


Abstract
Purpose: Circulating endothelial cells (CEC) comprise at least two distinct populations: bone marrow–derived circulating endothelial progenitors (CEP) and mature CECs derived from existing vasculature. We hypothesized that antiangiogenic agents may have differential effects on CEPs and mature CECs and that these changes may serve as a marker of biological activity.
Experimental Design: The effect of angiogenesis inhibitors on CECs was evaluated by flow cytometry after vascular endothelial growth factor (VEGF)–induced mobilization and in mice bearing Lewis lung carcinoma (LLC). Tumor angiogenesis was evaluated in parallel by immunohistochemistry.
Results: In nontumor-bearing mice, VEGF administration increased both mature CECs and CEPs. This increase was inhibited by the VEGF receptor 2 inhibitor ZD6474 as well as the VEGF inhibitor–soluble Flt-1. ZD6474 had no significant effect on CECs in the absence of exogenous VEGF stimulation. In contrast, LLC-bearing mice had an increase in mature CECs but not CEPs after 3 days of treatment with ZD6474. The increase in mature CECs was dose-dependent, accompanied by a decrease in tumor microvessel density, and preceded reduction in tumor volume. Treatment of LLC-bearing mice with the vascular targeting agent ZD6126 also increased mature CECs.
Conclusions: VEGF inhibitors can have differential effects on mature CECs and CEPs, and agents inhibiting tumor angiogenesis may cause a concomitant increase in mature CECs. This increase occurs in tumor-bearing but not in nontumor-bearing mice, suggesting that tumor endothelium is a potential source of mature CECs. Therefore, assessing both mature CECs and CEPs may provide insights into the mechanism of antiangiogenic agents and serve as an early surrogate marker of biological activity.
Angiogenesis plays a critical role in the growth and metastatic spread of tumors (1). Antiangiogenic agents targeting the vascular endothelial growth factor (VEGF) pathway, such as the monoclonal antibody bevacizumab, have shown promise for the treatment of a variety of malignancies, including lung, colorectal, and renal cell cancer (2–4). The clinical testing of these agents is currently hampered, however, by the lack of validated surrogate markers for measuring their biological effect on the targeted pathways and predicting early in the treatment course which patients are most likely to benefit (5). Such markers are needed because these drugs are expected to be primarily cytostatic rather than cytotoxic; thus, conventional means for assessing response to therapy based on tumor shrinkage may not accurately reflect the biological activity of these drugs or be useful for selecting an optimally effective dose. This could lead to the discontinuation of therapy before adequately assessing its potential therapeutic benefit. One approach is to directly assess biopsies of tumors taken pretreatment and posttreatment for angiogenesis or inhibition of specific proangiogenic pathways (i.e., VEGF receptor-2, VEGFR-2, signaling; refs. 6, 7). This method is invasive and not practical for monitoring changes over time. A suitable marker to assess the effect of antiangiogenic treatment would be noninvasive, applicable to multiple agents, and would be an early indicator of biological activity before gross changes in tumor size.
Circulating endothelial cells (CEC) have emerged as a potentially useful surrogate marker for several reasons. First, increased levels of CECs have been observed in cancer patients and have been associated with disease progression (8, 9). Second, CECs are known to be mobilized in response to VEGF both murine models (10, 11) and in humans (12, 13) and express VEGFR-2, the target for many angiogenesis inhibitors in development (14, 15). Third, increases in CECs have been observed in a variety of pathologic conditions in which vascular damage occurs, such as vasculitis (16), infection (17), septic shock (18), myocardial infarction (19), and sickle cell anemia (20).
At least two distinct populations of CEC have been identified: bone marrow–derived circulating endothelial progenitor (CEP) cells, which may contribute to pathologic neovascularization, and mature CECs, which are thought to be derived from mature vasculature. In addition to antigens identifying it as an endothelial cell [VEGFR-2 (Flk-1) and CD31 (platelet/endothelial cell adhesion molecule-1], CEPs are distinguished by the presence of one or more stem cell antigens on the cell surface such as CD117 or c-kit ligand receptor (21–23), Sca-1 (24), or in humans, CD133 (14). It is likely that additional subtypes of CEPs and mature CECs will be identified in the future. For example, a recent study suggests that within CEPs from human cord blood and adult peripheral blood, there may be a hierarchy in which one subpopulation, enriched in cord blood, has a particularly high proliferative potential (>100 population doublings) and the ability to form secondary and tertiary colonies (25).
Inhibition of CEPs has been observed in mice after treatment with the antiangiogenic agent endostatin (26–28) and cyclophosphamide given via a metronomic schedule (29), although in preliminary human studies an increase in total CECs has been observed after treatment with endostatin or the vascular targeting agent ZD6126 (30, 31). It is not yet clear whether these disparate results are due to methodologic differences in CEC measurement or because antiangiogenic agents can have different effects on distinct CEC populations.
In this study, we have investigated changes in mature CECs and CEPs after treatment with two agents that target tumor vasculature via distinct mechanisms. ZD6474 is a small molecule VEGFR-2 tyrosine kinase inhibitor that also has activity against EGFR and has been shown to inhibit angiogenesis and slow tumor growth in a broad range of murine models of cancer including Lewis lung carcinoma (LLC; ref. 32). It is currently undergoing clinical testing for patients with lung cancer and other types of solid tumors (33). ZD6126 is a tubulin-binding agent that is thought to selectively target inhibit tumor endothelium (34). CECs were investigated using two different models: a previously established model of VEGF-induced CEP mobilization and in mice bearing LLC tumors. Our results indicate that these agents may have differential effects on CEPs and mature CECs, and that in LLC-bearing mice, treatment-induced inhibition of tumor angiogenesis is accompanied by early increase in mature CECs.
Materials and Methods
Flow cytometry analysis of circulating endothelial cells. CECs in peripheral blood were evaluated using three- or four-color flow cytometry as previously described (27, 29). Red cell lysis was done using FACSLyse Solution (BD Biosciences, San Jose, CA), as per the manufacturer's directions. For experiments using two-color flow or in which apoptosis was evaluated, Ficoll gradient isolation (Sigma, St. Louis, MO) of peripheral blood mononuclear cells was done to remove red cells and platelets before incubation with antibodies. The following directly conjugated antibodies were used for detection of CECs and CEP cells in peripheral mouse blood: anti-mouse CD45-PerCP, Flk-1-PE (mouse VEGFR-2), CD31-APC (platelet/endothelial cell adhesion molecule-1), and CD117-FITC (c-kit receptor; all from BD Biosciences). For measurement of apoptosis, Annexin V-APC and 7-amino-actinomysin D (7AAD; Molecular Probes, Eugene, OR) were used as previously described (27). Flow cytometry was done using a FACSCalibur flow cytometer (BD Biosciences) and acquired data analyzed with FLOWJO flow cytometry analysis software (Treestar, Ashland, OR), with analysis gates designed to remove residual platelets and cellular debris. Between 50,000 and 100,000 events were typically counted for each mouse. As a positive control for detecting endothelial cells in blood, MS-1 cells, a transformed murine endothelial cell line isolated from pancreatic islets of C57BL/6 mice, were added to blood samples.
Vascular endothelial growth factor induced mobilization of circulating endothelial cells. All animal experiments were done in accordance with institutional animal care regulations.
Injections of recombinant adenovirus. Adenoviral injections were done as previously described (27). Briefly, FVB/NJ mice received and i.v. injection with combinations of Ad-VEGF, Ad-GFP (1 × 108 and 1 × 109 plaque-forming unit per mouse), and Ad-VEGF, Ad-sFLT (1 × 108 and 1 × 109 plaque-forming unit per mouse, respectively). Mice given injection with Ad-GFP (∼1 × 109 plaque-forming unit per mouse) served as control. Recombinant adenoviruses expressing VEGF (Ad-VEGF) and sFLT (Ad-sFLT) were obtained from Harvard Gene Therapy Initiative core (Harvard Institute of Medicine, Boston, MA). After 6 days, mice were sacrificed, and blood samples were retrieved and prepared for two-color flow cytometry to quantify CECs as described earlier. CECs/CEPs were assessed after 7 days after adenoviral injection, which was previously found to be the time of maximum CEC induction (35).
Injection of recombinant human vascular endothelial growth factor. 129/SVImJ mice (The Jackson Laboratory, Bar Harbor, ME) were injected with recombinant human VEGF (National Cancer Institute, Biological Resources Branch), 10 μg via i.p. injection, once daily for 5 days. This strain was chosen because it previously has been shown to have a robust response in the VEGF-induced corneal angiogenesis assay (36). Mice were anesthetized using isoflurane and blood collected by retroorbital puncture and anticoagulated using sodium citrate. Blood was kept at 4°C on ice until 150 μL of blood was analyzed by four-color flow cytometry to quantify CECs as described above.
Subcutaneous tumor model. LLC cells were grown in cell culture as previously described (37). Six- to 8-week-old C57/Bl6J mice were each injected s.c. with 6 × 106 LLC cells. Once tumor volumes reached an average of 400 mm3 (∼10 days), mice were randomized to vehicle by oral gavage (1% polysorbate 80) or treatment with ZD6474 (AstraZeneca, Alderley Park, Macclesfield, United Kingdom) at a dose of 10, 50, or 100 mg/kg by oral gavage for 3 days. Four to five mice were included in each group. Large tumors averaging ∼1,900 mm3 were randomized to oral treatment with vehicle (four mice) or treatment with ZD6126 (AstraZeneca; three mice) at 100 mg/kg daily. On day 3, mice were given isoflurane anesthesia and blood collected by retroorbital puncture and anticoagulated using sodium citrate (Sigma). Mice were then euthanized by cervical dislocation and tumors measured and harvested. Tumors were then cut into ∼200-mm3 pieces and immediately frozen in Tissue-Tek embedding medium (Sakura Finetek, Torrance, CA).
Immunohistochemistry. Immunohistochemical staining was carried out on 8-μm frozen sections. For CD31-Cy3 staining, section was dried for 2 to 3 hours at room temperature and circled with an Immedge Pen (Vector Laboratories, Burlingame, CA). Sections were immersed in 0.3% Triton X-100 PBS (T-PBS) and washed twice with PBS. Sections were then blocked for 1 hour with 5% goat serum (Jackson ImmunoResearch, West Grove, PA) in PBS + 0.3% Triton X-100 + 0.01% bovine serum albumin + 0.01% Thimerosal. This same solution without goat serum was used for dilution of antibodies. Incubation with primary antibody (rat anti-mouse CD31, 1:500, BD Biosciences) was carried out overnight. Slides were then washed with T-PBS and incubated with secondary antibody (Cy3-goat anti-rat 1:400, Jackson ImmunoResearch) for 4 to 5 hours. Slides were then washed with T-PBS and fixed with 4% paraformaldehyde for 15 minutes. The final washes with PBS were done before mounting with Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories) according to manufacturers instructions.
Quantification of tumor microvessel density. Tumor microvessel density was quantified for all treatment and control groups. At least 6 to 12 representative 40× fields of view were captured as epifluorescent digital images using a Spot digital camera (Spot Diagnostic Instruments, Sterling Heights, MI) attached to a Nikon TE-300 eclipse microscope. Only the red channel from each RGB image was exported from Adobe Photoshop (San Jose, CA). To calculate the microvessel density, area occupied by CD31-positive microvessels and total tissue area per section were quantified using Image J software (NIH, Bethesda, MD). Microvessel density was then calculated as a percentage of CD31 stained per tumor section (38). Tumor microvessel density was compared between all treatment groups and untreated/control mice.
Statistics. All statistical comparisons were done using SigmaStat software (Aspire Software International, Leesburg, VA).
Comparison of circulating endothelial cells. The number of CD45−, Flk+ (two-color analysis), or CD45−, Flk+, CD31+, CD117− (four-color analysis) mature CECs or CD45−, Flk+, CD31+, CD117+ CEPs detected for each mouse were expressed as a percentage of peripheral blood mononuclear cells detected for that mouse. Mean CEC and CEP values for each treatment group were then calculated and compared with control mice for that experiment. Applying means and t test for parametric data, or median and rank sum test for nonparametric data compared groups. Mean CEC and CEP level for each group was used to calculate correlation coefficients with dose of ZD6474 received.
Microvessel density. Treatment groups were compared by pooling the microvessel density values for each section within a treatment group and comparing the median values for each group with the rank sum test.
Results
Measurement of circulating endothelial cells by flow cytometry. CECs were assessed using flow cytometry on mouse peripheral blood using previously described methods (27, 29) modified as described below. In previous studies, CECs were identified based on their Flk1+CD45− phenotype (26). To further characterize different CEC populations, four-color flow cytometry was used to distinguish between four different cell surface markers including CD31 (platelet/endothelial cell adhesion molecule), a pan-endothelial marker, and CD117 (c-KIT), a stem cell marker present on CEPs but not mature CECs (21, 28, 29) as well as Flk1 and CD45. Figure 1 shows a representative analysis of CECs and CEPs. The initial analysis gate was used to exclude platelets and cellular debris (Fig. 1A). Hematopoeitic cells were excluded using CD45, whereas endothelial cells were contained in the CD45−Flk+ gate (Fig. 1B). These cells were further characterized using CD31 and CD117, with CEPs having phenotype CD45−Flk+CD31+CD117+ and mature CECs CD45−Flk+CD31+CD117− (Fig. 1C). More than 80% of CD45−Flk+ cells were also typically positive for the EC markers CD31+ (Fig. 1C) and MECA-32 (data not shown) providing further support for their endothelial phenotype. As a control, this panel reproducibly detected MS-1 cells, a murine endothelial cell line with a CEP phenotype, when added into murine peripheral blood (Fig. 1C). The number of CEPs and mature CECs in untreated mice was typically in the range of 0.2 to 0.5 and 5 to 10 cells per microliter of blood, respectively. Data from a representative experiment is shown in Table 1.

Strategy for flow cytometric detection of murine CECs. Whole blood (control) from mice and whole blood mixed with the murine endothelial cell line MS-1 cells (control + MS-1) was analyzed with four-color flow cytometry. A, initial gate used to exclude red cell and platelet debris. B, selection of CD45-PerCp–negative and Flk-PE-positive cells, which encompasses both mature CECs and CEPs. Inset, negative isotype controls for CD45-PerCP and Flk-PE. C, CECs are further characterized using CD31-APC and progenitor marker CD117-FITC. Inset, negative controls for CD31-APC and CD117-FITC. Note that the majority (>95%) of CD45−, Flk+ cells were also CD31+, confirming the circulating endothelial cell phenotype.
Strategy for flow cytometric detection of murine CECs. Whole blood (control) from mice and whole blood mixed with the murine endothelial cell line MS-1 cells (control + MS-1) was analyzed with four-color flow cytometry. A, initial gate used to exclude red cell and platelet debris. B, selection of CD45-PerCp–negative and Flk-PE-positive cells, which encompasses both mature CECs and CEPs. Inset, negative isotype controls for CD45-PerCP and Flk-PE. C, CECs are further characterized using CD31-APC and progenitor marker CD117-FITC. Inset, negative controls for CD31-APC and CD117-FITC. Note that the majority (>95%) of CD45−, Flk+ cells were also CD31+, confirming the circulating endothelial cell phenotype.
Levels of circulating endothelial cells measured by flow cytometry
| Cell phenotype | % PBMC ± SE | Cells/μL PB ± SE |
|---|---|---|
| Total CEC (CD45−Flk+) | 0.49 ± 0.12 | 8.20 ± 1.51 |
| Mature CEC (CD45−Flk+CD31+CD117−) | 0.39 ± 0.07 | 6.51 ± 0.98 |
| CEP (CD45−Flk+CD31+CD117+) | 0.02 ± 0.004 | 0.26 ± 0.08 |
| Cell phenotype | % PBMC ± SE | Cells/μL PB ± SE |
|---|---|---|
| Total CEC (CD45−Flk+) | 0.49 ± 0.12 | 8.20 ± 1.51 |
| Mature CEC (CD45−Flk+CD31+CD117−) | 0.39 ± 0.07 | 6.51 ± 0.98 |
| CEP (CD45−Flk+CD31+CD117+) | 0.02 ± 0.004 | 0.26 ± 0.08 |
NOTE: Data from representative experiment expressed as percentage of PBMC or absolute number of cells per μL of peripheral blood (C57/Bl6 mice, n = 5).
Abbreviations: PBMC, peripheral blood mononuclear cells; PB, peripheral blood.
Vascular endothelial growth factor–induced mobilization of circulating endothelial cells and circulating endothelial progenitors inhibited by ZD6474 in nontumor-bearing mice. Previously, it has been shown that CEPs can be mobilized by VEGF (11) and that CEP mobilization can be inhibited by angiogenesis inhibitors such as endostatin (27, 28) and angiostatin (39). To test the effects of ZD6474 on CEC mobilization, mice were injected with adenovirus encoding for VEGF (Ad-VEGF) or adenovirus expressing GFP (Ad-GFP) as previously described (27). Infection with Ad-VEGF led to a 3.6-fold increase in the number of CEC (Flk+CD45− peripheral blood mononuclear cells) compared with the Ad-GFP controls (Fig. 2A; P < 0.001 for comparison of control). Treatment with ZD6474 (100 mg/kg by gavage once daily) inhibited the VEGF-induced increase in CEC by 66%. As a control to verify that CEC mobilization was due to VEGF, adenoviral delivery of the VEGF inhibitor soluble Flt-1 also inhibited the VEGF-induced increase in CECs. To confirm these findings and further characterize whether the VEGF-induced increase was seen in both CECs and CEPs, we directly given human recombinant VEGF protein by i.p. injection (10 μg/d). VEGF induced a similar increase in both mature CECs and CEPs (Fig. 2B) as compared with vehicle-treated controls (P < 0.03 for comparison with controls). For both the CEP and mature CEC subgroups, there was a trend towards inhibition after treatment with ZD6474 and VEGF compared with VEGF alone. ZD6474 had no significant effect on CECs in vehicle-treated mice (Fig. 2B).
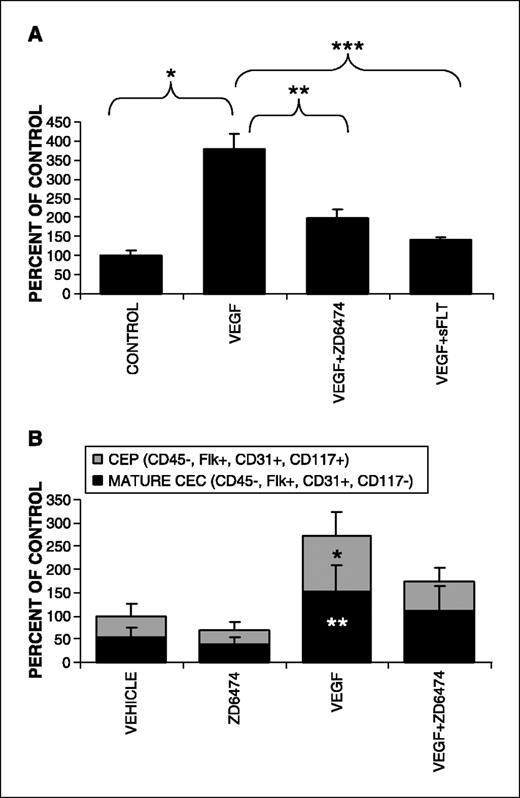
Treatment of mice with VEGF by two methods results in the mobilization of increased numbers of mature CECs and CEPs, which can be blocked with the antiangiogenic agents ZD6474 and sFlt. A, mice were injected with adenovirus encoding for VEGF165 (Ad-VEGF) or control adenovirus expressing GFP (Ad-GFP) as previously described (27). Oral cotreatment for 4 days with the antiangiogenic agent ZD6474 (100 mg/kg daily), or adenoviral-mediated delivery of the VEGF inhibitor-soluble Flt-1, was used to block VEGF-mediated CEC mobilization (*, P < 0.001; **, P = 0.02; ***, P < 0.001). B, mice were treated orally with ZD6474 (100 mg/kg daily) and sterile normal saline i.p., vehicle gavage (1% polysorbate 80) daily and recombinant human VEGF (10 μg i.p./d), the combination of VEGF and ZD6474, or saline i.p. and vehicle gavage for 5 days before analysis of peripheral blood for mature CECs and CEPs with four-color flow cytometry (*, P = 0.01 for CEPs; **, P = 0.03 for mature CECs compared with vehicle controls; P = 0.095 for CEP's VEGF compared with VEGF + ZD6474; P = 0.330 for CECs VEGF compared with VEGF + ZD6474).
Treatment of mice with VEGF by two methods results in the mobilization of increased numbers of mature CECs and CEPs, which can be blocked with the antiangiogenic agents ZD6474 and sFlt. A, mice were injected with adenovirus encoding for VEGF165 (Ad-VEGF) or control adenovirus expressing GFP (Ad-GFP) as previously described (27). Oral cotreatment for 4 days with the antiangiogenic agent ZD6474 (100 mg/kg daily), or adenoviral-mediated delivery of the VEGF inhibitor-soluble Flt-1, was used to block VEGF-mediated CEC mobilization (*, P < 0.001; **, P = 0.02; ***, P < 0.001). B, mice were treated orally with ZD6474 (100 mg/kg daily) and sterile normal saline i.p., vehicle gavage (1% polysorbate 80) daily and recombinant human VEGF (10 μg i.p./d), the combination of VEGF and ZD6474, or saline i.p. and vehicle gavage for 5 days before analysis of peripheral blood for mature CECs and CEPs with four-color flow cytometry (*, P = 0.01 for CEPs; **, P = 0.03 for mature CECs compared with vehicle controls; P = 0.095 for CEP's VEGF compared with VEGF + ZD6474; P = 0.330 for CECs VEGF compared with VEGF + ZD6474).
ZD6474 induces dose-dependent changes in tumor microvessel density and mature circulating endothelial cells in Lewis lung carcinoma–bearing mice. To explore whether changes in CECs might be a marker for antiangiogenic activity in tumor-bearing mice, we investigated the correlation between changes in CECs and inhibition of tumor angiogenesis as measured by microvessel density. We hypothesized that a VEGF pathway inhibitor could have differential effects on mature CECs and CEPs by increasing the shedding of fragile vessel wall–derived mature CECs but potentially inhibiting CEP mobilization. Previously, it has been shown that treatment with ZD6474 inhibits the growth of LLC in a dose-dependent manner by 61% to 79% after treatment for 2 weeks at doses ranging from 25 to 100 mg/kg (32). Consistent with this data, we observed a 65% inhibition in tumor growth after 2 weeks of treatment with ZD6474 at a dose of 50 mg/kg (data not shown). However, at this ZD6474 dose, tumor size did not differ significantly from controls until 7 days of treatment, at which time a 46% inhibition of tumor growth was observed (P = 0.04 for comparison with vehicle-treated controls). As we were interested in identifying markers of antiangiogenic activity that preceded gross changes in tumor size, we first assessed tumor vasculature after treatment of LLC-bearing mice with ZD6474 for 3 days at doses of 10, 50, and 100 mg per kg per day (n = 4-5 mice/group). We found a dose-dependent decrease in tumor microvessel density (Fig. 3; correlation coefficient = −0.316, P < 0.000001). Next, we examined whether changes in CECs also correlated with ZD6474 dosage after 3 days. A dose-dependent increase in CD45−, Flk+ CECs was also detected (correlation coefficient = 0.59, P < 0.01). The apoptotic fraction of the mobilized CEC was not significantly increased by treatment (data not shown). This dose-dependent increase was observed in mature CECs but not CEPs (Fig. 4B and C).

ZD6474 treatment of mice bearing established LLC tumors results in a dose-dependent inhibition of tumor angiogenesis. Mice (n = 5/group, mean tumor size ∼400 mm3) were treated with indicated doses of ZD6474 for 3 days before analysis. Tumors were harvested and frozen in ornithine carbamyl transferase before sectioning. Immunohistochemistry with anti-CD31 and Cy-3 was used to label endothelial cells. A, representative examples of epifluorescent images obtained from control and ZD6474 treated mice. B, microvessel density of LLC tumors after 3 days of treatment with increasing doses of ZD6474. Microvessel density was expressed as the percentage of CD31+ area per high-powered field. Analysis of 6 to 12 high-powered fields per tumor with ImageJ software (correlation coefficient = −0.316, P < 0.000001).
ZD6474 treatment of mice bearing established LLC tumors results in a dose-dependent inhibition of tumor angiogenesis. Mice (n = 5/group, mean tumor size ∼400 mm3) were treated with indicated doses of ZD6474 for 3 days before analysis. Tumors were harvested and frozen in ornithine carbamyl transferase before sectioning. Immunohistochemistry with anti-CD31 and Cy-3 was used to label endothelial cells. A, representative examples of epifluorescent images obtained from control and ZD6474 treated mice. B, microvessel density of LLC tumors after 3 days of treatment with increasing doses of ZD6474. Microvessel density was expressed as the percentage of CD31+ area per high-powered field. Analysis of 6 to 12 high-powered fields per tumor with ImageJ software (correlation coefficient = −0.316, P < 0.000001).

Dose-dependent rise in CECs in LLC-bearing mice treated with ZD6474. Mice bearing LLC tumors (n = 5/group, mean tumor size ∼400 mm3) were treated for 3 days with polysorbate 80 vehicle or one of three different doses of ZD6474 (10, 50, or 100 mg per kg per day). Mice were then anesthetized and blood was collected by retroorbital puncture before sacrifice. A, analysis of whole blood by flow cytometry using CD45-PerCP and Flk-PE to detect endothelial cells, and Annexin V-APC and 7-amino-actinomysin D to determine apoptotic fraction, as described earlier. Proportion of CD45−, Flk+ cells detected per correlates with dose of ZD6474 (*, P < 0.01 compared with control; correlation coefficient = 0.59, P = 0.006). Apoptotic fraction of CECs did not change significantly (data not shown). B, analysis of whole blood from same experiment using four-color flow cytometry with CD45-PerCP, Flk-PE, CD31-APC, and CD117-FITC. CEPs are distinguished from mature CECs by the presence of CD117, the c-kit ligand receptor, as has been shown previously (28). C, dose-dependent increase in mature CECs but not CEPs with ZD6474 dose (correlation coefficient 0.91, P = 0.09).
Dose-dependent rise in CECs in LLC-bearing mice treated with ZD6474. Mice bearing LLC tumors (n = 5/group, mean tumor size ∼400 mm3) were treated for 3 days with polysorbate 80 vehicle or one of three different doses of ZD6474 (10, 50, or 100 mg per kg per day). Mice were then anesthetized and blood was collected by retroorbital puncture before sacrifice. A, analysis of whole blood by flow cytometry using CD45-PerCP and Flk-PE to detect endothelial cells, and Annexin V-APC and 7-amino-actinomysin D to determine apoptotic fraction, as described earlier. Proportion of CD45−, Flk+ cells detected per correlates with dose of ZD6474 (*, P < 0.01 compared with control; correlation coefficient = 0.59, P = 0.006). Apoptotic fraction of CECs did not change significantly (data not shown). B, analysis of whole blood from same experiment using four-color flow cytometry with CD45-PerCP, Flk-PE, CD31-APC, and CD117-FITC. CEPs are distinguished from mature CECs by the presence of CD117, the c-kit ligand receptor, as has been shown previously (28). C, dose-dependent increase in mature CECs but not CEPs with ZD6474 dose (correlation coefficient 0.91, P = 0.09).
Vascular-targeting agent ZD6126 induces increases mature circulating endothelial cells in tumor-bearing mice. To investigate whether other types of antiangiogenic agents might also induce changes in CECs, we tested ZD6126, a vascular targeting agent thought to act selectively upon tumor endothelium (34, 40, 41). Because ZD6126 has been shown to preferentially target larger tumors, we analyzed mice bearing well-established LLC (mean size = 1,800 mm3). After treatment with ZD6126 (100 mg per kg per day orally by gavage), a 5-fold induction in mature CECs was observed (Fig. 5; P = 0.04). These CECs were predominantly (95%) mature CECs, although a small increase in CEPs was also observed.

Effects of vascular-targeting agent ZD6126 on CECs. Mice bearing LLC (mean size ∼1,800 mm3) were treated with ZD6126, an agent putatively specific for tumor vasculature (34), 100 mg/kg or vehicle control by oral gavage daily for 3 days. Mice were then anesthetized and blood was collected by retroorbital puncture. CECs were assessed by four-color flow cytometry as described above (*, P = 0.04; **, P = 0.02).
Effects of vascular-targeting agent ZD6126 on CECs. Mice bearing LLC (mean size ∼1,800 mm3) were treated with ZD6126, an agent putatively specific for tumor vasculature (34), 100 mg/kg or vehicle control by oral gavage daily for 3 days. Mice were then anesthetized and blood was collected by retroorbital puncture. CECs were assessed by four-color flow cytometry as described above (*, P = 0.04; **, P = 0.02).
Discussion
In this study, we have investigated changes in CECs and tumor vasculature after treatment with the VEGFR-2 inhibitor ZD6474 and the vascular targeting agent ZD6126. Our data provides evidence that antiangiogenic agents can have differential effects on distinct populations of CECs, causing a decrease in VEGF-mobilized CECs but an increase in mature CECs in LLC-bearing mice. The change in mature CECs were dose-dependent and were associated with a concomitant inhibition of tumor angiogenesis but preceded the inhibition of tumor growth, suggesting that mature CECs may be an early marker for antiangiogenic activity.
Prior studies in which CECs have been measured after treatment with antiangiogenic or anticancer agents have yielded disparate results. In murine studies, endostatin decreased the number of viable CEPs (27, 28), whereas cyclophosphamide either induced or inhibited CEPs depending on whether it was given by a conventional (every 21 days) or metronomic (every 6 days) dosing schedule (29). In preliminary clinical studies, CECs were observed to increase in cancer patients after treatment with the vascular targeting agent ZD6126 (31) and endostatin (30). These studies are difficult to compare, however, because CECs were measured by different methods and in some cases mature CECs were not distinguished from CEPs. Nevertheless, any model to explain these changes must account for the observations that CECs increased after treatment in some studies but decreased in others.
To further investigate the effect of angiogenesis inhibitors on CECs, we initially tested the ability of ZD6474 to prevent the VEGF-induced increase in CECs in nontumor-bearing mice. Consistent with earlier studies (11, 27, 42), VEGF given by adenoviral-mediated expression lead to a 3.6-fold increase in CECs (Fig. 2A). Treatment with ZD6474 or the VEGF-specific inhibitor-soluble sFlt (43) inhibited the CEC induction, confirming it to be a VEGF-specific effect. The change in specific CEC populations was further characterized after direct VEGF administration by i.p. injection. Increases were observed in both phenotypically mature CECs (CD45−, Flk+, CD31+, CD117−) and CEPs (CD45−, Flk+, CD31+, CD117+), both of which were partially inhibited by ZD6474 to a similar degree (Fig. 2).
VEGF may cause an increase in CEPs and mature CECs through several distinct mechanisms. VEGF is known to promote the mobilization of bone marrow–derived CEPs, which may subsequently differentiate into mature CECs (11, 42). This mobilization is mediated by VEGF binding to both VEGFR-1 and VEGFR-2, which may explain why the VEGF inhibitor sFlt-1 inhibited this mobilization to a greater degree than the VEGFR-2 inhibitor ZD6474 (Fig. 2). VEGF is also thought to promote survival by activating antiapoptotic pathways in both CEPs (44) and mature CECs, which have been sloughed off the vessel wall (45). Consistent with these observations, we have previously reported that the administration of adeno-VEGF decreased the fraction of apoptotic CECs by ∼70% compared with vector controls (27). Finally, VEGF may stimulate the proliferation of CEPs or mature CECs, although mature CECs seem to have only a limited proliferative capacity compared with CEPs (46).
In nontumor-bearing mice, the VEGF-induced increase of both mature CECs and CEPs was inhibited by ZD6474, but no significant change in either population was noted after ZD6474 treatment in control mice (Fig. 2B). By contrast, in tumor-bearing mice, ZD6474 led to a dose-dependent increase in mature CECs but no significant change in CEPs (Fig. 4B and C). The observation that mature CECs increased after treatment with ZD6474 in tumor-bearing mice but not in nontumor-bearing mice, suggests that the increase in mature CECs is due at least in part to presence of tumor and that ZD6474 has at least some degree of selectivity for tumor endothelial cells rather than endothelial cells from normal vasculature. Tumor endothelial cells are characterized by higher proliferation rates and greater dependence on growth factors for survival compared with normal endothelium and are therefore thought to be more vulnerable to damage induced by VEGF pathway inhibitors (47). If the increase in CECs was indeed due to sloughing of tumor endothelial cells, concomitant changes in tumor endothelium would be expected. To address this, we assessed the microvessel density of tumors from the same mice in which CECs were measured. As expected, we found a dose-dependent reduction in tumor microvessel density after 3 days of treatment with ZD6474 (Fig. 3B). Mature CECs but not CEPs increased in a dose-dependent manner that paralleled the decrease in microvessel density (Fig. 4C). It is worth noting that the changes in CECs preceded the inhibition of tumor growth, as differences in tumor size between treated and control mice were not observed until 7 days of treatment at a dose of 50 mg per kg per day. In the present study, CECs were only assessed at 3 days in the LLC model, although it is possible that changes in CECs may occur even earlier, or that other time points may be more predictive of antitumor efficacy. Nevertheless, these findings show that in the LLC model, both tumor microvessel density and mature CECs seemed early markers of antiangiogenic activity.
To address whether the increase in mature CECs was observed after treatment with agents that are thought to selectively affect tumor vasculature, we also tested the effect of the ZD6126. This tubulin-binding agent induces cytoskeletal changes that lead to the retraction and loss of tumor endothelium (34, 40). Treatment of LLC-bearing mice with ZD6126 (100 mg/kg daily) for 3 days induced an ∼5- fold increase in mature CECs after 3 days of treatment (Fig. 5).
These results show that agents targeting the tumor vasculature may induce an increase in mature CECs. Our data suggest that tumor endothelium is likely a source of at least some of these mature CECs, although we cannot rule out the possibility that the mature CECs may originate from other sources as well. This finding, coupled with the previously established finding that VEGF pathway inhibitors can inhibit the VEGF-induced mobilization of CEPs, leads us to propose the following model (Fig. 6). VEGF pathway inhibitors can have differential effects on mature CECs and CEPs, causing an inhibition of bone marrow–derived CEPs mobilized by VEGF but an increase in mature CECs reflecting an increase in sloughing of fragile, mature endothelium from tumor vasculature and potentially other sources as well.

Model of CECs in tumor-bearing animals. CEPs (gray ovals) are mobilized from the bone marrow in response to a variety of proangiogenic stimuli, such as VEGF, and contribute to tumor vasculature. Mature CECs may be derived, at least in part, from the shedding of fragile, mature endothelium from tumor vasculature into the circulation (black ovals). An antiangiogenic agent may have different effects on these two populations. For example, a VEGFR inhibitor could reduce VEGF-induced CEP mobilization but increase mature CEC shedding. These changes may serve as a marker of biological activity.
Model of CECs in tumor-bearing animals. CEPs (gray ovals) are mobilized from the bone marrow in response to a variety of proangiogenic stimuli, such as VEGF, and contribute to tumor vasculature. Mature CECs may be derived, at least in part, from the shedding of fragile, mature endothelium from tumor vasculature into the circulation (black ovals). An antiangiogenic agent may have different effects on these two populations. For example, a VEGFR inhibitor could reduce VEGF-induced CEP mobilization but increase mature CEC shedding. These changes may serve as a marker of biological activity.
This model provides a framework for reconciling seemingly contradictory results from prior studies. In murine models, different types of tumors vary in their ability to mobilize and recruit CEPs (48, 49). Consistent with this observation, our preliminary data suggests that in renal cell carcinoma patients with von Hippel-Lindau disease, a condition marked by dysregulation of the HIF pathway and high VEGF production, CEPs are present at markedly elevated levels compared with patients with other tumor types or noncancer patients.5
A. Norden-Zfoni, D. George, J.V. Heymach, unpublished data.
Grant support: NIH grants P01 CA45548 and P20 CA090578, Alberta Heritage Foundation for Medical Research Clinical Fellowship (P. Beaudry), Damon Runyon Cancer Research Foundation grant CI 24-04 and Damon Runyon-Lilly Clinical Investigator (J.V. Heymach), American Society for Clinical Oncology Young Investigator Award (J.V. Heymach), and American Association for Cancer Research-Amgen Research Fellowship (J.V. Heymach).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Acknowledgments
We thank Anat Norden-Zfoni for critically reviewing the article.