


Abstract
Upon a wide range of cellular stresses, p53 is activated and inhibits malignant transformation through the transcriptional regulation of its target genes related to apoptosis, cell cycle arrest, and DNA repair. However, its involvement in posttranslational modifications of proteins has not yet been well characterized. Here, we report the novel role of p53 in the regulation of protein citrullination. p53 transactivated peptidylarginine deiminase type 4 (PADI4) through an intronic p53-binding site. The PADI4 gene encodes an enzyme catalyzing the citrullination of arginine residues in proteins, and ectopic expression of p53 or PADI4 induced protein citrullination. In addition, various proteins were citrullinated in response to DNA damage, but knockdown of PADI4 or p53 remarkably inhibited their citrullination, indicating the regulation of protein citrullination in a p53/PADI4-dependent manner. We found that PADI4 citrullinated the histone chaperone protein, nucleophosmin (NPM1), at the arginine 197 residue in vivo under physiologic conditions. Citrullination of NPM1 by PADI4 resulted in its translocation from the nucleoli to the nucleoplasm, whereas PADI4 did not alter the localization of mutant NPM1 (R197K). Furthermore, ectopic expression of PADI4 inhibited tumor cell growth, and concordantly, the knockdown of PADI4 attenuated p53-mediated growth-inhibitory activity, demonstrating the significance of PADI4-mediated protein citrullination in the p53 signaling pathway.[Cancer Res 2009;69(22):8761–9]
Introduction
Mutations in the p53 gene are the most common genetic alteration observed in human cancers (1). Because ∼90% of them were detected within its DNA-binding domain (2, 3), the crucial function of p53 is considered as a sequence-specific transcription factor. Recently, extensive analyses were conducted to clarify the role of posttranslational modifications (PTM) in diverse cellular signaling pathways. Proteome analyses using two-dimensional gel electrophoresis showed that activated p53 altered the amounts of various protein spots without affecting their mRNA levels (4), suggesting that p53 could change the biological properties of proteins through modulation of PTMs. However, the role of p53 in the regulation of PTMs has not yet been elucidated.
Citrullination (also referred to as deimination) is one form of PTMs which converts an arginine residue in protein into a citrulline residue (5). This reaction is mediated by a Ca2+-dependent enzyme called peptidylarginine deiminase. Citrullination of proteins results in a loss of positive charge and causes a significant biochemical change. For example, PADI4 was shown to convert monomethyl-arginine residues of histone H3 and H4 to citrullines and negatively regulate gene expression (6, 7).
On the other hand, citrullinated peptides/proteins are recognized as non–self-proteins and subsequently activate immune systems. Citrullinated myelin basic protein in the brain was considered as a putative autoantigen associated with multiple sclerosis (8). Autoantibodies against citrullinated peptides derived from filaggrin or fibrin have been established as specific markers for rheumatoid arthritis (9–11). We have also revealed a functional haplotype of the PADI4 gene associated with rheumatoid arthritis (12). Hence, protein citrullination is now considered to play a pivotal role in various human diseases (13), but the role(s) of citrullination in carcinogenesis, if any, have not yet been reported.
Among five members belonging to the peptidylarginine deiminase family (14–16), we found PADI3 and PADI4 to be transactivated by ectopic expression of p53 in U373MG cells through our cDNA microarray analysis (17, 18). Therefore, we investigated the possible roles of p53 in the regulation of protein citrullination in this study.
Materials and Methods
cDNA microarray
cDNA microarray analysis was performed as described previously (19). Briefly, U373MG cells were infected with viral solutions and incubated at 37°C until the time of harvest. Polyadenylated RNA were isolated from U373MG cells using standard protocols. Each RNA sample was labeled and hybridized to a microarray consisting of 36,864 cDNA fragments. The results of the microarray analysis were deposited at the Gene Expression Omnibus database (accession no. GSE14953).4
Plasmid construction
Plasmids expressing PADI3 and PADI4 were kindly provided by Dr. Akari Suzuki (RIKEN, Yokohama, Japan). Catalytically inactive mutant PADI4 (D350A and D473A) was generated using the QuickChange site-directed mutagenesis kit (Stratagene). Primers are shown in Supplementary Table S1.
Cell culture and transfections
Each cell line was purchased from American Type Culture Collection, Lonza Biologics, Inc., or Japanese Collection of Research Bioresources. HCT116 (p53+/+ and p53−/−) cell lines were gifts from B. Vogelstein (Johns Hopkins University, Baltimore, MD). Cells were transfected with plasmids using FuGENE6 (Roche). Replication-deficient recombinant viruses Ad-p53 or Ad-LacZ, expressing p53 or LacZ, respectively, were generated and purified, as described previously (20, 21). U373MG cells were infected with viral solutions at various amounts of multiplicity of infection and incubated at 37°C until the time of harvest. For treatment with genotoxic stress, cells were continuously incubated with Adriamycin (ADR) for 2 h as indicated in the respective figure legends. Small interfering RNA (siRNA) oligonucleotides, commercially synthesized by Sigma Genosis, were transfected with LipofectAMINE 2000 reagent (Invitrogen) for 4 h. Sequences of oligonucleotides are shown in Supplementary Table S1. For treatment with calcium ionophore, cells were incubated with 5 μmol/L of A23187 (Calbiochem) for 1 h at 37°C.
Quantitative real-time PCR
Quantitative real-time PCR was conducted with the SYBR Green I Master on a LightCycler 480 (Roche). Primer sequences are indicated in Supplementary Table S1.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed using ChIP Assay kit (Upstate Biotechnology) as described previously (18). PCR amplifications of PADI4 intron 1, containing the consensus p53-binding sites, were performed on immunoprecipitated chromatin using the primers indicated in Supplementary Table S1.
Gene reporter assay
DNA fragments, including potential p53-binding sites of PADI4, BAX, FAS, or p21WAF1 were amplified and subcloned into the pGL3-promoter vector (Promega). Primers for amplification are shown in Supplementary Table S1. To make a series of mutant vectors, a point mutation “T” was inserted into the site of the 4th and the 14th nucleotide “C” and the 7th and the 17th nucleotide “G” of the consensus p53-BS using the QuickChange site-directed mutagenesis kit (Stratagene). Reporter assays were performed using the Dual Luciferase assay system (Promega) as described previously (18).
Antibodies
To develop antibodies against Cit197 NPM1, a NPM1 peptide (KKSICitDTPAKN) was chemically synthesized and used to immunize rabbits. The positive antisera were further purified by immune-affinity purification using a NPM1 Cit197 peptide. Polyclonal anti–modified citrulline antibody (17–347) was purchased from Upstate and the modification of citrulline residues was carried out according to the instructions of the supplier. Anti–β-actin monoclonal antibody (A5441) and anti-Flag monoclonal (F3165) antibody were purchased from Sigma. Anti-p53 monoclonal antibodies (OP140 and OP43) and anti-p21WAF1 monoclonal antibody (OP64) were purchased from Calbiochem. Anti-B23 (NPM1) polyclonal (sc-6013) and monoclonal (sc-32256) antibodies, anti-HA monoclonal (sc-7392) and polyclonal (sc-805) antibodies, anti-Myc monoclonal antibody (sc-40), anti-HSP90 monoclonal antibody (sc-13119), anti-PARP1 monoclonal antibody (sc-8007), and anti–glutathione S-transferase (GST) monoclonal antibody (sc-138) were purchased from Santa Cruz Biotechnology. Anti-PADI4 polyclonal antibody (ab50332) and anti-nucleolin polyclonal antibody (ab22758) were purchased from Abcam.
In vitro citrullination assay
GST-PADI4 and GST-NPM1 were generated by cloning of their coding sequences into pGEX6P-1 (Amersham). Proteins were expressed in Escherichia coli and purified on glutathione-Sepharose beads (Amersham) by standard methods. Deimination reactions were carried out using purified protein in 0.1 mol/L of Tris-HCl (pH 7.4), 5 mmol/L of DTT, and up to 1 mmol/L of CaCl2 at 37°C or using bead-bound substrate in 50 mmol/L of HEPES (pH 7.4), 2 mmol/L of DTT, 150 mmol/L of NaCl, 0.5% NP40, and 1 mmol/L of CaCl2 at 30°C. HEK293T cells were transfected with constructs expressing HA-tagged wild-type or mutant NPM1 (R13K, R101K, R142K, R197K, and R221K). At 48 h after transfection, cells were lysed in 50 mmol/L of HEPES (pH 7.4), 2 mmol/L of DTT, 150 mmol/L of NaCl, 0.5% NP40, and immunoprecipitated using an anti–HA-agarose (Sigma). The immunoprecipitated bead-bound protein was washed with lysis buffer and reactions were carried out as described above.
Immunoprecipitation
Cell extracts from HEK293T cells transfected with a plasmid encoding HA-PADI4 or U373MG cells infected with Ad-p53 were prepared by adding lysis buffer [50 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, and 0.5% NP40]. Lysates were precleared by incubation with protein G-Sepharose 4B (Zymed) and mouse IgG at 4°C for 1 h. Then, precleared cell extracts were incubated with anti-B23 (NPM1) antibody (sc-32256) and protein G-Sepharose 4B at 4°C for 2 h. The beads were washed four times with 1 mL of ice-cold lysis buffer and immunoprecipitated proteins were released from the beads by boiling in sample buffer for 2 min.
Western blot analysis
To prepare whole cell extracts, cells were collected and lysed in chilled radioimmunoprecipitation assay buffer [50 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% NP40] for 30 min on ice and centrifuged at 16,000 × g for 15 min. Samples were subjected to SDS-PAGE and immunoblotting using a standard procedure. Blots of deiminated proteins were treated with medium for chemically modifying citrulline residues at 37°C for 3 h and then modified citrulline residues were detected with a modified citrulline-specific antibody (anti-MC). Immunocytochemistry was performed as described previously (18).
Mass spectrometric analysis
Each sample was loaded onto SDS-PAGE followed by CBB-R250 staining. The excised protein bands were reduced in 10 mmol/L of tris (2-carboxyethyl)phosphine with 50 mmol/L of ammonium bicarbonate for 30 min at 37°C and alkylated in 50 mmol/L of iodoacetamide with 50 mmol/L of ammonium bicarbonate for 45 min in the dark at 25°C. Porcine trypsin (Promega) was added for a final enzyme to protein ratio of 1:20. The digest was conducted at 37°C for 16 h and then desalted on an Oasis HLB cartridge (Waters). The resulting peptide mixture was separated on a 75 μm × 150 mm L-column (Chemicals Evaluation and Research Institute, Japan) using a 30-min linear gradient from 5.4% to 29.2% acetonitrile in 0.1% formic acid with a total flow of 200 nL/min. The eluting peptides were ionized by electrospray ionization and analyzed by a QSTAR Elite QqTOF system (Applied Biosystems/MDS Sciex). Peptide tandem mass spectrometry (MS/MS) analyses were acquired in an information-dependent mode using the Analyst QS software 2.0 acquisition features (rolling collision energy and dynamic exclusion; Smart Exit). Peak list files generated by the Protein Pilot version 1.0 software (Applied Biosystems) using default parameters were exported to a local MASCOT version 2.2.03 (Matrix Science) search engine for protein database searching, in which an additional modification file “citrullination” [H(−1) N(−1) O] was set for a variable modification. All MS/MS analyses indicating citrullinated peptide fragments were reconfirmed manually.
Purification of nucleoli
Nucleoli were purified using a procedure described previously (22). In brief, cells were resuspended in 15 volumes of a hypotonic buffer [10 mmol/L Tris-HCl (pH 7.4), 10 mmol/L NaCl, and 1 mmol/L MgCl2] and incubated on ice for 30 min. Then Nonidet P40 (Roche) was added (final concentration of 0.3%) and homogenization was performed with seven strokes through a 21-gauge needle. Nuclei were collected by centrifugation at 1,200 × g for 5 min and resuspended in 10 volumes of 0.25 mol/L sucrose containing 10 mmol/L of MgCl2. The supernatant containing the cytoplasmic fraction was harvested for further analyses. Nuclei were then purified by centrifugation at 1,200 × g for 10 min through a 0.88 mol/L sucrose cushion containing 0.05 mmol/L of MgCl2. Purified nuclei were resuspended in 10 volumes of 0.34 mol/L sucrose containing 0.05 mmol/L of MgCl2 and sonicated on ice. Nucleoli were then purified from the resulting homogenate by centrifugation at 2,000 × g for 20 min through a 0.88 mol/L sucrose cushion containing 0.05 mmol/L of MgCl2. The supernatant containing the nucleoplasmic fraction devoid of nucleoli was harvested for further analyses. Purified nucleoli were resuspended in 0.34 mol/L of sucrose containing 0.05 mmol/L of MgCl2 for further analyses.
Cell proliferation/cell death assay
Colony formation assays were carried out in six-well culture plates. Cells transfected with pCAGGS/PADI4 or mock plasmid were cultured in the presence of geneticin (Invitrogen) for 2 wk. Colonies were stained with crystal violet (Sigma) and scored using ImageJ software. U373MG cells or U-2 OS cells were transfected with siRNA oligonucleotide designed to suppress the expression of PADI4 or EGFP (control) 6 h before treatment with adenoviral vector or ADR. Apoptotic cells or viable cells were quantified by fluorescence-activated cell sorting analysis or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide analysis as described previously (18).
Results
DNA damage induces protein citrullination in a p53/PADI4-dependent manner
To validate the results of our previous cDNA microarray analyses, we analyzed the expression of PADIs in U373MG cells infected with adenovirus vector expressing either p53 (Ad-p53) or LacZ (Ad-LacZ; refs. 17, 18). As a result, we found that both PADI3 and PADI4 were transactivated by p53 (Fig. 1A), indicating the possible roles of p53 in the regulation of PTMs.

Regulation of protein citrullination by p53. A, quantitative PCR analysis of five PADI isozymes mRNA in U373MG cells infected with Ad-p53 or Ad-LacZ at a multiplicity of infection of 8. β2-Microglobulin was used for the normalization of expression levels. Columns, relative expressions of PADI isozymes; bars, SD (n = 2). B, U373MG (p53 mutant) cells were infected with adenovirus expressing p53 or lacZ at a multiplicity of infection of 20. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti–modified citrulline (MC), anti-p53, anti-p21WAF1, or anti–β-actin antibody. *, exogenously expressed p53. Arrowheads, citrullinated proteins. C, U-2 OS cells were treated with ADR at the indicated doses (left). At 6 h after transfection of each siRNA, U-2 OS cells were treated with 2 μg/mL of ADR for 2 h (right). siEGFP was used as a control. At 36 h after ADR treatment, whole cell extracts were subjected to immunoblotting with anti-MC antibody.
Regulation of protein citrullination by p53. A, quantitative PCR analysis of five PADI isozymes mRNA in U373MG cells infected with Ad-p53 or Ad-LacZ at a multiplicity of infection of 8. β2-Microglobulin was used for the normalization of expression levels. Columns, relative expressions of PADI isozymes; bars, SD (n = 2). B, U373MG (p53 mutant) cells were infected with adenovirus expressing p53 or lacZ at a multiplicity of infection of 20. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti–modified citrulline (MC), anti-p53, anti-p21WAF1, or anti–β-actin antibody. *, exogenously expressed p53. Arrowheads, citrullinated proteins. C, U-2 OS cells were treated with ADR at the indicated doses (left). At 6 h after transfection of each siRNA, U-2 OS cells were treated with 2 μg/mL of ADR for 2 h (right). siEGFP was used as a control. At 36 h after ADR treatment, whole cell extracts were subjected to immunoblotting with anti-MC antibody.
To examine the involvement of p53 in protein citrullination, we infected U373MG cells with Ad-p53 or Ad-LacZ. Subsequent Western blot analysis using an antibody against a chemically modified form of citrulline (23) revealed multiple additional citrullinated proteins in the cells infected with Ad-p53 (Fig. 1B). To activate endogenous p53, we treated U-2 OS osteosarcoma cells with ADR and observed several citrullinated proteins in cells treated with ADR in a dose-dependent manner (Fig. 1C). Similar results were obtained in ADR-treated HCT116 cells (Supplementary Fig. S1). In addition, treatment of siRNA designed to suppress p53 expression (sip53) remarkably inhibited the citrullination of these proteins (Fig. 1C), implicating that DNA damage could induce protein citrullination in a p53-dependent manner.
PADI4 is a mediator of DNA damage–induced protein citrullination
Next, we treated U373MG cells with siRNA designed to suppress PADI3 or PADI4 expression (siPADI3 or siPADI4) prior to Ad-p53 treatment (Supplementary Fig. S2). Treatment of the cells with siPADI4 almost completely inhibited the citrullination of the 37-kDa protein in Ad-p53–infected U373MG cells, whereas that with siEGFP or siPADI3 revealed no effect on citrullination (Fig. 2A). Furthermore, introduction of PADI4 in HEK293T cells induced citrullination of various proteins including the 37-kDa protein, whereas that of PADI3 or catalytically inactive mutant-PADI4 (D350A and D473A; ref. 24) induced weak or no protein citrullination (Fig. 2B). We then treated U-2 OS cells with ADR and found the induction of PADI4 mRNA (Fig. 2C), in concordance with the results of protein citrullination. To investigate p53-dependent activation of PADI4 expression, we treated HCT116 p53+/+ and HCT116 p53−/− cells with ADR and found the induction of PADI4 only in HCT116 p53+/+ cells in an ADR dose-dependent manner (Fig. 2C). The results of Western blot analysis also revealed the increased expression of PADI4 protein in Ad-p53–infected U373MG or ADR-treated U-2 OS cells, respectively (Fig. 2D). Taken together, we suspect that PADI4 is a key mediator of p53-induced protein citrullination.
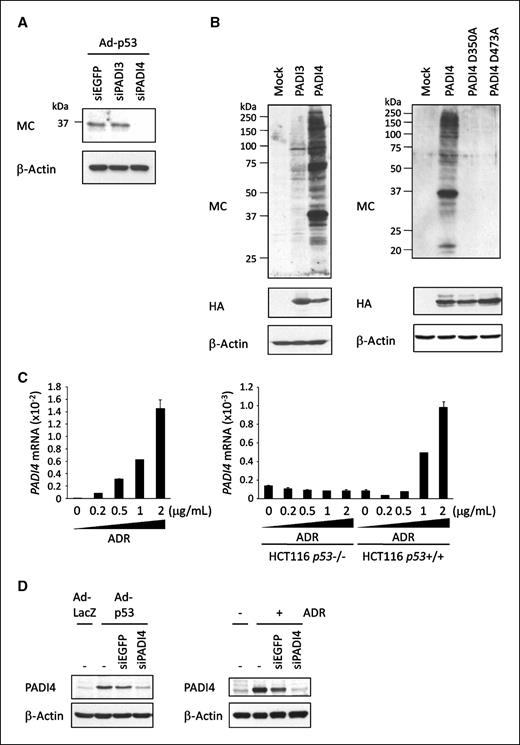
PADI4 induces protein citrullination. A, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-p53. At 36 h after infection, whole cell extracts were subjected to immunoblotting using anti-MC antibody. siEGFP was used as a control. B, HEK293T cells were transfected with plasmid expressing PADI3, PADI4, mutant PADI4, or mock. At 36 h after transfection, whole cell extracts were subjected to immunoblotting using anti-MC antibody. C, quantitative PCR analysis of PADI4 expressions at 36 h after treatment with ADR in U-2 OS cells (left) and in HCT116 p53+/+ and HCT116 p53−/− cells (right). Columns, mean; bars, SD (n = 2). B2M was used for normalization of PADI4 expression levels. D, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-LacZ or Ad-p53 (left) and U-2 OS cells were treated with 2 μg/mL of ADR for 2 h (right). siEGFP was used as a control. At 36 h after infection or treatment, whole cell extracts were subjected to immunoblotting with anti-PADI4 antibody.
PADI4 induces protein citrullination. A, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-p53. At 36 h after infection, whole cell extracts were subjected to immunoblotting using anti-MC antibody. siEGFP was used as a control. B, HEK293T cells were transfected with plasmid expressing PADI3, PADI4, mutant PADI4, or mock. At 36 h after transfection, whole cell extracts were subjected to immunoblotting using anti-MC antibody. C, quantitative PCR analysis of PADI4 expressions at 36 h after treatment with ADR in U-2 OS cells (left) and in HCT116 p53+/+ and HCT116 p53−/− cells (right). Columns, mean; bars, SD (n = 2). B2M was used for normalization of PADI4 expression levels. D, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-LacZ or Ad-p53 (left) and U-2 OS cells were treated with 2 μg/mL of ADR for 2 h (right). siEGFP was used as a control. At 36 h after infection or treatment, whole cell extracts were subjected to immunoblotting with anti-PADI4 antibody.
PADI4 is a target of p53
To further investigate whether PADI4 is a direct transcriptional target of p53, we surveyed genomic sequences of the PADI4 gene on chromosome 1p36.13 and identified two potential p53-binding sequences (p53BS-A and p53BS-B) within the first intron (Fig. 3A). ChIP assays using U373MG cells that were infected with either Ad-p53 or Ad-LacZ indicated the binding of p53 protein to the genomic fragment including p53BS-A (Fig. 3B). We subsequently analyzed the promoter/enhancer activity of the DNA fragment including p53BS-A and p53BS-B (p53BS-AB) using a reporter assay and found that wild-type p53 significantly enhanced luciferase activity (Supplementary Fig. S3). The DNA fragment containing the base substitutions within BS-A completely diminished the enhancement of luciferase activity (Fig. 3C), indicating that p53 directly regulates PADI4 expression through p53-responsible element, p53BS-A in intron 1.

PADI4 is a direct transcriptional target of p53. A, genomic structure of the PADI4 gene. Black boxes, locations and relative sizes of 16 exons. White boxes, locations of potential p53-binding sites (p53BS-A and p53BS-B) in a p53-binding region (p53BS-AB). Comparison of each p53BS to the consensus p53 binding sequence. R, purine; W, A or T, Y, pyrimidine. Identical nucleotides to the consensus sequence are written in capital letters. The underlined cytosine and guanine were substituted for thymine to examine the specificity of each p53-binding site. B, ChIP assay was performed using U373MG cells that were infected with Ad-p53 (lane 1 and lanes 3−5) or Ad-LacZ (lane 2). DNA-protein complexes were immunoprecipitated with an anti-p53 antibody (lanes 2 and 3) followed by quantitative PCR analysis. Input chromatin represents a small portion (2%) of the sonicated chromatin before immunoprecipitation (lane 1). Immunoprecipitates with an anti-Flag antibody (lane 4) or in the absence of an antibody (lane 5) were used as negative controls. Ct values of immunoprecipitated samples were normalized to the corresponding value for input. Columns, mean; bars, SD (n = 2). C, results of luciferase assay of p53BS-AB with or without substitutions at either of the p53BS fragments. Luciferase activity is indicated relative to the activity of mock vector with SDs (n = 2).
PADI4 is a direct transcriptional target of p53. A, genomic structure of the PADI4 gene. Black boxes, locations and relative sizes of 16 exons. White boxes, locations of potential p53-binding sites (p53BS-A and p53BS-B) in a p53-binding region (p53BS-AB). Comparison of each p53BS to the consensus p53 binding sequence. R, purine; W, A or T, Y, pyrimidine. Identical nucleotides to the consensus sequence are written in capital letters. The underlined cytosine and guanine were substituted for thymine to examine the specificity of each p53-binding site. B, ChIP assay was performed using U373MG cells that were infected with Ad-p53 (lane 1 and lanes 3−5) or Ad-LacZ (lane 2). DNA-protein complexes were immunoprecipitated with an anti-p53 antibody (lanes 2 and 3) followed by quantitative PCR analysis. Input chromatin represents a small portion (2%) of the sonicated chromatin before immunoprecipitation (lane 1). Immunoprecipitates with an anti-Flag antibody (lane 4) or in the absence of an antibody (lane 5) were used as negative controls. Ct values of immunoprecipitated samples were normalized to the corresponding value for input. Columns, mean; bars, SD (n = 2). C, results of luciferase assay of p53BS-AB with or without substitutions at either of the p53BS fragments. Luciferase activity is indicated relative to the activity of mock vector with SDs (n = 2).
Identification of nucleophosmin as a substrate of PADI4
Nucleophosmin (NPM1), a ubiquitously-expressed nucleolar protein, was previously reported to be possibly citrullinated with treatment of calcium ionophore in HL-60 cells (14). The molecular weight of NPM1 was almost identical to that of the 37-kDa citrullinated protein (Supplementary Fig. S4). Therefore, we suspect that NPM1 is one of the candidate substrates for PADI4. To examine whether PADI4 could directly interact with and citrullinate NPM1, cell extracts from PADI4-transfected HEK293T cells or Ad-p53–infected U373MG cells were immunoprecipitated with anti-NPM1 antibody or control mouse IgG. Subsequent Western blot analyses indicated the binding of endogenous NPM1 with exogenous or endogenous PADI4 (Fig. 4A). To investigate in vivo citrullination of NPM1, we transfected HEK293T cells with plasmid expressing wild-type or mutant PADI4. Then, endogenous NPM1 protein was purified with anti-NPM1 antibody and subjected to Western blotting with anti–modified citrulline antibody. As a result, we found citrullination of NPM1 in wild-type PADI4-transfected cells although not in mock-transfected or mutant PADI4–transfected cells (Fig. 4B). Moreover, knockdown of NPM1 with siRNA against NPM1 (siNPM1) reduced the amount of the 37 kDa citrullinated protein in ADR-treated U-2 OS cells (Fig. 4C). Then we purified recombinant NPM1 and PADI4 proteins, both fused to GST. We incubated GST-NPM1 with GST-PADI4 for 1 hour at 37°C with different concentrations of Ca2+ and found that PADI4 citrullinated NPM1 under intracellular Ca2+ concentrations of 10−7 mol/L in vitro (Fig. 4D). These data clearly indicated that PADI4 citrullinates NPM1 under physiologic conditions.
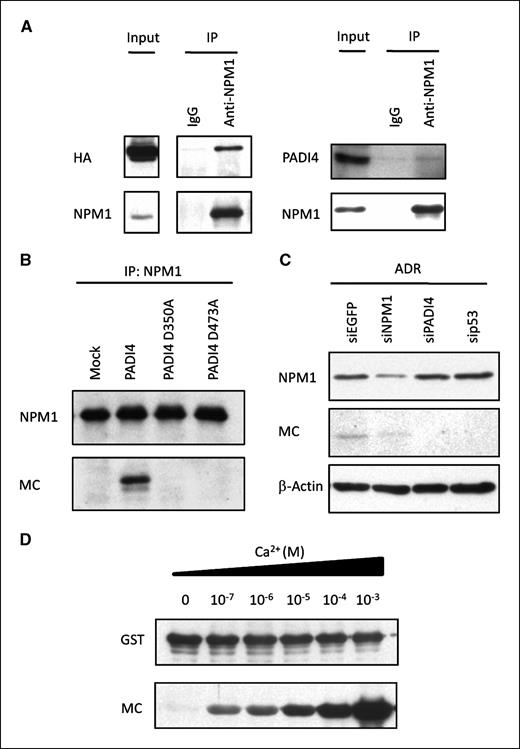
NPM1 is a substrate of PADI4. A, interaction of NPM1 and PADI4. Cell extracts from HEK293T cells that were transfected with plasmid expressing HA-PADI4 were immunoprecipitated using anti-NPM1 antibody or mouse IgG, followed by immunoblotting with anti-HA or anti-NPM1 antibody (left). Cell extracts from U373MG cells that were infected with Ad-p53 were immunoprecipitated using anti-NPM1 antibody or mouse IgG, followed by immunoblotting with anti-PADI4 or anti-NPM1 antibody (right). B, HEK293T cells were transfected with each plasmid indicated. Cell extracts were immunoprecipitated using anti-NPM1 antibody, followed by immunoblotting with anti-NPM1 or anti-MC antibody. C, U-2 OS cells were transfected with each siRNA 6 h prior to treatment with ADR. siEGFP was used as a control. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti-NPM1 or anti-MC antibody. D, in vitro citrullination analyses of NPM1. Recombinant NPM1 was incubated with GST-PADI4 in the presence of indicated concentrations of CaCl2 at 37°C for 1 h, followed by immunoblotting with anti-GST or anti-MC antibody.
NPM1 is a substrate of PADI4. A, interaction of NPM1 and PADI4. Cell extracts from HEK293T cells that were transfected with plasmid expressing HA-PADI4 were immunoprecipitated using anti-NPM1 antibody or mouse IgG, followed by immunoblotting with anti-HA or anti-NPM1 antibody (left). Cell extracts from U373MG cells that were infected with Ad-p53 were immunoprecipitated using anti-NPM1 antibody or mouse IgG, followed by immunoblotting with anti-PADI4 or anti-NPM1 antibody (right). B, HEK293T cells were transfected with each plasmid indicated. Cell extracts were immunoprecipitated using anti-NPM1 antibody, followed by immunoblotting with anti-NPM1 or anti-MC antibody. C, U-2 OS cells were transfected with each siRNA 6 h prior to treatment with ADR. siEGFP was used as a control. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti-NPM1 or anti-MC antibody. D, in vitro citrullination analyses of NPM1. Recombinant NPM1 was incubated with GST-PADI4 in the presence of indicated concentrations of CaCl2 at 37°C for 1 h, followed by immunoblotting with anti-GST or anti-MC antibody.
PADI4 citrullinates NPM1 at the arginine 197 residue in vivo
To identify the citrullinated site(s) of NPM1, we immunopurified endogenous NPM1 proteins from PADI4-transfected HEK293T cells. Following liquid chromatography MS/MS analysis, we identified the citrullination of NPM1 at the 197th arginine residue (data not shown). Then we constructed plasmid expressing mutant NPM1 (R197K NPM1) and introduced HEK293T cells. Wild-type and mutant-NPM1 protein were purified from cell extracts and subjected to in vitro citrullination assay. Concordant with the results of liquid chromatography MS/MS analysis, substitution of the 197th arginine residue caused a significant reduction of protein citrullination by GST-PADI4 (Fig. 5A).
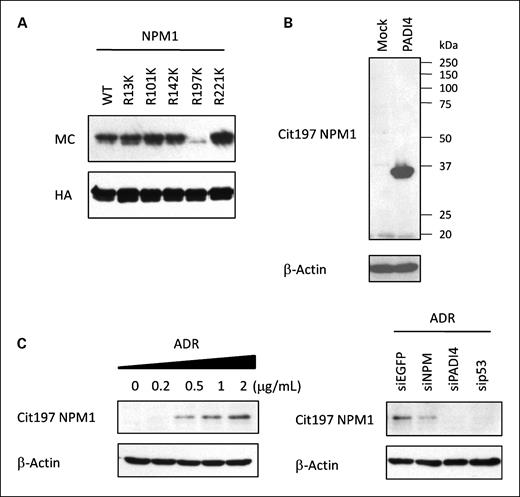
In vivo citrullination of NPM1. A, immunopurified wild-type or mutant NPM1 proteins were incubated with GST-PADI4 in the presence of 1 mmol/L of CaCl2 at 30°C for 1 h, followed by immunoblotting with anti-MC antibody. B, HEK293T cells were transfected with each plasmid indicated and subjected to immunoblotting with anti-citrullinated NPM1 antibody (Cit197 NPM1). C, U-2 OS cells were treated with ADR for 2 h. Each siRNA was transfected at 6 h prior to treatment with 2 μg/mL of ADR (right). siEGFP was used as a control. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti-citrullinated NPM1 antibody (Cit197 NPM1).
In vivo citrullination of NPM1. A, immunopurified wild-type or mutant NPM1 proteins were incubated with GST-PADI4 in the presence of 1 mmol/L of CaCl2 at 30°C for 1 h, followed by immunoblotting with anti-MC antibody. B, HEK293T cells were transfected with each plasmid indicated and subjected to immunoblotting with anti-citrullinated NPM1 antibody (Cit197 NPM1). C, U-2 OS cells were treated with ADR for 2 h. Each siRNA was transfected at 6 h prior to treatment with 2 μg/mL of ADR (right). siEGFP was used as a control. At 36 h after treatment, whole cell extracts were subjected to immunoblotting with anti-citrullinated NPM1 antibody (Cit197 NPM1).
To examine the in vivo citrullination of NPM1 at the 197th arginine residue, we raised an antibody that could recognize citrullinated NPM1 using a peptide corresponding to amino acids 193 to 202. Antibody titers and specificity were shown by ELISA using citrullinated peptide (Cit197: KKSI_Cit_DTPAK) and noncitrullinated peptide (Arg197: KKSI_R_DTPAK; Supplementary Fig. S5). Western blotting using these antibodies clearly indicated the citrullination of NPM1 in PADI4-transfected HEK293T cells (Fig. 5B) as well as ADR-treated U-2 OS cells in a p53/PADI4-dependent manner (Fig. 5C). These findings strongly implied that cellular DNA damage could induce the citrullination of NPM1 at the 197th arginine residue.
Crucial role of PADI4 in a p53 downstream pathway
Protein citrullination reduces the positive charge of proteins and may subsequently influence their structure and function. NPM1 was shown to continuously shuttle between the nucleus and the cytoplasm and translocate from the nucleoli to the nucleoplasm as a response to DNA damage (25). Therefore, we investigated the effect of protein citrullination on subcellular localization of NPM1. Immunocytochemical analysis revealed that cotransfection of NPM1 with PADI4 altered the subcellular localization of NPM1 from the nucleoli to the nucleoplasm in HEK293T cells (Fig. 6A). Moreover, coexpression of p53 with NPM1 in H1299 cells changed subcellular localization of NPM1 from the nucleoli to the nucleoplasm (Supplementary Fig. S6A). In contrast, PADI4 did not induce the translocation of nucleolin (nucleolar protein) as well as mutant NPM1 (R197K; Supplementary Fig. S6B and S6C). To validate these findings, we transfected either wild-type or mutant PADI4 into HEK293T cells and separated the cells into three fractions—cytoplasm, nucleoli, and nucleoplasm—using sucrose gradient centrifugation (22). The results of Western blot analysis clearly indicated the translocation of NPM1 from the nucleoli to the nucleoplasm in cells transfected with wild-type PADI4, whereas NPM1 was dominantly located at nucleoli in the cells transfected with mock or mutant-PADI4 plasmid (Fig. 6B). The expressions of PARP1 (nuclear protein) and HSP90 (cytoplasmic protein) were used as quality and quantity controls. These findings clearly indicated that the p53-PADI4 pathway would regulate subcellular localization of NPM1.
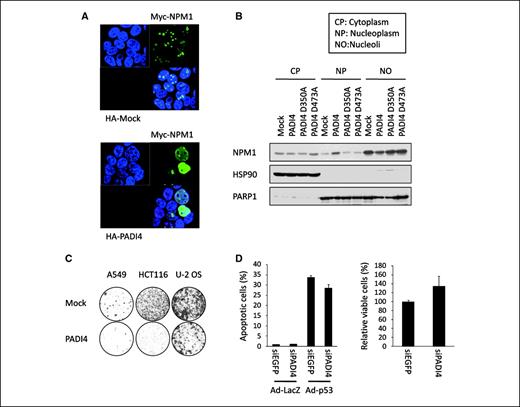
Role of citrullination on NPM1 function. A, subcellular distribution of NPM1 and PADI4 proteins was examined by immunocytochemistry. At 36 h after transfection with plasmid expressing myc-NPM1 and HA-PADI4, HEK293T cells were fixed and stained with anti-myc antibody (Alexa Fluor 488) and anti-HA antibody (Alexa Fluor 594). B, HEK293T cells were transfected with each plasmid indicated, and subcellular localization (cytoplasm, nucleoplasm, and nucleoli) of NPM1 protein was analyzed by Western blot analysis. PARP1 (nuclear protein) and HSP90 (cytoplasmic protein) were used as quality and quantity controls. C, cells were transfected with plasmid expressing PADI4 or mock plasmid, and colony formation assay was performed. The cells were cultured in the presence of geneticin (0.6, 0.5, and 0.4, mg/mL for A549, HCT116, and U-2 OS cells, respectively) for 2 wk. D, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-p53 or Ad-LacZ (left). siEGFP was used as a control. Proportions of apoptotic cells were indicated as percentage of sub-G1 fractions in fluorescence-activated cell sorting analysis with SDs (n = 2). At 6 h after transfection of each siRNA, U-2 OS cells were treated with 2 μg/mL of ADR (right). Cell viability was examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Columns, ratio of cells transfected with siEGFP; bars, SD (n = 4).
Role of citrullination on NPM1 function. A, subcellular distribution of NPM1 and PADI4 proteins was examined by immunocytochemistry. At 36 h after transfection with plasmid expressing myc-NPM1 and HA-PADI4, HEK293T cells were fixed and stained with anti-myc antibody (Alexa Fluor 488) and anti-HA antibody (Alexa Fluor 594). B, HEK293T cells were transfected with each plasmid indicated, and subcellular localization (cytoplasm, nucleoplasm, and nucleoli) of NPM1 protein was analyzed by Western blot analysis. PARP1 (nuclear protein) and HSP90 (cytoplasmic protein) were used as quality and quantity controls. C, cells were transfected with plasmid expressing PADI4 or mock plasmid, and colony formation assay was performed. The cells were cultured in the presence of geneticin (0.6, 0.5, and 0.4, mg/mL for A549, HCT116, and U-2 OS cells, respectively) for 2 wk. D, at 6 h after transfection of each siRNA, U373MG cells were infected with Ad-p53 or Ad-LacZ (left). siEGFP was used as a control. Proportions of apoptotic cells were indicated as percentage of sub-G1 fractions in fluorescence-activated cell sorting analysis with SDs (n = 2). At 6 h after transfection of each siRNA, U-2 OS cells were treated with 2 μg/mL of ADR (right). Cell viability was examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Columns, ratio of cells transfected with siEGFP; bars, SD (n = 4).
Finally, we assessed the possible role of PADI4 in the p53-dependent growth-suppressive pathway. We conducted colony formation assays using several cancer cell lines and found that ectopic expression of PADI4 significantly inhibited the growth of cancer cells (A549, HCT116, and U-2 OS cells; Fig. 6C). We then treated U373MG cells with either siPADI4 or siEGFP prior to infection with Ad-p53 and found that the proportion of apoptotic cells was decreased in siPADI4-treated cells (Fig. 6D). Knockdown of PADI4 also inhibited the ADR-induced growth suppression in U-2 OS cells (Fig. 6D). Taken together, our findings clearly showed that PADI4 would play critical roles in a p53 downstream pathway.
Discussion
Although a number of p53 targets related to cell cycle arrest and apoptosis have been characterized, the role of p53 in the regulation of PTMs has not yet been well clarified. Here, we revealed that DNA damage induces the citrullination of various proteins in a p53/PADI4-dependent manner. We also showed that PADI4 citrullinates NPM1 at arginine 197 residue and regulates subcellular localization of NPM1. Moreover, our findings revealed that knockdown of PADI4 attenuated the growth-suppressive effect of p53. Considering that PADI4 is a direct target of p53, PADI4 would be one of the important mediators in the p53-signaling pathway.
In several hematologic malignancies, the NPM1 locus on chromosome 5q35 is frequently translocated and forms oncogenic fusion proteins such as NPM-ALK, NPM-RARa, and NPM-MLF1 (26). NPM1 is also overexpressed in various solid tumors such as colorectal and bladder cancers, and its overexpression is associated with tumor progression (27, 28). On the other hand, mutations of the NPM1 gene were found in about one-third of adult de novo acute myelogenous leukemias causing aberrant cytoplasmic expression (29). Thus, the role of NPM1 in carcinogenesis still remains unclear and controversial. NPM1 is essential for the development of the mouse embryo (30) and is also implicated in multiple functions including ribosomal biogenesis (31), centrosome duplication (32), and regulation of tumor suppressor pathways such as p53 (33) and CDKN2A (34). At the nucleoli, NPM1 is involved in rRNA transcription, pre-rRNA processing, and ribosome subunit assembly which are essential for cell growth and maintenance of cellular homeostasis (35). Because PADI4 induced the nucleoli/nucleoplasm translocation of NPM1, the growth-suppressive effect mediated by the p53/PADI4 pathway would be attributed to the failure of ribosome biogenesis.
Furthermore, we showed the physiologic regulatory mechanism of protein citrullination. Most previous studies assessing the enzymatic activities of PADIs were conducted under nonphysiologic conditions using calcium ionophore or at higher calcium concentrations. Although activated PADI4 with calcium ionophore was shown to induce NPM1 citrullination in HL-60 cells similar to our findings (14), these results are not likely to reflect the physiologic function of PADI4. To our knowledge, this is the first report demonstrating the role of protein citrullination under physiologic conditions.
Taken together, our findings clearly revealed the novel role of p53 in the regulation of protein citrullination. Although further functional analyses are essential to fully characterize the p53/PADI4-mediated protein citrullination in carcinogenesis, our findings should shed light on the novel function of p53 that can directly or indirectly modulate the biological properties of various proteins through protein citrullination.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Grant support: Japan Society for the Promotion of Science and Ministry of Education, Culture, Sports, Science and Technology of Japan (no. 18687012; K. Matsuda). C. Tanikawa is a JSPS Research Fellow.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank A. Suzuki for providing plasmid and helpful discussion, and K. Makino and A. Takahashi for technical assistance.