





Abstract
Background The primary analysis of the INBUILD trial showed that in subjects with progressive fibrosing interstitial lung diseases (ILDs), nintedanib slowed the decline in forced vital capacity (FVC) over 52 weeks. We report the effects of nintedanib on ILD progression over the whole trial.
Methods Subjects with fibrosing ILDs other than idiopathic pulmonary fibrosis, who had ILD progression within the 24 months before screening despite management deemed appropriate in clinical practice, were randomised to receive nintedanib or placebo. Subjects continued on blinded randomised treatment until all subjects had completed the trial. Over the whole trial, mean±sd exposure to trial medication was 15.6±7.2 and 16.8±5.8 months in the nintedanib and placebo groups, respectively.
Results In the nintedanib (n=332) and placebo (n=331) groups, respectively, the proportions of subjects who had ILD progression (absolute decline in FVC ≥10% predicted) or died were 40.4% and 54.7% in the overall population (hazard ratio (HR) 0.66, 95% CI 0.53–0.83; p=0.0003) and 43.7% and 55.8% among subjects with a usual interstitial pneumonia (UIP)-like fibrotic pattern on high-resolution computed tomography (HRCT) (HR 0.69, 95% CI 0.53–0.91; p=0.009). In the nintedanib and placebo groups, respectively, the proportions who had an acute exacerbation of ILD or died were 13.9% and 19.6% in the overall population (HR 0.67, 95% CI 0.46–0.98; p=0.04) and 15.0% and 22.8% among subjects with a UIP-like fibrotic pattern on HRCT (HR 0.62, 95% CI 0.39–0.97; p=0.03).
Conclusion Based on data from the whole INBUILD trial, nintedanib reduced the risk of events indicating ILD progression.
Abstract
In patients with fibrosing ILDs other than IPF who had shown progression of ILD within the prior 2 years, events indicating further progression occurred frequently. Over a 16-month period, nintedanib reduced the risk of such events versus placebo. https://bit.ly/3yiZXnS
Introduction
Nintedanib, an intracellular inhibitor of tyrosine kinases, inhibits processes fundamental to the progression of lung fibrosis [1]. Randomised placebo-controlled trials demonstrated that nintedanib reduces the rate of progression of interstitial lung disease (ILD) over 52 weeks in patients with idiopathic pulmonary fibrosis (IPF) [2], systemic sclerosis-associated ILD (SSc-ILD) [3] and chronic fibrosing ILDs with a progressive phenotype [4], resulting in the regulatory approval of nintedanib for the treatment of these conditions.
The INBUILD trial enrolled subjects with chronic fibrosing ILDs other than IPF who met criteria for progression of ILD within the previous 2 years despite management deemed appropriate in clinical practice [4]. Over 52 weeks, nintedanib reduced the rate of decline in forced vital capacity (FVC) (mL per year) versus placebo by 57% in the overall population and by 61% in subjects with a usual interstitial pneumonia (UIP)-like fibrotic pattern on high-resolution computed tomography (HRCT) (the co-primary analysis populations) [4]. In subgroup analyses, no heterogeneity was detected in the effect of nintedanib versus placebo on the rate of FVC decline across diagnostic subgroups [5]. As subjects continued to receive blinded randomised treatment until the last subject had completed the trial, most subjects received trial medication for >52 weeks. Here, we report the effects of nintedanib versus placebo on time-to-event end-points assessing progression of ILD over the whole INBUILD trial.
Methods
Subjects
The design of the INBUILD trial (ClinicalTrials.gov: NCT02999178) has been described and the protocol is publicly available [4]. Briefly, eligible subjects had a fibrosing ILD other than IPF, diagnosed by the investigator according to their usual practice, reticular abnormality with traction bronchiectasis (with or without honeycombing) of >10% extent on HRCT, FVC ≥45% predicted and diffusing capacity of the lung for carbon monoxide (DLCO) 30– <80% predicted. Subjects met at least one of the following criteria for ILD progression within the 24 months before screening, despite management deemed appropriate in clinical practice: relative decline in FVC ≥10% predicted; relative decline in FVC 5– <10% predicted and worsened respiratory symptoms; relative decline in FVC 5– <10% predicted and increased extent of fibrosis on HRCT; or worsened respiratory symptoms and increased extent of fibrosis on HRCT. These criteria were determined by the investigator with no central adjudication. Subjects taking azathioprine, cyclosporine, mycophenolate mofetil, tacrolimus, rituximab, cyclophosphamide or oral glucocorticoids >20 mg·day−1 were not enrolled. Initiation of these medications was allowed after 6 months of the trial in cases of deterioration of ILD or autoimmune disease.
Trial design
Subjects were randomised to receive nintedanib 150 mg twice daily or placebo, stratified by fibrotic pattern on HRCT (UIP-like fibrotic pattern or other fibrotic patterns) based on central review [4]. For each subject, the trial consisted of two parts: Part A, which comprised 52 weeks of treatment, and Part B, a variable treatment period beyond week 52 during which subjects continued to receive blinded randomised treatment until all the subjects had completed the trial. Subjects who discontinued treatment were asked to attend all visits as planned, including an end-of-treatment visit and a follow-up visit 4 weeks later. Subjects who were still on treatment at the end of Part B were eligible to enter an open-label extension study (INBUILD-ON; ClinicalTrials.gov: NCT03820726). The final database lock took place after all subjects had completed the follow-up visit or had entered INBUILD-ON (figure 1).

INBUILD trial design [4]. R: randomisation 1:1 stratified by high-resolution computed tomography pattern (usual interstitial pneumonia-like fibrotic pattern or other fibrotic patterns). #: visits occurred every 16 weeks until end of treatment; ¶: first database lock took place after the last subject had completed the week 52 visit; +: final database lock took place after all patients had completed the follow-up visit or entered the open-label extension study (INBUILD-ON).
Analyses
In the overall population and in subjects with a UIP-like fibrotic pattern on HRCT, we assessed the effects of nintedanib on time to the following: absolute and relative declines in FVC ≥5% predicted, absolute and relative declines in FVC ≥10% predicted, death, ILD progression (absolute decline in FVC ≥10% predicted) or death, and acute exacerbation of ILD (defined in [4]) or death, and acute exacerbations of ILD were not adjudicated. Analyses were based on time to first event. Analyses were based on a log-rank test (stratified by UIP-like fibrotic pattern or other fibrotic patterns on HRCT in analyses in the overall population), and a Cox proportional hazards model, stratified by the same factor, was used to derive the hazard ratio (HR) and 95% confidence interval. Adverse events, reported irrespective of causality, in the overall population were coded using preferred terms in the Medical Dictionary for Regulatory Activities (MedDRA) and are presented descriptively. Analyses were based on subjects who received at least one dose of trial medication. Analyses were pre-specified except for the analyses of time to absolute and relative declines in FVC % predicted.
Results
Subjects
In the overall population, 663 subjects received nintedanib (n=332) or placebo (n=331); 412 subjects (62.1%) had a UIP-like fibrotic pattern on HRCT (n=206 in each treatment group). The baseline characteristics of the patient population have been described [4, 5]. In the overall population, the mean±sd age was 65.8±9.8 years, FVC was 69.0±15.6% predicted, DLCO was 46.1±13.6% predicted, and the most frequent ILD diagnoses were chronic hypersensitivity pneumonitis (26.1%) and autoimmune ILDs (25.6%) (supplementary table S1). Over the whole trial, mean±sd exposure to trial medication was 15.6±7.2 and 16.8±5.8 months in the nintedanib and placebo groups, respectively; 34.3% and 30.2% of subjects in these groups, respectively, prematurely discontinued trial medication. The median follow-up time for the time-to-event end-points was ∼19 months.
Time-to-event end-points
The proportions of subjects with an absolute decline in FVC ≥5% predicted were 65.4% and 79.5% in the nintedanib and placebo groups, respectively, in the overall population (HR 0.67, 95% CI 0.56–0.81; nominal p<0.0001) and 66.5% and 81.6%, respectively, in subjects with a UIP-like fibrotic pattern on HRCT (HR 0.64, 95% CI 0.51–0.80; nominal p<0.0001) (table 1). The proportions with a relative decline in FVC ≥5% predicted were 73.8% and 86.1% in the nintedanib and placebo groups, respectively, in the overall population (HR 0.71, 95% CI 0.60–0.84; nominal p<0.0001) and 73.8% and 86.4%, respectively, in subjects with a UIP-like fibrotic pattern on HRCT (HR 0.69, 95% CI 0.55–0.86; nominal p=0.0006) (table 1).
Time to absolute and relative declines in forced vital capacity (FVC) ≥5% predicted or ≥10% predicted using data up to the final database lock in the INBUILD trial
The proportions of subjects with an absolute decline in FVC ≥10% predicted were 34.3% and 48.3% in the nintedanib and placebo groups, respectively, in the overall population (HR 0.64, 95% CI 0.50–0.81; nominal p=0.0002) and 37.4% and 48.1%, respectively, in subjects with a UIP-like fibrotic pattern on HRCT (HR 0.69, 95% CI 0.51–0.93; nominal p=0.0138) (table 1). The proportions with a relative decline in FVC ≥10% predicted were 48.5% and 66.8% in the nintedanib and placebo groups, respectively, in the overall population (HR 0.63, 95% CI 0.51–0.77; nominal p<0.0001) and 49.0% and 68.0%, respectively, in subjects with a UIP-like fibrotic pattern on HRCT (HR 0.61, 95% CI 0.47–0.79; nominal p=0.0001) (table 1).
In the overall population, 36 subjects (10.8%) in the nintedanib group and 45 subjects (13.6%) in the placebo group died (HR 0.78, 95% CI 0.50–1.21; nominal p=0.26) (figure 2a). Among subjects with a UIP-like fibrotic pattern on HRCT, 25 subjects (12.1%) in the nintedanib group and 36 subjects (17.5%) in the placebo group died (HR 0.66, 95% CI 0.40–1.10; nominal p=0.11) (figure 2b).
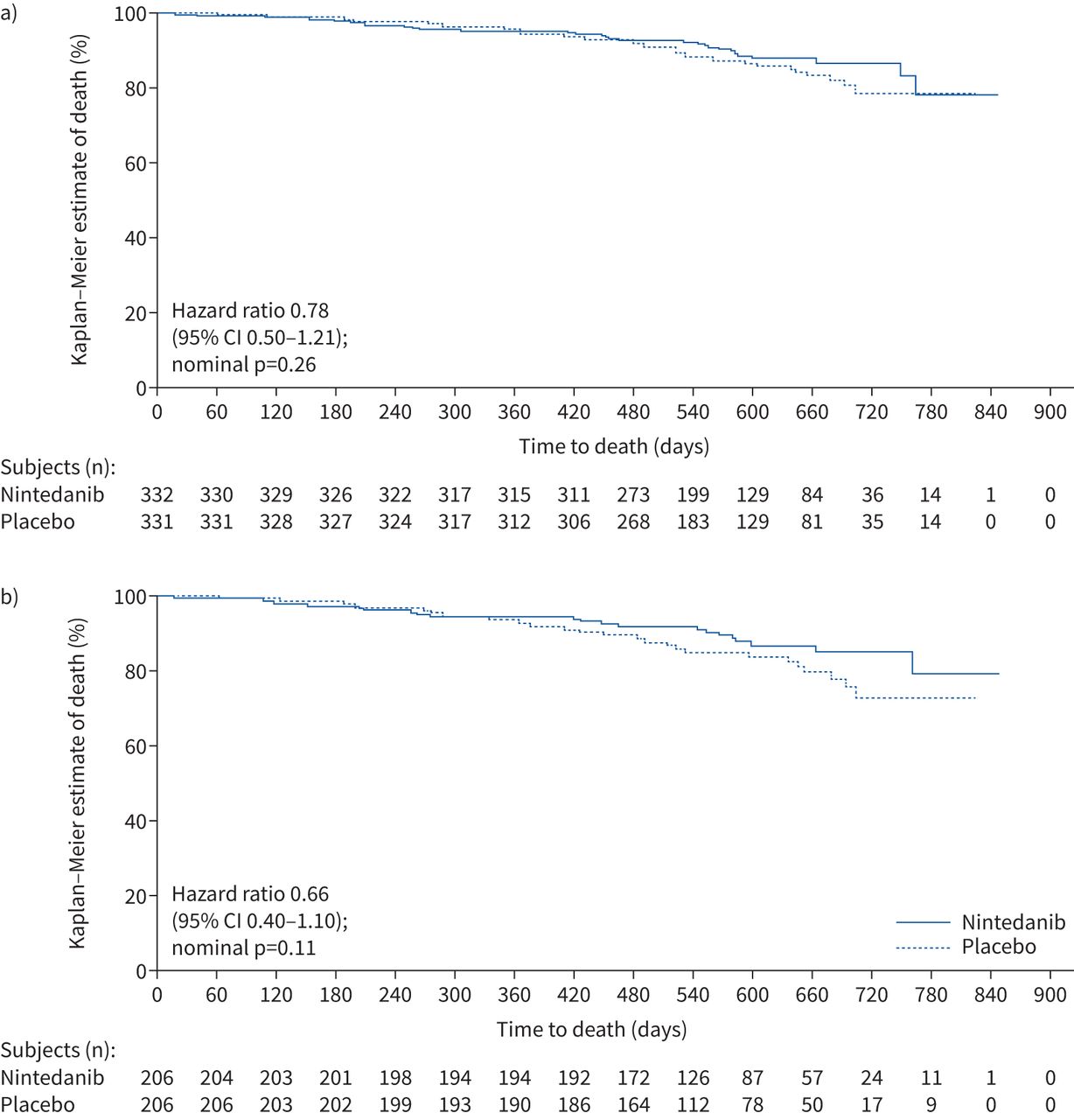
Kaplan–Meier estimates of time to death in a) the overall population and b) subjects with a usual interstitial pneumonia-like fibrotic pattern on high-resolution computed tomography in the INBUILD trial.
In the overall population, 134 subjects (40.4%) in the nintedanib group and 181 subjects (54.7%) in the placebo group had ILD progression (absolute decline in FVC ≥10% predicted) or died (HR 0.66, 95% CI 0.53–0.83; p=0.0003) (figure 3a). Among subjects with a UIP-like fibrotic pattern on HRCT, 90 subjects (43.7%) in the nintedanib group and 115 subjects (55.8%) in the placebo group had ILD progression or died (HR 0.69, 95% CI 0.53–0.91; nominal p=0.009) (figure 3b).

Kaplan–Meier estimates of time to progression of interstitial lung disease (ILD) or death in a) the overall population and b) subjects with a usual interstitial pneumonia-like fibrotic pattern on high-resolution computed tomography in the INBUILD trial.
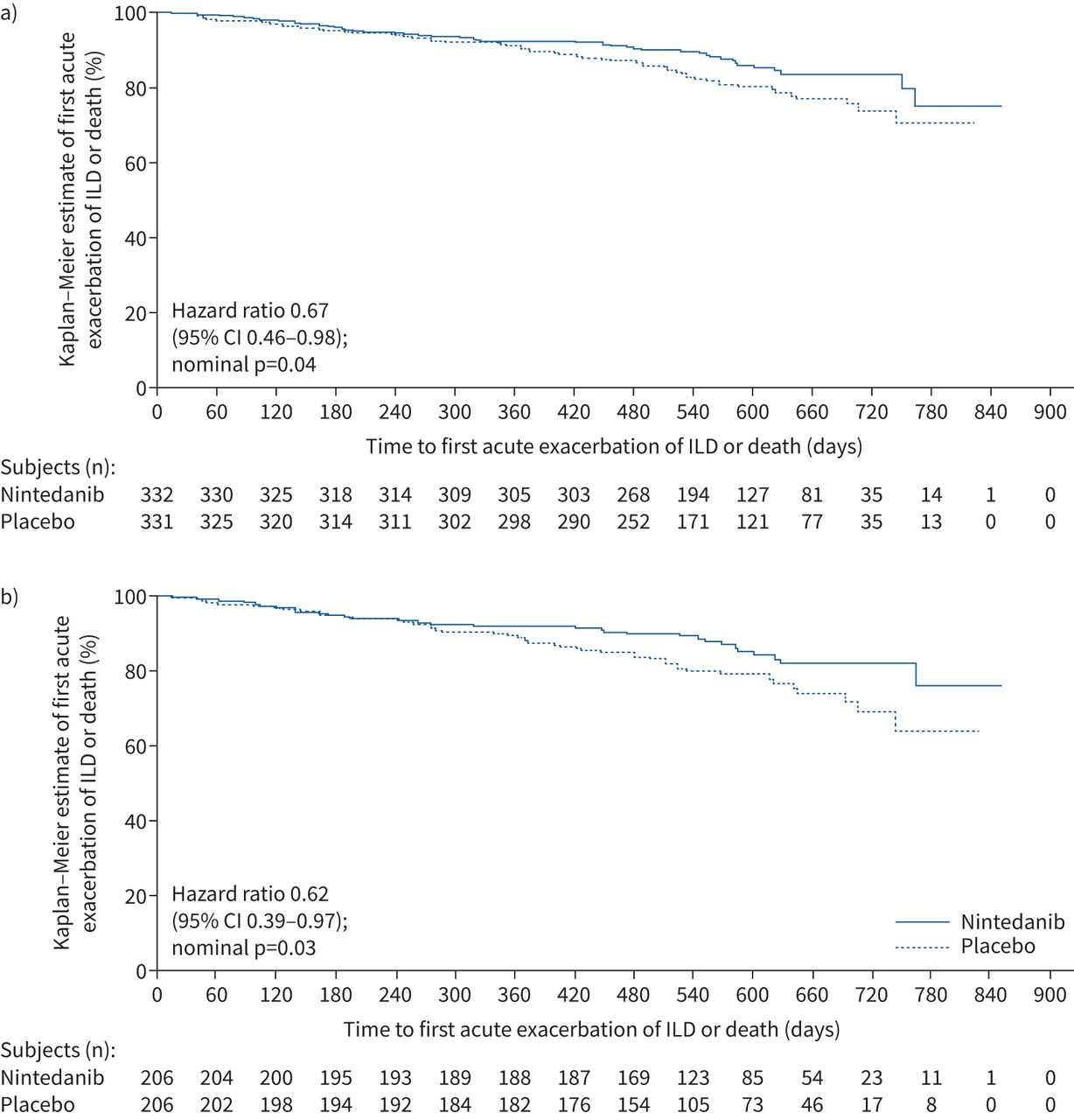
In the overall population, 46 subjects (13.9%) in the nintedanib group and 65 subjects (19.6%) in the placebo group had an acute exacerbation of ILD or died (HR 0.67, 95% CI 0.46–0.98; nominal p=0.04) (figure 4a). Among subjects with a UIP-like fibrotic pattern on HRCT, 31 subjects (15.0%) in the nintedanib group and 47 subjects (22.8%) in the placebo group had an acute exacerbation of ILD or died (HR 0.62, 95% CI 0.39–0.97; nominal p=0.03) (figure 4b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier estimates of time to first acute exacerbation of interstitial lung disease (ILD) or death in a) the overall population and b) subjects with a usual interstitial pneumonia-like fibrotic pattern on high-resolution computed tomography in the INBUILD trial.
Adverse events
In the overall population, diarrhoea was the most frequent adverse event (136.4 events per 100 patient-years in the nintedanib group and 23.0 events per 100 patient-years in the placebo group) (table 2). Nausea, vomiting, decreased appetite and weight decrease were also more frequently reported in the nintedanib group than in the placebo group (table 2). Adverse events led to permanent discontinuation of trial medication in 73 subjects (22.0%) and 48 subjects (14.5%) in the nintedanib and placebo groups, respectively. In the nintedanib and placebo groups, respectively, 131 subjects (39.5%) and 20 subjects (6.0%) had at least one dose reduction and 128 subjects (38.6%) and 41 subjects (12.4%) had at least one treatment interruption to manage adverse events. Serious adverse events occurred in 147 subjects (44.3%) and 164 subjects (49.5%) in the nintedanib and placebo groups, respectively (table 3).
Rates per 100 patient-years of the most frequent adverse events in the overall population of the INBUILD trial
Rates per 100 patient-years of the most frequent serious adverse events in the overall population of the INBUILD trial
Adverse events of increase in alanine aminotransferase (ALT) and increase in aspartate aminotransferase (AST) were more common in subjects treated with nintedanib than placebo (table 2). Based on laboratory tests, elevations in ALT and/or AST to ≥3 times the upper limit of the normal range were observed in 47 subjects (14.2%) in the nintedanib group and six subjects (1.8%) in the placebo group. For most of these cases (43 out of 47 subjects in the nintedanib group and six out of six subjects in the placebo group), these elevations returned to within the normal range spontaneously with continued treatment or after dose reduction or treatment interruption. One subject in each treatment group had concurrent elevations in liver enzymes and bilirubin that met criteria for Hy's law.
Discussion
These analyses, based on the longest duration of follow-up in the INBUILD trial (median ∼19 months), show that in patients with fibrosing ILDs that have progressed within the 2 years prior to enrolment, events indicating further progression of ILD occurred frequently. In the placebo group, 55% of subjects in the overall population and 56% of subjects with a UIP-like fibrotic pattern on HRCT had progression of ILD (absolute decline in FVC ≥10% predicted) or died. These findings support previous analyses showing that over 52 weeks, subjects who received placebo in the INBUILD trial had a clinical course similar to patients with untreated IPF, irrespective of the underlying ILD diagnosis or the fibrotic pattern on HRCT [6]. Our observations lend further support to the concept that there are patients with progressive fibrosing ILDs who can be managed based on their similarity in longitudinal clinical behaviour despite treatment considered appropriate in clinical practice [1, 7, 8].
While there is no established definition for progressive fibrosing ILD, a number of sets of criteria have been proposed based on a multifaceted approach including measurement of decline in FVC [8–10]. Our observations demonstrate that the inclusion criteria used in the INBUILD trial identified patients with progressive ILD who were at high risk of further progression and mortality. This does not mean that patients do not need to receive an accurate diagnosis of ILD (this remains essential to ensure that systemic and environmental causes of ILD can be addressed), but highlights the importance of prompt identification of patients whose ILD is progressing so that they can be managed appropriately.
A decline in FVC of >10% predicted has consistently been associated with mortality in studies across fibrosing ILDs [11–14]. Previous analyses of data from the INBUILD trial have shown that over 52 weeks, a relative decline in FVC of >10% predicted was associated with a more than three-fold increase in the risk of death, both in the overall population and in subjects with a UIP-like fibrotic pattern on HRCT [6]. Over the whole trial, nintedanib reduced the risk of a relative decline in FVC of ≥10% predicted by 37% compared with placebo in the overall population and by 39% in subjects with a UIP-like fibrotic pattern on HRCT.
Acute exacerbations are a recognised feature of the natural history of IPF [15]. While there is no established definition for an acute exacerbation of ILDs other than IPF, there is increasing evidence that acute exacerbations of ILD occur in a proportion of patients with such ILDs and are associated with high mortality [16, 17]. Over the whole INBUILD trial, 20% of subjects in the placebo group had an acute exacerbation of ILD or died. Nintedanib reduced the risk of an acute exacerbation or death by 33% versus placebo in the overall population and by 38% in those with a UIP-like fibrotic pattern on HRCT, similar to the reduction in the risk of acute exacerbations observed with nintedanib in patients with IPF [18].
The adverse event profile of nintedanib over the whole INBUILD trial was consistent with that observed over the first 52 weeks [4], with no new safety signals observed. The adverse event profile of nintedanib in the INBUILD trial was consistent with that observed in subjects with IPF [2, 19] and SSc-ILD [3, 20]. Gastrointestinal adverse events, particularly diarrhoea, were the most common adverse events. Elevations in liver enzymes were more common in subjects treated with nintedanib than placebo, but most normalised spontaneously or after dose reduction or treatment interruption.
In conclusion, analyses based on the data from the whole INBUILD trial show that in patients with chronic fibrosing ILDs that had shown progression within the 2 years prior to enrolment, events indicating further progression of ILD occurred frequently. Nintedanib slowed the progression of ILD, with an adverse event profile that was consistent with that observed over 52 weeks.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary table S1 ERJ-04538-2020.Supplement
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-04538-2020.Shareable
Acknowledgements
We thank the patients who participated in this trial. Writing support was provided by Elizabeth Ng and Wendy Morris (FleishmanHillard, London, UK), which was contracted and funded by Boehringer Ingelheim. The authors were fully responsible for all content and editorial decisions, were involved at all stages of development, and provided their approval on the final version. The authors did not receive payment for development of this article. Boehringer Ingelheim was given the opportunity to review this article for medical and scientific accuracy as well as intellectual property considerations.
Footnotes
This study is registered at ClinicalTrials.gov with identifier number NCT02999178. To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all authors access to all relevant material, including participant-level clinical study data, and relevant material as needed by them to fulfil their role and obligations as authors under the ICMJE criteria. Furthermore, clinical study documents (e.g. study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer-reviewed journal and if regulatory activities are complete and other criteria met per the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data: https://trials.boehringer-ingelheim.com. Prior to providing access, documents will be examined, and, if necessary, redacted and the data will be de-identified to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Clinical study reports and related clinical documents can also be requested via the link: https://trials.boehringer-ingelheim.com. All requests will be governed by a document sharing agreement. Bona fide, qualified scientific and medical researchers may request access to de-identified, analysable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a data sharing agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Researchers should use the https://trials.boehringer-ingelheim.com link to request access to study data.
A plain language summary of this article is available at: https://globalmedcomms.com/respiratory/flaherty/PLSforINBUILD.
Author contributions: K.R. Flaherty, A.U. Wells, V. Cottin, A. Devaraj, Y. Inoue, L. Richeldi, S.L.F. Walsh, M. Kolb, S. Stowasser, R. Schlenker-Herceg and K.K. Brown were involved in the design of the study. R-G. Goeldner was involved in data analysis. All authors were involved in the interpretation of the data and in the writing and critical review of the article.
Conflict of interest: K.R. Flaherty reports grants and personal fees from Boehringer Ingelheim and Roche/Genentech; and personal fees from Bellerophon, Blade Therapeutics, Celgene, FibroGen, Respivant, Sanofi Genzyme and Veracyte.
Conflict of interest: A.U. Wells reports personal fees from Blade Therapeutics, Boehringer Ingelheim and Roche.
Conflict of interest: V. Cottin reports personal fees and nonfinancial support from Actelion and Roche/Promedior; grants, personal fees and nonfinancial support from Boehringer Ingelheim; and personal fees from AstraZeneca, Bayer/MSD, Bristol-Myers Squibb, Celgene, Fibrogen, Galapagos, Galecto, Novartis, Sanofi and Shionogi.
Conflict of interest: A. Devaraj reports personal fees from Boehringer Ingelheim, Galapagos, Galecto, GlaxoSmithKline and Roche.
Conflict of interest: Y. Inoue reports other from Asahi Kasei, Boehringer Ingelheim, Galapagos, Roche, Savara, Inc., Shionogi and Taiho; and grants from the Japan Agency for Medical Research and Development and Ministry of Health, Labour and Welfare of Japan.
Conflict of interest: L. Richeldi reports grants and personal fees from Boehringer Ingelheim and Roche; and personal fees from Asahi Kasei, Biogen, Bristol-Myers Squibb, Celgene, CSL Behring, FibroGen, ImmuneWorks, Nitto, Pliant Therapeutics, Promedior, Respivant and Toray.
Conflict of interest: S.L.F. Walsh reports grants and personal fees from Boehringer Ingelheim; and personal fees from Bracco, Galapagos, Open Source Imaging Consortium, Roche and Sanofi.
Conflict of interest: M. Kolb reports grants and personal fees from Boehringer Ingelheim, Pieris, Prometic, Roche; personal fees from Algernon, AstraZeneca, GlaxoSmithKline, Indalo and Third Pole, Inc.; and other from the European Respiratory Society.
Conflict of interest: D. Koschel reports personal fees and nonfinancial support from Boehringer Ingelheim; and personal fees from Roche.
Conflict of interest: T. Moua has nothing to disclose.
Conflict of interest: S. Stowasser is an employee of Boehringer Ingelheim International GmbH.
Conflict of interest: R-G. Goeldner is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG.
Conflict of interest: R. Schlenker-Herceg is an employee of Boehringer Ingelheim Pharmaceuticals, Inc.
Conflict of interest: K.K. Brown reports personal fees and nonfinancial support from Boehringer Ingelheim; grants from the National Heart, Lung, and Blood Institute; personal fees from Biogen, Blade Therapeutics, DevPro Biopharma, Dispersol, Galapagos, Galecto, Huitai Biomedicine, Humanetics, Lifemax, Lilly, Pliant, Third Pole Therapeutics and Theravance; and other from the Open Source Imaging Consortium.
Support statement: The INBUILD trial was funded by Boehringer Ingelheim International GmbH. Funding information for this article has been deposited with the Crossref Funder Registry.
- Received December 16, 2020.
- Accepted July 24, 2021.
- Copyright ©The authors 2022.
This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions{at}ersnet.org
References










