





Abstract
Current guidelines recommend adding a long-acting inhaled β2-agonist (LABA) to inhaled corticosteroids (ICS) in patients with uncontrolled asthma. This study evaluated the novel, once-daily LABA vilanterol trifenatate (VI) in asthma patients who remained symptomatic despite existing ICS therapy.
The study involved a randomised, double-blind, placebo-controlled trial of VI (3, 6.25, 12.5, 25 and 50 μg), administered once daily in the evening by dry powder inhaler for 28 days, in asthma patients aged ≥12 yrs symptomatic on current ICS therapy. The primary end-point was trough (24 h post-dose) forced expiratory volume in 1 s (FEV1); secondary end-points were weighted mean FEV1, peak expiratory flow (PEF), symptom-/rescue-free 24-h periods, and safety.
A significant relationship was observed between VI dose and improvements in trough FEV1 (p=0.037). Statistically significant increases in mean trough FEV1, relative to placebo, were documented for VI 12.5–50 μg (121–162 mL; p≤0.016). Dose-related effects of VI were observed on weighted mean (0–24 h) FEV1, morning/evening PEF, and symptom-/rescue-free 24-h periods. All doses of VI were well tolerated with low incidences of recognised LABA-related adverse events (tremor 0–2%; palpitations 0–2%; glucose effects 0–1%; potassium effects 0–<1%).
Once-daily VI 12.5–50 μg resulted in prolonged bronchodilation of at least 24 h with good tolerability in asthma patients receiving ICS. Based on the overall efficacy and adverse event profile from this study, the optimum dose of VI appears to be 25 μg.
Asthma is a chronic inflammatory disorder of the airways involving several inflammatory cells and multiple mediators that can cause periodic airflow obstruction, which is characteristic of the disease [1, 2]. Asthma is associated with substantial burden to the patient and healthcare systems [3], particularly when control is poor [4]. With sustained controller treatment, patients may be maintained free of symptoms and other clinical features of asthma for prolonged periods [5]. However, evidence from cross-sectional surveys suggests that a high proportion of patients remain uncontrolled despite controller therapy [6–8].
Inhaled corticosteroids (ICS) are considered the cornerstone of controller asthma therapy and treatment guidelines recommend the addition of a long-acting inhaled β2-agonist (LABA) to ICS for those patients not adequately controlled on ICS [3]. This combination approach improves lung function and reduces asthma symptoms, rescue medication use and the number of exacerbations compared with ICS alone [9]. Thus, asthma control is achieved in a greater proportion of patients [5, 10, 11]. Currently available combination therapies, such as those that contain the LABAs salmeterol or formoterol, require twice-daily administration due to their duration of action, but treatment adherence remains a problem in chronic use [6]. This may be improved by the use of a single combination inhaler instead of two [12, 13], but further benefit might be provided by reducing the dosing frequency required for maintenance of control to once daily.
Vilanterol trifenatate (VI; GW642444M) is a LABA with inherent 24 h activity that is being developed as a once-daily clinical treatment for asthma in combination with fluticasone furoate, a novel ICS also active for 24 h [14]. Experimental models have shown that VI is potent and, compared with salmeterol, has a faster onset and longer duration of action [15, 16]. VI is also highly selective for the β2 receptor with >1,000-fold greater selectivity for this receptor than for the β1 and β3 receptors [16].
The aim of the present study was to evaluate the efficacy, dose responsiveness, duration of effect, and safety of adding VI (dosed once daily in the evening) to ICS therapy in patients with asthma who remained symptomatic on ICS alone. Some of the results of this study have been previously reported in abstract form [17, 18].
METHODS
Setting
A multinational, multicentre, randomised, double-blind, placebo-controlled, dose-ranging study conducted between December 2007 and September 2008.
Patients
Patients were aged ≥12 yrs with asthma [19] first diagnosed ≥6 months prior to screening, with reversibility to salbutamol (400 μg; increase in baseline forced expiratory volume in 1 s (FEV1) of ≥12% and ≥200 mL), pre-bronchodilator FEV1 of ≥40% to ≤90% of predicted [20], and maintained on a stable dose of an ICS for ≥4 weeks prior to screening. Complete inclusion and exclusion criteria are provided in the online supplementary material, appendix S1.
The study was approved by local ethics review committees and conducted in accordance with the Declaration of Helsinki and good clinical practice guidelines. All patients gave written informed consent.
Interventions
After a 14-day run-in, patients were randomised to receive VI (GlaxoSmithKline Clinical Trials Supplies, Ware, UK) 3 μg, 6.25 μg, 12.5 μg, 25 μg or 50 μg, or placebo, administered once daily in the evening for 28 days via a single-step activation dry powder inhaler. VI was dosed in the evening as it is being developed as the LABA component of a new once-daily ICS/LABA combination. The ICS component of the combination, fluticasone furoate, exhibits noninferior efficacy when dosed once daily in the evening compared to twice-daily dosing [21], and as such VI was dosed in the evening to mimic the planned time of dosing of the ICS/LABA combination. The follow-up period was 7 days. Patients continued on their stable maintenance ICS throughout the study from screening through to follow-up. Short-acting β2-agonists (replaced by rescue salbutamol metered-dose inhalers at screening) were permitted, but not 6 h prior to or during clinic visits.
Patients visited the clinic on days 1, 7, 14 and 28 for FEV1 measurements. On days 1 and 28, serial FEV1 measurements were made pre-dose and at various intervals up to 24 h post-dose. Peak expiratory flow (PEF), symptom and rescue medication data were recorded daily in an electronic diary.
Randomisation and masking
The central randomisation schedule was generated by the sponsor using a validated computerised system (RandAll). Patients were randomised using Registration and Medication Ordering System (RAMOS), an automated, interactive telephone-based system that was used by the investigator or designee to register the patient, randomise the patient and receive medication assignment information. Prior to randomisation, patients were stratified by baseline % predicted FEV1 (≥40% to ≤65% and >65% to ≤90%) with a 1:1 (approximate) allocation using a randomly permuted block size of six.
Patients and investigators were blinded to treatment assignment, and the placebo and VI formulations were indistinguishable.
Outcome measurements
The primary efficacy end-point was change from baseline in trough FEV1 (defined as the mean of the evening pre-bronchodilator FEV1 values obtained 23 and 24 h after dosing) at day 28.
Secondary end-points were: change from baseline in weighted mean 24 h serial FEV1 on days 1 and 28; change from baseline in daily morning and evening PEF averaged over days 1–28; change from baseline in percentage of symptom-/rescue-free 24 h periods during the 28 day treatment period; and difference in post-salbutamol FEV1 between 24 h after dosing on days 1 and 28, between screening and 24 h after dosing on day 1, and between screening and 24 h after dosing on day 28.
The proportion of patients obtaining both ≥200 mL and ≥12% increase from baseline in FEV1 was calculated over 0–24 h on days 1 and 28. The change over 0–4 h was an “other” end-point, while the change after 4–24 h was a post hoc analysis.
Safety evaluation
Safety was assessed by monitoring adverse events (AEs) and serious AEs (SAEs), worsening asthma/exacerbations, laboratory parameters, vital signs, 12-lead ECG and potassium and glucose levels.
Statistical analysis
All efficacy analyses were pre-specified in the intent-to-treat population. It was estimated that 594 patients (99 per group) would be needed to provide 97% power (two-sided α=0.05) to detect a dose–response effect of 200 mL improvement in FEV1 per 50 μg of VI, assuming a standard deviation of 430 mL (GlaxoSmithKline; data on file).
The primary analyses were performed using SAS software (SAS Institute Inc., Cary, NC, USA) in a step-wise approach. First, a dose–response test at day 28 was performed. If statistically significant, pair-wise testing of each dose of VI versus placebo was performed using an ANCOVA model adjusted for baseline FEV1, country amalgamation, age, sex, baseline % pred FEV1 stratum and treatment group, using a last observation carried forward approach.
Serial FEV1 on days 1 and 28 was analysed using a repeated measures model adjusting for baseline and treatment factors. QTc interval was calculated by Fridericia formula (QTcF). Other statistical analyses are described in appendix S2 of the online supplementary material.
Further information
Further methodological details can be found in appendix S2 of the online supplementary material.
RESULTS
Study population
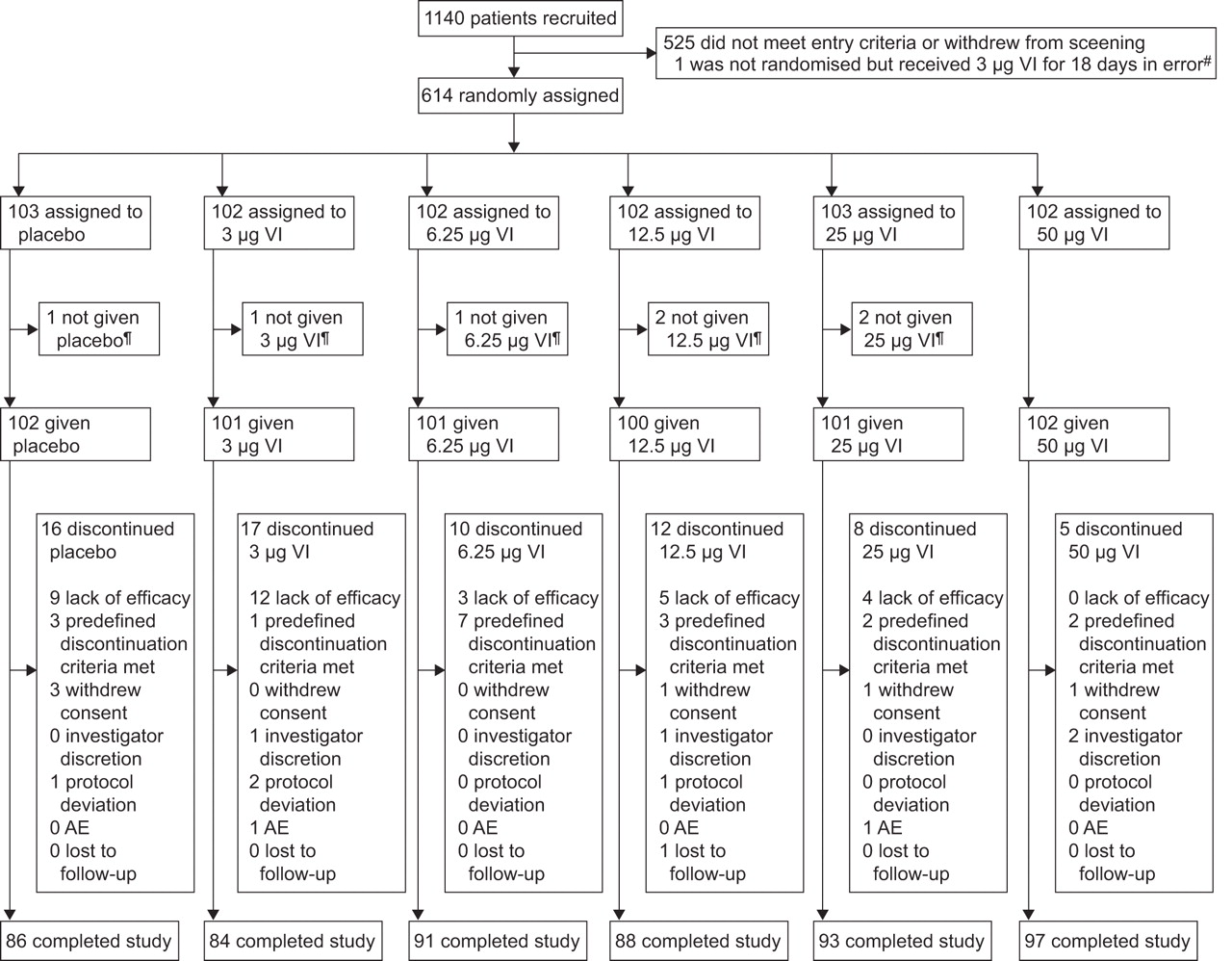
Of 1,140 patients screened, 614 underwent randomisation and 539 completed the study (fig. 1). The main reasons for withdrawal were lack of efficacy (n=33) and meeting predefined discontinuation criteria (n=18).

Patient enrolment and completion of the study. AE: adverse event; VI: vilanterol trifenatate. #: data from this patient were not included in the efficacy and safety summary tables, but safety data were collected to ensure there were no safety issues; ¶: seven patients were randomised in error but did not receive study drug.
The demographic and baseline clinical characteristics of patients in the six treatment groups are shown in table 1. Duration of asthma history was similar across treatment groups (≥10 yrs in 64–74% of patients) and most (67%) patients had a history of atopy. Per cent predicted pre-bronchodilator FEV1(65–68%), absolute reversibility (518–563 mL), and per cent reversibility (24–27%) in FEV1 were also comparable across the six groups. Mean exposure to study medication was 26–28 days and mean overall reported compliance to treatment was high (99–107%). During the study, fluticasone propionate (32–44%) and budesonide (33–48%) were the most frequently used maintenance ICS therapies across the six study arms; beclometasone-equivalent doses in each treatment group (mean±sd) were as follows: placebo: 814.4±537.5 μg; VI 3 μg: 698.3±405.53 μg; VI 6.25 μg: 747.6±467.68 μg; VI 12.5 μg: 736.3±473.29 μg; VI 25 μg: 736.4±411.78 μg; and VI 50 μg: 709.8±517.24 μg.
Efficacy
The mean change from baseline in trough (23–24 h post-dose) FEV1 at day 28 in the placebo group was 147 mL. There was a significant relationship between the dose of VI and trough FEV1 response (p=0.037, excluding placebo). Direct comparisons of each VI dose versus placebo showed statistically significant improvements in trough FEV1 for 12.5 μg, 25 μg and 50 μg VI, but not for the lower doses of 3 μg and 6.25 μg (table 2; fig. 2). Results in the per-protocol population were consistent with those in the intent-to-treat population (data not shown).

Adjusted mean change from baseline in trough forced expiratory volume in 1 s (mL) versus placebo at day 28 (intent-to-treat population). Error bars represent 95% confidence intervals. VI: vilanterol trifenatate.
Serial FEV1 data on days 1 and 28 (fig. 3) show an onset of action at the first measurement time point (15 min) and a sustained duration of response over the 24 h of observation, for all doses of VI. The ratio of peak post-dose FEV1 (over the first 4 h) versus trough FEV1 on day 28 was approximately one in all VI groups and placebo (the ratio varied between 1.04 and 1.06, standard deviation varied between 0.053 and 0.091). The change from baseline in weighted mean for 24 h serial FEV1 (average area under the curve minus baseline) was statistically significant for all doses of VI versus placebo on both days 1 and 28 (except for the 6.25 μg dose on day 1). A dose-related effect of VI was observed on both days 1 and 28, with greatest improvements versus placebo seen for the 25 μg (day 1 and day 28 data, respectively: 193 mL and 165 mL) and 50 μg (215 mL and 172 mL) doses (fig. 3).

Change from baseline serial forced expiratory volume in 1 s (FEV1) adjusted treatment differences from placebo (mL) from a repeated measures model (a, c) and adjusted treatment differences from placebo of weighted mean 24 h serial FEV1 (b, d) on (a, b) day 1 and (c, d) day 28 (intent-to-treat population). a) se: 44 mL at 4 h (all doses); 46 mL (25 μg) and 47 mL (all other doses) at 16 h; 46 mL at 24 h (all doses). c) se: 48 mL (25 μg, 50 μg), 49 mL (6.25 μg, 12.5 μg), and 50 mL (3 μg) at 0 h; 46 mL (50 μg), 48 mL (3 μg), and 47 mL (all other doses) at 4 h; 49 mL (25 μg, 50 μg), 50 mL (6.25 μg, 12.5 μg), and 51 mL (3 μg) at 16 h; 48 mL (50 μg), 49 mL (6.25 μg, 25 μg), and 50 mL (3 μg, 12.5 μg) at 24 h. Error bars represent 95% confidence intervals. VI: vilanterol trifenatate.
The study design also allowed for patients to be stratified according to baseline lung function (upper strata >65% to ≤90%; lower strata ≥40% to ≤65% pred FEV1). In the upper strata absolute mean reversibility ranged from 542.3 mL to 616.7 mL and from 22.4% to 26.4%. In comparison, mean absolute reversibility was slightly lower across the treatment groups (447.4 mL to 542.8 mL) in the lower strata, while mean per cent reversibility was slightly higher (26.4–31.1%). In the upper strata there was little evidence of a dose response at doses of 12.5 μg or greater; there appeared to be a reasonable response to the 3 μg dose that was not evident at the 6.25 μg dose. In contrast, in the lower strata (table 2), there was some evidence of a dose-related effect ranging from no effect with 3 μg (-49 mL difference relative to placebo) to a 139 mL increase with the 50 μg dose.
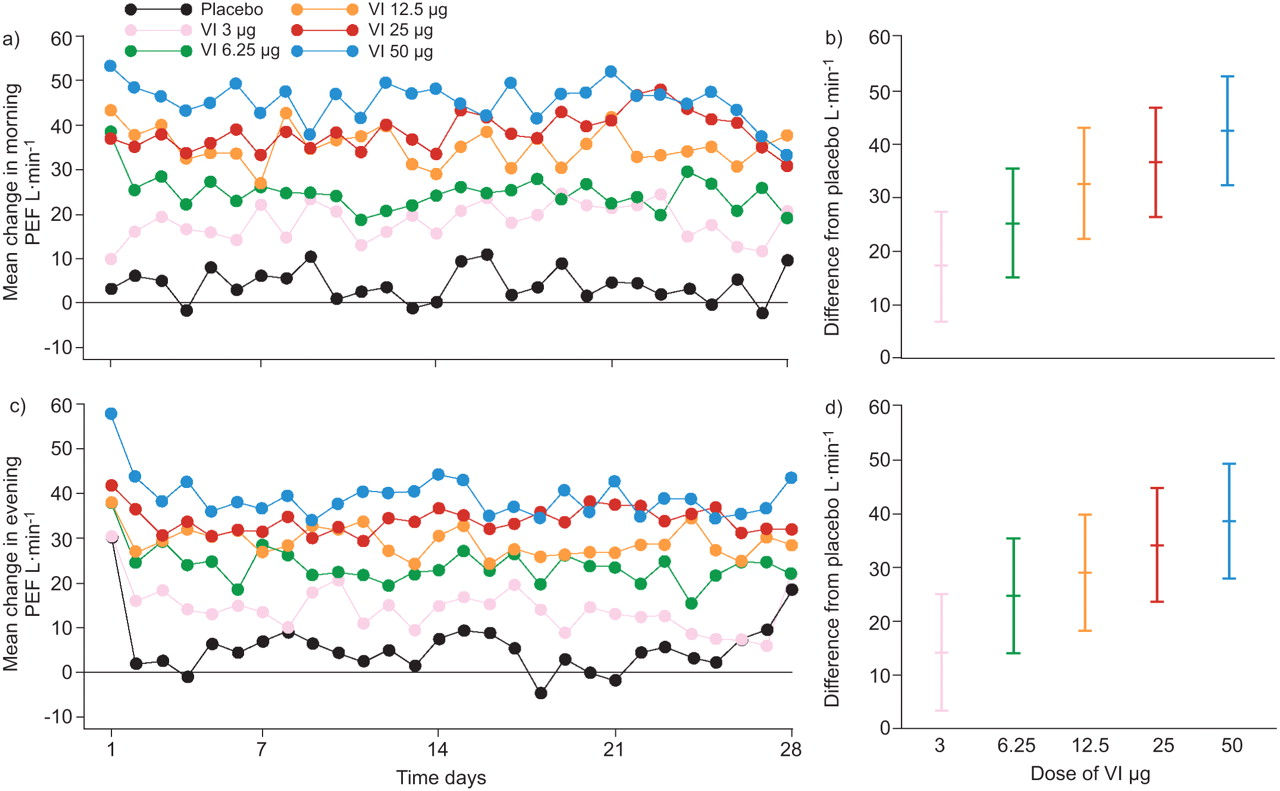
All doses of VI increased morning and evening PEF in a dose-dependent fashion (fig. 4). Morning and evening PEF averaged over the 28-day treatment period were significantly greater than placebo for all doses of VI (fig. 4), with the greatest increases recorded for 12.5 μg, 25 μg and 50 μg VI, respectively, in morning PEF (32.3 L·min−1, 36.2 L·min−1, 42.1 L·min−1) and evening PEF (28.5 L·min−1, 33.6 L·min−1, 38.0 L·min−1; all p<0.001).

Daily mean change in peak expiratory flow (PEF; L·min−1) from baseline (a, c) and adjusted treatment differences from placebo of change from baseline in PEF over days 1–28 (b, d) for (a, b) morning and (c, d) evening PEF (intent-to-treat population). VI: vilanterol trifenatate. Error bars represent 95% confidence intervals.
VI increased the percentage of symptom-free 24 h periods versus placebo by 8–22% in a dose-dependent manner; the effect was statistically significant for all doses of VI, except 3 μg (table 3). VI also dose-dependently increased the percentage of rescue-free 24 h periods versus placebo by 11–28% (p<0.05 for all doses), with the greatest increases seen with the 25 μg dose (table 3).
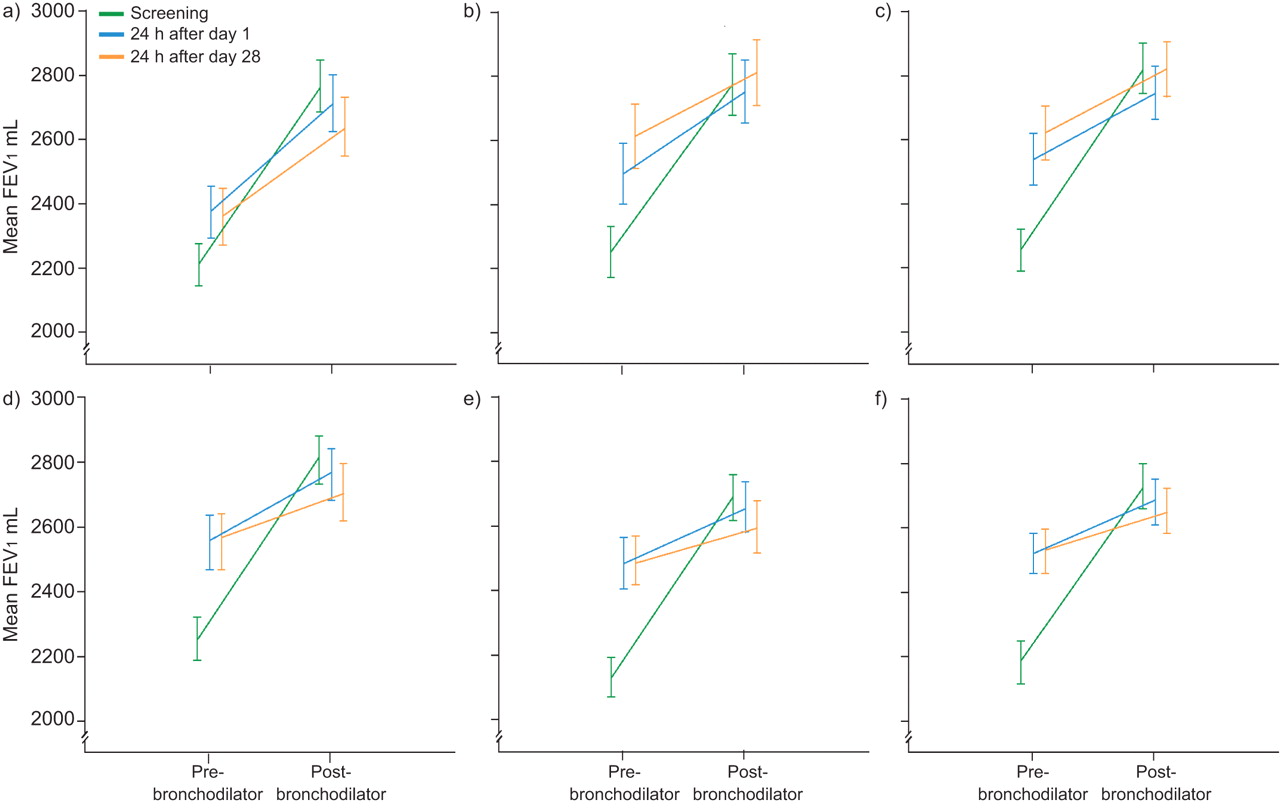
Comparable improvements in absolute FEV1 post-salbutamol at screening and on day 1 were observed in all treatment arms and this effect was not attenuated by 28 days of therapy with any dose of VI (fig. 5). There were no statistically significant differences between time points or active treatment and placebo (p>0.05; online supplementary table S1).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Response to salbutamol 24 h after administration of placebo or each dose of vilanterol trifenantate (VI) for 1 day or 28 days (absolute values) (intent-to-treat population). a) Placebo; b) VI 3 μg; (c) VI 6.25 μg; d) VI 12.5 μg; e) VI 25 μg; f) VI 50 μg. FEV1: forced expiratory volume in 1 s. Error bars represent standard errors.
The proportion of patients obtaining ≥200 mL and ≥12% increase from baseline in FEV1 (0–4 h) on days 1 and 28 increased with VI dose (online supplementary fig. S1). When measured over 0–24 h, >50% of patients in the 25 μg and 50 μg VI groups maintained the ≥200 mL and ≥12% improvement in FEV1 for most time points.
Safety and tolerability
The incidence of AEs during treatment with VI was not dose related and was similar to placebo (table 4). Moreover, no SAEs were reported in any treatment group. No deaths or hospitalisations were reported. Cough was reported by four patients (two 3 μg VI, one 12.5 μg VI, one 25 μg VI; all considered unrelated to treatment by the study investigator). There was a low incidence of LABA class-related AEs. Tremor was reported by two patients (both received 6.25 μg VI; one occurred on day 1 and one on day 6; one event was considered to be possibly treatment-related). Palpitations considered to be possibly treatment-related were reported by four patients (one placebo, two 3 μg VI, one 6.25 μg VI). One patient reported impaired glucose tolerance (12.5 μg VI; considered to be possibly treatment related) and one patient had increased blood glucose levels (50 μg VI; considered to be unrelated to treatment). No AEs of low potassium were reported. The incidence of asthma exacerbations was low and similar across treatment groups, with the greatest proportions reported for placebo (4%) and 3 μg VI (7%) compared with 0–3% in the other treatment groups.
No trends were observed for haematology, clinical chemistry, urinalysis values, or vital signs over time for any dose of VI, including glucose and potassium differences, and QTcF differences (online supplementary material, appendix S3). There was no evidence of a statistically significant difference in weighted mean change in pulse rate versus placebo 0–4 h after dosing for any group at any time point (mean differences from placebo -1.8 to +1.8 beats per minute (bpm)), except for the 50 μg VI group on day 28 (+2.2 bpm; p=0.047; which was below the predefined level of clinically relevant concern defined as 6 bpm).
DISCUSSION
This report presents clinical data for inhaled VI, administered once daily in the evening to asthma patients receiving maintenance ICS therapy. VI dose-dependently improved trough FEV1 (the primary end-point), showing a sustained duration of action of at least 24 h at doses ≥12.5 μg. Significant improvements were also observed on the end-points of trough FEV1 analysed by baseline FEV1, weighted mean (0–24 h) FEV1, morning and evening PEF, and symptom-free and rescue-free 24 h periods. All doses of VI were well tolerated.
The wide range of doses used in this study permit the dose–response profile of VI to be assessed. Doses were selected based on unpublished data from earlier trials, from which it was predicted that the 3 μg dose would have no or little effect and the 50 μg dose would likely cause maximal achievable bronchodilation. Indeed, a significant dose-related effect on lung function could be documented. This was further supported by the results showing that the 3 μg dose of VI resulted in no significant effect, the 12.5 μg dose caused intermediate effects, and the bronchodilator responses with 25 μg and 50 μg VI were more substantial. However, the two higher doses of VI caused similar improvement in lung function, suggesting that a maximal response to VI had been reached in this study population, representing the dose–response curve plateau for FEV1. Overall, slightly more pronounced effects were observed with VI 25 μg versus 12.5 μg, for example on end-points of morning and evening PEF, as well as symptom-free and rescue-free 24 h periods. The dose response was also assessed in the two strata; however, caution should be applied when interpreting the results of the individual strata as the study was not powered to detect treatment differences within each stratum. What is evident is the overlap in response to individual doses in each stratum. There is no indication of a different dose being optimal in upper or lower strata.
The maintained significant effect of VI on the primary end-point of trough FEV1, as measured 24 h after the previous dose, confirms a 24 h duration of effect of VI on lung function in asthma patients concomitantly being treated with maintenance ICS. The serial FEV1 profiles also provide evidence of the 24 h duration of VI at 12.5 μg doses or higher, as the offset of effect from peak to trough are parallel to placebo treatment. This time course suggests that VI exerts a prolonged bronchodilatory effect and, as such, could be included in a once-daily treatment regimen in combination with an ICS for asthma. However, further studies are needed in order to determine how the duration of bronchodilation with VI compares with that of the older LABAs salmeterol and formoterol [22–24].
Statistically significant increases in FEV1 were observed at the first time point measured (15 min after inhalation) and maximum effect was documented at 3–4 h on day 1, and within 1–2 h on day 28. Furthermore, the evidently sustained immediate bronchodilation of an inhaled short-acting β2-agonist during regular treatment with VI further argues against tolerance to the immediate bronchodilation of a β2-agonist used as rescue medication, as post-salbutamol FEV1values were similar between screening and day 28 in all VI treatment groups, despite the increase in pre-bronchodilator FEV1observed as early as day 1 with VI therapy. The current study was not designed to directly assess the detailed onset of action of VI or tolerance to the clinical effect at the level of the β2-receptor. However, the overall results argue that tolerance to the bronchodilator response of a short-acting β2-agonist is not apparent in this large asthma study, in which patients were concomitantly treated with ICS.
All doses of VI were well tolerated, with no SAEs reported in any treatment group, and AEs were not related to the dose of VI. Inhalation of β2-agonists is often associated with predictable effects such as tremor, hypokalaemia, increased heart rate and increased QTcF intervals, which were observed to a minor extent with VI in this study. A minimal, not clinically relevant, effect on pulse rate was observed (increase of 2 bpm) with the highest dose of VI (50 μg), which would also be expected with other inhaled β2-agonists at such a high dose. Our data support the overall conclusion that doses of VI up to 50 μg have limited side-effects. Clearly, the influence of VI on rare events such as asthma-related worsening or mortality [25, 26] cannot be addressed in a study of this nature and therefore must be monitored in future long-term trials.
In selecting the optimal dose of a therapy, the aim is to attain maximal efficacy at the lowest possible dose, to avoid side-effects, and to maintain highest possible therapeutic ratio. In the development of previous LABAs, increasing doses of salmeterol (50 μg, 100 μg and 200 μg) resulted in dose-related effects on lung function, but the highest dose produced significant effects on blood pressure, heart rate and tremor [27], making the two lower doses more suitable for further clinical development. In studies with formoterol, although 6 μg, 12 μg, and 24 μg showed incremental dose-related efficacy on FEV1[28], the lowest dose, which was effective and devoid of side-effects, was selected for further development, again to attain highest possible therapeutic ratio. These efficacy data, together with the absence of dose-related AEs in this study, suggests that 12.5 μg and/or 25 μg may be suitable doses for further development as part of a fixed-dose combination therapy in adult asthma patients.
This study was powered to determine the dose-related effect of VI on trough FEV1 and was sufficiently large to determine the frequency of recognised side-effects. The placebo effect on FEV1 was substantial, which is often seen in large parallel group studies in asthma. The reasons for the pronounced placebo effect are unclear, but may be related to the timing of the trough FEV1measurement, as evening lung function tends to be higher due to the circadian variation that is observed in asthma patients [22, 29, 30]. Another possible effect is the “trial effect” of patients in the placebo arm increasing adherence to their maintenance ICS therapy and thus achieving greater than expected lung function. Unfortunately, as no data were collected on adherence to maintenance therapy, this can only be postulated. Importantly, significant dose-related effects of VI could be documented despite this placebo response.
In conclusion, regular once-daily treatment with VI was well tolerated and resulted in a prolonged duration of bronchodilation of at least 24 h at doses of 12.5 μg and greater, with a favourable therapeutic ratio at doses of 12.5 μg and 25 μg, with the greatest benefit seen at the 25 μg dose. These findings in patients receiving maintenance ICS therapy suggest VI can be developed as a once-daily LABA in combination with a once-daily ICS for the treatment of asthma. Such once-daily combination therapies have the potential to improve adherence to therapy in patients taking long-term inhaled therapy.
Acknowledgments
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. We thank all patients who took part in the study, all of the investigators at the 88 centres, and S. Baggen (GlaxoSmithKline, Zeist, the Netherlands), who assisted in the management of the study and helped to address data queries. Editorial support in the form of development of a draft outline in consultation with the authors, development of a manuscript first draft in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing, and graphic services was provided by D. Cutler at Gardiner-Caldwell Communications, Macclesfield, UK and was funded by GlaxoSmithKline. Colour printing charges were paid for by GlaxoSmithKline.
Footnotes
This article has supplementary material available from www.erj.ersjournals.com
Support Statement
The study was funded by GlaxoSmithKline (study number B2C109575).
Clinical Trial
This study is registered at www.clinicaltrials.gov with identifier number NCT00600171.
Statement of Interest
Statements of interest for all authors of this manuscript can be found at www.erj.ersjournals.com/site/misc/statements.xhtml
- Received July 15, 2011.
- Accepted January 8, 2012.
- ©ERS 2012
REFERENCES










