


Abstract
Patients with inflammatory bowel disease (IBD) have a higher risk of developing colitis-associated-cancer (CAC); however, the underlying processes of disease progression are not completely understood. Here, the molecular processes of inflammation-driven colon carcinogenesis were investigated using IL10-deficient mice (IL10 KO). IL10 KO mice were euthanized after development of colitis and dysplasia. IHC was performed for markers of colitis-induced DNA damage (CIDD): oxidative DNA lesions (8-oxoG), double-strand breaks (DSB; γH2AX). and DSB repair. MSI, LOH (Trp53, Apc), and global methylation (CIMP) were assessed on microdissected tissue. Comet assay for DNA damage, immunofluorescence, and immunoblotting were performed on intestinal organoids from wild-type (WT) and IL10 KO mice. Sequential biopsies and surgical specimens from IBD and CAC patients were used for IHC analysis. Severity of inflammation correlated with number of dysplasia. 8-oxoG and γH2AX-positive cells were significantly increased in inflamed and dysplastic areas along with activation of DSB repair. The amount of positively stained cells strongly correlated with degree of inflammation (8-oxoG: R = 0.923; γH2AX: R = 0.858). Neither CIMP, MSI nor LOH was observed. Enhanced DSBs in IL10 KO organoids were confirmed by comet assay and increased expression of γH2AX. Human clinical specimens exhibited significantly higher γH2AX and 8-oxoG in IBD, dysplasia, and CAC compared with normal mucosa. These data indicate that inflammation-driven colon carcinogenesis in IL10 KO mice and IBD patients is associated with oxidative DNA damage and overt presence of DSB. Mol Cancer Res; 16(4); 634–42. ©2018 AACR.
Introduction
Inflammatory bowel disease (IBD), a chronic inflammation of the gut, is linked to a high risk of developing colitis-associated cancer (CAC). The molecular mechanisms involved in colitis-associated carcinogenesis are incompletely understood. Development of CAC shows mechanistic differences to sporadic colorectal cancer despite shared mechanisms such as chromosomal instability, microsatellite instability, and CpG-island hypermethylation (CIN, MSI, and CIMP, respectively; ref. 1). In contrast to the adenoma–carcinoma sequence of sporadic colorectal cancer, CAC lesions are often multifocal throughout the colonic mucosa, and progression is associated with certain mutations, chromosomal aberrations (such as change in number), and clonal expansion of those (2, 3).
Chronic inflammation is considered to drive carcinogenesis by oxidative stress (4–6). The upregulated production of reactive oxygen and nitrogen species (ROS and RON, respectively) causes cellular damage to lipids, proteins, and nucleic acids due to an outrun of scavenging potential. As a consequence, nucleotides can be oxidized (such as guanine to 8-oxoG), resulting in base transversion (GC-TA), abasic sites, and DNA single- and double-strand breaks (7–12). For cellular integrity, DNA double-strand breaks (DSB) can be fatal, and their repair is initiated by the cellular DNA damage response (DDR) consisting of a multiprotein signaling cascade (13, 14). An early event in DSB response is phosphorylation of the H2A subtype histone H2AX at ser-139 (also known as γH2AX) by the phosphatidylinositol 3-like kinases ATM, ATR, and DNA-PK (15, 16). Phosphorylation of H2AX spreads from the site of a DSB into both directions, resulting in initiation and maintenance of DDR. Hence, γH2AX is a common marker for DSB formation (17, 18). Through indirect as well as direct interactions with proteins like BRCA1 and MRE11 with its BRCT domain, γH2AX recruits several DDR proteins at DSBs and activates DNA repair either by homologous recombination (HR) or nonhomologous end joining (NHEJ; ref. 19). NHEJ is a repair process in which the two DNA ends are religated without the need for a homolog, a pathway that puts DNA integrity above repair fidelity potentially leading to significant DNA alterations, such as mismatches, deletions, and even CIN (20, 21). In contrast, HR utilizes the sister chromatid as a template strand to reinstate a correct DNA sequence.
IL10 KO mice have been extensively used as a model for IBD and also CAC. These mice develop spontaneous enterocolitis and concomitant colitis-associated neoplasia (22). Interestingly, the mechanism underlying the inflammation-driven carcinogenesis remains unclear. In a previous study, neither MSI or CIN nor k-RAS, p-53, or APC gene mutations were identified in tumors from IL10 KO mice (23). Our aim was to challenge the molecular pathway of colitis-associated carcinogenesis in IL10 KO mice. Here, we identified colitis-induced DNA damage (CIDD) and induction of error-prone NHEJ as drivers of dysplasia in IL10 KO mice, independent of MSI and CIMP. Human samples exhibited a similar CIDD with disease progression from IBD to low-grade dysplasia (LGD), high-grade dysplasia (HGD), and CAC.
Materials and Methods
Animal experiments
C57BL/6 IL10 KO mice were a kind gift from Dr. Terrence Barrett (University of Kentucky, Lexington, KY) and bred under specific pathogen-free (SPF) conditions. For experiments, 98 mice were conventionally housed at the Biomedical Research Facility at the Medical University of Vienna (Vienna, Austria). Animals were kept for 27 weeks; a group of 10 animals was euthanized one week after transfer to non-SPF and used as inflammation-free control samples. After 1 week of conventional housing, 43 mice received piroxicam (Sigma-Aldrich) in their chow (C1000, Altromin) at a concentration of 200 ppm for 7 days. Body weight and health status were checked at least once a week. Animals were euthanized in case of significant weight loss (>20% within 1 week) or symptoms of severe inflammation like bloody diarrhea and rectal prolapse. Animal studies were performed under the Austrian Animal Experiments Regulation and approved by the animal experiment committee in accordance with the institutional good scientific practice guidelines (BMWF-66.009/0099_II/3b/2011).
Histologic analysis and inflammation score
Intestines were dissected, flushed with PBS, and coiled up as Swiss rolls followed by 10% formalin treatment as described previously (24). Hematoxylin and eosin (H&E) staining was performed on paraffin-embedded sections from mouse intestinal tissue (25). Independent histologic analysis was carried out by 2 scientists, one of which was a pathologist, scoring the number of tumors, the number of HGDs, and the grade of inflammation (26). Human samples were selected from endoscopic biopsies or surgical specimens of patients with IBD and CAC. Written informed consent was obtained from patients. The study was approved by the local ethics committee (EC number: 1354/2012) and complying with international and institutional guidelines for good scientific practice.
IHC
The human samples were obtained from the Clinical Institute of Pathology at Medical University of Vienna. IHC of formalin-fixed paraffin-embedded slides was performed as described previously (27) with primary antibodies against 8-oxoG, γH2AX, MRE11, KU70, BRCA1, Nrf2, and p53 (Supplementary Table S1; Supplementary Methods). Briefly, slides were deparaffinized with 2 cycles of xylol and concomitant rehydration with descendent alcohol series. Heat-induced antigen retrieval was carried out either with citrate buffer (pH 6) or with Tris EDTA (pH 9) for nuclear proteins. Normal goat serum buffer was used as blocking solution, followed by optional M.O.M. (Vector Labs) blocking and subsequent primary antibody incubation (4°C overnight). Staining was performed using biotinylated antibody, Avidin–Biotin–HRP Complex (Vectastain, PK-6100), and diaminobenzidine. Slides were counterstained with hematoxylin (Merck 5174).
Laser capture microdissection
Tissue sections mounted on PEN (polyethylene naphthalate) slides (Leica Microsystems) were dissected using the LMD6000 laser microdissection microscope (Leica Microsystems) to obtain samples from noninflamed, inflamed, and dysplastic intestinal epithelium. DNA was extracted from the microdissected samples using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer's instructions.
Loss of heterozygosity and MSI analysis
Loss of heterozygosity (LOH) was examined for Trp53 (chromosome 11; 42,8cM) and Apc (chromosome 18; 18,5cM) with 3 loci-aligning dinucleotide PCR amplifications, which were selected from the ABI prism mouse mapping primer list (Supplementary Methods). For Trp53, the primer pairs were specific for segments D11mit86 (25.1cM), D11mit4 (36.1cM), and D11mit285 (49.2cM). Apc markers were specific for segments D18mit12 (9.8cM), D18mit177 (14.2cM), and D18mit91 (19.7cM). For amplification the Multiplex PCR Kit (Qiagen) was used. All primers were labeled with fluorescent dye (FAM, NED, VIC, Supplementary Table S2). The PCR reaction consisted of 7.5 μL master mix, 1.5 μL Solution Q, 0.5 μL per primer (6 primers/reaction), and a DNA amount of 5-10 ng. For MSI analysis, reagents were combined prior to analysis: 8.6 μL formamide, 0.4 μL GeneScan 500 LIZ size standard (Applied Biosystems), and 0.4 μL PCR product followed by a denaturation step at 95°C and immediately cooled on ice. Carried out under following PCR conditions: 15 minutes at 95°C, 35 cycles at 94°C for 30 seconds; 60°C for 30 seconds, and 72°C for 30 seconds; followed by 72°C for 10 minutes. Analysis was done by comparing noninflamed, inflamed, and dysplastic tissue using GeneMapper Software 4.0. To additionally analyze the microsatellite status, 4 dinucleotide and 2 polyA repeat markers were used (Supplementary Table S3) and handled and analyzed as described previously (28). All experiments were carried out on a Genetic Analyzer 3500 (Applied Biosystems).
Methylation analysis
To determine the global DNA methylation status, DNA from microdissected tissue was converted using EpiMark Bisulfite Conversion Kit (New England Biolabs) according to the manufacturer's protocol (Supplementary Methods).
TUNEL staining
To assess the number of apoptotic cells in mouse tissue samples TUNEL staining (TdT-mediated dUTP–biotin nick end labeling) was performed according to manufacturer's protocol, using the In Situ Cell Death Detection Kit (Roche). See Supplementary Methods for more details.
Isolation of intestinal organoids and Western blotting
Small intestinal and large bowel organoids (SIO and LBO, respectively) were prepared from IL10 KO and wild-type (WT) mice (29). Organoids were cultured in Matrigel with a passage every 3 to 5 days in a 1:2 ratio. Treatment of organoids was carried out with TNFα (10 ng/mL, eBioscience BMS301), IL6 (10 ng/mL, eBioscience BMS341), recombinant murine IL10 (rmIL10, 10 ng/mL, Preprotech, 210-10), rmIL22 (10 ng/mL, eBioscience 14-8221-63), and sulforaphane (10 μmol/L, Sigma-Aldrich S4441) for 24 hours or with H2O2 (100 μmol/L) for 1 hour. For Western blotting, organoids were washed and resuspended in 1 mL PBS and transferred into a precooled Eppendorf tube, followed by centrifugation at 400 × g for 5 minutes at 4°C. Supernatant was discarded and the washing step was repeated. The cell pellet was lysed in RIPA buffer (on ice for 10 minutes), followed by a short sonication and subsequent cooling on ice for 30 minutes. Using Bradford assay, protein concentration was measured and 20 μg of protein was used for SDS-PAGE. Immunoblotting was performed on a PVDF membrane. The protein bands were visualized with IRDye coupled anti-rabbit or anti-mouse antibodies (either or both mouse/rabbit; LI-COR) and scanned on Odyssey imager (LI-COR Biotechnology).
Immunofluorescence microscopy
For immunofluorescence (IF), SIOs and LBOs were fixed in 4% paraformaldehyde at room temperature for 30 minutes and blocked in horse serum with 0.1% Triton X-100 for 1 hour. Samples were incubated with primary antibody in 1% BSA/PBS overnight at 4°C, then washed and incubated with secondary antibody (Alexa Fluor 488) for 1 hour at room temperature. Then, samples were washed, dried, mounted with DAPI-containing medium (Vector Laboratories), and imaged on an Olympus BX51 microscope. Evaluation of γH2AX staining was carried out using ImageJ (Supplementary Methods).
Alkaline single-cell gel electrophoresis assay (comet assay)
Statistical analysis
Means of normal distributed data were compared using unpaired Student t test; nonparametric Mann–Whitney U test was used for nonnormally distributed data. Tumor incidence and inflammation analysis were tested with Pearson χ2 test. Correlation between grade of inflammation and expression of certain markers was assessed by Spearman rank correlation. Two-sided ANOVA with Dunnet post hoc analysis was used to compare all subgroups together. If P values were equal or less than 0.05, they were considered as statistically significant (*, P ≤ 0.05; **, P < 0.01; ***, P < 0.001). GraphPad Prism 6.01 software (GraphPad Software) was used to analyze all data.
Results
Inflammation and tumor incidence in IL10 KO mice
IL10 KO mice were used as model to investigate the mechanism underlying inflammation-driven carcinogenesis. To induce a high degree of inflammation and synchronize inflammation onset (and secondary tumorigenesis), half of the mice were treated with piroxicam for 1 week (26, 32). Distal inflammation and dysplasia were assessed by colonoscopy after 5 months, at which point piroxicam-fed animals exhibited more signs of inflammation and higher number of dysplastic or hyperplastic lesions compared with untreated IL10 KO mice (Fig. 1A). Animals were euthanized after 27 weeks, and intestines were prepared for further analysis. Histologic inflammation scoring revealed an average inflammation grade of 2.86 ± 0.63 [95% confidence interval (CI), 2.66–3.06] with a prevalence of 100% for the piroxicam-fed mice (n = 43), and 1.8 ± 0.81 (95% CI, 1.55–2.04) with a prevalence of 84.1% for the group without piroxicam (n = 45, P < 0.001). Tumor counting showed a total number of 13 HGD lesions and 4 invasive carcinomas were found in untreated IL10 KO. Piroxicam-fed IL10 KO mice had 25 HGDs (P < 0.05) and 6 invasive carcinomas (Fig. 1B; Supplementary Fig. S1A; Supplementary Table S4). The severity of inflammation correlated to the number of dysplastic lesions (R = 0.728, P < 0.01; Fig. 1C) in piroxicam-treated mice. Because of the low number of affected mice in the untreated group, all following experiments were carried out with samples from piroxicam-treated mice only.

Inflammation and disease progression in IL10 KO mice. A, Results of colonoscopy indicating percentage of dysplastic and inflamed lesions in investigated mice (IL10 KO: n = 20, IL10 KO piroxicam n = 9). Exemplary colonoscopy pictures showing noninflamed, inflamed, and dysplastic areas of the murine large bowel. B, Percentage of mice with histologically determined high-grade dysplastic lesions and invasive carcinomas. C, Piroxicam-fed mice exhibit higher grades of inflammation (t test, two-tailed; ***, P < 0.001) compared with IL10 KO mice fed with normal chow. The number of dysplastic lesions was positively correlated to the grade of inflammation (Spearman ρ, R = 0.701 for control and R = 0.728 for piroxicam group; **, P < 0.01).
Inflammation and disease progression in IL10 KO mice. A, Results of colonoscopy indicating percentage of dysplastic and inflamed lesions in investigated mice (IL10 KO: n = 20, IL10 KO piroxicam n = 9). Exemplary colonoscopy pictures showing noninflamed, inflamed, and dysplastic areas of the murine large bowel. B, Percentage of mice with histologically determined high-grade dysplastic lesions and invasive carcinomas. C, Piroxicam-fed mice exhibit higher grades of inflammation (t test, two-tailed; ***, P < 0.001) compared with IL10 KO mice fed with normal chow. The number of dysplastic lesions was positively correlated to the grade of inflammation (Spearman ρ, R = 0.701 for control and R = 0.728 for piroxicam group; **, P < 0.01).
Absence of MSI, LOH, and CIMP in IL10 KO mice
To examine the molecular pathways leading to colorectal carcinogenesis in IL-KO mice, we analyzed MSI, LOH, and CIMP in DNA from microdissected samples (noninflamed, inflamed, and dysplastic). Fragment analysis was performed for MSI and LOH analysis using certain markers. Three markers, each aligning the Trp53 or Apc gene, were investigated for LOH, whereas 6 markers were additionally utilized for MSI analysis (25). Of all analyzed dysplastic samples, LOH could not be detected at any marker when comparing dysplastic or inflamed tissue to noninflamed mucosa samples. No peak loss was observed, and calculated peak ratios revealed no change. Fragment analysis also revealed no change at any of the six microsatellites in any of the dysplastic or inflamed samples compared with noninflamed tissue (Supplementary Fig. S2A and S2B). These data indicate that neither LOH at Trp53 or Apc nor MSI is involved in colorectal tumor development in this CAC model.
To assess the CIMP, the murine B1 element was analyzed using primers specific for the methylated or unmethylated sequence. Tumor DNA from dysplasia-bearing mice was bisulfite converted and compared with converted noninflamed tissue samples as control. Quantification of the target sequences revealed no significant change in the levels of methylated or unmethylated CIMPs (Supplementary Fig. S2C), indicating that global methylation was not affected as well.
CIDD increases with mucosal inflammation and neoplastic transformation, resulting in DSBs and associated DDR
As the common molecular pathways of sporadic colorectal carcinogenesis were not affected, we further tested for the presence of oxidative stress and DDR. The formation of DNA lesions, such as 8-oxoG, due to generation of ROS and NOS, is considered as a marker of oxidative stress. We examined dysplastic, inflamed, and noninflamed samples for 8-oxoG lesions and assessed the ratio of positive stained cells/per crypt at ×400 magnification (Fig. 2, left). A significant correlation was detected between the inflammation grade and the presence of 8-oxoG–positive cells (R = 0.923; P < 0.01), indicative for a mucosal increase in oxidative DNA damage with the degree of intestinal inflammation. This effect was also observed with disease progression, as there was a significant increase in 8-oxoG–positive cells in dysplastic tissue (Fig. 2, right).

Oxidative stress is upregulated with grade of inflammation and disease progression in IL10 KO, resulting in increased DSBs and upregulated DDR. Left, staining for γH2AX (showing DSBs), 8-oxoG (oxidative stress), MRE11 (DDR), BRCA1 (representative for HR), and KU70 (representative for NHEJ) on noninflamed, inflamed, and dysplastic samples. Right, inflammation grading (grade 0–4) and disease progression. γH2AX-positive nuclei are significantly increased in inflamed (***, P < 0.001) and dysplastic samples (***, P < 0.001), indicating increase in DSBs with disease severity, predominantly in the basal crypt region and the intermediate zone. Disease progression shows increase of DSBs from noninflamed to inflamed to dysplastic tissue. The number of DSBs significantly correlates with the grade of inflammation (R = 0.858, P < 0.001). An increase in 8-oxoG–positive cells is seen with increase in disease severity. Disease progression from noninflamed to inflamed to dysplastic tissue shows a significant increase in oxidative DNA damage (***, P < 0.001). A significant positive correlation is seen between grade of inflammation and 8-oxoG–positive cells (R = 0.923, P < 0.01). An increase of DDR and DNA repair mechanism is detected in the crypts of inflamed and in dysplastic samples. IRS of DDR and DNA repair proteins indicate significant increase with disease severity (**, P < 0.01; ***, P < 0.001) and strong positive correlation with grade of inflammation (MRE11: R = 0.785, P < 0.01; BRCA1: R = 0.921, P < 0.01, KU70: R = 0.851, P < 0.01). Statistical analysis was done with ANOVA using Dunnet post hoc analysis and Spearman ρ for correlation analysis.
Oxidative stress is upregulated with grade of inflammation and disease progression in IL10 KO, resulting in increased DSBs and upregulated DDR. Left, staining for γH2AX (showing DSBs), 8-oxoG (oxidative stress), MRE11 (DDR), BRCA1 (representative for HR), and KU70 (representative for NHEJ) on noninflamed, inflamed, and dysplastic samples. Right, inflammation grading (grade 0–4) and disease progression. γH2AX-positive nuclei are significantly increased in inflamed (***, P < 0.001) and dysplastic samples (***, P < 0.001), indicating increase in DSBs with disease severity, predominantly in the basal crypt region and the intermediate zone. Disease progression shows increase of DSBs from noninflamed to inflamed to dysplastic tissue. The number of DSBs significantly correlates with the grade of inflammation (R = 0.858, P < 0.001). An increase in 8-oxoG–positive cells is seen with increase in disease severity. Disease progression from noninflamed to inflamed to dysplastic tissue shows a significant increase in oxidative DNA damage (***, P < 0.001). A significant positive correlation is seen between grade of inflammation and 8-oxoG–positive cells (R = 0.923, P < 0.01). An increase of DDR and DNA repair mechanism is detected in the crypts of inflamed and in dysplastic samples. IRS of DDR and DNA repair proteins indicate significant increase with disease severity (**, P < 0.01; ***, P < 0.001) and strong positive correlation with grade of inflammation (MRE11: R = 0.785, P < 0.01; BRCA1: R = 0.921, P < 0.01, KU70: R = 0.851, P < 0.01). Statistical analysis was done with ANOVA using Dunnet post hoc analysis and Spearman ρ for correlation analysis.
Oxidation of DNA bases can lead to single- and DSBs due to creation of abasic or complex DNA damage sites by several 8-oxoGs in close proximity (7, 33). To assess the level of DSBs in colitis and dysplasia, we stained for phosphorylated histone H2AX, yH2AX, an early event in DSB response. Dysplastic, inflamed, and noninflamed samples were evaluated according to the ratio of positive nuclei per crypt at ×400 magnification. The amount of positively stained nuclei was significantly increased in inflamed (P < 0.001) and dysplastic tissue (P < 0.001) compared with noninflamed tissue. The increased staining was predominant in basal crypt region and in the intermediate zone (Fig. 2, left). In addition, the increase in DSBs positively correlated to the degree of inflammation (R = 0.858, P < 0.001; Fig. 2, right)
If not repaired, DSBs are hazardous for cell survival. In fact, phosphorylation of H2AX initiates DDR and DSB repair. To assess the exact DDR mechanism, samples were stained for MRE11, representing the MRN complex (MRE11–RAD50–NBS1) that induces the DSB response. Furthermore, it interacts with DSB repair proteins, such as KU70 [which together with KU80 binds to DSB ends and is required for NHEJ and BRCA1 (involved in HR)], which were further analyzed. Dysplastic samples were stained and compared with inflamed and noninflamed samples. An increase of positively stained nuclei in inflamed (P < 0.01) and dysplastic tissue compared with noninflamed for each MRE11, BRCA1, and KU70 (P < 0.001) was observed, indicating an upregulation of DDR for both NHEJ and HR (Fig. 2, left). Also, a positive correlation existed between grade of inflammation and rate of positive nuclei (MRE11: R = 0.785, P < 0.01; BRCA1: R = 0.921, P < 0.01, KU70: R = 0.851, P < 0.01; Fig. 2, right). These data support the notion that in contrast to MSI or CIN, DSBs are the major oxidative DNA lesions within the mucosal proliferative zone with induction of both HR and NHEJ, which likely affects DNA fidelity in intestinal stem cells.
As a consequence of DSBs, damaged cells can either survive (after DNA repair) or undergo apoptosis. To test whether the lesions are associated with apoptosis, TUNEL staining was performed. We did not detect an increase in apoptotic cells in inflamed and dysplastic crypts (Supplementary Fig. S3), indicating that the DSBs did not trigger apoptosis.
Intestinal IL10 KO organoids exhibit increased DSBs
To validate the in vivo findings observed in IL10 KO mice, an ex vivo model of intestinal organoids from WT and IL10 KO mice was used. To examine whether DNA damage could be induced in organoids, they were treated with either proinflammatory cytokines TNFα and IL6 (for 24 hours) or with oxidative stress inducer hydrogen peroxide (H2O2, for 1 hours). γH2AX was increased upon treatment with TNFα, IL6, or H2O2. When compared with WT, phosphorylated H2AX was higher in untreated IL10 KO organoids corroborating the observations in IL10 KO mice (Fig. 3A). An increase in spontaneous DSBs upon IL10 deficiency was further confirmed by comet assay and modified FPG comet assay (which detects oxidative DNA damage by strand break induction at oxidized DNA bases) of these SIOs (Fig. 3B). Immunofluorescence confirmed higher basal levels of nuclear γH2AX in IL10 KO organoids (Fig. 3C), again implying ongoing DSBs in IL10 KO organoids compared with WT. Treatment with H2O2 further increased DSBs, whereas IL10 treatment reverted the amount of DSBs in IL10 KO organoids compared with untreated, presumably due to a reduction in cellular levels of ROS. IL22 treatment showed no effect on γH2AX (Fig. 3C and D). One of the main cellular sensors of oxidative stress is Nrf2, which coordinates the expression of antioxidant proteins (34, 35). In fact, aberrant cytoplasmic accumulation of Nrf2 was observed in IL10 KO organoids compared with WT (Supplementary Fig. S4A). Activating Nrf2 using sulforaphane did not change the amount of γH2AX found in LBOs (Supplementary Fig. S4B). We further investigated the level of STAT3 phosphorylation after treatment with sulforaphane, IL10, or IL22. Activation of Nrf2 with sulforaphane showed no profound effect on STAT3 phosphorylation between WT and IL10 KO LBOs. IL22 treatment resulted in the strongest STAT3 phosphorylation in both WT and IL10 KO organoids (Supplementary Fig. S4C). These data suggest increased basal levels of DSBs in IL10 KO organoids, likely due to an excessive presence of ROS and reduced scavenging potential indicated by cytoplasmic accumulation of Nrf2, a condition that can be attenuated by IL10 treatment.

Increased DSBs in IL10 KO intestinal organoids. A, Western blot analysis of proteins from small intestinal- and large bowel organoids (WT, IL10 KO) treated with proinflammatory cytokines TNFα, IL6 (24 hours), and H2O2 (1 hour) showed increase of γH2AX after proinflammatory treatment, profoundly in IL10 KO organoids. Actin and tubulin were used as loading control. B, DSBs in WT and IL10 KO organoids were analyzed using comet assay, with and without FPG. FPG treatment induces further breaks at oxidized DNA bases, thereby detecting oxidative DNA damage (t test, two-tailed, ***, P < 0.001). IL10 KO organoids exhibited more DSBs and oxidative DNA damage than WT organoids, in classic comet assay and FPG-comet assay, respectively. C, IF with antibody against γH2AX (green) and DAPI to visualize nuclei in WT and IL10 KO organoids. DSBs are increased in IL10 KO organoids, whereas treatment with 10 ng/mL rmIL10 reduced the amount of positive γH2AX foci in IL10 KO organoids compared with untreated IL10 KO SIOs. D, Western blot analysis of LBOs treated with either rmIL10 or rmIL22, showing a reduction in γH2AX after rmIL10 treatment.
Increased DSBs in IL10 KO intestinal organoids. A, Western blot analysis of proteins from small intestinal- and large bowel organoids (WT, IL10 KO) treated with proinflammatory cytokines TNFα, IL6 (24 hours), and H2O2 (1 hour) showed increase of γH2AX after proinflammatory treatment, profoundly in IL10 KO organoids. Actin and tubulin were used as loading control. B, DSBs in WT and IL10 KO organoids were analyzed using comet assay, with and without FPG. FPG treatment induces further breaks at oxidized DNA bases, thereby detecting oxidative DNA damage (t test, two-tailed, ***, P < 0.001). IL10 KO organoids exhibited more DSBs and oxidative DNA damage than WT organoids, in classic comet assay and FPG-comet assay, respectively. C, IF with antibody against γH2AX (green) and DAPI to visualize nuclei in WT and IL10 KO organoids. DSBs are increased in IL10 KO organoids, whereas treatment with 10 ng/mL rmIL10 reduced the amount of positive γH2AX foci in IL10 KO organoids compared with untreated IL10 KO SIOs. D, Western blot analysis of LBOs treated with either rmIL10 or rmIL22, showing a reduction in γH2AX after rmIL10 treatment.
γH2AX as marker of CIDD in human IBD
To evaluate the presence of CIDD in IBD and CAC, we also examined γH2AX in colonic tissue sections from biopsies and surgical specimens of patients with normal mucosa, IBD, LGD, HGD, CAC, CAC-adjacent tissue, colorectal cancer, and familial adenomatous polyposis samples (FAP; Fig. 4A). 8-oxoG staining was used to evaluate levels of oxidative stress in normal, IBD, and CAC samples. Of all 52 analyzed samples, sequential specimens from 6 patients were available (Fig. 4B, top and colored bars). They all exhibited a sequential increase in γH2AX from LGD or HGD to CAC. Biopsies identified as LGD already show distinct γH2AX-positive areas, with a threshold above 20% positive cells. This is also true for IBD surveillance biopsies of patients, who later developed a LGD or HGD. The CAC-adjacent tissue exhibited specific crypts, solely positive for γH2AX, potentially marking crypts indefinite for dysplasia. (Fig. 4A). The highest level of γH2AX was observed in LGD, HGD, and CAC samples (P < 0.001) as well as in colorectal cancer (P < 0.01; Fig. 4C). Interestingly, the staining for p53 revealed a similar pattern as seen in HGD and CAC-γH2AX staining with nuclear accumulation of p53 (Fig. 4B/D). Quantifying the marker for oxidative stress revealed already overt presence of 8-oxoG in IBD samples, with almost 70% positive nuclei (P < 0.001) and also in CAC (P < 0.001; Fig. 4E). These findings show overt CIDD, marked by 8-oxoG and γH2AX, and its involvement in the sequence from IBD to LGD to HGD to CAC.
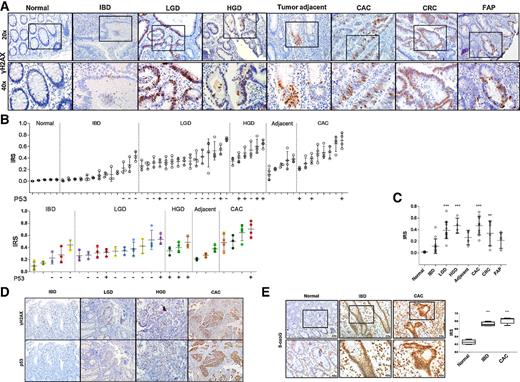
γH2AX as a marker of CIDD in patients with IBD. A, Exemplary pictures of γH2AX staining of human samples (normal, IBD, LGD, HGD, tumor adjacent, CAC, colorectal cancer, FAP) indicating increase of DSBs with disease progression and severity. B, Analysis of patient samples stained for γH2AX; each bar represents a patient; colored bars indicate the same patient with available sequential biopsy and surgical samples. Profound increase of γH2AX-positive nuclei marking DSBs is seen in LGD, HGD, and CAC samples. Below the x-axis, corresponding p53 staining (D) is shown as either – (nuclei p53 negative) or + (nuclei p53 positive). C, Cumulative analysis of γH2AX staining showing significantly higher levels for CAC, HGD, LGD (***, P < 0.001) and colorectal cancer (**, P < 0.01) samples compared with normal mucosa. E, IHC for 8-oxoG in human colon samples from healthy subjects, IBD, and CAC patients, showing overt presence of oxidized DNA damage in IBD and CAC samples. IRS of 8-oxoG levels are significantly increased in IBD (***, P < 0.001) and CAC (***, P < 0.001). Statistical analysis was done with two-sided ANOVA using Dunnet post hoc analysis.
γH2AX as a marker of CIDD in patients with IBD. A, Exemplary pictures of γH2AX staining of human samples (normal, IBD, LGD, HGD, tumor adjacent, CAC, colorectal cancer, FAP) indicating increase of DSBs with disease progression and severity. B, Analysis of patient samples stained for γH2AX; each bar represents a patient; colored bars indicate the same patient with available sequential biopsy and surgical samples. Profound increase of γH2AX-positive nuclei marking DSBs is seen in LGD, HGD, and CAC samples. Below the x-axis, corresponding p53 staining (D) is shown as either – (nuclei p53 negative) or + (nuclei p53 positive). C, Cumulative analysis of γH2AX staining showing significantly higher levels for CAC, HGD, LGD (***, P < 0.001) and colorectal cancer (**, P < 0.01) samples compared with normal mucosa. E, IHC for 8-oxoG in human colon samples from healthy subjects, IBD, and CAC patients, showing overt presence of oxidized DNA damage in IBD and CAC samples. IRS of 8-oxoG levels are significantly increased in IBD (***, P < 0.001) and CAC (***, P < 0.001). Statistical analysis was done with two-sided ANOVA using Dunnet post hoc analysis.
Discussion
Patients with ulcerative colitis (UC) are at risk for developing colorectal cancer. The molecular events of inflammation-driven carcinogenesis are incompletely understood and differ from sporadic colorectal cancer as well as familial cancer syndromes. Although oxidative DNA damage is considered the driving force of mutagenesis in this setting, the actual molecular DNA lesion and the associated DNA repair pathway have not been investigated. Here, we studied these molecular events in IL10-deficient mice, a well-established model of CAC. We confirm that MSI, LOH, and CIMP were not observed in these mice (23). Instead, oxidative DNA lesions (i.e., 8-oxoG) were found in the crypts of IL10 KO mice and were associated with upregulation of DSB response and repair proteins (like γH2AX, MRE11, BRCA1, and KU70). Ex vivo, IL10 KO organoids displayed spontaneous DSBs. Human sections also exhibited an increase in 8-oxoG in IBD and CAC. γH2AX was induced in LGD, HGD, tumor adjacent, and CAC tissue. Therefore, we believe that DSBs are the core molecular lesion in CIDD and inflammation-driven carcinogenesis. Activation of the DSB repair pathway promotes cell survival and potential accumulation of mutations leading to CAC.
Both, RONs and ROS contribute to DNA damage (6, 36, 37). In fact, 8-oxoG increased with the grade of inflammation and was present in dysplastic lesions of IL10 KO mice (38, 39). Similar γH2AX and DSB repair proteins increased with the grade of inflammation and dysplasia without inducing apoptosis. Induction of DSBs were previously reported for UC, gastric cancer, and in ATM-deficient mice upon dextran sodium sulfate (40–43). Compared with HR, NHEJ is considered error prone because of the direct ligation of broken DNA ends without a homologous template. NHEJ is active in all cell-cycle states and appears to be more effective than HR, yet results in more DNA deletions and translocations (44). Our data suggest that besides HR, NHEJ is also active and a possible mutational driver in this setting. In the absence of alteration in other examined molecular mechanisms (MSI, LOH, CIMP), this seems to be a predominant pathway of carcinogenesis in IL10 KO mice. Abundance of CIDD together with error-prone repair might predispose to accumulation and expansion of premutagenic lesion. In addition, crypt cells under inflammatory stress have higher proliferation rates and are therefore under constant replication stress, which is another cause of increased DSBs (8, 45).
In the ex vivo model, IL10-deficient organoids displayed spontaneous DSBs without the presence of inflammatory cells or cytokines, a condition that could be reverted by the addition of recombinant IL10. In parallel, Nrf2 was found mainly in its aberrant cytoplasmic location. As Nrf2 elicits its ROS scavenging function as a transcription factor, cytoplasmic accumulation indicates an impaired activity, thereby showing a diminished potential to reduce ROS in IL10 KO organoids. Remarkably, Nrf2 was shown to facilitate HR and its activation protects from DNA. On the contrary, inhibition of Nrf2 activity led to increase in γH2AX and reduced DSB repair (46, 47). Using sulforaphane as an Nrf2 activator, we could not detect a change in DSB levels in IL10 KO organoids. Such an effect has been postulated for skin after UV radiation or in human lymphocytes after contact with DNA-damaging substances (48, 49). The abrogated Nrf2 signaling in IL10 KO mice possibly enhances intracellular ROS, DNA damage, and NHEJ. The overt presence of ROS is likely further sustained as foci of γH2AX were shown to trigger and maintain proinflammatory cytokine secretion (50). Our experiments showed no overt phosphorylation of STAT3 or a difference in its phosphorylation levels between WT- and IL10 KO organoids, after Nrf2 activation or cytokine treatment. The effect of IL10 itself might be attributable to its ability to regulate oxidative homeostasis (51). Nevertheless, further research is needed to elucidate potential interaction and crosstalk between Nrf2 and the IL10–STAT3 signaling cascade. IL10 itself is known to regulate oxidative homeostasis (51).
Our mouse and ex vivo findings were verified in human tissue section. The longitudinal analysis of IBD samples revealed a similar pattern of CIDD as observed in IL10 KO mice. An increase in DSBs was seen in LGD, HGD, and CAC samples compared with normal biopsies. Samples from IBD patients showed at least 20% γH2AX-positive nuclei (per crypt), if they developed LGD in later stages. We also examined p53 expression, considered as one of the first-hit targets in the sequela of IBD to CAC and having a critical role in DDR (52, 53). The areas with nuclei positive for p53 were mostly seen in HGD and CAC samples and were associated with γH2AX-positive areas. Although a normal p53 function cannot be ruled out, given the fact that these are already progressed neoplasias, it is likely that the observed nuclear accumulation of p53 indicates a nonfunctional mutated protein (54, 55). Hence, this creates a condition of cellular damage and survival resulting in carcinogenesis.
Here, we propose γH2AX as a marker for CIDD and disease progression for surveillance endoscopy in IBD. Evaluating H2AX activation was already tested in various malignancies such, as gastric, lung, and cervical cancer, and it is discussed as a potential marker for cancer progression as well as for radiation and chemotherapy response (45, 56–58). In IBD, the risk of developing CAC is heterogeneous and relates to duration, extent, and severity of disease. γH2AX staining of biopsies from surveillance endoscopy may help separating those individuals at risk from those without. More studies are needed before we can use this biomarker for adjusting surveillance intervals or selecting patients who benefit most from chemoprevention or surgery.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
Conception and design: V. Khare, G. Paul, C. Gasche
Development of methodology: V. Khare, F. Ferk
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): A. Frick, V. Khare, G. Paul, M. Lang, S. Knasmüller, G. Oberhuber
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A. Frick, V. Khare, M. Lang
Writing, review, and/or revision of the manuscript: A. Frick, V. Khare, M. Lang, A. Beer, G. Oberhuber, C. Gasche
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): A. Beer, C. Gasche
Study supervision: V. Khare, C. Gasche
Other (conducted SCGE assay and evaluated the slides): F. Ferk
Acknowledgments
This study was supported by the Federal Ministry of Economy, Family and Youth and the National Foundation for Research, Technology and Development and in part by the Austrian Science Fund (P27681 to C. Gasche). IL10 KO mice were gratefully provided by Dr. Terrence A. Barrett and Jeffrey B. Brown, Northwestern University, Feinberg School of Medicine, Chicago, IL.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.