Article Text

Abstract
Background Somatic mosaicism is being increasingly recognised as an important cause of non-Mendelian presentations of hereditary syndromes. A previous whole-exome sequencing study using DNA derived from peripheral blood identified mosaic mutations in DICER1 in two children with overgrowth and developmental delay as well as more typical phenotypes of germline DICER1 mutation. However, very-low-frequency mosaicism is difficult to detect, and thus, causal mutations can go unnoticed. Highly sensitive, cost-effective approaches are needed to molecularly diagnose these persons. We studied four children with multiple primary tumours known to be associated with the DICER1 syndrome, but in whom germline DICER1 mutations were not detected by conventional mutation detection techniques.
Methods and results We observed the same missense mutation within the DICER1 RNase IIIb domain in multiple tumours from different sites in each patient, raising suspicion of somatic mosaicism. We implemented three different targeted-capture technologies, including the novel HaloPlexHS (Agilent Technologies), followed by deep sequencing, and confirmed that the identified mutations are mosaic in origin in three patients, detectable in 0.24–31% of sequencing reads in constitutional DNA. The mosaic origin of patient 4's mutation remains to be unequivocally established. We also discovered likely pathogenic second somatic mutations or loss of heterozygosity (LOH) in tumours from all four patients.
Conclusions Mosaic DICER1 mutations are an important cause of the DICER1 syndrome in patients with severe phenotypes and often appear to be accompanied by second somatic truncating mutations or LOH in the associated tumours. Furthermore, the molecular barcode-containing HaloPlexHS provides the sensitivity required for detection of such low-level mosaic mutations and could have general applicability.
- Genetics
- Molecular genetics
- Paediatric oncology
Statistics from Altmetric.com
Introduction
Mosaicism arises following the acquisition of a de novo mutation during post-zygotic development, resulting in an individual with two populations of cells that are genetically distinct.1 Mosaicism is being increasingly recognised as the cause of a diverse range of sporadic albeit likely genetic clinical disorders, the aetiology of which was previously unknown.1–4 This is largely attributed to improved genomic sequencing technologies that have provided better ability to detect genetic changes in subpopulations of cells. Despite this, detecting low-level mosaicism is still challenging.
The DICER1 syndrome or pleuropulmonary blastoma (PPB) familial tumour and dysplasia syndrome (OMIM #601200) is typically caused by heterozygous germline mutations in DICER1, which encodes a small RNA endoribonuclease responsible for processing hairpin precursor miRNAs into mature miRNAs that in turn post-transcriptionally regulate expression of target messenger RNAs. Since studies began on DICER1 in 2009,5 >140 heterozygous germline mutations have been published.6 DICER1 syndrome is associated with a predisposition to several rare phenotypes including PPB, cystic nephroma (CN), ovarian Sertoli–Leydig cell tumour (SLCT), multinodular goitre (MNG), nasal chondromesenchymal hamartoma (NCMH), pineoblastoma, pituitary blastoma (PitB) and other rare conditions.6 DICER1 syndrome has autosomal-dominant inheritance with variable penetrance and may present from infancy through adolescence and occasionally later.6 Using whole-exome sequencing, Klein et al7 have recently reported mosaic missense mutations in DICER1, affecting the RNase IIIb metal-ion binding domain, in peripheral blood DNA from two infants; allele frequencies were 21% and 28%, respectively. Each infant had extensive bilateral lung cysts consistent radiographically with cystic PPB, developed bilateral Wilms tumour (in one child in large kidneys with underlying renal dysmorphology) and had global developmental delays and various overgrowth stigmata. Klein et al used ‘Global delay, Lung cysts, Overgrowth and Wilms (GLOW)’ syndrome to describe this phenotype. Brief mention of mosaic DICER1 mutations also appears in one abstract.8
Here, we describe our detailed molecular investigation of four children with multiple primary tumours consistent with the DICER1 syndrome phenotype, but in whom germline DICER1 mutations had not been detected by Sanger sequencing or multiplex ligation-based probe amplification (MLPA) assay9 of genomic DNA isolated from peripheral blood or saliva.
Materials and methods
Details of sample acquisition, gDNA extractions, Sanger sequencing and the MLPA assay are presented in the online supplementary material methods.
Targeted captures and next-generation sequencing
We interrogated both tumour and normal tissues for evidence of somatic mosaicism using a standard HaloPlex targeted capture10 (Agilent Technologies, Santa Clara, USA), an in-house Fluidigm Access Array (Fluidigm, San Francisco, California, USA), and a novel development of the HaloPlex assay that incorporates molecular barcodes for high-sensitivity sequencing as a custom design (HaloPlexHS).
In brief, the HaloPlexHS targeted capture method is specifically designed to identify low allele frequency variants through the attachment of a 10-nucleotide-long molecular barcode to the captured sample DNA molecules. High sensitivity is supported by the capture of up to eight different restriction fragments per targeted base in the region of interest (figure 1). In our case, >75% of the targeted bases were covered by at least four probes (see online supplementary figures S1A-B and S2). During downstream analysis of the sequencing data, molecular barcode sequence data are used to collapse reads originating from the same sample molecule, which improves base calling accuracy and allows for accurate quantification of the mutant allele fraction within each sample as it excludes possible PCR amplification bias. The design used in this study captures 499 kb and encompasses the full DICER1, DROSHA, AGOII, TRBP2 and DGCR8 loci, all miRNA-processing-associated genes (see online supplementary figure S1).6 ,11 We will be pleased to make the design of this array available; please contact the authors for further details. We processed three gDNA samples from different sites from each child for a total of 12 samples (excluding controls), 11 of which were non-tumourous. The sequencing data were generated on the Illumina HiSeq2500 sequencer using the 150 bp paired-end sequencing protocol across four rapid flow-cell lanes. The depth of coverage achieved for each sample is depicted in online supplementary figure S1C and D. We also used the standard HaloPlex targeted capture system with a similar probe design (see online supplementary figure S2), but which does not incorporate molecular barcodes to facilitate the removal of duplicate reads. We sequenced a total of 28 gDNA samples using the Illumina MiSeq sequencer, 17 of which were non-tumourous and 11 were tumour derived. Lastly, we used a custom design Fluidigm Access Array that selectively captures all exons and exon–intron boundaries of DICER1 (described previously)12 to allow for cross-platform validation of our findings. Following capture with Fluidigm, we sequenced 22 normal gDNA samples and 15 tumour gDNA samples using the Illumina MiSeq for a total of 37 samples (see online supplementary figure S3).

Graphical representation of the HaloPlexHS design principle. The HaloPlexHS captures up to eight different restriction fragments per targeted base in the region of interest, ensuring that the great majority of target region is ultimately covered. During hybridisation, each sample is given a unique index sequence (green), allowing for pooling of up to 96 samples per sequencing lane. A degenerate molecular barcode sequence (red) is also incorporated during hybridisation, which makes it possible to track individual target amplicons during sequence analysis and to remove duplicate reads if necessary.
Inspection of suspected mosaic mutations
Initial analysis of the suspected mosaic missense mutations in the HaloPlexHS data was performed using the SureCall software V.3.0 (beta version), provided by Agilent Technologies. Sequencing reads were aligned to the human genome (hg19) and the duplicate reads were removed. The relative frequency of the four bases at the position of each mutation were manually inspected and recorded. We analysed the standard HaloPlex data in the same way using the then publically available SureCall software (V.2.0.7.0) and analysed the Fluidigm-generated data using the Integrative Genomics Viewer software (IGV V.2.3).
Calculation of threshold of detection in the HaloPlexHS data
We calculated the percentage of false nucleotides at the position of the respective mosaic DICER1 RNase IIIb mutations in each of the samples by dividing the number of reads containing an aberrant base by the total number of reads, multiplied by 100 (eg, blood DNA from patient 2 contained 4 of 9650 reads with a false T allele, or 0.04%). Using all samples with >100 reads covering the region of interest, we calculated the median number of false reads to be 0.04% (range 0–0.35%). By studying the distribution of false positive reads at the positions of the four RNase IIIb mutations, we were able to calculate the total number of false reads. We then took the fourth quintile of this distribution (0.06%) as the cut-off, below which we regarded all mutation calls as false positive. To be conservative, we considered the threshold of detection to be 0.1%.
Bioinformatic methods
The standard HaloPlex and Fluidigm-generated data sets were separately analysed using our custom bioinformatics pipeline as follows: raw paired-end reads were trimmed using Trimmomatic V.0.3313 to a minimum length of 30 nucleotides. Illumina Truseq adapters were removed in palindrome mode. A minimum Phred quality score of 30 was required for the 3′ end. Single end reads as well as paired-end reads failing previous minimum quality controls were discarded. Individual read groups were aligned, using bwa V.0.7.12, with default parameters,14 to the UCSC hg19 reference human genome from Illumina iGenomes website.15 Aligned reads from multiple read groups belonging to the same sample were indexed, sorted and merged using Sambamba V.0.5.4,16 a faster implementation of the Samtools algorithms.17 ,18
Various quality control parameters were used, including depth of coverage, based on metrics collected for each sample using bedtools V.2.24.019 and aggregated using custom Python V.2.7.9 codes. We applied GATK V.3.3.0 base quality score recalibration and indel realignment.20 As amplification duplicates were not removed, we also added ‘-nt NONE’ parameter to change the corresponding default down-sampling behaviour.
We performed SNP and INDEL discovery and genotyping across each cohort of samples simultaneously using standard hard filtering parameters according to GATK Best Practices recommendations.21 ,22 In addition to HaplotypeCaller algorithm, we also used UnifiedGenotyper in separate runs. Samtools’ new multiallelic calling model (-m parameter), as implemented in bcftools V.1.2, was also used.23 All variants were annotated with functional prediction using snpEFF V.3.6.24 Additionally, functional annotation of variants found in two public databases (ie, NCBI dbSNP V.14225 and dbNSFP V.2.826 ,27) were added using SnpSift, as part of the same software package.28
HaloPlexHS data set was analysed using the SureCall software V.3.0 (beta version). All variants obtained from all three data sets were inserted into a Gemini database,29 aggregated and selected according to snpEFF predictions. Finally, they were manually validated against read alignments using IGV software V.2.3.30 ,31
Comparison of locus coverage and percentage of homozygosity
The average coverage of the five targeted loci was calculated using GATK's DepthOfCoverage tool (see online supplementary figure S4). The total number of SNPs per locus was extracted from a Gemini database loaded with only the HaplotypeCaller variants (see above for details). The percentage of homozygosity was then calculated using the number of homozygous alternate or reference SNPs for the DICER1 locus or the other four targeted loci.
Results
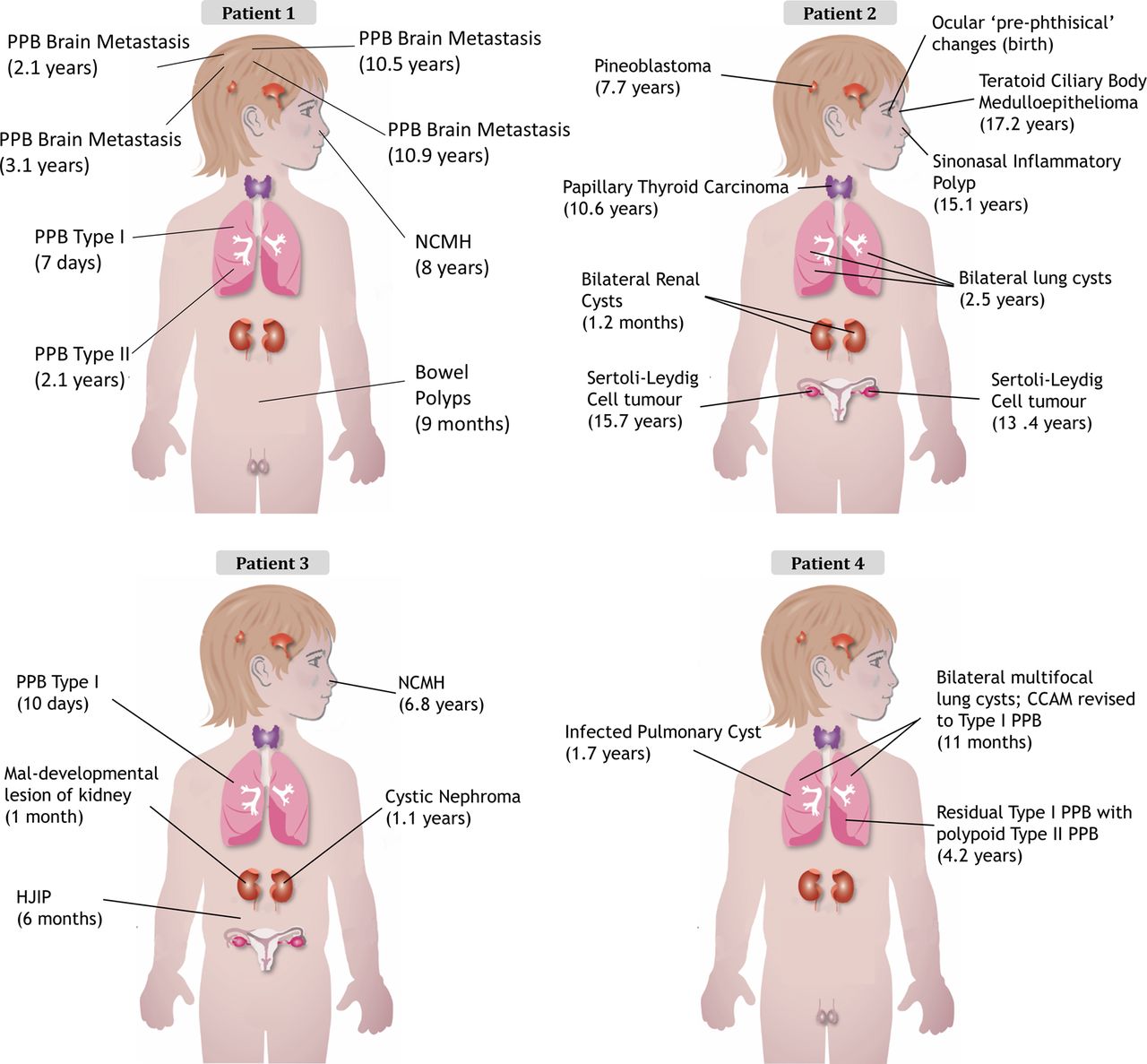
The clinical presentation of each child is outlined in table 1 and figure 2 with further details available in the online supplementary data and figures S5 and S6. The clinical cases of both patients 132 and 233 have been previously described. The family history for all four children was unremarkable (see online supplementary figure S5).
Clinical summary
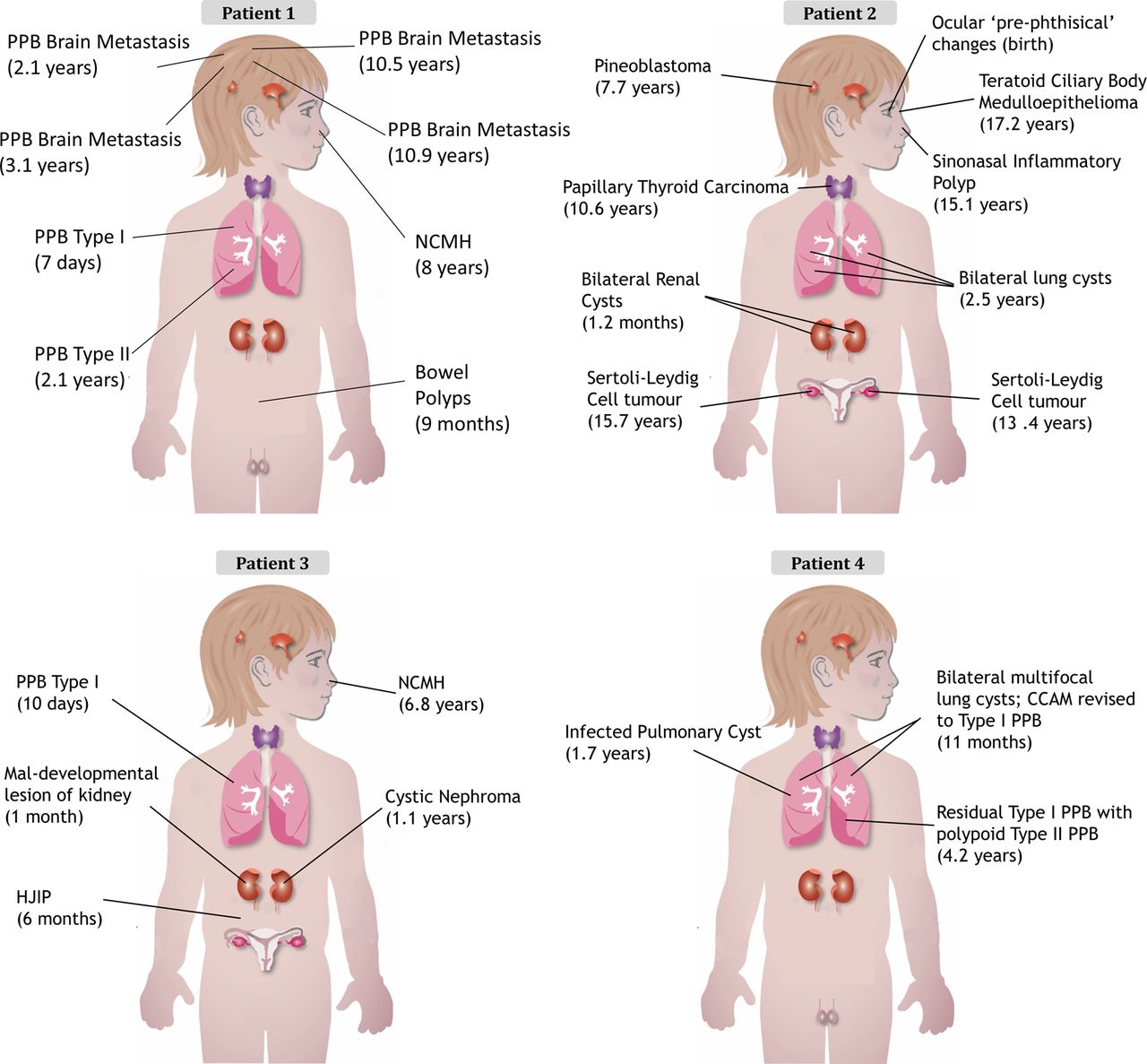
Diagnosis and (age at diagnosis) for each child's tumour. CCAM, congenital cystic adenomatoid malformation; HJIP, hamartomatous juvenile intestinal polyps; NCMH, nasal chondromesenchymal hamartoma; PPB, pleuropulmonary blastoma.
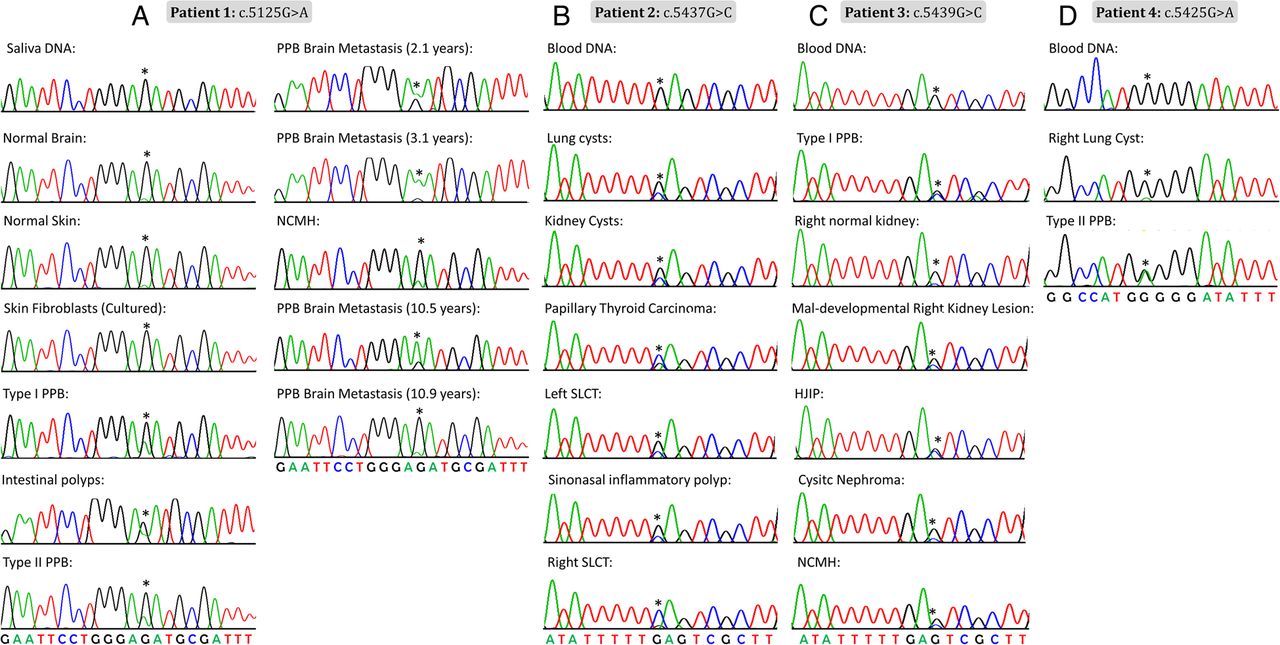
In most individuals with clinical features suggestive of the DICER1 syndrome, germline truncating mutations in DICER1 are accompanied by specific somatic ‘hotspot’ missense mutations occurring within the sequence encoding the RNase IIIb domain.6 ,12 ,34 Our initial Sanger sequencing efforts did not identify causal germline mutations in DICER1, but we reasoned that if the tumours contained a somatic DICER1 mutation, the disease presentation could be due to an occult germline mutation in DICER1 or in a closely related gene. We thus undertook Sanger sequencing of gDNA extracted from multiple tumour samples from each patient to determine whether they harboured a somatic DICER1 mutation. We observed the same missense mutation within the sequence encoding the DICER1 RNase IIIb domain in multiple tumours from each patient (patient 1: c.5125G>A [p.D1709N], number of tumours sequenced (n)=8; patient 2: c.5437G>C [p.E1813Q], n=6; patient 3: c.5439G>C [p.E1813D], n=5; and patient 4: c.5425G>A [p.G1809R], n=2) (figure 3). There are a diverse range of missense mutations reported to occur at these particular hotspot locations,6 with 215 reports of such hotspot mutations in the literature, 115 of which were confirmed somatic and the great majority of the rest are presumed likely somatic. They mostly affect 1 of 11 nucleotides within the sequence encoding the DICER1 RNase IIIb domain.6 We, therefore, thought it very unlikely that the same hotspot mutation would occur in all independent disease foci in each child, raising suspicion of somatic mosaicism for the identified DICER1 mutations.

Chromatograms showing the mosaic DICER1 mutation (indicated by an asterisk) in multiple tissue samples. (A) Patient 1 (c.5125G>A), (B) patient 2 (c.5437G>C), (C) patient 3 (c.5439G>C) and (D) patient 4 (c.5425G>A). HJIP, hamartomatous juvenile intestinal polyps; NCMH, nasal chondromesenchymal hamartoma; PPB, pleuropulmonary blastoma; SLCT, Sertoli–Leydig cell tumour.
We confirmed a mosaic distribution of the respective mutations in patients 1–3 using the three targeted-capture platforms: the mutant allele fraction was significantly higher in tumour samples and was detected at low levels in multiple normal tissues from the three patients. The average mutant allele frequency in non-tumour samples ranged from 0% to 13.58% in normal tissues from patient 1 (12.77–60.80% in tumour samples); from 0% to 1.33% normal in tissues from patient 2 (0–99.41% in tumours) and from 0% to 31.73% in normal tissues from patient 3 (6.55–91.89% in tumours). The numbers of sequencing reads and the percentage of mutant and wildtype alleles per patient sample are indicated in table 2, online supplementary table S1 and figure 4A–D. Comparing the four technologies used, it is clear that the HaloPlexHS data offers greater precision, given the ability to remove duplicate reads and in so doing increase the base calling accuracy, as described above. We considered 0.1% to be the threshold for detection, below which all mutant alleles detected were considered false positive (see table 2 and the ‘Materials and methods’). Of particular interest is patient 2, c.5437G>C, where in saliva DNA, using HaloPlexHS, we identified 5 of 1972 reads with the C allele (0.25%), whereas only one read was seen for a T allele at this position (0.05%). This T allele is clearly a false positive read. By contrast, in blood DNA, we identified 4 of 9650 reads (0.04%) for both the C and T alleles (table 2), suggesting that both the C and the T alleles are false positives. From this result, we conclude that the c.5437G>C mutation is present in saliva DNA, but not in blood DNA. The c.5437G>C mutation was also identified at low levels in urine (table 2).
Number of reads containing mutant versus wildtype base at position of interest

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mutant allele frequencies, second somatic DICER1 mutations and loss of heterozygosity (LOH). Bar graph indicating the mutant allele fraction detected in multiple tissues from (A) patient 1, (B) patient 2, (C) patient 3 and (D) patient 4. The percentage mutant base fraction (red fill) to wildtype base fraction (grey fill) at the position of interest is indicated for each sample. Green background shading (Left) indicates samples processed using the HaloPlex Standard or Fluidigm Access Array, and blue shading (Right) indicates samples processed using the HaloPlexHS technology. The percentages given for samples sequenced using the HaloPlex Standard or Fluidigm Access Array are averages of all successful runs. Samples designated with an asterisk are tumours found to carry second somatic, likely truncating DICER1 mutations (see online supplementary table S1 for details). (E–H) Bar graphs illustrating evidence of LOH in tumour samples from patient 1 (E) and patient 2 (F) and lack of LOH in a representative tumour from patient 3 (G) and patient 4 (H). In (E) and (F), there is a notable increase in the number of heterozygous to homozygous SNPs in the tumour samples relative to the germline, which is indicative of LOH. This shift is not evident in tumours from patient 3 (G) and patient 4 (H). CN, cystic nephroma; NCMH, nasal chondromesenchymal hamartoma; PPB, pleuropulmonary blastoma; SLCT, Sertoli–Leydig cell tumour.
We hypothesised that, in the setting of a mosaic DICER1 RNase IIIb mutations, we might discover second somatic mutations outside of the RNase IIIb domain, which initiate two-hit tumourigenesis as seen in most DICER1-related tumours. All three data sets were analysed as described in the ‘Materials and methods’, an outline of which is presented in online supplementary figure S4. We identified individually distinct second somatic likely deleterious DICER1 mutations in patient 2's left ovarian SLCT (c.4626_4626delG; p.Q1542Hfs*18) and sinonasal inflammatory polyp (c.4458_4458delA; p.K1486Nfs*4), in patient 3's NCMH (c.4651_4652insTGCT; p.E1551Vfs*7) and in patient 4's type II PPB, which arose in a pre-existing lung cyst (c.1966C>T; p.R656*) (see online supplementary figure S6A). Each of these second somatic mutations was validated via Sanger sequencing and is predicted to prematurely truncate the DICER1 protein (see online supplementary table S1 and figure 4). Furthermore, we detected loss of heterozygosity (LOH) in one of patient 1's PPB brain metastases and in three additional lesions from patient 2 (follicular thyroid carcinoma, right SLCT and kidney cysts). Evidence of LOH in the tumours is supported by both quantitative and qualitative analyses, as presented in figure 4E–H, online supplementary figure S7 and table S2. In online supplementary figure S7A, LOH is evident in the tumours from patient 1, column 8, and in patient 2, columns 6 and 7, where there is a visible reduction in the average coverage of the DICER1 locus relative to the respective germline samples. Similarly, in online supplementary figure S7B, there is a visible increase in the per cent SNP homozygosity for tumours occurring in patient 1, column 7, and patient 2, columns 5 and 6, which is indicative of LOH.
Despite the time interval between the cancer diagnosis and blood sampling, it is possible that we may be detecting traces of circulating tumour DNA in blood samples or infiltrating tumour cells in normal tissue sampled from areas adjacent to tumours. In addition to collecting tissues that are less likely to contain contaminating tumour DNA (eg, hair and saliva), we also wanted to carry out additional analyses to explicitly determine whether we were indeed picking up contaminating tumour DNA. Three of the above exonic somatic mutations were not detected at all in germline samples in the HaloPlexHS data set. The remaining somatic mutation found in patient 4's type II PPB was detected at 0.04% in blood DNA, well below the 0.1% threshold for likely real mutations (see ‘Materials and methods’), and we are thus able to establish that the mosaic mutation-containing alleles detected in the non-tumorous samples were not derived from infiltrating tumour cells or circulating tumour DNA. The identified second somatic mutations and the number of alternate and wildtype alleles per patient sample are summarised in online supplementary table S3.
Discussion
With the use of a targeted approach, combined with deep and ultra-deep sequencing, we detected low-level DICER1 mutant allele fractions in three patients exhibiting mosaicism for the detected mutations. The fourth case is also likely to be a mosaic for DICER1. These mosaic missense mutations were localised to ‘hotspots’ within the sequence encoding the DICER1 RNase IIIb domain and have been shown to selectively reduce 5p miRNA processing.34–36 We also discovered likely pathogenic second somatic mutations or LOH in tumours from all four patients, thus showing that the two-hit model applied to the tumours we studied (table 2 and online supplementary table S1).
The exact developmental stage at which the mosaic mutations were acquired has not been accurately determined, but given the presence of the mutant allele in tissue samples from all three germ layers, we suspect that the mutations occurred prior to gastrulation.1 ,37 The mosaic origin of patient 4's mutation remains to be unequivocally established (table 2, online supplementary table S1 and figures 3 and 4). Without additional normal samples from both the lung(s) and other distant normal sites, we were not able to determine whether (a) the child is a somatic mosaic with an undefined, yet limited distribution of the mutation; (b) mosaicism is present but is confined to the lungs; or (c) the two lung lesions separately acquired the c.5425G>A ‘hit’ by chance. In the latter case, the 6.9% mutant allele frequency in the reactive lung tissue would have to be attributed to cancer cells that were not obviously present on detailed histopathological examination (see online supplementary figure S8), and therefore, we do not favour this explanation. Moreover, the detection of a second somatic truncating mutation in the PPB type II sample (see online supplementary table S1) and the absence of any further extrapulmonary DICER1-related lesions in this person support the hypothesis that somatic mosaicism is present but is confined to the lungs. In this case, acquisition of the missense mutation would have occurred much later during embryonic development than in the other three cases.
Of note, mosaic DICER1 mutations in our cases and the two previously described cases7 are localised to the sequence encoding the RNase IIIb domain. We have also identified additional likely pathogenic second somatic mutations or LOH in the tumours. These findings strongly suggest that the molecular paradigm of multi-organ mosaic RNase IIIb mutations followed by second ‘hits’ in other regions of DICER1 is precisely the reverse of typical, now well-described DICER1 molecular events in which somatic RNase IIIb mutations follow inactivating germline mutations.6 This mosaic paradigm affecting the highly critical RNase IIIb residues may explain the apparently severe phenotype seen in three of our cases as well as the severity of Klein's GLOW syndrome cases and their overgrowth and developmental delay. Although having several diseases, our cases manifested typical DICER1 phenotypes, and none had overgrowth or developmental delay. One of Klein's cases had renal dysmorphology, and patient 3 had both CN and contralateral microscopic renal medullary maldevelopment characterised by increased loose mesenchyme, disorganised collecting system and dilated lymphatic vessels, which have not been previously described (see online supplementary data). Both of Klein's patients developed bilateral Wilms tumour; unilateral Wilms and bilateral disease in paired organs are a known feature of DICER1 syndrome.38–41 Rather than comprising a new syndrome, we are inclined to believe that multi-organ mosaic RNase IIIb mutations result in an unusually severe overall DICER1 phenotype, within which the pleiotropy typical of DICER1 disease may occasionally result in overgrowth or developmental delay. Identification and analysis of additional mosaic cases may clarify this ambiguity.
Non-RNase IIIb mosaic mutations are likely to exist, and we predict that the phenotype caused by non-RNase IIIb mosaic mutations would be less severe than those caused by mosaic mutations directly affecting the DICER1 RNase IIIb domain, and therefore, such mutations may be more likely to go undetected. There has been one additional reported instance of a de novo germline DICER1 mutation (c.5125G>C; p.G1709H) affecting a metal-ion binding residue within the RNase IIIb domain.12 The child is severely affected: he presented at birth with a PitB, extensive multifocal bilateral lung cysts and bilateral renal cystic masses. The c.5125G>C mutation was seemingly heterozygous in lymphocyte gDNA, but extensive investigations to confirm or rule out mosaicism were not possible.12 The inference from these data is that both mosaic and non-mosaic germline missense mutations affecting exons encoding the metal-ion binding domain of DICER1 underpin a particularly severe disease phenotype and may induce a large number of disease foci per child, depending on the specific tissue distribution of the mutation (patient 4 might exemplify this more limited yet significant mutation distribution). In support of this, we recently identified a paternally inherited novel heterozygous germline DICER1 mutation, c.5441C>T (p.S1814L), in a girl who developed an SLCT and MNG before the age of 13 years (Wu et al, unpublished data). This mutation, although located within the RNase IIIb domain, does not directly affect one of the critical catalytic or metal-binding residues within this domain (eg, residues 1705, 1709, 1810 and 181342 ,43). The less severe phenotype exhibited by this child may possibly be related to the ‘sparing’ of the above-mentioned metal-ion binding residues. It is notable that no inherited germline DICER1 mutations at a nucleotide encoding a metal-ion binding residue have been reported.
Cancer susceptibility syndromes such as familial adenomatous polyposis (FAP) and the neurofibromatoses (NF) are also associated with a mosaic origin of the causative mutations. In these conditions, the disease course of the mosaic form is reported to be milder than the inherited, non-mosaic presentation.44–46 For children with mosaic DICER1 mutations affecting the RNase IIIb domain, the disease appears to be more severe (including earlier onset, greater number of disease foci and greater range of phenotypes) than in the more typical autosomal-dominant forms. This disparity may be attributed to the nature of the mutations required to initiate tumourigenesis in DICER1 syndrome—typically a first-hit truncating germline mutation occurs in any protein-encoding region and a second ‘hit’ specifically affects the RNase IIIb domain. Such combinations are likely to be rare since it appears the selected second hit nearly always affects a very limited number of nucleotides encoding the RNase IIIb metal ion-binding domains. In contrast, in the DICER1 mosaicism reported here, the initial ‘hit’ is the acquisition of a missense RNase IIIb hotspot mutation. The second likely truncating mutation occurs anywhere across the gene (see online supplementary table S1), and is therefore, stochastically more likely to occur than a RNase IIIb mutation. Thus, we postulate that the combination of the specific effects of the RNase IIIb mutation and widespread inactivating second hits accounts for more severe clinical manifestations in these children. Therefore, we predict that non-RNase IIIb mosaic DICER1 mutations, like mosaic mutations in FAP and NF, will cause a disease phenotype that is milder than both the autosomal-dominant form and that caused by RNase IIIb mosaic DICER1 mutations.
The importance of identifying the causative mutations in these children is several fold: unaffected parents who have an affected child may want to know the risk of recurrence in future pregnancies. Furthermore, the affected children themselves may want to know the probability of transmission to future offspring. Understanding the genetic cause and the mechanism underlying the phenotype provides information that can be used to ascertain such risk. Even in heterozygous germline DICER1 mutation carriers, screening for DICER1-related conditions is problematic, as discussed elsewhere.6 Mosaicism further complicates such considerations because the distribution of a somatic mutation would be difficult to determine, but it should be borne in mind that these children may be at increased risk compared with other DICER1 mutation-positive children. In the cases reported here, transmission of the RNase IIIb mutations to the next generation seems unlikely, although in the future, testing of sperm or ova or fetal genetic testing might be considered.
Several high-sensitivity sequencing methods are currently being applied to discover low-frequency mutations, each with its own advantages and disadvantages. The most prominent so far are the PCR-based Safe-SeqS47 and the Molecular Inversion Probe-based smMIP method.48 Whole-genome sequencing with higher than average depth of coverage has also been used to identify de novo mutations that occurred post-zygotically, as was recently reported by Acuna-Hidalgo et al.49 Despite these technical advances, detecting low-level mosaicism is still challenging. Low-level mosaic mutations fall below the threshold of sensitivity for many sequencing methods, and other more sensitive technologies are costly and, therefore, may not be practical in either the research or clinical setting. In our hands, the novel HaloPlexHS target enrichment system containing molecular barcodes provided the sensitivity required for detection of mutant allele fractions as low as 0.24%. We found the HaloPlexHS to be an economically feasible platform. It is suitable for covering entire genomic regions (in our case, 499 kb), but currently 5 Mbp is possible, which is in contrast to PCR product and Molecular Inversion Probe–based methods. Additional advantages of HaloPlexHS over the smMIP method include a much lower DNA input requirement (∼50 ng) and the redundancy in the HaloPlexHS probe design allows for the vast majority of targeted bases to be covered by at least four probes, ensuring high coverage, but without increasing the cost of the capture. HaloPlexHS is likely to be broadly applicable to other situations where mosaicism can occur but yet remain undetected by currently available technologies. The design implemented can be easily adopted by other investigators interested in identifying mutations in DICER1 and other genes encoding the components of the miRNA processing machinery. We also demonstrate the utility of the HaloPlexHS in FFPE-derived DNA. Our findings suggest that the targeted ultra-deep next-generation sequencing of the DICER1 locus is a useful technique for the identification of mosaic DICER1 mutations.
In summary, by using a new high-sensitivity mutation detection system, we demonstrate that mosaic DICER1 RNase IIIb missense mutations are an occasional and important genetic cause of the DICER1 syndrome in patients presenting with multiple primary tumours associated with the syndrome, but for tumour initiation, they often appear to be accompanied by second somatic truncating non-RNase IIIb DICER1 mutations or LOH.
Acknowledgments
The authors dedicate this publication to the memory of Sean Callahan. We thank Charmian Cher, Josh Zhiyong Wang and Martin Angers (Agilent Technologies) for their assistance with the HaloPlexHS design and capture and Dr Elizabeth Perlman and Dr Michael McDermott for their assistance with pathology review.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors LdK performed the DICER1 hotspot sequencing, initial Fluidigm data analysis and mutation validation and wrote the manuscript. LdK, YCW and BR performed the HaloPlexHS capture. TR and DB performed computational analysis. NS performed the initial constitutional gDNA sequencing and MLPA analysis. MW sequenced the tumours of patient 3 and provided some concepts used in the discussion. EW conducted sample acquisition and associated administration work. CS, SMJH, JHMM, JMvH, AHMB, DAP, AR, KLD, TT, JC and CE referred patients, provided samples and collected clinical information. LF created the design of the HaloPlexHS. DB-DS provided expert pathology opinion. JRP reviewed the diagnostic images, collected clinical information and edited the manuscript. JR and WDF designed the study and edited the manuscript. All authors read and approved the final manuscript.
Funding This research was made possible thanks to the support of the Alex's Lemonade Stand Foundation grant to WDF, the Genome Canada Science Technology Innovation Centre, the Compute Canada Resource Allocation Project wst-164-ab and the Genome Innovation Node grants to JR, and the Vanier Canada Graduate Scholarship to LdK.
Competing interests LF is an employee of Agilent Technologies.
Patient consent Depending on the ages of the participants at the time of recruitment, eligible relatives signed a consent form in accordance with the IRB protocol or participants themselves provided written informed consent.
Ethics approval The study was approved by the Institutional Review Board of the Faculty of Medicine of McGill University, Montreal, Quebec, Canada (no. A12-M117-11A).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Sequencing data are available upon request. Please contact the corresponding author.