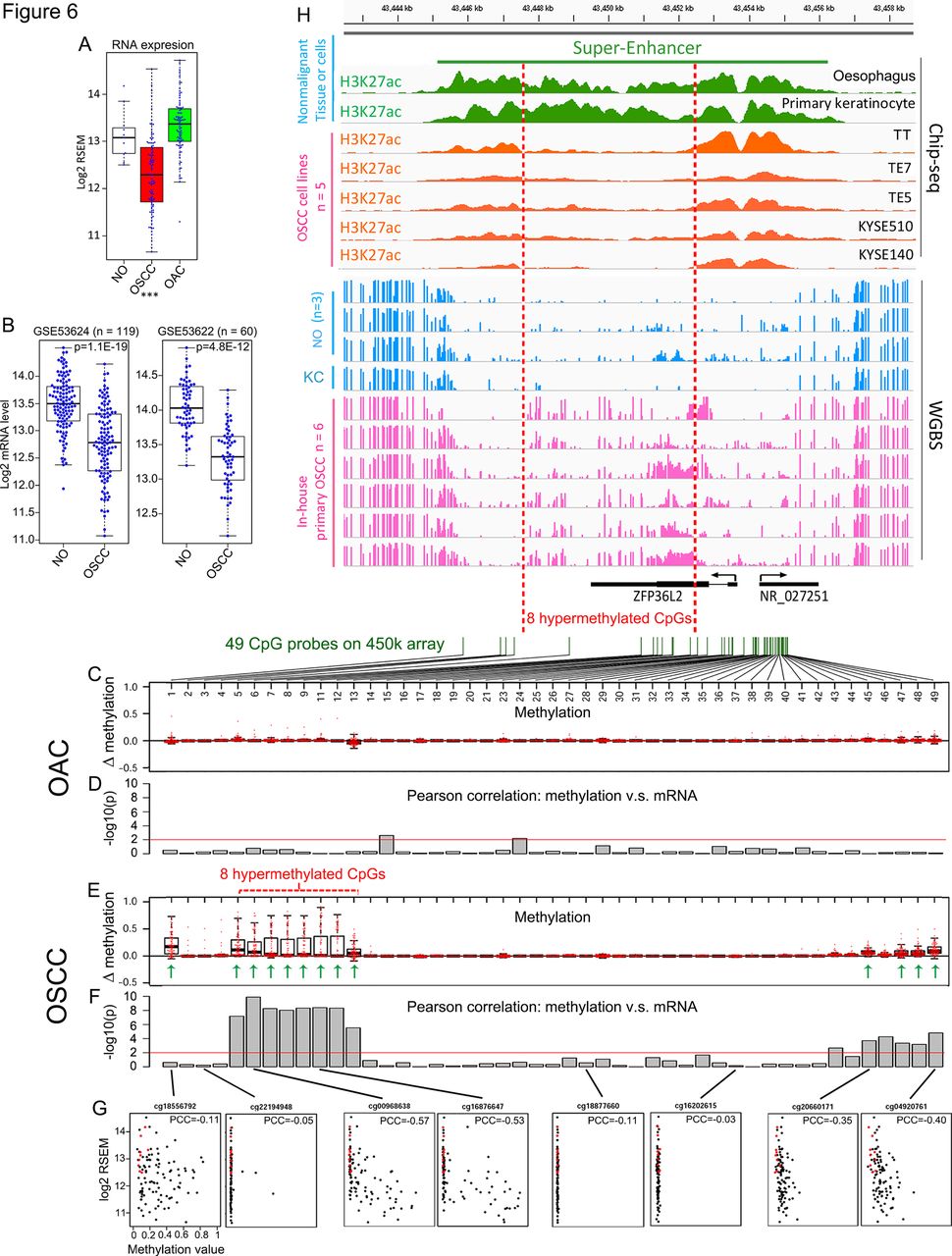
Enhancer hypermethylation and reduced expression of ZFP36L2 in OSCC. (A) ZFP36L2 mRNA level in TCGA non-malignant oesophagus (NO), OSCC and OAC samples. ***, p<0.001 comparing OSCC to NO (t-test). (B) ZFP36L2 mRNA level was retrieved from two different RNA array results comparing matched OSCC samples and their adjacent NO. (C and E) Box plots showing the increase of methylation values (tumour-normal) in either OAC or OSCC samples relative to NO tissues across 47 out of 49 CpG probes (two had no values) in ZFP36L2 loci and flanking regions. (D and F) Column plots showing the significance of Pearson correlation test between the methylation value of each CpG and ZFP36L2 mRNA level in either OAC or OSCC samples. Red lines in (D) and (F) denote significant level (p=0.01). (G) Scatter plots showing Pearson correlation of representative CpGs. Red dots denote NO samples, black dots denote tumour samples. (H) IGV snapshot displaying hypermethylation of ZFP36L2 enhancer in OSCC samples (pink tracks) compared with NO epithelium or primary keratinocytes (KC, blue tracks) measured by WGBS assay. Associated histone modifications in NO and primary keratinocytes (green tracks) and OSCC cell lines (orange tracks) are shown, and a super-enhancer region is highlighted. All the ChIP-seq tracks are in the same scale (0–8) and so are the WGBS tracks (0–1). OAC, oesophageal adenocarcinoma; OSCC, oesophageal squamous cell carcinoma; TCGA, The Cancer Genome Atlas. PCC, pearson correlation coefficient; WGBS, whole-genome bisulfite sequencing.
We also found several significant associations between mutational signatures and survival. Notably, OSCC individuals positive for signature 16-like mutational profile had a significantly worse survival rate, and this association was still significant under the multivariate Cox regression model (figure 3B). Signature 1 exhibited a protective effect for both patients with OSCC and OAC, although this was not significant under Cox regression (figure 3B).
Tumour-suppressive functions of CUL3 and ZFP36L2 in OSCC
As a number of novel SMGs were identified in the present study, we next explored their biological significance through functional approaches. We chose to focus on OSCC since our group has access to more functional models and experience in this subtype.3 9 30 31 The biological relevance of all four novel OSCC SMGs (ATF5, PTCH1, CUL3, ZFP36L2) until now, have been unknown in this cancer, and they were selected for further investigations. As an initial screen, knockdown experiments using pooled siRNAs (online supplementary table 4) were performed in four wild type cell lines with relatively high endogenous expression of these four candidates. We noted that depletion of either CUL3 or ZFP36L2 (but not ATF5 or PTCH1) consistently enhanced the proliferation of OSCC cells (online supplementary figure 7). Importantly, the antiproliferation effect of these two genes appeared to be specific to OSCC cells but not OAC cells (online supplementary figure 8); thus, we focused on characterising these two SMGs in OSCC in greater detail.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
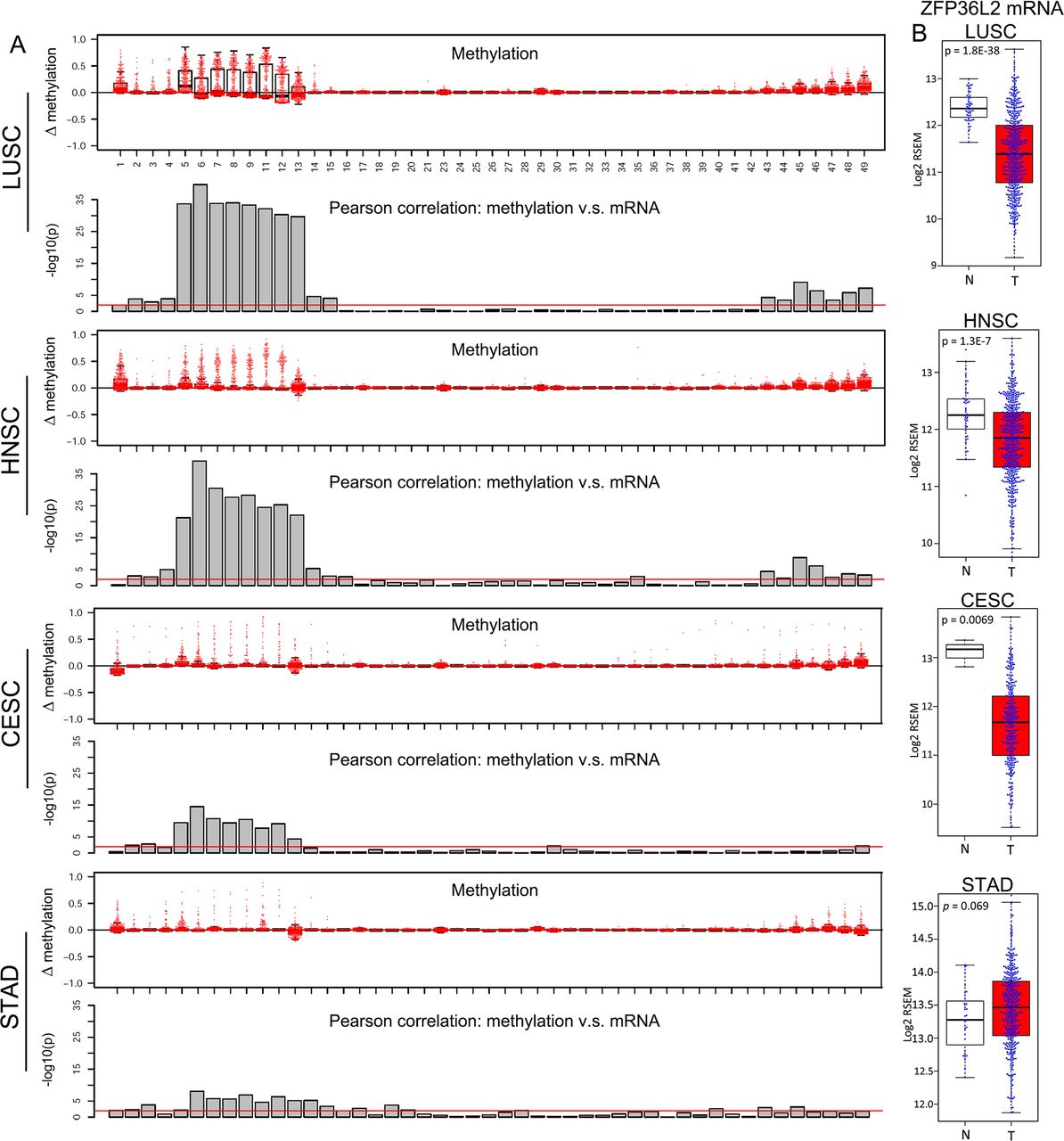
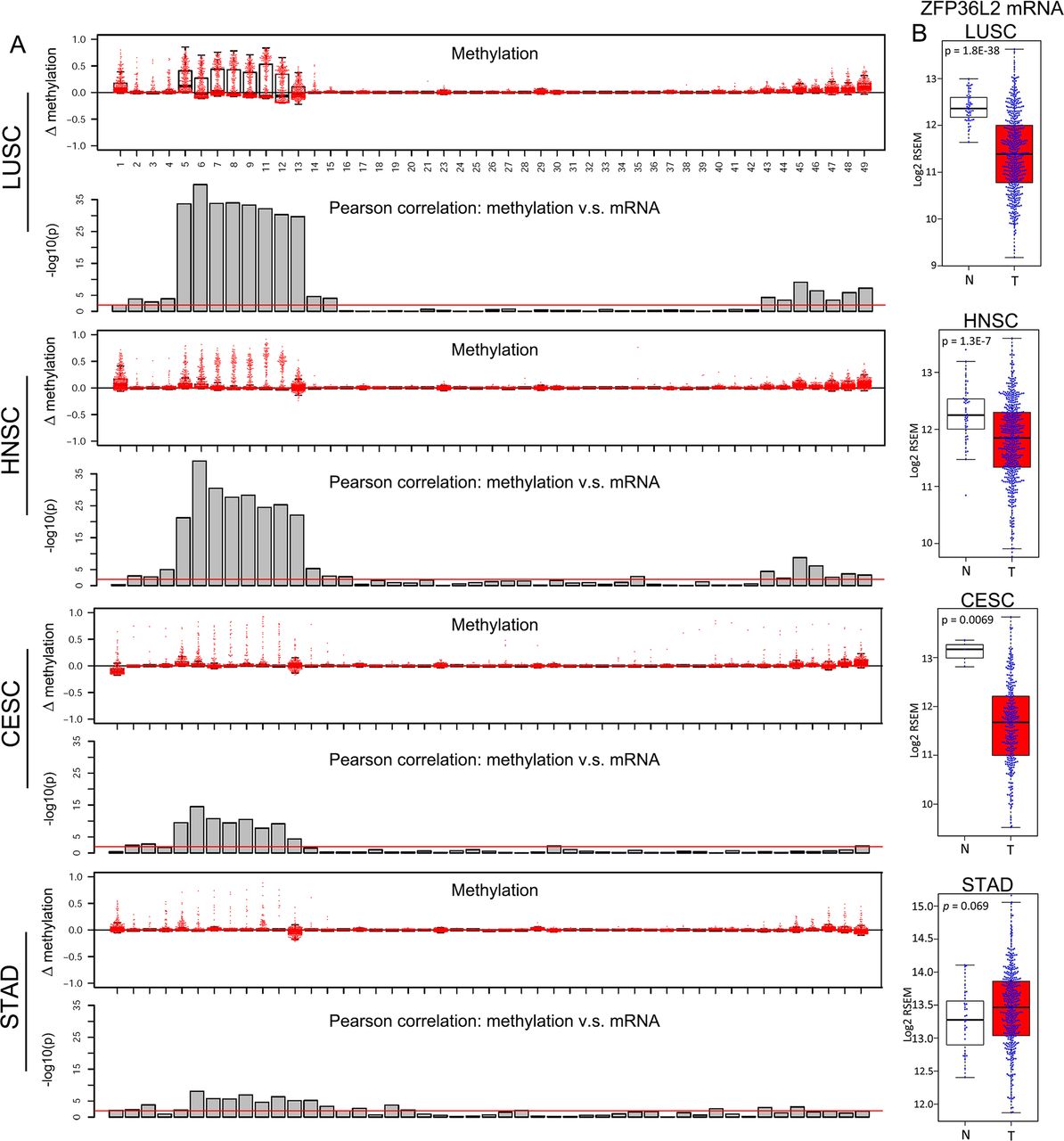
Lineage-specific ZFP36L2 epigenetic silencing in squamous cell carcinoma (SCC) samples. (A) Box plots showing the increase of methylation value (tumour-normal) in indicated cancers relative to their matched non-malignant tissues across the same set of CpG probes as in figure 6C. Column plots showing the significance of Pearson correlation test between the methylation value of each CpG and ZFP36L2 mRNA level in indicated cancer types. Red line denotes p=0.01. (B) Bar plots comparing ZFP36L2 mRNA expression between matched non-malignant tissues (N) and tumour samples (T) in indicated cancer types. LUSC, squamous cell cancer of the lung; HNSC, squamous cell cancer of the head and neck; STAD, stomach adenocarcinoma.
Consistent with the cell proliferation results, silencing of either CUL3 or ZFP36L2 promoted foci formation of different OSCC cells. Interestingly, cell migration was also increased on loss of these two SMGs (figure 4A–C). Additional single siRNAs were used to confirm the on-target effects generated by pooled siRNAs (online supplementary figure 9). In gain-of-function assays, ectopic expression of either gene suppressed both cell proliferation and migration (figure 4D,E). Further, ectopic expression of either CUL3 or ZFP36L2 significantly repressed xenograft growth in mice, with ZFP36L2 showing a stronger inhibitory effect (figure 4F). Together, these results strongly suggest a tumour-suppressive property of both CUL3 and ZFP36L2 in OSCC cells.
Mutation and deletion of CUL3 activate both Wnt-β-catenin and NRF2 signallings in OSCC and other SCCs
As the core scaffolding protein in Cullin3-RING complex, CUL3 interacts with many different BTB (for BR-C, ttk and bab) domain-containing factors to regulate the turnover of substrate proteins. One of the well characterised BTB-containing factors is KEAP1, which targets the transcriptional factor NRF2 (encoded by NFE2L2) for degradation, thereby regulating oxidative processes.32 Oxidative stress contributes to the pathogenesis of many malignancies, and mutations in both KEAP1 and NFE2L2 are driver events in a variety of carcinomas, including OSCC but not OAC.6 33 However, somatic mutations targeting CUL3 in human cancers have not been highlighted as often. In our analysis, both CUL3 and NFE2L2 were significantly mutated in OSCC samples but not in OAC; and as expected, mutations in the CUL3/KEAP1/NFE2L2 pathway were mutually exclusive (online supplementary figure 10A). Notably, interrogation of TCGA copy number data sets revealed focal homozygous deletions in the CUL3 locus in OSCC (figure 5A). Examination for both CUL3 genomic mutations and deletions across all TCGA-profiled tumours found that these genetic lesions were highly enriched in SCCs, including LUSC, HNSC, CESC and BLCA, relative to other cancer types (figure 5A,B). A total of 44% of CUL3 mutations were truncating occurring throughout the gene body (figure 5B), a characteristic pattern for loss of function mutations. The genomic deletions led to decreased transcript abundance across all types of SCCs (figure 5C), strongly suggesting CUL3 as tumour suppressor in squamous cell malignancies, and supporting our earlier functional results. We next investigated the signalling transductions regulated by CUL3 in OSCC cells. As expected, NRF2 pathway was prominently controlled by CUL3, as both NRF2 protein itself and its canonical target genes (ABCC3, PRDX1) were upregulated on depletion of CUL3 (figure 5D,E, online supplementary figure 10B). Moreover, CUL3 inactivation by either mutation or deletion was significantly associated with increased NRF2 protein expression in patients with TCGA HNSC(figure 5F). Since CUL3 has recently been shown to mediate the ubiquitination and degradation of Dishevelled, thereby restraining Wnt-β-catenin pathway in 293T and glioma cells,34 35 we asked whether CUL3 suppressed β-catenin activity in OSCC cells. Notably, CUL3 knockdown increased β-catenin protein level, with concordant changes of Wnt-β-catenin downstream factors, such as c-Myc, Cyclin D1 and p27 (figure 5D). Further supporting this regulation, CUL3 mutation or deletion were significantly associated with the upregulation of β-catenin protein in both OSCC and HNSC cohorts (figure 5F). This inhibitory effect on both NRF2 and β-catenin signalling by wild type CUL3 was confirmed by overexpression experiments (figure 5D,E,online supplementary figure 10B). The therapeutic potential of targeting either Wnt-β-catenin or NRF2 pathways in OSCC cells has been suggested, but in vitro assays inhibiting either one by itself showed modest antineoplastic effects at best.36 37 Indeed, we also observed that OSCC cells were insensitive to either ICG001 (a small-molecule chemical inhibiting β-catenin and CBP) or trigonelline (a natural compound binding and inhibiting NRF2) (online supplementary figure 10C). As CUL3 exhibited robust antiproliferative function through dual suppression of both β-catenin and NRF2 activities, we hypothesised that combinational targeting of both transcription factors might produce synergistic effects. Although a synergistic antitumour effect was not achieved in short-term cell proliferation assay (online supplementary figure 10C), such effect was found in long-term foci formation assay (figure 5G). Together, these results indicate a potential strategy for OSCC treatment by dual targeting Wnt-β-catenin and NRF2 signallings (figure 5H).
DNA hypermethylation of ZFP36L2 super-enhancer is associated with its silencing in OSCC
As a member of an RNA-binding protein family, ZFP36L2 has a CCCH zinc finger domain and binds to the adenylate-uridylate (AU)-rich element in the 3’UTR (untranslated region) of target mRNAs promoting their decay.38 ZFP36L2 has been shown to regulate erythroid differentiation39 and B cell quiescence.40 Interestingly, ZFP36L2 appears to have opposing roles in different tumour types. For example, ZFP36L2 inhibited T cell leukaemia in mice,41 but it enhanced the malignancy of pancreatic adenocarcinoma cells.42 However, the biological relevance of ZFP36L2 in most human cancers is unknown.
Considering the low incidence of ZFP36L2 mutations in OSCC (~2%), we explored alternative genomic aberrations. Unlike CUL3, no significant genetic deletions involving ZFP36L2 were discovered in TCGA samples (data not shown). Intriguingly, we observed frequent and significant downregulation of ZFP36L2 mRNA in OSCC, but not OAC samples, compared with non-malignant oesophageal (NO) mucosa (figure 6A). This downregulation was corroborated in two independent RNA microarray data sets comparing OSCC samples to matched adjacent NO (figure 6B).
We next asked whether ZFP36L2 was silenced epigenetically in these tumours by examining the methylation β values of all 49 CpG probes spanning ZFP36L2 gene locus from TCGA HM450 array data (Infinium HumanMethylation450). By plotting the methylation change between NO mucosa and oesophageal tumours, we found frequent hypermethylation of 13 CpG loci in OSCC samples (figure 6E, green arrows), but not OAC samples (figure 6C). For each CpG, we next calculated a Pearson correlation coefficient between its methylation β value and ZFP36L2 mRNA level, across either OSCC or OAC samples (figure 6D,F,G). Importantly, all but one of the 13 CpGs uniquely hypermethylated in OSCC were inversely correlated with ZFP36L2 expression, indicative of epigenetic silencing (figure 6F,G). In contrast, no such correlation existed in OAC (figure 6D).
Both the hypermethylation (figure 6E) and correlation (figure 6F) analyses indicated that a set of eight CpGs spanning a region ~1.5 kb downstream of the ZFP36L2 promoter to ~1 kb downstream of its 3’UTR (bracketed in figure 6E) were most prominently associated with ZFP36L2 epigenetic silencing. Unfortunately, this distal region is sparsely covered by HM450 array, which by design has more dense coverage of gene promoter regions. In order to better define this epigenomic regulation with higher resolution, we performed whole-genome bisulfite sequencing (WGBS) in six primary OSCC and two NO mucosa samples (figure 6H). Because DNA methylation changes at functional regulatory elements often co-occur with alterations of histone modifications,43 we also performed H3K27ac ChIP-seq (predictive of both active promoters and enhancers) analysis on five OSCC cell lines and compared that against NO mucosa from the Roadmap Epigenomics Consortium (Methods). Consistent with HM450 array data, ZFP36L2 promoter (TSS ±1 kb) was generally unmethylated in healthy oesophagus and weakly methylated in OSCC samples (figure 6H). In contrast, all six OSCC samples had hypermethylation throughout a ~5 kb region downstream of the ZFP36L2 promoter, covering the eight hypermethylated CpG regions discovered by HM450, and extending it by ~1.5 kb (figure 6H, between red dotted lines). Notably, ChIP-seq results revealed that this genomic section and its flanking region was covered with extensive H3K27ac, forming a super-enhancer44 in healthy oesophagus. The presence of a super-enhancer may indicate an important role for ZFP36L2 in oesophageal or squamous cell biology, since super-enhancer-assigned genes are enriched in processes regulating cell identity and/or specialised cellular function.45 Importantly, H3K27ac modification was markedly diminished in OSCC cells in the same region that both gained DNA methylation (figure 6H, between red dotted lines) and correlated with ZFP36L2 silencing (figure 6F,G). Together, these data strongly suggest that a gain in DNA methylation within this super-enhancer results in the epigenetic silencing of ZFP36L2 in many OSCC cases.
Epigenetic silencing of ZFP36L2 in all types of SCC
We explored whether ZFP36L2 super-enhancer hypermethylation occurred in other forms of SCCs, first using WGBS data. In LUSC, we confirmed that the ZFP36L2 super-enhancer region had a hypermethylation pattern similar to OSCC in two out of four tumour cases, but was unmethylated in two normal lung epithelium samples (online supplementary figure 11). Although no OAC WGBS results are publicly available, we analysed STAD, as it displays a molecular resemblance to OAC.6 This super-enhancer region was unmethylated in all four STAD samples and one normal gastric sample, suggesting this epigenetic silencing event might be specific to the squamous lineage. Indeed, we noted that ZFP36L2 was unmethylated and had a similar super-enhancer in primary keratinocytes, a type of squamous cell (figure 6H).
We next used TCGA HM450 data to analyse additional SCC types (figure 7). Both the pattern of super-enhancer hypermethylation and its correlation to ZFP36L2 silencing were strikingly similar across all types of SCCs, but not STAD (figure 6C–G and figure 7A). Finally, ZFP36L2 mRNA expression was significantly reduced in all SCCs, but not in STAD (figure 7B), suggesting that this epigenetic silencing is a pan-SCC phenomenon. Given that increased DNA methylation can prevent the regulation of some transcription factors on targeted DNA elements,46–48 these data together indicate that hypermethylation may decrease the binding of transcription factor(s) to ZFP36L2 super-enhancer and maintain its silencing in SCC.
Discussion
Oesophageal cancer is a common malignancy without effective therapy. Several previous studies investigated the genomic differences between OSCC and OAC subtypes.6 14 49 50 A number of subtype-specific genetic aberrations have been established as driver events, such as the OSCC-specific amplification of SOX2 and TP63 and mutations of NOTCH1 and ZNF750, and the OAC-specific deletion of SMAD4 and mutations of KRAS and ARID1A. However, since these studies were based on small numbers of patients14 and gene-panel sequencing49 or single nucleotide polymorphism (SNP)-array profiling,50 many important genomic differences between OSCC and OAC have not been robustly assessed, such as moderate-frequency to low-frequency SMGs and mutational signatures, along with their clinical associations.
Based on a harmonisation and careful re-analysis of 1048 whole-exome sequenced cases, this study represents the largest oesophageal cancer genome analysis thus far, enabling a comprehensive comparison of the genomic features between OSCC and OAC. The present data show that these two subtypes are molecularly distinct and reveal several new distinguishing molecular features including (1) novel SMGs and differences in clonal composition; (2) subtype-specific mutational signatures and a signature-based prognostic biomarker; and (3) subtype-specific inactivation of a novel tumour suppressor, ZFP36L2, by both genetic and epigenetic mechanisms.
A total of nine previously undescribed SMGs were discovered in the present analysis, and all of them exhibited subtype specificity (four in OSCC and five in OAC). However, one should keep in mind that even a subtle increase of the rate of sequencing artefacts and/or SNPs in mutation calling may make true SMGs harder to be distinguished from the background, leading to false-negative results. Therefore, additional SMGs might have been missed by the present approach. Nevertheless, our result expands the current understanding of driver events in oesophageal cancer, provides a catalogue of candidate genes for future characterisation, and reinforces the notion that OSCC and OAC are completely distinct at the molecular level. We confirmed and, more importantly, provided further evidence that OSCC is more similar to several other squamous-cell-derived cancers than it is to OAC.
Two out of four OSCC-specific SMGs (CUL3 and ZFP36L2) displayed strong antiproliferation function in OSCC but not in OAC cells. While we did not perform additional functional screens here, the other two SMGs (PTCH1 and ATF5) may play critical roles in other processes such as cell migration, invasion and epithelial-mesenchymal transition. Using detailed biochemical, cellular and animal assays, we further confirmed both CUL3 and ZFP36L2 as lineage-specific tumour suppressors in OSCC. Interestingly, both were also found to be inactivated through additional mechanisms besides single-nucleotide variations (CUL3 through homozygous deletion, and ZFP36L2 through epigenetic silencing of a cell-type specific enhancer). Together, these findings provide an important molecular foundation for further understanding the pathogenesis of these two distinct oesophageal cancers, and the SCC biology in general.
References
Footnotes
D-CL, HQD, J-JX and AM contributed equally.
Contributors D-CL and HQD conceived and devised the study. D-CL, HQD and BPB designed experiments and analysis. HQD, J-JX, AM, Y-YJ, L-WD, J-ZH and X-EX performed the experiments. D-CL, HQD, AM, TCS, CL and BPB performed bioinformatic and statistical analysis. D-CL, HQD, J-JX, BPB and HPK analysed the data. J-JH, M-RW, L-YX and E-ML contributed reagents and materials. D-CL, BPB and HPK supervised research and wrote the manuscript.
Funding This research is supported by the National Research Foundation Singapore under its Singapore Translational Research Investigator Award (NMRC/STaR/0021/2014) and administered by the Singapore Ministry of Health’s National Medical Research Council (NMRC), the NMRC Centre Grant awarded to National University Cancer Institute, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives to HPK. D-CL was supported by the National Center for Advancing Translational Sciences UCLA CTSI Grant UL1TR000124, CURE: DDRC DK P30 41301 Pilot and Feasibility Award and Tower Cancer Research Foundation. This work is also partially funded by the NIH/NHGRI grant R01HG006705 to BPB, and SOCCI developmental funds to HQD.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. The funding statement has been updated.