Article Text

Abstract
Distinguishing between hereditary and inflammatory neuropathy is usually straightforward on clinical grounds with the help of a family history. There are nevertheless cases where the distinction is less clear. The advent of molecular genetics has in the past several years aided confirmatory diagnosis for an increasing proportion of patients with genetic neuropathy. Various reports have described associations of Charcot-Marie-Tooth disease with a suspected or confirmed inflammatory neuropathy occasionally responding to immunotherapy. Possible predisposition to an inflammatory component was suggested in a subset of patients. Such reports have, however, been relatively few in number, suggesting the rarity of such associations and of such a predisposition if it exists. There have been a number of publications detailing clinical presentations suggestive of inflammatory neuropathy in patients with a known or later proven genetic aetiology, and subsequently felt to be part of the phenotype rather than representing an association. A number of genetically mediated multisystemic diseases with neuropathy have otherwise been reported as mimicking chronic inflammatory demyelinating polyneuropathy (CIDP). The most common example is that of familial amyloid polyneuropathy, of particular concern for the clinician when misdiagnosed as CIDP, in view of the therapeutic implications. We review the literature on reported associations, mimics and misdiagnoses of hereditary and inflammatory neuropathy and attempt to determine a practical approach to the problem in clinical practice using clinical features, electrophysiology, histopathology and targeted early genetic testing. The issue of attempting immunomodulatory therapy is discussed in view of the published literature.
- NEUROPATHY
- GENETICS
- PERIPHERAL NEUROPATHOLOGY
- NEUROPHYSIOL, CLINICAL
Statistics from Altmetric.com
Introduction
Hereditary and inflammatory neuropathies represent disorders of primarily different pathophysiological bases which first require early identification in view of the therapeutic benefit that the latter group may derive. Establishing a genetic diagnosis has also become important in view of novel potential therapeutic avenues, avoidance of inappropriate immunomodulatory therapies, as well as genetic counselling.
A number of cases of associations of genetic and inflammatory neuropathies have been reported in recent years. Genetic susceptibility to inflammatory damage has frequently been postulated. Besides co-occurrence, coincidental or not, several cases of patients with hereditary neuropathies confirmed by molecular studies have been described as presenting with features suggestive of an acquired inflammatory process. Other genetically mediated multisystemic diseases have been reported as misdiagnosed and inappropriately treated as chronic inflammatory demyelinating polyneuropathy (CIDP).
Methods
We conducted a systematic literature review from a Medline search up to February 2016 on reports of mentioning association, mimics or misdiagnoses implicating genetic and inflammatory neuropathy. Only papers published in English and French were considered. Our search methodology followed the standard guidelines for systematic literature reviews outlined in the PRISMA statement.1 Abstracts were excluded. We used as search MeSH terms: ‘Charcot-Marie-Tooth’, ‘CMT’, ‘genetic’, ‘hereditary’, ‘inflammatory’, ‘hereditary motor and sensory neuropathy’, ‘HMSN’, ‘acute inflammatory demyelinating polyradiculoneuropathy’, ‘acute inflammatory demyelinating polyneuropathy’, ‘chronic inflammatory demyelinating polyradiculoneuropathy’, ‘chronic inflammatory demyelinating polyneuropathy’, ‘CIDP’, ‘Lewis-Sumner syndrome’, ‘Multifocal Acquired Demyelinating Sensory and Motor Neuropathy’, ‘MADSAM’, ‘Guillain-Barré syndrome’, ‘GBS’, ‘HNPP’, ‘hereditary neuropathy with liability to pressure palsies’; ‘multifocal motor neuropathy’, ‘MMN’, ‘corticosteroid’, ‘intravenous immunoglobulins’, ‘familial amyloid polyneuropathy’, ‘FAP’, ‘mitochondrial’, ‘mitochondrial neurogastrointestinal encephalomyopathy’, ‘MNGIE’. On the basis of results of initial searches, we also included in further searches, ‘sensory ataxic neuropathy with dysarthria and ophthalmoplegia’, ‘SANDO’, ‘Tangier's disease’ and ‘Refsum's disease’.
Results
We retrieved 63 articles which had descriptions of patients with hereditary neuropathy in association or in combination with inflammatory neuropathy or, alternatively, with initial misdiagnosis.
Literature linking inflammatory and hereditary neuropathy
Published reports are here detailed by Charcot-Marie-Tooth disease (CMT) subtype after a brief description of the initial pregenetic era historical reports. Cases of confirmed molecular diagnosis are summarised in table 1.
Clinical reports of associations, mimics and misdiagnoses of genetically confirmed cases of Charcot-Marie-Tooth disease, per subtype, with inflammatory neuropathy (literature 1966—December 2015)
In 1982, Dyck et al2 initially reported seven patients with hereditary sensory and motor neuropathy who responded to corticosteroid treatment. A further case was reported with described response to immunosuppressive therapy.3 Antoine et al4 reported in 1989 two patients with clinical CMT who developed a steroid-responsive CIDP. Joy and Oh5 reported in 1989 a case of tomaculous neuropathy with an acute and recurrent picture. Three childhood cases of CMT were also reported with CIDP.6 Vital et al7 described in 1992 two children with demyelinating CMT who presented with inflammatory features on nerve biopsy. More recently, in 2003, Odaka et al8 reported a 30-year-old patient with genetically unconfirmed sporadic early-onset axonal CMT who developed an axonal, anti-GM1 antibody positive Guillain-Barré syndrome (GBS) after Haemophilus influenzae infection.
Familial forms of inflammatory neuropathy, without known or suspected underlying CMT, have otherwise also been described. CIDP has been described in two siblings.9 A number of cases of familial GBS have been reported.10–16 Some have described similar human leucocyte antigen (HLA) typing in affected participants,11 ,13 raising the possibility of genetic susceptibility. Whether some of these descriptions could relate to undiagnosed genetic neuropathy is possible.
CMT1A
Acute and subacute inflammatory demyelinating polyneuropathy
Revealing CMT1A
An acute inflammatory demyelinating polyneuropathy (AIDP) was reported in association with CMT1A by Malandrini et al17 in a 15-year-old girl with acute-onset paraesthesiae and weakness, pes cavus, quadriparesis and areflexia. Electrophysiology revealed demyelination with dispersion. Cerebrospinal fluid (CSF) examination showed raised protein of 0.9 g/L. Plasma exchanges led to improvement. A sural biopsy showed onion-bulb formations and prominent endoneurial lymphocytes and macrophages. Genetic testing confirmed CMT1A.
Münch et al18 described a case of severe post-infectious GBS in a patient with pes cavus, thin legs without weakness. His brother had a mild sensory and motor neuropathy. Nadir occurred within 24 h and intubation and mechanical ventilation were required. CSF protein was raised at 0.71 g/L. Electrophysiology was demyelinating. Intravenous immunoglobulins (IVIg) were effective. Genetic testing proved CMT1A.
Desurkar et al19 reported two children presenting with AIDP who were subsequently diagnosed with CMT1A. The paternal grandmother of one of the children, whose illness was preceded by diarrhoea, had been investigated for CIDP was, however, subsequently found to have CMT1A. The child had an electrophysiologically severe demyelinating neuropathy and IVIg was partially helpful. The second child reportedly had a post-Mycoplasma pneumoniae AIDP with consistent electrophysiology and a raised CSF protein of 1.8 g/L. She had dysmorphic features and a positive family history. Genetic testing confirmed CMT1A. IVIg resulted in full recovery within 8 weeks.
In known CMT1A Vital et al20 described a previously asymptomatic patient with CMT1A presenting acutely with ataxia and paraesthesiae, responding to IVIg. Electrophysiology was demyelinating and, to a degree, heterogeneously. CSF protein was normal. Histology revealed few endoneurial inflammatory cells and several figures of macrophage-induced demyelination, GBS was reported in an elderly patient with previously asymptomatic CMT1A by Peters et al.21 CMT was suspected on the basis pes cavus and hammer toes with homogeneously reduced motor conduction velocities. IVIg had no effect in the short term but slow improvement followed over a year.
Mazzeo et al22 reported a patient with known CMT1A who developed postdiarrhoeal subacute inflammatory demyelinating polyradiculoneuropathy, revealed by massive nerve hypertrophy. Back pains and distal weakness progressing over 7 weeks resulted in impaired mobility. CSF protein was raised at 1.5 g/L and electrophysiology demonstrated motor velocities of 9–18 m/s and acute distal denervation on EMG. Lumbosacral MRI showed hypertrophy of nerve roots with gadolinium enhancement which settled on repeat scanning 4 months later. Response to IVIg was good with functional recovery and regression of pain.
Chronic inflammatory demyelinating polyradiculoneuropathy
Revealing CMT1A
CIDP was described in a patient with suspected, then subsequently confirmed, CMT1A in a context of discovery of HIV positivity in a 42-year-old man.23 Electrophysiology revealed severe homogeneous motor slowing in the arms (14–16 m/s) with marked secondary axonal loss. CSF protein was 0.74 g/L with 7 lymphocytes/mm3. Sural nerve biopsy showed epineurial perivascular lymphocytic infiltrates and figures of macrophage-induced demyelination. Therapeutic response to intravenous steroids produced reversion of sensory symptoms but distal lower limb weakness partially persisted.
Nakai et al24 described a 2-year-old boy who had early-onset CMT1A with unexpected inflammatory histopathological features. Despite no description of a rapid/stepwise decline, this led to a trial of corticosteroid treatment which produced no benefit. The authors raised the possibility of the pathological changes being due to early biopsy rather than indicative of inflammatory disease.
Seven patients with CMT1A and an associated inflammatory component were reported by Ginsberg et al.25 This series consisted of four females and three males of mean age 39.1 years (range 18–69). All had a stepwise deterioration with acute/subacute deterioration. Six had positive sensory symptoms. Conduction block was detected in 3/7, CSF protein was raised in 4/7 and all had inflammatory infiltrates on nerve histology. Four had a favourable therapeutic response, two to steroids and two to IVIg.
Ben Yousef-Turki et al26 described a 15-year-old boy who developed within 6 months gait instability, upper limb weakness and tremor, losing the ability to walk within a year. Examination revealed areflexic quadripaesis with ataxia. Electrophysiology showed a severe demyelinating neuropathy with marked dispersion. CSF protein was of 1 g/L. Histological findings were described as those of a ‘hypertrophic neuritis’. Family history revealed that two siblings and the mother were affected and genetic testing confirmed CMT1A. A remarkable response to IVIg, followed by oral corticosteroid therapy, occurred with recovery of complete independence. Subsequent relapses also responded to treatment.
Kume et al27 described a 48-year-old patient with loss of finger dexterity and hand pains. Examination showed pes cavus, distal atrophy, moderate weakness of the intrinsic hand and distal lower limb muscles and hyporeflexia. Family history revealed bilateral pes cavus in the father and son. CSF protein was elevated (0.98 g/L). MRI revealed hypertrophy of the brachial and lumbar plexuses with marked gadolinium enhancement which persisted despite immunotherapy. Genetic testing confirmed underlying CMT1A. The authors described the ‘modified F-ratio’, an electrophysiological index, as suggesting a CIDP with proximal-predominant demyelination. After immunotherapy, pains settled and a marked reduction of the modified F-ratio was observed suggesting resolution of radicular inflammation.
With known CMT1A Propensity for the presence of autoantibodies to PMP22 and complement was shown in nerve biopsy specimens of 12 patients with CMT1A with stepwise progression from a cohort of 55 participants by Gabriel et al.28 Five of these 12 cases were sporadic. One patient had an IgM paraprotein, and three had a history of improvement with immunosuppression. One had worsened significantly during each pregnancy. The authors suggested that these patients may have an immunological susceptibility to an inflammatory demyelinating neuropathy or, alternatively, that the inherited neuropathy may expose myelin antigens, or that the 17p duplication may contain genes that modify the immune response in some patients.
Marques et al29 reported the case of an 18-month-old child with CMT1A who developed unexpectedly rapidly progressive weakness and disability. Electrophysiology showed non-uniform slowing. Nerve biopsy showed no inflammation. The patient responded to IVIg with improved gait, ability to climb stairs and reduction of timed 10 m walk from 20 to 10 s. She subsequently became IVIg-dependent.
Hereditary neuropathy with liability to pressure palsies
Crum et al30 described one patient with simultaneous multiple mononeuropathies after gastrointestinal and respiratory infections. A paternal uncle was reported as having had GBS. Electrophysiology suggested hereditary neuropathy with liability to pressure palsies (HNPP). Histology showed tomaculae with, however, one small collection of mononuclear inflammatory cells around a small epineurial blood vessel. CSF was normal. Genetic testing demonstrated the 17p11.2 deletion. Recovery was spontaneous and although the authors had considered AIDP and vasculitis in the differential, they felt their patient had a ‘fulminant’ form of HNPP rather than an association with inflammatory disease.
Le Forestier et al31 reported a patient presenting with a 15-year history of recurrent polyradiculoneuropathy. Electrophysiology showed demyelination only at entrapment sites and CSF protein was raised at 0.90 g/L. Corticosteroids were said to produce ‘minimal improvement’. After successive episodes, a sural biopsy showed tomaculae which prompted genetic testing confirming HNPP.
Degos et al32 described a 31-year-old woman who presented with a clinical diagnosis of postviral AIDP. CSF protein was 0.56 g/L. The disease was considered as mild and left untreated and subsequently recovered. Electrophysiology was suggestive of HNPP and confirmatory genetic testing was obtained.
Pou Serradell et al33 described, among 31 participants from 6 families with HNPP, 12 who presented with acute paralysis. Although not specifically discussed, the differential appeared to include AIDP and vasculitic neuropathy. Treatment was not described. Several participants had a progressive demyelinating polyneuropathy but whether any had a picture of typical CIDP was uncertain.
Korn-Lubetzki et al34 described a family with inflammatory demyelinating polyneuropathy and HNPP. The same authors had 8 years earlier reported two members of the same family (father and daughter) with inflammatory neuropathy.35 The latest report described another daughter aged 24 years who, over weeks, developed right leg sensory impairment, areflexia and distal lower limb weakness preventing independent walking. The CSF study showed no abnormality. Electrophysiology showed prolonged distal motor latencies, moderately slowed motor conduction velocities and slowed sensory velocities. The diagnosis was felt to be CIDP. Treatment with oral steroids resulted in stabilisation followed by improvement/normalisation of strength and reflexes. The father, who developed a chronic inflammatory neuropathy after surgery, had on sural nerve biopsy loss of myelinated fibres, remyelination but no inflammatory features or tomacula. The other daughter had a clinical course suggestive of AIDP, which occurred 2 weeks after serology-proven EBV infection. All three participants were previously healthy and none had reported trauma or nerve compression. Genetic testing confirmed PMP22 deletion.
Remiche et al36 reported a case of CIDP associated with HNPP, 5 weeks after influenza A H1N1 vaccination in a 54-year-old female with diabetes. Distal pain with proximal and distal weakness of the 4 limbs progressively worsened over 3 months from onset. Examination revealed pes cavus, global MRC grade 4 weakness and hyporeflexia. Electrophysiology showed diffuse demyelination including outside entrapment sites. CSF protein was 0.74 g/L with normal cell count. The patient improved remarkably on IVIg to which she became dependent. Genetic testing performed in view of the electrophysiology showed PMP22 deletion.
Vrinten et al37 described a 35-year-old female with neuropathic pain in the right leg and recurrent weakness and sensory loss in the legs resolving spontaneously after weeks. Examination showed mild asymmetrical proximal and distal weakness and sensory loss with hyporeflexia/areflexia. Electrophysiology demonstrated bilateral ulnar motor conduction blocks at the elbows, diffusely prolonged distal motor latencies and absent F-waves in the legs. A diagnosis of CIDP was made. The patient responded favourably to IVIg, but genetic testing subsequently confirmed HNPP. An ‘n-of-one’ double blind randomised controlled trial was described, this patient demonstrating benefit of IVIg on weakness and pain. The authors concluded that the patient had HNPP and associated CIDP.
The latest report by Shah et al38 recently described a case of CIDP mimicking HNPP in a 43-year-old male. Symptoms were most evident at entrapment sites, electrodiagnostics showed features of acquired demyelination and genetic testing was negative. The patient was described as improving on IVIg. This report is excluded from table 1 as was an unusual case of reversed misdiagnosis of CIDP for HNPP.
X-linked CMT
Tabaraud et al reported a case of a 71-year-old woman with progressive distal weakness, paraesthesiae and areflexia. CSF was normal and electrophysiology revealed non-uniform motor slowing and dispersion with absent sensory responses.39 A sural nerve biopsy showed demyelination without inflammation. A trial with steroids, followed by IVIg, failed to help. Subsequent genetic testing revealed a mutation in GJB1 (nonsense GAG to TAG at position 367, leading to a premature end of protein synthesis.
Ginsberg et al25 reported in their 2004 series one patient with pain and numbness spreading from 1 to 3 limbs over a year. Examination showed dysmorphic features and symmetrical weakness and profound large fibre sensory loss. Brain MRI was normal as was CSF protein. Motor velocities were homogeneously slowed, with no conduction blocks. A Cys173Arg mutation was found in the GJB1coding region. Sural nerve biopsy showed prominent clumps of Schwann cells with numerous perivascular epineurial T cells also present in the perineurium. A dramatic but transient improvement of pain and sensory symptoms was obtained with IVIg. Steroids were unhelpful.
Ryan and Jones described a 13-year-old boy presenting with hand tremor accentuated by stress and physical activity.40 Examination showed bilateral pes cavus, intermittent upper limb action tremor and hypoactive reflexes. Over 2 years, the tremor worsened and ankle reflexes were lost. CSF protein was raised (1.07 g/L) with mild lymphocytic pleocytosis. Electrophysiology showed conduction block and dispersion and F-wave delay. IVIg produced an initial benefit of handwriting and fine motor skills which was unsustained. Genetic testing revealed a novel 13-bp deletion in the coding region of GJB1 (c.318_330del).
Mitchell et al described three patients with initially suspected inflammatory neuropathy who were subsequently found to have GJB1 mutations.41 In one, the diagnosis was thought to be one of CIDP on the basis of demyelinating physiology and raised CSF protein (0.78 g/L). In another, presence of motor conduction block with near normal sensory studies suggested a multifocal motor neuropathy (MMN). In the third case, a slowly progressive picture and demyelinating physiology had suggested CMT1. PMP22 duplication was absent. A superimposed CIDP was, however, suggested electrophysiologically by mild dispersion/conduction block in one nerve. CSF protein was normal. In all three patients, immunomodulatory therapy (IVIg; IVIg and azathioprine; steroids, IVIg, azathioprine and plasma exchanges, respectively) was objectively unsuccessful, although there was a mild subjective effect on sensory symptoms described in the first case. The authors suggested that GJB1 mutations may lead to the erroneous diagnosis of inflammatory neuropathy and that X-linked CMT requires consideration in unresponsive CIDP or MMN.
Sakaguchi et al42 reported a 15-year-old male with a novel GJB1 mutation who presented with left hemiparesis. MRI demonstrated hyperintensity in T2 and reduced diffusion in the posterior centrum semiovale and splenium of the corpus callosum, which resolved in days. Electrophysiology otherwise showed non-uniform slowing and dispersion. CIDP with central involvement was suspected and IVIg attempted with a degree of improvement of strength. The patient's mother was described as presenting at the age of 34 years with dysaethesiae and left foot drop. Since electrophysiology was similarly suggestive of an acquired neuropathy, she was diagnosed with CIDP and treated with IVIg. Her sensory symptoms disappeared after four courses, although motor function was unaltered. Both patients had a novel single pair base deletion: c.397delT in the GJB1 gene. Other cases of CMT1X with central involvement have been reported, although without unusual peripheral physiology to indicate radiculoneuropathic inflammatory disease.43 ,44
Finally, Miki et al45 described three members of a family with CMT1X who responded to IVIg. Two brothers aged 32 and 27 years experienced subacute deterioration of their neuropathy. Electrophysiology showed relatively homogeneous motor velocity slowing, with conduction block and dispersion. CSF protein was described as raised but only at 44 mg/mL in the older brother. Both experienced objective improvement of strength and CMT scores with IVIg and, in the older brother, improved electrophysiology. Their younger sister aged 24 years had experienced a slowly progressive course. She had a demyelinating neuropathy but no features suggestive of an inflammatory cause. IVIg was described as resulting in improvement of toe movements only in her case. All three siblings were found to have a novel GJB1 c.227T[G (p.L76R) missense mutation. The authors concluded that some patients with CMT1X may be susceptible to inflammatory neuropathy.
CMT due to Myelin Protein Zero mutations
GBS revealing MPZ mutation
A novel Myelin Protein Zero (MPZ)mutation was described in a 44-year-old patient with acute-onset back pains and diffuse pararaesthesiae, lasting for 2 months.46 This was a (c.431delT) in exon 3, resulting in a frameshift mutation within the transmembrane domain of MPZ (Leu144fs). Electrophysiology done months after the episode was demyelinating with homogeneously reduced motor velocities, without conduction blocks/dispersion and with absent sensory responses. The patient declined lumbar puncture and nerve biopsy. A sensory GBS was postulated as a possible trigger of the episode, although another explanation may have been an alcohol binge reported by the patient 3 days before onset.
CIDP revealing MPZ mutation
Donaghy et al47 reported a family with CMT due to a MPZ mutation which presented with a partially steroid-responsive neuropathy. The proband, aged 58 years, developed severe late-onset tetraparesis impairing walking and upper limb function. CSF protein was raised at 0.72 g/L without pleocytosis. Electrophysiology revealed diffuse motor slowing with a single conduction block. Steroids were started and resulted in marked although transient improvement. Subsequent treatment with IVIg, plasma exchanges and immunosuppressants was unsuccessful. Other affected family members had late-onset positive sensory symptoms including pain, with or without mild distal weakness. Importantly, none of the affected participants had pes cavus or other reported dysmorphic features. Electrophysiology showed moderate slowing without conduction blocks. One of the proband's brothers underwent a sural nerve biopsy which mainly showed axonal degeneration without inflammation. Sequencing of the MPZ gene revealed a heterozygous change at codon 99 (ATC to ACC) resulting in an isoleucine to threonine substitution.
Watanabe et al48 reported a 37-year-old female with late-onset, asymmetrical, stepwise weakness, limited to distal muscles. CSF protein was mildly raised at 0.65 g/L and electrophysiology demonstrated demyelination with severe secondary axonal loss. Histology showed severe demyelination and onion-bulb formation without inflammation. A previously-reported heterozygous MPZ mutation was detected (Arg98His). Steroid therapy was described as producing a mild degree of improvement of distal strength and motor conduction velocity.
Laurà et al49 reported six Italian patients with late-onset (45–55 years), rapidly progressive axonal neuropathy, which was found to be associated with the same MPZ heterozygous mutation (208 C>T in exon 2) causing Pro70Ser substitution. Importantly, proximal lower limb involvement was present in 4 and the disease was sporadic in one, raising the possibility of an inflammatory neuropathy. Treatment with steroids and immunosuppressants proved ineffective in the sporadic case but was not attempted in the other participants.
HSAN I
Houlden et al50 described a previously asymptomatic 60-year-old man who developed a rapidly progressive painful neuropathy resulting in wheelchair-dependence. Electrophysiology showed a demyelinating neuropathy with conduction blocks and dispersion. CSF protein was raised at 1.93 g/L and nerve biopsy showed axonal loss without inflammation. He was diagnosed with CIDP but subsequently underwent confirmatory genetic testing for an SPTLC1 mutation (Cys133Trp). He failed to respond to IVIg, steroids and plasma exchange but was convincingly described as responding to cyclophosphamide with only persistent sensory signs and distal weakness.
CMT4C
Houlden et al51 described, from five families with SH3TC2 mutations, one subject with a 20-year history of a superimposed inflammatory neuropathy. This patient had interestingly developed, after stopping steroids for ulcerative colitis, rapid decline in neurological function. Electrophysiology showed motor demyelination with temporal dispersion and conduction block. CSF protein was 1.06 g/L. She responded to IVIg for 12 years but this was not sustained beyond and steroids and immunosuppressants were unhelpful.
CMT4J
Zhang et al52 reported rapidly progressive asymmetrical weakness in two siblings unaccompanied by sensory symptoms despite sensory electrophysiological as well as pathological involvement. The patients were compound heterozygotes for mutations of FIG4, Arg183Stop in exon 6 and Ile41Thr in exon 2. Electrophysiology was severely demyelinating with also severe widespread denervation of proximal as well as distal muscles in keeping with an anterior horn cell process. Immunotherapy was not described. However, with the reported picture, particularly in the absence of upper motor neuron signs, an inflammatory neuropathy, although not discussed by the authors, appeared possible. We decided to include this paper in this review for that reason.
Nicholson et al53 described three participants with late-onset CMT4J, with asymmetric proximal weakness. Neurophysiology and pathology were consistent with a severe demyelinating neuropathy with secondary axonal loss. An initial diagnosis of CIDP was made in 2, one failing to respond to IVIg and the other described as benefiting from oral corticosteroids. No further details were provided.
Cottenie et al54 described a patient with demyelinating CMT who developed rapidly progressive proximal and distal weakness in a single limb over 2 years. Electrophysiology showed an asymmetrical picture with conduction block/dispersion. Nerve biopsy performed 12 years earlier had shown severe demyelination without inflammation. Treatment with corticosteroids produced no benefit. No other immunomodulatory therapies were used. FIG4 sequencing showed the Ile41Thr point mutation in exon 2 and an 8-bp deletion in exon 8 causing the frameshift protein truncation mutation p.K278YsX5, confirming CMT4J.
Literature linking inflammatory neuropathy to multisystemic genetic disorders with neuropathic expression
Familial amyloid polyneuropathy
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a rare systemic disease due to endoneurial amyloid deposits.55 ,56 It is of autosomal dominant transmission due to a point mutation of the transthyretin (TTR) gene. The Val30Met mutation, found worldwide, is the most common TTR mutation, although the Thr60Ala mutation is the most common in the UK57 There have been several reports of FAP misdiagnosed as CIDP.
One report described a 40-year-old female patient with progressive multifocal upper limb demyelinating neuropathy leading to an initial diagnosis of the Lewis-Sumner syndrome.58 IVIg failed to help. A sural nerve biopsy demonstrated amyloid deposition. TTR sequencing revealed a valine122isoleucine mutation.
Planté-Bordeneuve et al59 reported misdiagnosis for CIDP in 18/90 patients with non-familial TTR-FAP. Two-thirds received high doses of IVIg and corticosteroids. Causes of misdiagnosis were symmetric sensory symptoms and lack of attention given to autonomic features, raised CSF protein and electrophysiology.
Cappellari et al60 described from eight patients with known TTR mutations, two with an initial misdiagnosis of chronic inflammatory neuropathy. A compatible phenotype backed by demyelinating physiology and raised CSF protein contributed to misdiagnosis, athough pain, dysautonomia and systemic involvement eventually occurred in the majority.
A further report described three patients with subsequently confirmed FAP due to Val30Met mutations, presenting with a clinical/electrophysiological picture indistinguishable from CIDP.61 Two received immunotherapy without benefit.
Koike et al62 described a series of 15 Japanese patients with Val30Met FAP of late onset (>49 years), for whom the initial diagnosis was one of CIDP in 8. In retrospect, all had lower limb pains at initial presentation. Autonomic neuropathy was, however, not the chief complaint in any. CSF protein was raised in nearly 50% and demyelinating electrophysiology reported in about a third. Pathologically, amyloid deposits in sural nerve specimens were found in 12/15 cases.
Adams et al63 reported a high frequency of an ataxic variant of FAP, particularly in a non-Met30 variant, due to Tyr77 TTR mutation. Similarly, rapidly progressive ataxia was the predominant feature in another reported series of 15 patients with FAP of variable genetic basis.64 Since sensory including ataxic CIDP variants are not uncommon, these descriptions appear highly relevant to the issue of misdiagnosis. Other reported phenotypes in the French FAP population have been multifocal upper limb neuropathy and pure motor neuropathy, raising the issue of the differential with the Lewis-Sumner syndrome and MMN.63 ,65
Gibani et al66 described a patient with a progressive disabling neuropathy, achalasia and weight loss who received a diagnosis of CIDP on clinical grounds and in view of mild electrophysiological axonal neuropathy and, importantly, T-lymphocyte infiltration on sural biopsy. Multiple treatments including IVIg, steroids and immunosuppressants were attempted without success. Colonic biopsy review demonstrated amyloid deposition and genetic testing showed the Val30Met mutation.
More recently, Mariani et al,67 in a large genotype-phenotype correlation study, demonstrated predominant early large fibre involvement in non-Met30 cases with frequent electrophysiological demyelination and raised CSF protein. The European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) clinical and electrodiagnostic criteria for CIDP68 were met in no less than 40% of Val107 cases and 24% in late Met30 cases and histopathological signs of segmental demyelination/remyelination found in nearly a third.
Mitochondrial diseases
Uncini et al69 initially described in 1994 a case of demyelinating neuropathy with dispersion consistent with an acquired aetiology, in the context of a mitochondral cytopathy with multiple mitochondrial deletions presenting with ophthamoplegia, leucoencephalopathy, myopathy and gastrointestinal dysfunction.
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal-recessive disorder caused by mutation in the gene encoding thymidine phosphorylase.70 Clinically, MNGIE is characterised by gastrointestinal dysmotility, major weight loss, ophthalmoparesis, ptosis, mitochondrial myopathy, asymptomatic central nervous system white matter abnormalities, and sensory and motor neuropathy. In the majority, the neuropathy has mixed axonal and demyelinating features.71
Bedlack et al71 described five patients with MNGIE in whom the neuropathy had prominent demyelinating features. All were initially misdiagnosed with CIDP, and three treated with immunomodulatory drugs. Clinical features suggestive of CIDP included symmetric weakness progressing over at least 2 months, hyporeflexia, fluctuating course (in 2), CSF protein elevation and demyelinating electrophysiology. Demyelination/remyelination was found on nerve biopsy in 2. All but one had ocular or gastrointestinal signs. Abnormal thymidine levels or thymidine phosphorylase activity and confirmatory genetic testing was obtained in all.
Garone et al72 reviewed the records of 102 patients with MNGIE and described 14% as presenting with peripheral neuropathy. CIDP had been erroneously diagnosed in 6, with demyelinating physiology and raised CSF protein levels in the majority. All developed gastrointestinal features. Pupe et al73 described a 36-year-old male with MNGIE mimicking CIDP. However, the clinical picture as detailed appeared highly suggestive of MNGIE with electrophysiology and histopathology being compatible with CIDP. Filosto et al74 also described a male patient with distal paraesthesiae, and global lower limb weakness without gastrointestinal symptoms but with accompanying ptosis, extraocular weakness and deafness. Electrophysiology showed demyelination confirmed histologically and CSF protein was 3.17 g/L. IVIg, steroids and cyclophosphamide were unhelpful. Biochemical and genetic testing were confirmatory of MNGIE. Scarpelli et al75 reported one male patient who had a demyelinating sensory and motor neuropathy described as a ‘CIDP-like phenotype’. Clinical aspects, however, appeared incompatible with CIDP with ptosis, extraocular muscle weakness and sensorineuronal deafness.
Tangier disease
Tangier disease is a rare autosomal-recessive disorder caused by mutations in the adenotriphosphate binding cassette transporter-1 gene (ABCA1),76 characterised by an abnormal lipoprotein profile. Clinical findings include hepatosplenomegaly, enlarged yellow tonsils and peripheral neuropathy in approximately 50% of patients.
Théaudin et al77 reported a case of Tangier disease in a 15-year-old girl with subacute onset demyelinating asymmetric neuropathy with upper limb conduction block mimicking the Lewis-Sumner syndrome. CSF protein was normal. IVIg did not help. A suggestive abnormal lipid profile was later obtained. Genetic testing confirmed a homozygous mutation of the ABCA1 gene (in exon 14 (c.1759C>T), Arg587Trp). Zyss et al78 reported four cases of Tangier disease with a slowly progressive upper limb neuropathy and facial diplegia. Two patients displayed lower limb involvement. Electrophysiology showed demyelination in all patients in at least one nerve. In one, conduction blocks were detected in two nerves. Onset was asymmetrical in one patient who was first misdiagnosed with the Lewis-Sumner syndrome.
Discussion
In the case of CMT, the first observation from this review is the relatively small number of reports contrasting with its high prevalence as the most common hereditary neuromuscular disorder, reaching 1 in 1200 in recent studies.79 We have found 68 descriptions with confirmed genetic testing (31 CMT1A, 8 with clear descriptions for HNPP, 11 for CMTX1, 11 for MPZ mutations, 1 with HSAN I, 1 with CMT4C and 5 with CMT4J). Despite the frequently hypothesised genetic susceptibility to inflammatory involvement, this suggests a chance association. Furthermore, some reports describe an infectious trigger, indicating a probably simple coincidental occurrence. Several are, in addition, unconvincing for a superimposed inflammatory neuropathy, based on clinical description, electrophysiology, CSF protein and treatment-unresponsiveness. Frequency of described associations are higher in CMT1A, in keeping with the higher prevalence of this subtype. A definite therapeutic response is a major supportive criterion for a positive diagnosis of CIDP.68 Whereas for PMP22-mediated disease (CMT1A, HNPP) a beneficial therapeutic effect was described in 18/23 treated cases (78.3%), in X-linked CMT, on the other hand, no convincing motor response to immunomodulatory therapy was reported in the majority (7/11; 63.7%). There was also considerable heterogeneity of therapies tried, as well as definitions of treatment-response, making reliable comparative analyses impossible. As a result of treatment inefficacy, it was suggested, although unproven, that the different phenotypes described resulted directly from the GJB1 mutation rather than due to an associated inflammatory process.41 Reports on MPZ mutations frequently lacked detail and the presence of a definite inflammatory component appeared uncertain in most.46 ,47 ,49 A therapeutic response was reported to steroids, albeit transiently in 2/3 treated participants, with the response being very minimal in one.48 In the other rarer forms of CMT, a treatment response was described in 3/5, but detailed descriptions were limited.50–54
Greater susceptibility to inflammation remains a possibility in a very small subset of participants, as suggested in one study with CMT1A.28 Interestingly, a recent review of the genetics of GBS and CIDP suggest their associations with disease severity rather than predisposition.80 It is conceivable that this may suggest that most or all CMT participants undergo an associated inflammatory process, but, which in only the few harbouring additional genetic predisposition, subsequently becomes symptomatic. In this regard, the recent review by Martini and Willison brings further insight into the possible inflammatory features present in nerves affected by genetic dysfunction, suggesting that although inflammatory mechanisms may be implicated in their pathogenesis, these are different to the processes involved when CIDP is superimposed.81 Only in the latter case may treatment be effective.
Clinically, besides stepwise or sudden onset of more rapid progressive decline, the presence of pain and positive sensory symptoms as well as proximal weakness represents red flag signs for an inflammatory component in CMT. In this regard, identifying the ‘DADS’ (‘distal acquired demyelinating symmetric’) form of CIDP may be especially challenging in the setting of CMT.68 ,79
The use of investigations searching for inflammatory features provides interesting information from reported cases. We summarise in table 2 the features of typical hereditary and inflammatory neuropathy and the main findings to look for in suspected overlap cases. Figure 1 demonstrates the features of motor block and dispersion in a case of demyelinating CMT with subsequently associated CIDP. In reported cases with CMT1A, 9/17 (52.9%) showed conduction block and/or dispersion. Interestingly, one case of HNPP and one with MPZ mutation showed these features, as opposed to 9/11 (81.8%) of CMT1X and 3/7 (42.9%) of patients with the rarer CMT subtypes. There are, however, a number of issues with such findings in relation to genetic neuropathy subtype. First, in CMT1X, heterogeneous slowing as well as conduction blocks and dispersion have also been described without a suspected or final diagnosis of superimposed CIDP in several reports.40 ,82 ,83 Second, in HNPP, conduction block and dispersion are importantly not unexpected at compression sites. Third, other reports, not directly relevant to, and therefore not included in this review, also describe similar acquired demyelinating-like electrophysiological features in other CMT subtypes.84 ,85 However, we believe the relative scarcity of published literature indicates that electrophysiology still probably remains specific to routinely distinguish between genetic and acquired neuropathies. Also, other indices such as the Terminal Latency Index and Modified F-ratio, normal in CMT1A but abnormal in CIDP or antimyelin associated glycoprotein (MAG) neuropathy, may be useful in the electrodiagnostic process.86 Our clinical experience suggests that values >30% at distal levels, as in recently published criteria for CIDP,68 are appropriate cut-offs for conduction block and dispersion in demyelinating CMT such as CMT1A, as are not generally found in typical disease. We have found that cut-offs used at proximal levels including >50% at Erb's point for conduction block may, however, not be reliable in demyelinating CMT (Y A Rajabally, unpublished data).
Suggested distinguishing features between hereditary and inflammatory neuropathy and main findings indicative of overlap
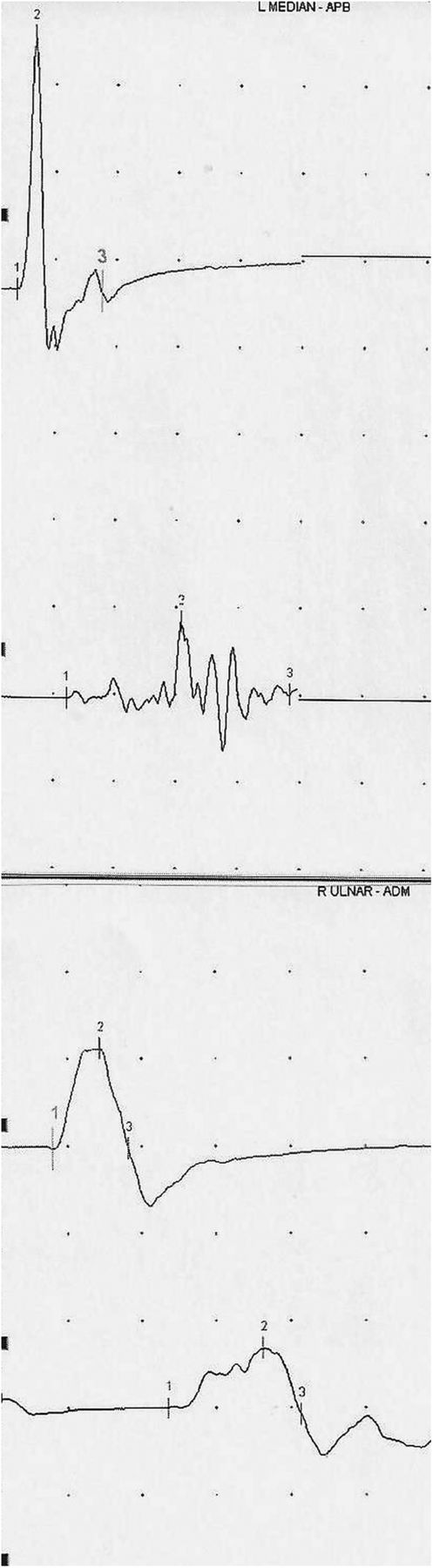
Conduction block and temporal dispersion in the left median and right ulnar motor nerves at the forearm in a patient with demyelinating CMT and clinically suspected superimposed CIDP. CMT, Charcot-Marie-Tooth disease; CIDP, chronic inflammatory demyelinating polyneuropathy.
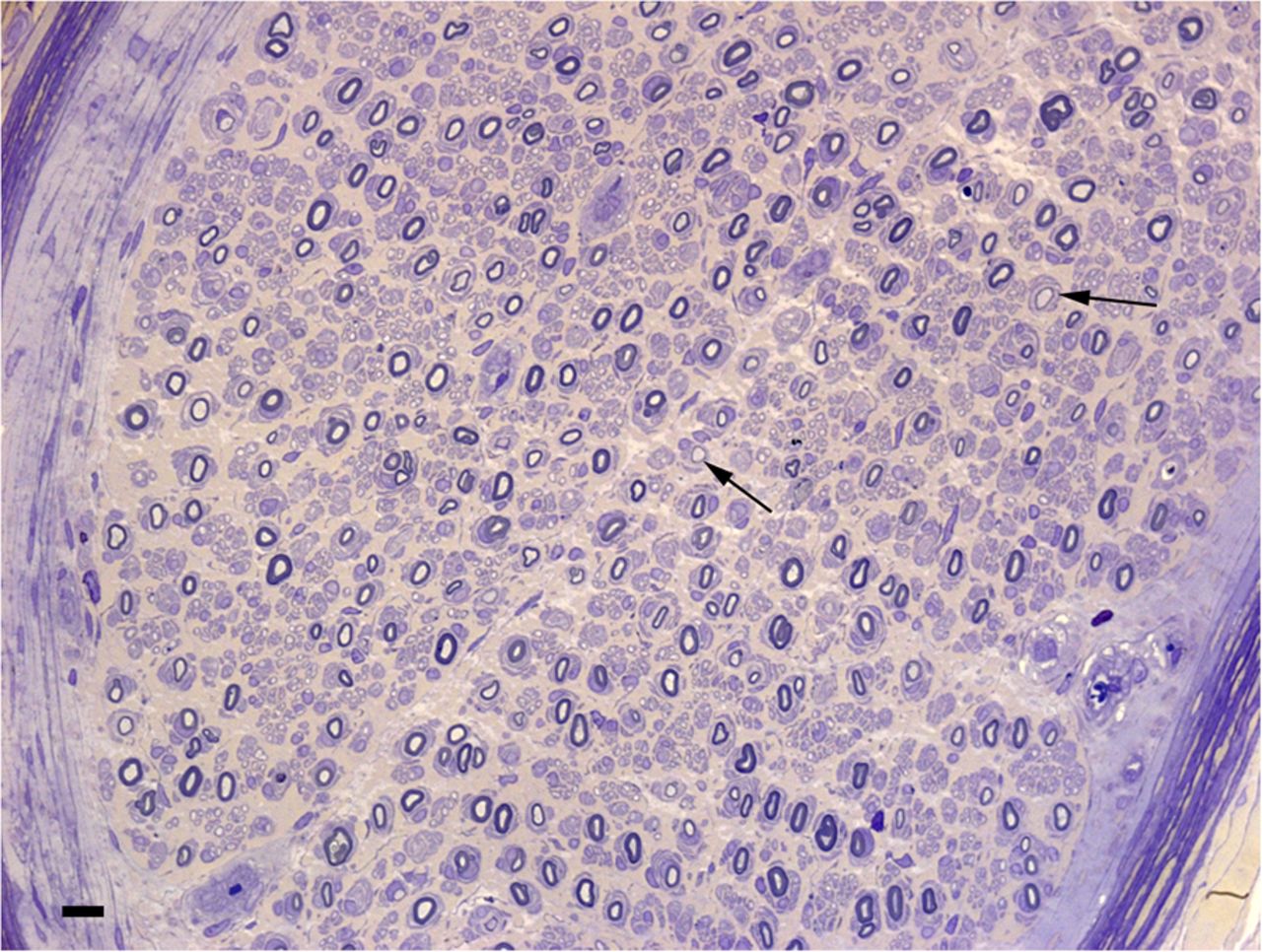
CSF protein levels may be helpful as indicated by the cases reviewed, of which 20/27 (75%) benefited diagnostically. Moderate CSF protein elevations have, however, been described in CMT.87 ,88 Whether these patients may actually have had a CIDP-like course cannot be excluded, although it is plausible that CSF protein is raised in typical CMT, particularly of the demyelinating type and through various mechanisms including nerve root hypertrophy and impaired CSF flow.88 Nerve pathology is of generally established limited benefit in the diagnosis of CIDP.89 In the context of suspected, underlying or associated genetic neuropathy, the existing literature suggests that the situation is similar. Findings may occasionally be confirmatory of associated inflammatory pathology and therefore very useful, as demonstrated in figures 2 and 3. Only four reports on a total of 12 patients (33.3%), with interestingly 10 analysed in only two neuropathology laboratories, were described as showing overt inflammatory features. Whether these numbers reflect the pathological expertise required in this setting is possible. The usefulness of MR neurography in differentiating CMT1A from CIDP is not established. Greater nerve enlargement may actually occur in demyelinating genetic neuropathy,90 implying clearly little utility in determining the presence of superimposed inflammation. It is possible that greater enhancement with gadolinium may be present with inflammatory involvement.91 ,92 Nerve enlargement is also reported on ultrasonography with a possibly greater number of nerve enlargement sites in case of inflammatory disease.93 The practical usefulness of ultrasonography in this setting, however, remains equally unproven.

Resin section of the sural nerve in a 19-year-old boy with CMT1A (patient 7 in Ginsberg et al.25 There are onion-bulb formations around many of the myelinated fibres. Some fibres have inappropriately thin myelin sheaths (arrows). Bar=10 μm. Reproduced with permission from Oxford University Press. CMT, Charcot-Marie-Tooth disease.

{kind=link}
{kind=link}
{kind=link}
Frozen section from a patient with CMT1A with associated CIDP (patient 7 in Ginsberg et al.25 Same biopsy with antibodies to CD4 showing small collections of positive cells in the endoneurium (arrow) and epineurium. Bar=20 μm. Reproduced with permission from Oxford University Press. CMT, Charcot-Marie-Tooth disease; CIDP, chronic inflammatory demyelinating polyneuropathy.
The existing literature suggests that misdiagnosis of FAP for CIDP can represent a significant practical problem. Treatment-unresponsiveness, severe autonomic dysfunction, weight loss and a history of carpal tunnel syndrome should be regarded as red flags. Although currently rarely used, early TTR sequencing may be appropriate in selected cases. Misdiagnosis of MNGIE or Tangier's disease for CIDP appears to occur when insufficient consideration is given to associated clinical features and electrophysiological demyelination is overemphasised as a marker of CIDP.
In conclusion, CMT in its more common forms does not appear a likely predisposing factor to inflammatory neuropathy. Next generation sequencing offering panel testing, although limited by its own challenges, may, however, in future shed further light for specific CMT subtypes or possibly demonstrate forms with inflammatory-like phenotypes.94 In cases of stepwise progression/rapid decline, electrophysiology remains a useful tool. The CSF study may be helpful, although it may be appropriate to use higher cut-offs, such as, arbitrarily, >1 g/L, to define abnormal protein levels, unless the clinical context is highly suggestive, in which case we consider that a cut-off >0.50 g/L may remain adequate. Histopathology may occasionally contribute depending on the availability of a highly specialised laboratory. From the therapeutic perspective, immunomodulation appears justified in case of rapid deterioration, particularly but not only when stepwise, even when an inflammatory component is unproven. Even in possible CIDP-like forms of CMT such as CMT4J, we believe that the contradictory literature and paucity of reports make it difficult to deny a trial of treatment in suspected cases. In the presence of red flags or treatment refractoriness, a high index of suspicion is required, particularly for late-onset FAP, and early consideration for a tissue diagnosis and genetic testing is warranted. We recommend that new cases are published in future. This may allow for a better understanding of the overlap between hereditary and inflammatory neuropathy and ultimately improve management of such challenging patients.
Acknowledgments
The authors are grateful to Professor Jean-Michel Vallat (Department of Neurology, University Hospital Dupuytren, Limoges, France) for his invaluable opinion and advice on this review. The authors thank Dr. Rosalind King and Professor Lionel Ginsberg (Departments of Neuropathology and Neurology, Royal Free Hospital, UK) for kindly providing figures 2 and 3.
References
Footnotes
Contributors YAR, DA and SA were involved in the literature review and drafting manuscript. PL was involved in the revising manuscript for important intellectual content.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.