Article Text

Abstract
Rapid eye movement (REM) sleep behaviour disorder (RBD) is characterised by complex motor enactment of dreams and is a potential prodromal marker of Parkinson's disease (PD). Of note, patients with PD observed during RBD episodes exhibit improved motor function, relative to baseline states during wake periods. Here, we review recent epidemiological and mechanistic findings supporting the prodromal value of RBD for PD, incorporating clinical and electrophysiological studies. Explanations for the improved motor function during RBD episodes are evaluated in light of recent publications. In addition, we present preliminary findings describing changes in the activity of the basal ganglia across the sleep–wake cycle that contribute to our understanding of RBD.
- PARKINSON'S DISEASE
- EMG
- MOTOR CONTROL
- MULTISYSTEM ATROPHY
- SLEEP DISORDERS
Statistics from Altmetric.com
As in every other disease, so here, the earlier the remedies are resorted to, the greater will be the probability of success.
James Parkinson, 1817
REM sleep behaviour disorder
Background
Rapid eye movement (REM) sleep behaviour disorder (RBD) is categorised as a parasomnia, the group of sleep disturbances that is characterised by the occurrence of atypical motor, behavioural or cognitive events, occurring at any time between transitioning to sleep and waking. Other parasomnias include somnambulism, recurrent isolated sleep paralysis and sleep-related hallucinations. In RBD, patients, typically men aged 60–70 years, do not experience the electromyographic (EMG) atonia that accompanies physiological REM sleep.1 The pathological lack of atonia is termed REM sleep without atonia (RSWA).2 The lack of atonia is associated with complex motor enactment of vivid, often frightening dreams, that can result in sleep-related injury (SRI) to patients as well as to their sleeping partners. One study of 41 patients with RBD reported that 46% of patients injured themselves and 34% injured their sleeping partners during RSWA episodes.3 During motor enactment, patients may sing, laugh, punch, kick or cry out. Falling out of bed is a common cause of SRI, often resulting in contusions, with the potential for more serious events, including traumatic brain injuries and subdural haematomas in severe cases. The presence of RBD can have a significant negative impact on quality of life, as repeated SRI can cause anticipatory anxiety for, and fear of, falling asleep.4 Modifying sleeping arrangements is the only RBD treatment recommended by the American Academy of Sleep Medicine (AASM), while pharmacological interventions with clonazepam and melatonin are suggested, but carry risks or are not as well supported, respectively.5 Evidence for several therapies was graded using the Oxford Centre for Evidence-based Medicine Levels of Evidence by a specially convened AASM task force in 2010, with only modification of sleeping arrangements meeting criteria for the highest treatment recommendation, Level A.5 Reflecting these findings, many uncontrolled patients sleep separate from their partners and move mattresses to the floor to decrease risk of falls. Some resort to more inventive methods, including wearing oven mitts over their hands to prevent scratching or fully restraining themselves to prevent bolting out of bed. Officially recognised as a medical diagnosis in 1990, the diagnostic criteria and understanding of RBD are still evolving.
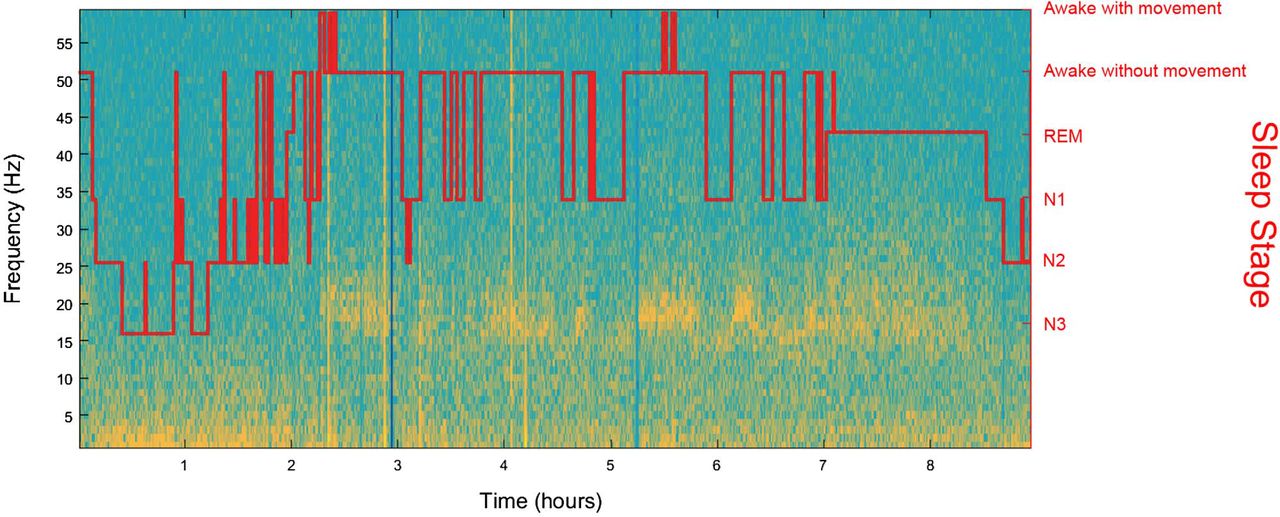
Published in March 2014, the most recent iteration of the International Classification of Sleep Disorders (ICSD) lists 10 core categories of parasomnias, in which only RBD requires a polysomnograph (PSG) for diagnosis.2 PSG is a commonly performed, multimetric, diagnostic test used to identify sleep-associated illnesses such as RBD: in the setting of a clinical sleep laboratory, physiological information, including EEG, electro-oculogram (EOG), electromyogram (EMG) and pulse oximetry, are collected over the course of a night's sleep. These physiological recordings are then scored per well-established AASM criteria, with diagnoses based on quantifiable objective data. PSG results are displayed as a stair plot with each plateau corresponding to the wake, REM, N1, N2 or N3 state, and transitions between states are represented as vertical lines. A representative stair plot is shown in figure 1 (red line). RBD is formally defined by the presence of dream enactment behaviour (DEB) and RSWA. Although patient history or bed partner interview may confirm DEB, the diagnosis of RSWA—and therefore RBD—rests on the presence of EMG abnormalities during REM sleep.2 ,6 A single-question screen for RBD, the REM Sleep Behaviour Disorder Single-Question Screen (RBD1Q), has demonstrated impressive sensitivity (93.8%, with 95% CI 90.0 to 96.2) and specificity (87.2%, with 95% CI 82.4 to 90.8), when compared to gold-standard PSG diagnosis, but the RBD1Q is not part of recognised diagnostic criteria.7

Representative patient data of LFP recordings from the STN during sleep for an individual participant. Heat map depicts spectral power overlaid with timestamp matched PSG output. LFPs were recorded 3 weeks post unilateral implantation into the STN for the treatment of PD. Patient recorded from the OFF medication and OFF stimulation state. LFP power is greatest in areas with warmer colours relative to cooler colours. Left y-axis represents LFP frequencies. PSG stair plot described by right y-axis, corresponding to LFP signal according to time on x-axis. LFP, local field potential; PD, Parkinson's disease; PSG, polysomnograph; STN, subthalamic nucleus.
Because of the requirement for PSG confirmation to establish the diagnosis of RBD, the true prevalence of RBD is unclear, but estimates range from 0.38% to 0.5% according to DSM-V. EMG abnormalities necessary for the diagnosis of RBD are defined as an excess of muscle tone and/or phase twitch during REM sleep. Specific scoring guidelines as to what constitutes ‘abnormal’ EMG findings are moving from qualitative to quantitative criteria.6 Although RBD episodes may vary widely in duration, findings suggest the majority last <2 min, and occur at a frequency of <1 per week.3 Within the RBD, diagnosis are more nuanced classifications of subclinical, clinically probable, idiopathic and secondary RBD, with recent efforts to characterise ‘prodromal’ RBD. Secondary RBD refers to the concurrent presence of another neurological condition such as a PD.2 ,6 However, idiopathic RBD (iRBD) occurs in the absence of such conditions and is known to precede PD and other neurodegenerative synucleinopathies such as Lewy body dementia (LBD) or multiple system atrophy (MSA), by as much as 50 years with a well-documented average of 5–10.8–10 Generally, 50–75% of patients who present with iRBD will go on to develop PD within 10 years of iRBD diagnosis.8–10 In 2014, Iranzo et al8 reported that 90.9% of 174 patients diagnosed with iRBD, developed a defined neurodegenerative syndrome within 14 years. This powerful finding holds promise for diagnosing PD earlier in the disease course.
PD and prodromal value
Well-established clinical criteria are used to diagnose PD, but due to heterogeneous presentation and the existence of closely related disorders, diagnosis can be challenging. Pathognomonic for PD is the presence of intraneuronal α-synuclein protein accumulation, but the risk-to-benefit calculus currently supports diagnosis based on clinical criteria rather than confirmatory brain biopsy.11 When the aetiology of PD cannot be attributed to specific genetics or strong familial history, postmortem neuropathological confirmation is required to establish the diagnosis of PD with rigour.12 Recent evidence suggests that α-synuclein may be found in peripheral tissues and skin allowing for less invasive confirmation, but validation is required.13 The absence of a simple, validated, in vivo, diagnostic test for PD poses a major obstacle to treatment and advancing research, as therapies and outcomes can be very different for PD compared to closely related disorders, such as MSA. Effective early diagnosis must allow the identification of PD among an even larger cohort of related neurodegenerative and neuropsychiatric disorders. Early diagnosis will be crucial for instituting treatments that can halt, slow or reverse disease course. Current treatments, such as levodopa and deep brain stimulation (DBS), can help manage motor symptoms, but are not proven to affect disease course. However, these treatments are currently implemented sometimes after motor symptom onset, when neurodegeneration has already led to 50–70% loss of dopaminergic neurons within the substantia nigra pars compacta (SNc). Appreciably altering disease course from this late-stage degeneration has been unsuccessful, leading to efforts to reliably diagnose PD at earlier stages. Longitudinal PD studies have attempted to recognise markers that consistently manifest before motor symptoms, identifying declines in working memory, visuospatial processing and bradyphrenia, beginning ∼5 years prior to motor deterioration. However, these signs are not specific to PD and difficult to disentangle from cognitive decline associated with other aetiologies.12 Historically, the lack of unambiguous, prodromal features specific to PD has posed a major barrier to effective treatment, but in the last 20 years several potential prodromal biomarkers have been recognised.
The identification of PD prodromal biomarkers is largely due to a reconsideration of the importance of nonmotor symptoms (NMS) in PD pathogenesis and management. Interest in NMS has arisen through the realisation of their cumulative burden on patient mood, daily activities and quality of life. In 2010, the American Academy of Neurology released a 10-point quality measure recommendation, with half of these points pertaining to NMS, and citing gaps in assessment despite the strong association between NMS and quality of life.14 Following this recommendation, NMS are now recognised to present in predictable constellations correlating with younger or older PD age-of-onset (defined as onset before 45 years or after 65 years, respectively). Specifically, anosmia, constipation, urinary urgency, nocturia, hallucinations and components of RBD are generally more common in older-age onset PD, while young-onset PD may be more associated with restless leg and excessive sweating.15 Along with these observations came the realisation that many NMS manifest several decades prior to PD-related motor symptoms, shifting their clinical value from treatment targets to potential prodromal markers.11 Among NMS, anosmia, constipation and sleep disturbances, such as RBD, are the most extensively characterised. Considering these findings, in 2015, the International Parkinson and Movement Disorder Society (MDS) released research diagnostic criteria for the prodromal phase of PD.16 Among the PD risk factors are male sex, exposure to pesticides and family history, while prodromal markers range from olfactory loss to orthostatic hypotension. Using these criteria retrospectively to assess a patient cohort (N=488), Mahlknecht et al17 found a 5-year 60% positive predictive value for incident PD. Traits found to be most predictive included family history, substantia nigra hyperechogenicity, olfactory loss and symptomatic hypotension. Because of the retrospective nature of this study, costly and time-consuming examinations included in the MDS criteria that would potentially have improved predictive accuracy—such as PSG-determined iRBD—were not assessed.
Investigations into the MDS prodromal criteria have led to the finding that iRBD has particularly high sensitivity and specificity for conversion to synucleinopathies, in contrast to other prodromal markers such as constipation and anosmia. Further investigations have found biological bases for the high rate of iRBD-to-PD conversion. Imaging studies have identified significant changes in brainstem white matter in iRBD patients compared to controls, and specifically in brain regions affected by PD, including the substantia nigra and olfactory cortex.18 Additional work has implicated brainstem regions, including the sublaterodorsal nucleus, laterodorsal tegmental, pericoerulear, pedunculopontine nuclei (PPN) and ventrolateral reticulospinal tracts, in RBD pathogenesis.1 ,19 A single-photon emission CT (SPECT) and transcranial sonography imaging study conducted by Iranzo et al,20 followed 43 patients with iRBD over a period of 2.5 years, with 27 patients displaying either reduced dopamine (DA) transporter uptake or hyperechogenicity of the substantia nigra—both physiological changes seen in early PD—and 8 of 27 patients developing a neurodegenerative disease within 2.5 years (5 of 8 developed PD). Patients with iRBD who did not present with abnormal imaging findings did not go on to develop PD or related disorders. The ability to distinguish those patients with iRBD who will ultimately develop synucleinopathies is of obvious importance. The connection between RBD and PD has been further strengthened by the recent finding of PD-associated memory impairments in individuals with iRBD.21 These findings support the notion that iRBD is a prodromal PD biomarker—not only because of linear conversion, but also because of associated functional and behavioural changes prior to the onset of PD motor symptoms.
While the utility of iRBD in prodromal identification of PD is increasing substantiated, it should be noted that although iRBD has shown a high rate of conversion to synucleinopathies, only a minority of newly diagnosed PD cases show RBD.22 This is likely reflective of the heterogeneous aetiology of PD, with iRBD indicative of some subpopulation of all mechanisms of PD development. Although this indicates that iRBD is not a prodromal marker for all types of PD, it also provides a means to differentiate between subtypes of PD, which may have implications for treatment.
Insights into PD pathogenesis
The value of iRBD in elucidating PD pathogenesis stems from a detailed understanding of the brainstem: functional deficits can be tied to pathologies at well-defined anatomical locations without the need for imaging. The resolution of this understanding is high for sleep–wake cycling, as recently evidenced by successful optogenetic control of the REM phenomenon via modulation of the dorsal medulla GABAergic neurons in rodents.23 Studies characterising the location of RBD deficits point to locus coeruleus (LC) and related areas as lesion sites. Since RBD appears well in advance of symptomatic dopaminergic neurodegeneration of SNc, it is possible that this relationship describes the caudal-to-rostral directionality of a larger disease process. The Braak Staging Model describes such a timeline, proposing that constipation and anosmia present even before the sleep disturbances of PD, as a consequence of the more peripheral location their respective nervous system control centres.11 Clinical evidence from patients with iRBD supports Braak's hypothesis that neurodegeneration via α-synuclein misfolding may arise in the periphery and progress centrally up through to the cortex.11 The timeline of onset of idiopathic anosmia to RBD to PD does not describe all patients; however, raising the possibility that for each pattern of prodromal symptom presentation, there is a corresponding, distinct PD aetiology. Possibly, neuroprotective interventions will not be uniform in their impact across these groups. Because of the relatively high rates of conversion of iRBD into PD, patients with iRBD are well suited to aid in the development, and to assess the efficacy, of neuroprotective therapies. Recently published work by Postuma et al24 quantified the number of iRBD patients needed to prove significant reduction in neurodegeneration assuming a therapy provided a modest HR of 0.5, finding that a sample size of fewer than 80 patients would be sufficient.
Insights into movement processing
Evaluations of patients with RBD-PD have provided insights into the underlying neural mechanism of movement. Improvements in patient movement and speech during RBD episodes have been reported, relative to awake periods.3 ,25 In 2007, De Cock et al reported on 53 patients with RBD-PD, with 100% of bed partners reporting improvement of at least 1 component of motor function during RBD episodes. These authors suggest that the levodopa-like improvement in motor symptoms during RBD may be attributable to primary motor cortex activity bypassing the inhibitory role of the subthalamic nucleus (STN) and eventually transmitting to lower motor neurons.3 Ictal SPECT imaging has supported the hypothesis that the basal ganglia are bypassed during RBD episodes, as well.26 In 2011, De Cock et al published a study evaluating RBD in patients with MSA, reporting that patients with MSA similarly improved motor control during RBD episodes. In contrast to PD, patients with MSA respond to levodopa for only 2–3 years—if at all—suggesting that motor improvement during an RBD episode is not mediated through DA signalling, or at least not exclusively.25 Transient RBD-associated improvements in motor function bear a resemblance to another movement-related phenomenon—that of paradoxic kinesis (PK). During PK, spontaneous, transient motor improvement accompanies intense stimuli. Certain behavioural paradigms have been found to reproducibly provoke PK-like improvements in motor control, allowing researchers to evaluate this phenomenon in the clinical setting.27
Application of these paradigms to patients undergoing DBS surgery is particularly interesting because it is possible to have implanted patients perform tasks while recording the neuronal activity of nuclei such as STN or globus pallidus internus (GPi). Activity is measured using electrophysiological signals, including single-unit potential and local field potential (LFP). LFPs are oscillations thought to represent the aggregate discharges from neurons within the immediate vicinity of recording sites.28 ,29 LFP can be recorded through the same macroelectrode contacts used to deliver stimulation via DBS. Recordings are most often performed intraoperatively, but can be collected chronically, years after surgery.29 ,30 Analysis of LFP recordings has led to the identification of a number of electrophysiological findings correlated with PD states and symptoms, and has elucidated aspects of basal ganglia function. For example, the β-band (13–30 Hz) of the LFP spectrum has greater power in the OFF medication and OFF stimulation states. In comparison, the converse has been reported for γ (60–90 Hz) power. β is often regarded as ‘antikinetic’, because it is present when patients are in an OFF state or at rest, and decreases in spectral power during motor planning and execution. Conversely, γ is considered ‘prokinetic’ because its spectral power increases with the initiation of movement relative to rest. This information has led to clinically useful advances in PD patient care and DBS device design.31 Using LFP recordings in conjunction with PK-inducing behavioural paradigms allows clinicians to examine whether known DA-related changes in basal ganglia activity can explain PK-related improvements in movement. Reporting on such a study in 2012, Anzak et al27 were unable to connect PK to DA, supporting the presence of a DA-independent modifying circuit. Candidates for this DA-independent modifying circuit include the brainstem reticular-activating system and PPN, with recently reported recording from PPN (n=1) during a PK task supporting this hypothesis.32 These reports have helped highlight the relative contributions of dopaminergic and non-dopaminergic control on movement processing, largely arising from analysis of patients with RBD-PD.
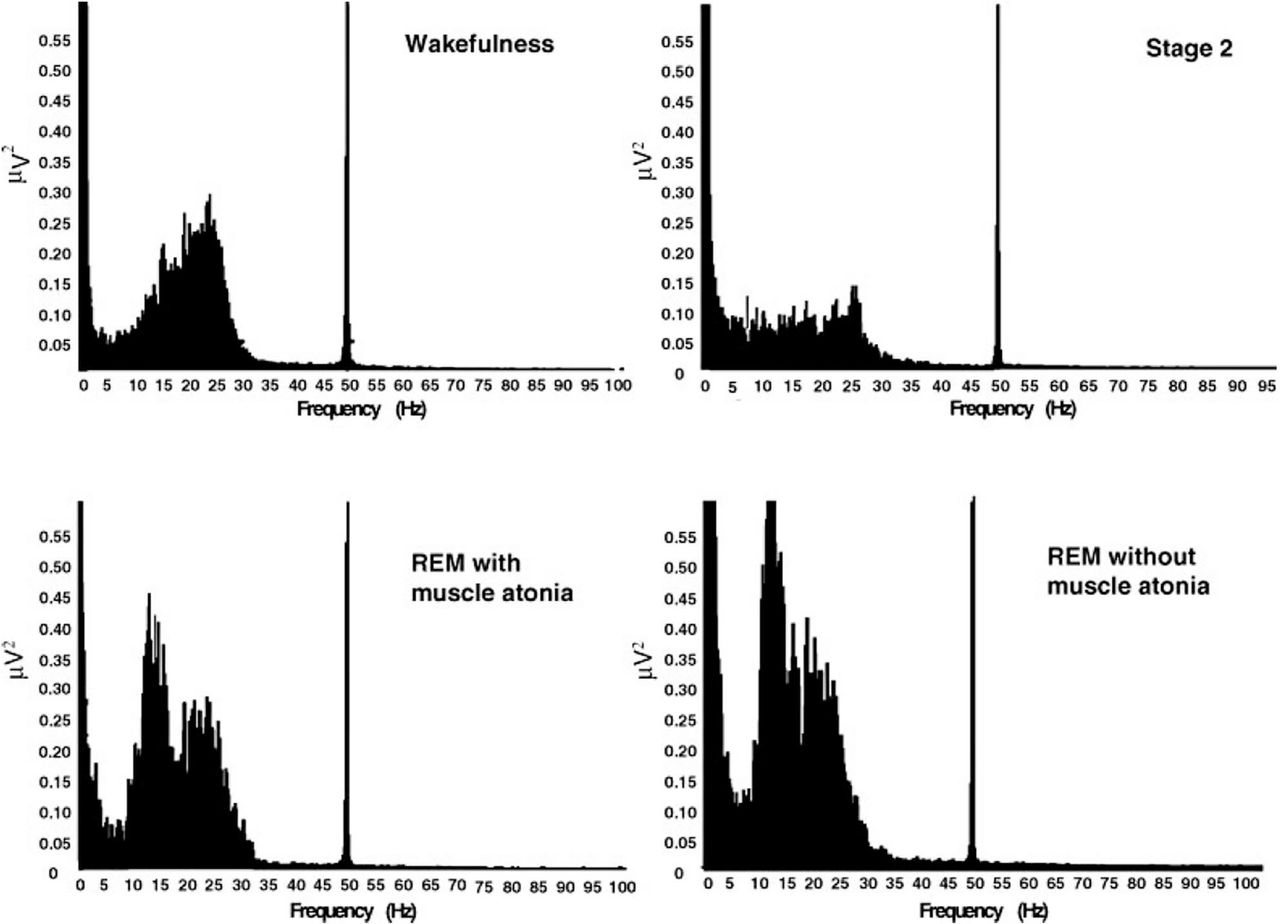
LFP recordings from STN and/or PPN in patients experiencing an RBD episode could prove an invaluable contribution to this field. Few reports to date have attempted to provide characterisation of changes in the activity of deep brain structures relative to arousal state. To the best of our knowledge, only two publications describe deep brain LFP spectral changes with respect to sleep stage.33 ,34 In 2009, Urrestarazu et al33 reported the first characterisation of STN activity with respect to sleep stage, identifying 3–8-s long segments in 3 of 10 patients as periods of RSWA, based on EMG of limb muscles (figure 2). β-Band during REM was found by these investigators to be comparable or higher than awake periods, and significantly greater than in other sleep stages (figure 3). Comparing RSWA segments to atonic segments during REM sleep, the authors found power in the β-band to be greater during RSWA segments (figure 2). Although RBD was not explicitly assessed, the increase in β during RSWA identified by Urrestarazu et al would likely also be present during RBD episodes. These findings might support the existence of a non-DA-modifying circuit because if dopaminergic control at the level of STN and related structures were solely responsible for PD motor symptoms, we would expect to see a worsening of motor function during RSWA/RBD, rather than the reported improvements, because of the known increase in β during REM and RSWA. As acknowledged by the authors, conclusions from this study are limited because video recordings corresponding to the RSWA periods did not demonstrate clear patient movement, raising questions about the diagnosis of RSWA. Furthermore, specifics on how the diagnosis of RSWA was determined, beyond the presence of an abnormal EMG, were not delineated. At present, the data provided in the Urrestarazu et al publication are most rigorous description of STN LFP spectral changes over the sleep–wake cycle available. Additional reports on this subject would likely have a high impact on the understanding of STN function as well as RBD.

Representative STN single-channel spectral analysis during wakefulness and different sleep stages. REM sleep further characterised by the presence or absence of muscle atonia. Figure taken with permission from Urrestarazu et al.33 Copyright © 2000–2016 by John Wiley & Sons, or related companies. All rights reserved. STN, subthalamic nucleus.

Group analysis of 10 PD participants bilaterally implanted in the STN. β-power during wakefulness and different sleep stages as recorded by STN electrode and EEG quantified. There is a significant decrease in β-band from ‘OFF’ wakefulness to stage 2 (p<0.0001). STN β-power during REM is slightly higher than during wakefulness (p=0.002, n=30), and significantly higher than during stages 2 and 4 (p<0.0001, n=30). Differences between the results analysing all STN channels and the STN channels with the best β-peaks were not statistically significant. Figure taken with permission from Urrestarazu et al.33 Copyright © 2000–2016 by John Wiley & Sons, or related companies. All rights reserved. PD, Parkinson's disease; PSG, polysomnograph; STN, subthalamic nucleus.
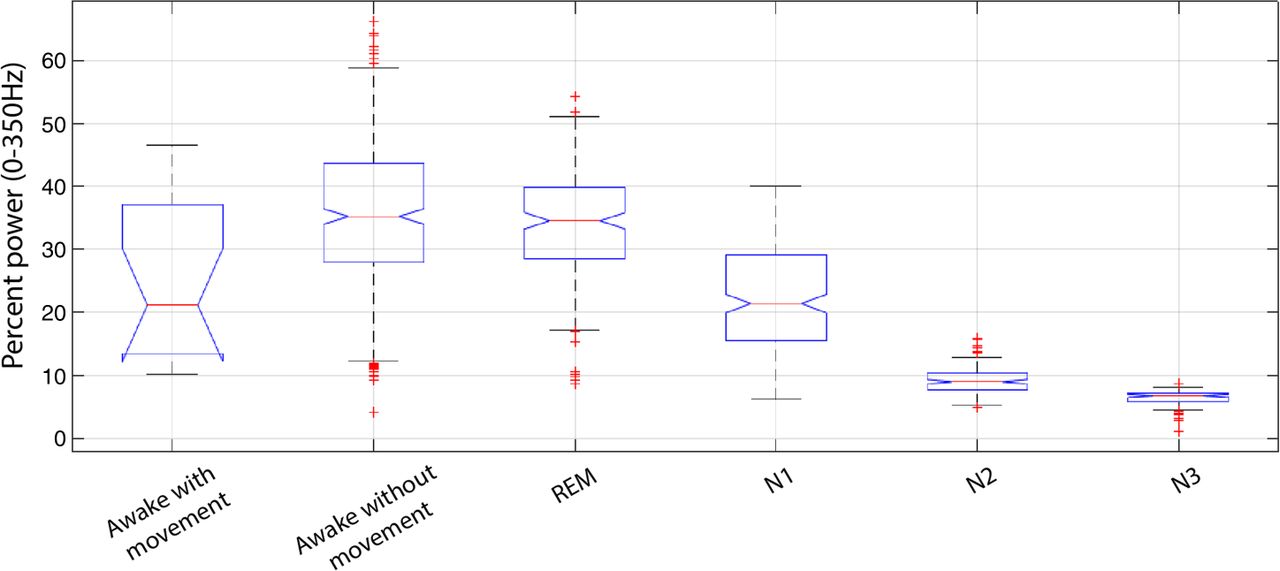
The cost and challenges of performing LFP recordings from awake, implanted patients during an operative procedure are not trivial, but expanding the duration of a DBS surgery to capture recordings during sleep is neither feasible nor ethical. Alternatively, capturing sleep cycle LFP data from patients chronically implanted with a research device that stimulates and records (Medtronic PC+S) is technically feasible, but currently consumes significant battery life. Nonetheless, detailed characterisation of LFP spectral changes across sleep stages would provide an idea of the predictive value of basal ganglia LFP in relation to sleep–wake states. Components of such an analysis of unilateral STN LFP recordings from 10 PD participants, with concurrent overnight PSG captured during a full night of sleep, are shown in figures 1, 4 and 5. DBS surgery was performed as previously described, and data were collected using an XLTEK-EMU40 system.30 ,35 ,36 The following montage was used for sleep evaluation according to the 2007 American Academy of Sleep Medicine guidelines: F3-C3, P3-O1, F4-C4 and P4-O2, LEOG-O2, REOG-O1 and chin EMG.37 ,38 In brief, our analysis supports the finding that changes in STN activity tightly correlate with PSG-determined sleep–waking transitions and with specific sleep stages (figure 1). Of note, in figure 1, the β-band (13–30 Hz) appears to reflect dynamic changes in spectral power across sleep–waking and REM–NREM transitions. Frequency-specific analysis shows that changes in β are greatest in the lower half of the frequency range (figure 4). Furthermore, quantitative analysis of β-power across awake and sleep states demonstrates greater power during REM compared to other sleep stages within participant (figure 5). This relationship is also observed in preliminary group analysis (n=10). Median values for per cent of total spectral power (0–350 Hz) along with 25% (Q1) and 75% (Q3) quartiles for group analysis (n=10) are as follows (median, Q1, Q3): REM (29%, 20%, 38%), N1 (18%, 12%, 25%), N2 (8%, 6%, 11%) and N3 (5%, 4.5%, 6.5%).

Spectral power of STN LFP β-band (13–30 Hz) across PSG-determined sleep–awake states for individual participant. REM β-power closer in magnitude to wake states than other sleep stages. LFP, local field potential; PSG, polysomnograph; STN, subthalamic nucleus.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notched box plot quantifying spectral power in STN LFP β-band (13–30 Hz) across PSG-determined sleep–awake states for individual participant (same participant as in figure 4). Midbox horizontal lines represent the median, with the notch representing the 95% CI for the median. The lower and upper extremes of the boxes represent 25% (Q1) and 75% (Q3) quartiles, respectively, with the difference between Q1 and Q3 representing the IQR. Lower boxplot whisker extends to (Q1–IQR) and upper boxplot whisker extends to (Q3+IQR). LFP, local field potential; PSG, polysomnograph; STN, subthalamic nucleus.
Shortcomings
Although iRBD has strong epidemiological and increasingly convincing mechanistic evidence supporting its use as a PD biomarker, there are some caveats to consider. RBD almost exclusively affects male patients (nearly 90%) over the age of 50. The strong bias towards presentation in men may represent underdiagnosis in women—due perhaps to a less obvious presentation of DEB in women, or gender-based discrepancies in reporting or history taking—but alternative explanations to this male predominance include a sex-linked genetic aetiology. Reports indicating that PD NMS might be attributable to endocrine dysfunction led to the hypothesis that RBD, with its high proportion of male patients, is caused by testosterone deficiency.39 However, in 2007, Chou et al40 assayed testosterone levels in patients with PD+RBD versus patients with PD, reporting no significant differences. Regardless, the discrepancy between iRBD and PD patient demographics suggests that the utility of tracking iRBD-to-PD conversion would extend only to a subset of patients with PD.
Additionally, although iRBD most often converts to PD, it can also precede the diagnosis of LBD and MSA. This lack of specificity is not unique to RBD, as other prodromal PD features such as anosmia or constipation are also known to be associated with multiple diagnoses, including Alzheimer's disease. Even a hypothetical biomarker based on blood or cerebrospinal fluid (CSF) would likely lack the resolution to differentiate between synucleinopathies as a consequence of the shared α-synuclein hallmark. Ultimately, the prodromal value of iRBD in establishing the diagnosis of PD will require an appreciation for what distinguishes the individual synucleinopathies from one another, but still holds significant clinical utility as a prodromal marker for synucleinopathies in general.
Conclusions
Mounting evidence from cohort, electrophysiological and animal work strongly supports the role of iRBD as a clinically significant prodromal marker for PD, or closely related Parkinsonian syndromes such as LBD or MSA. The greatest barrier to harnessing this knowledge to improve patient outcomes is timely and clinically rigorous RBD diagnosis, hampered by the current need for PSG-confirmed RSWA. Raising patient awareness for the significance of RBD in predicting a subsequent Parkinsonian disorder could enhance self-monitoring and sleeping partner monitoring and reporting. Less expensive and less intrusive methods, aimed at distinguishing which patients with iRBD are likely to convert to synucleinopathies, need to be developed. In parallel, animal model and clinical electrophysiological experiments aimed at deciphering STN/GPi spectral activity during RBD episodes are needed to further elucidate the role of a non-dopaminergic movement processing circuit. This may lead to novel pharmaceutical and/or DBS targets for movement disorders, while contributing to our understanding movement processing.
References
Footnotes
Acknowledgements The authors thank Drs Urrestarazu and Artieda for allowing us to use of figures from their 2009 publication ‘β Activity in the Subthalamic Nucleus During Sleep in Patients with Parkinson's Disease’.
Contributors AT conceived the manuscript with significant input from co-authors. NFI and JAT provided assistance with processing data depicted in figures 1,4 and 5 DSK and JT evaluated the accuracy and value of statements concerning RBD and PD, and AA provided overall edits.
Funding This work was supported by National Science Foundation (CBET-1343548) and Medtronic.
Competing interests None declared.
Patient consent Obtained.
Ethics approval University of Minnesota Institutional Review Board.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement Preliminary data presented is being prepared for a manuscript.