





Adipose tissue plays an important role in regulating female fertility, owing to not only its energy stores but also the endocrine actions of secreted adipokines. As one of the adipokines, adiponectin is almost exclusively secreted from the fat, and its circulating concentration is paradoxically reduced in obesity. Although recent studies implied a purported positive role of adiponectin in ovarian functions, definitive in vivo evidence has been sorely lacking. We have consistently observed subfertility in female adiponectin null mice and therefore postulated a protective role of adiponectin in ovarian functions. Female adiponectin null mice displayed impaired fertility, reduced retrieval of oocytes, disrupted estrous cycle, elevated number of atretic follicles, and impaired late folliculogenesis. Analysis of their sera revealed a significant decrease in estradiol and FSH but an increase in LH and testosterone at proestrus. In addition, we found marked reduction of progesterone levels at diestrus, a significant decrease in LH receptor expression as well as in the number of GnRH immunoreactive neurons. Adiponectin deficiency also altered the peak concentrations of LH surge and led to lower expression of Cytochrome P450 family 11 subfamily A member 1 (P450scc), an enzyme critical for progesterone synthesis, as well as an increase in BCL2 associated X, apoptosis regulator and Insulin like growth factor binding protein 4 in atretic follicles. These physiological and molecular events were independent of insulin sensitivity. Thus, we have revealed a novel mechanism linking adiponectin and female fertility that entails regulation of reproductive hormone balance and ovarian follicle development.
Clinical and epidemiological studies have shown that body weight, particularly adiposity, is intimately involved in the regulation of female reproductive functions. For example, approximately 50% of polycystic ovarian syndrome (PCOS) women are overweight or obese (1), whereas underweight women with poor fat stores tend to have oligomenorrhea (2). Adipose tissue is an endocrine organ that can secrete a series of adipokines, one of which is adiponectin (also known as Acpr30,a 30-kDa protein member of the TNF-α family, adipocyte complement-related protein). Unlike other adipokines, circulating level of adiponectin is negatively correlated with body mass index and abdominal fat accumulation (3). Adiponectin regulates a variety of metabolic processes (4), including insulin sensitivity, lipid synthesis, antiatherogenesis, and antiinflammation. These actions mainly depend on its capability of binding to 2 widely distributed G protein-coupled receptors, AdipoR1 and AdipoR2, to exert pleiotropic effects of adiponectin (5).
Adiponectin and adiponectin receptors have been detected in granulosa cells (GCs) (6, 7), follicular fluid, oocytes, corpus luteum (8, 9), and theca cells (7), thus raising the possibility of adiponectin regulating ovarian functions. Indeed, a recent study has found that, albeit subject to the same in vitro fertilization procedure, the nonobese women who conceived had significantly higher serum adiponectin concentrations than the nonobese women who did not conceive (10). In large animal studies, the level of all circulating adiponectin isoforms has been found significantly reduced in the female pigs with subfertility (11). Several studies in humans have shown higher circulating levels of adiponectin in women than in men, indicating that ovarian hormones may in turn regulate adiponectin production (12, 13).
The studies about the potential roles of adiponectin in regulating ovarian functions have produced some controversial results. For example, a physiologically relevant dose of human recombinant adiponectin (10 μg/mL) has been shown to increase IGF-1-induced progesterone (P4) secretion in cultured hen preovulatory follicle GCs (7). Similarly, a separate study reported that adiponectin could stimulate the expression of the genes associated with follicle development and production of steroid synthetic enzymes through activating MAPK-ERK1/2 pathway in cultured porcine GCs (8). In contrast, a different group has reported the inhibitory effects of adiponectin on steroidogenesis in cultured bovine theca cells, particularly in the presence of LH and IGF-1 (14). In addition, a negative effect of adiponectin on GnRH secretion was also shown in GT1–7 cells (15). Thus, in vivo studies using adiponectin-deficient models are sorely needed to precisely define the functions of adiponectin in modulating ovarian follicle development and gonadal hormone production. In fact, although breeding the colonies of adiponectin knockout mice, we have consistently observed their low birth rate and small number of pups per birth. Using the female knockout model, we tested the hypothesis that adiponectin plays a protective role in female fertility and ovarian functions, particularly with respect to the regulation of follicle development and the activity of hypothalamic-pituitary-gonadal axis. Through such studies, we hope to establish a new molecular linkage between adiponectin and ovarian follicle development and to reveal a novel pathogenic basis for the women who suffer infertility.
Materials and Methods
Experimental animals
The adiponectin knockout mice in C57BL/6 background (kindly provided by Dr Philipp E. Scherer) were crossed with wild-type (WT) mice to generate F1 generations. The resulting heterozygotic mice were crossed to produce female litters carrying 3 genotypes of adiponectin: WT, heterozygotic mutation (APN+/−), and homozygotic mutation (APN−/−). All mice were bred in house and fed ad libitum using regular mouse chow. All procedures were carried out strictly in accordance to the guidelines by the Nanjing Medical University Animal Care and Use Committee.
Genotyping and DNA extraction
The adiponectin allele was screened by PCR amplification of tail genomic DNA samples isolated with a QIAGEN DNeasy kit (QIAGEN). The genotype of adiponectin (NM_009605) knockout mice were confirmed with 3 PCR primers, Reverse primer 5′-GGA CCC CTG AAC TTG CTT CAC-3′, Forward primerWT 5′-TTC AAT TCC CAG CAC CCA CAG TAA-3′, and Forward primerko 5′-GAA TGG GCT GAC CGC TTC CTC GTG-3′. The resulting PCR fragment(s) for WT adiponectin will show only 1 band of 228 bp, 2 bands of 228 and 480 bp for adiponectin heterozygotic deletion, and only 1 band of 480 bp for adiponectin homozygotic deletion.
Fertility assessments
Female mice (8 wk of age) of different genotypes were mated with proven fertile C57BL/6 male mice for a period of 28 weeks (16). The time intervals to each litter and litter size for each pair were recorded. The mean numbers of total litters per female in the 28-week period were also calculated.
Superovulation
Female mice at 32 days of age were induced to ovulate by ip injections of 7.5-IU pregnant mare serum gonadotropin (PMSG) (T = 0, where T, time), followed by 7.5 IU of human chorionic gonadotropin (hCG) (T = 48 h), and were killed 14 hours later (T = 62 h). After hormonal treatment, the mice were sacrificed and the ovaries with the oviduct ampullae dissected out. The cumulus mass surrounding ovulated oocytes were flushed out and dissociated from cumulus cells using 0.1% hyaluronidase in M2 medium. The number of oocytes was counted under the microscope.
Hematoxylin-eosin staining and serial sections
One of the bilateral ovaries was fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned on a microtome at a thickness of 5 μm. After stained with hematoxylin and eosin, all follicles composed of primordial, primary, preantral, antral, and atretic follicles were counted in every twelfth section with a distance of approximately 60 μm (17), 8–10 sections per ovary were evaluated. “Primordial” follicles contain an intact oocyte with a visible nucleolus surrounded by a single layer of fusiform-shaped GCs. “Primary” follicles consist of an intact enlarged oocyte with a visible nucleolus and a single layer of cuboidal GCs. “Preantral” follicles contain an oocyte with a visible nucleolus and more than 1 layer of GC or thecal cells with no visible antrum. “Antral” follicles possess more than 1 layer of GCs with a visible different size antrum. Follicles are considered atretic if they contain either a degenerating oocyte, disorganized GCs, pyknotic nuclei, shrunken GCs, or apoptotic bodies (18). Only the follicles with a visible nucleolus in the oocyte are counted to avoid duplication in the counting of the follicles (19). The total number of follicles comprise the primordial, primary, preantral, antral, and atretic follicles. All above follicle types are expressed as percentages of the total number of follicles. The other ovary was dissected and snap frozen in liquid nitrogen for further analysis.
Vaginal cytology and estrous examination
Vaginal smears were obtained by vaginal lavage between 8 and 10 am every day for a period of 35 days. After transferred to a dry glass slide, the smear mixture was air dried and stained with toluidine blue stain before evaluated under the optical microscope. Different stages of estrous cycle (diestrus, metestrus, estrus, and proestrus) were determined with a previously described protocol, and primarily based on the presence or absence of leukocytes, cornified epithelial, and nucleated epithelial cells (20).
Hormone determinations
Blood samples were obtained on days of diestrus and proestrus. All blood samples were stored at −80°C until use. None of the samples were previously thawed. FSH, estradiol (E2), P4, and testosterone (T) were analyzed with ELISA kits (Cusabio). The intra- and interassay coefficients of variation are both less than 15% for FSH, E2, P4, and T. LH was determined by ELISA (Jiancheng) with an intraassay coefficient of variation of less than or equal to 10% and an interassay coefficient of variation of less than or equal to 12%. Serum adiponectin and insulin concentrations were measured using mouse adiponectin and insulin ELISA kits (EZMADP-60K and EZRMI-13K; Millipore) with the sensitivity of 0.2 ng/mL, respectively. The intra- and interassay coefficients of variation for adiponectin were in the range of 3.8%–5.6% and 6.8%–10.8%, respectively. The intra- and interassay coefficients of variation for insulin were 3.22% and 6.95%, respectively.
Immunostaining
Mouse ovarian tissues were routinely fixed in 10% neutral-buffered formalin, dehydrated through an alcohol series, and subsequently embedded in paraffin. Five-micrometer sections were dewaxed in xylene, rehydrated, and subjected to incubation in 3% H2O2 to inhibit endogenous peroxidase. Antigen retrieval was performed in 10 mmol/L citric acid buffer (pH 6.0). The tissue sections were then blocked with 10% normal goat serum and incubated with primary antibodies overnight at 4°C. The primary antibodies were specifically against LH receptor (LHR) (Santa Cruz Biotechnology), AdipoR1 (Abcam), and AdipoR2 (Abcam). For the negative control, nonimmune rabbit serum was used in place of the LHR antibody. After visualization with liquid diaminobenzidine (DAB), the tissue sections were counterstained with hematoxylin. Immunohistochemical results were assessed by Image Proplus Analysis Software 6.0.
Immunocytochemistry for GnRH
Mice were anesthetized with 2% chloral hydrate (ip) and perfused (iv) via the ascending aorta with 0.01M PBS 50 mL followed by 4% paraformaldehyde 150 mL. Brain tissues were fixed overnight in 4% paraformaldehyde before transferred sequentially into 15% and 30% sucrose solution until they settled. Brains sections (40 μm thick) were sectioned coronally. GnRH immunohistochemistry was performed as previously described (21). Ten sections obtained from the level of the organum vasculosum of the laminae terminalis/preoptic area (POA) per brain were incubated in blocking solution for 60 minutes and then were treated with a mouse monoclonal anti-GnRH antibody (MAB5456; Millipore) overnight at 4°C. The sections were incubated with a biotinylated secondary antibody (A21210; Abbkine) before reacting with a standard avidin-biotin complex reaction with Ni-3, 3′5, 5′ DAB (Chromagen kit number PK6100; Vector Laboratories). All sections containing GnRH immunoreactive neurons were identified under a light microscope (Nikon E100) and were counted by 3 different observers. The number of GnRH+ cells in 10 sections of POA were added to obtain the value for each brain tissue.
Terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphates nick end-labeling (TUNEL) assay
Analysis of apoptotic cells (containing fragmented DNA) in the 3 groups was performed by the TUNEL method (In Situ Cell Death Detection kit POD; Roche Diagnostics) according to the manufacturer’s instructions. Paraffin-embedded tissue sections (4 μm) of mouse ovary were deparaffinized and treated with 30% H2O2/methanol to block endogenous peroxidase activity. After washing with PBS, the slides were incubated with 20-μg/mL proteinase K (Sigma) in 10mM Tris-HCl (pH 7.4) at room temperature for 30 minutes. After washing with PBS, the positive control sample (but not experimental samples) was treated with deoxyribonuclease (5 μg/mL) for 15 minutes at 4°C. Then, all tissue samples were rinsed in PBS and incubated with TUNEL reaction mixture at 37°C for 30 minutes. The tissue was treated in POD (converter peroxidase) for 30 minutes at 37°C. Subsequently, tissues were rinsed in PBS and incubated in DAB (POD substrate) for 10 minutes. The following steps were the same as the method of immunohistochemistry. The sections were observed under the microscope. Immunohistochemical results were assessed by Image-Pro Plus analysis software 6.0.
Measurement of LH surge
Ovaries were removed from the mice anesthetized by the ip injection of chloral hydrate (400 mg/kg). Briefly, the anesthetized animal’s ventral surface was shaved and cleaned, and the ovaries were dissected out through midline incisions in the skin and abdominal musculature. After removal of ovary, the muscle was sutured and the skin closed with sterile wound clips. Treatment with E2 was as following. 1) ovariectomized (OVX) mice received sc capsules (Silastic brand; Dow Corning) containing 0.625-mg E2 in sesame oil (Sigma-Aldrich Corp) at the time of OVX. 2) On the seventh day after OVX, the mice were given the sc injection of E2 dissolved in sesame oil at various doses of 0, 0.1, 5, 20, 100, 200, and 400 mg/kg. The mice were anesthetized with pentobarbital (3 mg/100 mL, ip) (22) before collecting blood samples (100 μL per time) at 1–5 pm by an automatic blood sampling system (DraQ; EICOM) as described previously (23). An equal volume (100 μL) of heparinized saline (CP Pharmaceuticals Ltd) was replaced through the atria cannula after each blood collection. Plasma was separated by centrifugation and stored at −80°C until assay.
Total RNA extraction, cDNA synthesis, and quantitative real-time PCR
Ovary RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacture’s protocol, total RNA (500 ng) was reverse transcribed to cDNA with the qRT Super Mix II (Vazyme). The mRNA level of genes (LHR, Bax, IGFBP-4, and CYP11A1) were measured using SYBR Green Master Mix (Applied Biosystems). The primers were, for LHR (M87571), 5′-CTTATACATAACCACCATACCAG-3′ and 5′-ATCCCAGCCACTGAGTTCATTC-3′; for Bax (L22472), 5′-ACCAGCTCTGAACAGATCATG-3′ and 5′-TGGTCTGGATCCAGACAAG-3′; for IGFBP-4 (NM_008341), 5′-CGTCCTGTGCCCCAGGGTTCCT-3′ and 5′-GAAGCTTCACC CCTGTCTTCCG-3′; for CYP11A1 (NM_019779 XM_975231), 5′-AGATCCCTTCCCCTGGTGACAATG-3′ and 5′-CGCATGAGAAGAGTATCGACGCATC-3′; and for GAPDH (NG_007073), 5′-GTACAAGGAGAACCAAGCAACGAC-3′ and 5′-GTGCCGTCTTTC ATTACACAGGAC-3′. Quantitative real-time PCR analysis was performed using a PCR System (StepOnePlus; Applied Biosystems) with GAPDH as the internal control. The PCR conditions were 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, 60°C for 20 seconds, and 72°C for 20 seconds. All reactions were performed in triplicates. A corrected Ct (δCt) was calculated by subtracting the GAPDH Ct from the sample Ct. Relative differences were calculated according to the formula: fold change = 2 (sample δCt − control δCt).
Western blot analysis of LHR
Ovarian samples were extracted with the Radio-Immunoprecipitation Assay buffer. Protein lysates were resolved by SDS-PAGE. Protein marker (PS11) was bought from GeneON. After transblotting, the membrane was incubated with the LHR antibody at a 1:1000 dilution followed by the secondary antibody at a 1:5000 dilution. ECL-PLUS Western blotting kit (Beyotime) was applied for detection. The intensity of the visualized band was analyzed using Image Lab 4.0 analysis software from Bio-Rad.
Statistical analysis
Data were analyzed by one-way ANOVA (followed by Tukey post hoc test) or t test (for 2 groups) using GraphPad Prism 5.0 (GraphPad Software). All results were expressed as mean ± SEM. P < .05 was regarded as indicative of statistical significance.
Results
Impaired fertility of adiponectin-deficient female mice
This study was initiated as a result of consistent observations that adiponectin null mice produced significantly lower number of pups relative to the WT mice after they were mated with each other (data not shown). To address this issue, we focused on the reproductive ability of the female mice. Adiponectin deficiency was confirmed through PCR analysis of tail gDNA samples (Supplemental Figure 1A), Western blot analysis, and ELISA of serum adiponectin (Supplemental Figure 1, B and C). Serum adiponectin concentrations in the heterozygous mice were approximately 50% of that in WT mice, and became undetectable in the adiponectin null mice.
The female mice of all genotypes were continuously mated with WT C57/BL6 males of 8–10 weeks of age for 28 weeks. Haploid deficiency of adiponectin resulted in a downward trend in the number of offspring compared with WT mice, although the difference did not reach statistical significance. Homozygotic mutation of adiponectin gene led to a significant reduction in litter sizes compared with control group (17.8 ± 3.7 vs 30.3 ± 3.5, P < .05) (Figure 1), suggesting a significant decline in female fertility in the knockout mice.
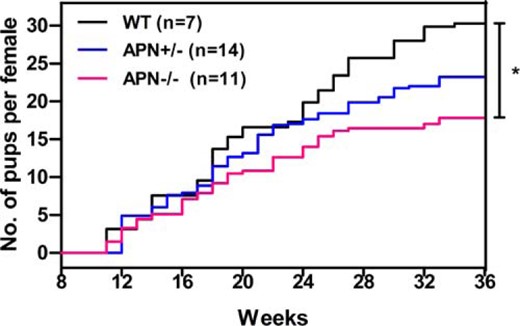
Adiponectin homozygotic mutation induces subfertility in female mice. Comparison of the cumulative number of pups per female mouse was observed in the 28-week period. WT female (black line, n = 7), APN+/− female (blue line, n = 14), and APN−/− female (red line, n = 11); *, P < .05, APN−/− vs WT mice. Data were represented as mean ± SEM. Significant differences were determined using one-way ANOVA analysis, followed by Tukey’s post hoc test.
Ovulation defects in adiponectin-deficient mice
To understand the cause(s) underlying fertility paucity as a result of adiponectin deficiency, we examined the development of ovarian follicles. Adiponectin haploid deficiency (APN+/−) led to significant reduction in the number of oocytes after ovulation induction of PMSG/hCG at the age of 32 days (21.0 ± 2.1 vs 28.8 ± 1.8 in WT, P < .05) (Figure 2). However, the decrease of ovulated oocytes became very pronounced in the null mice (APN−/−) (5.4 ± 1.6 vs 28.8 ± 1.8 in WT, P < .001) as well as contrast to that in the APN+/− mice (5.4 ± 1.6 vs 21.0 ± 2.1 in APN+/−, P < .001) (Figure 2). It should be noted that all the female mice selected for the ovulation studies had very similar body weight, thus making it unlikely that the observed decline in oocyte numbers was related to body weight changes or nutritional status (Supplemental Figure 2C).
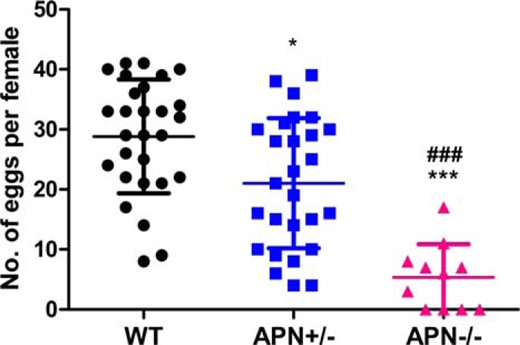
Ovulated oocytes are down sharply in the APN−/− mice. Thirty-two-day-old female mice of WT (n = 28), APN+/− (n = 27), and APN−/− (n = 11) were stimulated with PMSG/hCG, then oocytes were collected and calculated from the 3 genotype mice after superovulation; *, P < .05, APN+/− vs WT mice; ***, P < .001, APN−/− vs WT mice; ###, P < .001, APN−/− vs APN+/− mice. Data were represented as mean ± SEM. Significant differences were determined using one-way ANOVA analysis, followed by Tukey’s post hoc test.
Abnormal follicle development in adiponectin null mice
In light of the defective ovulation in adiponectin null mice, we analyzed the histology of ovaries from 4-month-old mice to evaluate the development of follicles (Figure 3A). In WT mice, all stages of follicles were clearly visible, and the oocytes bore complete zona pellucida and neatly lined GCs. However, female adiponectin null mice clearly showed a substantial increase in atretic follicles and markedly reduced number of preantral and antral follicles. Specifically, the percentages of atretic follicles in the homozygotic mutant mice was increased (19.3 ± 0.8% vs 8.5 ± 0.4% in WT, P < .001) (Figure 3B), which was in sharp contrast to that in the WT mice. The difference in atretic follicles between adiponectin haploid-deficient and null mice also reached statistical significance (19.3 ± 0.8% in APN−/− vs 13.0 ± 2.2% in APN+/−, P < .05) (Figure 3B). Figure 3B also showed that the percentages of preantral (13.6 ± 0.9% vs 23.4 ± 2.2% in WT, P < .01) and antral follicles (8.4 ± 0.5% vs 13.0 ± 0.5% in WT, P < .01) were reduced in adiponectin null mice. The percentages of primordial and primary follicles were essentially identical among all 3 genotypes. All of the ovaries selected in this experiment had similar weight (Supplemental Figure 2D). In addition, there were no changes in serum insulin levels relative to those of WT littermate controls (Supplemental Figure 2A). Glucose tolerance tests also revealed essentially the same glucose-disposal curves and insulin sensitivity (Supplemental Figure 2B). We also noted that adiponectin receptors (R1 and R2) were all expressed in theca cells, interstitial cells, and to a lesser extent in GCs and oocytes (Supplemental Figure 3), with approximately similar levels in WT and knockout mice.
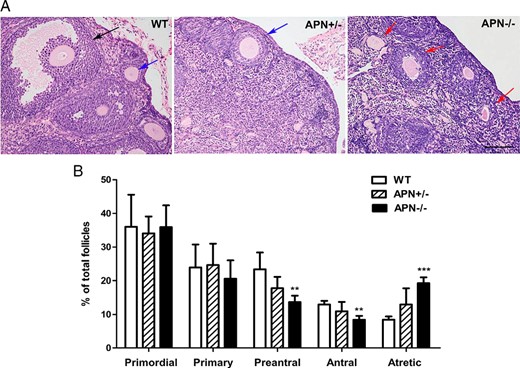
Morphological changes in ovaries of adiponectin homozygotic mutation mice. A, Histology of ovaries from WT, APN+/−, and APN−/− mice at 4 months of age was representatively photographed at magnification ×200. Scale bar, 100 μm. Antral follicle (black arrow), preantral follicle (blue arrow), atretic follicle (red arrow). B, Percentages of primordial, primary, preantral, antral, and atretic follicles were evaluated; **, P < .01’ ***, P < .001, APN−/− vs WT mice; #, P < .05; APN−/− vs APN+/− mice. Data were represented as mean ± SEM, n = 5. Significant differences were determined by one-way ANOVA, followed by Tukey’s post hoc test.
Abnormal estrous cycles in adiponectin-deficient mice
Because the estrous cycle of mice is closely associated with ovarian follicular development and hormonal changes, we performed daily vaginal smears to monitor estrous cycle and the duration in each stage of estrous cycle. The WT mice displayed 4 stages, including diestrus, metestrus, estrus, and proestrus, in every estrous cycle for 4–6 days. However, adiponectin-deficient (APN+/−, APN−/−) females exhibited ostensibly abnormal estrous cycles, which was particularly prominent in adiponectin null mice (Figure 4A). The number of estrous cycles of adiponectin null mice was significantly decreased during the 35 days of observation relative to WT (4.2 ± 0.2 vs 6.6 ± 0.2 cycles in WT, P < .001) (Figure 4B) and adiponectin heterozygotic mice (4.2 ± 0.2 vs 5.8 ± 0.2 cycles in APN+/−, P < .001) (Figure 4B). The heterozygotic females (APN+/−) spent longer days in diestrus (15.8 ± 1.3 vs 10.6 ± 0.5 d in WT, P < .05) (Figure 4C), whereas the null-deficient mice (APN−/−) presented significant changes in all 4 stages of estrous cycle that were characterized by extended periods of diestrus (22.4 ± 1.5 vs 10.6 ± 0.5 d in WT, P < .001) and shortened time of proestrus (5.0 ± 0.7 vs 8.6 ± 0.2 d in WT, P < .01), estrus (4.8 ± 0.7 vs 10.6 ± 0.5 d in WT, P < .001), and metestrus (2.8 ± 0.5 vs 5.6 ± 0.5 d in WT, P < .05) (Figure 4C). Even after comparing the results of 2 different genotypes, null (APN−/−) mice still spent significantly fewer days in proestrous (5.0 ± 0.7 vs 7.4 ± 0.6 d in APN+/−, P < .05) and estrous (4.8 ± 0.7 vs 8.0 ± 1.0 d in APN+/−, P < .05) phases and more days in diestrous phase (22.4 ± 1.5 vs 15.8 ± 1.3 d in APN+/−, P < .01) (Figure 4C) when compared with heterozygosis mutant (APN+/−) mice.
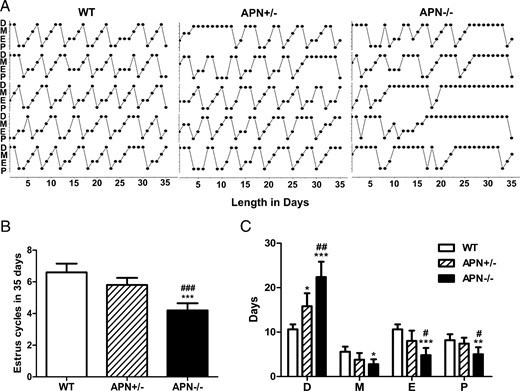
The estrous cycle is significantly interrupted in adiponectin null mice. A, Distribution of 4 stages of estrous cycle in 3 4-month-old genotype mice in 35 days; D, diestrus; M, metestrus; E, estrus; P, proestrus. B, Bar graphs show estrous cycles of the 3 groups in 35 days; ***, P < .001, APN−/− vs WT mice; ###, P < .001, APN−/− vs APN+/− mice. C, Length of each estrous cycle stage in WT, APN+/−, and APN−/− mice during 35 days; *, P < .05; **, P < .01; ***, P < .001, APN−/− vs WT mice; #, P < .05; ##, P < .01, APN−/− vs APN+/− mice. Data were represented as mean ± SEM; n = 5. Significant differences were determined by one-way ANOVA, followed by Tukey’s post hoc test.
Hormonal dysregulation in adiponectin-deficient mice
At proestrus, circulating levels of E2 (78.7 ± 11.6 vs 116.9 ± 6.9 pg/mL, P < .05) (Figure 5A) and FSH (10.4 ± 0.4 vs 12.3 ± 0.3 ng/mL, P < .05) (Figure 5B) were significantly decreased, whereas LH (5.4 ± 0.2 vs 4.4 ± 0.3 ng/mL, P < .05) (Figure 5C) levels were significantly increased in adiponectin null mice relative to the WT controls. However, the null mice (APN−/−) exhibited lower P4 levels than the WT at diestrus but not proestrus (13.8 ± 2.3 vs 23.5 ± 2.0 ng/mL, P < .05) (Figure 5D). Interestingly, a distinct increase of plasma T concentration was observed in APN−/− mice at proestrus (3.7 ± 0.9 vs 0.3 ± 0.3 ng/mL, P < .01) (Figure 5E). Contrary to the results at proestrus, the levels of E2, FSH, LH, and T in diestrus were very similar among all 3 genotypes (Figure 5, A–C and E). In addition, no difference in serum sex hormone-binding globulin levels was observed among the 3 genotypes at proestrus or diestrus (data not show).
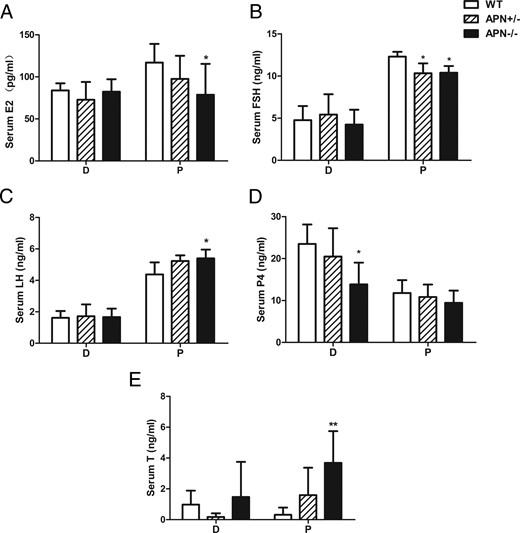
Adiponectin deletion affects the production of sex hormones. A–E, At 4 months of age, serum levels of E2, FSH, LH, P4, and T in WT, APN+/−, and APN−/− mice at D (diestrus) and P (proestrus); *, P < .05; **, P < .01, vs WT mice. Data were represented as mean ± SEM, n = 7. Significant differences were determined by one-way ANOVA, followed by Tukey’s post hoc test.
GnRH immunoreactive cells in adiponectin null mice
We further evaluated whether the drop of FSH was due to any potential defect of GnRH production. GnRH neurons were identified as brown, bipolar, and clear nuclei neurons that were present primarily in the medial POA (Figure 6A). At diestrus, the numbers of GnRH immunoreactive cells were similar among all 3 genotypes. In contrast, at proestrus, the number of GnRH+ neurons was prevented the increase relative to that of WT (117 ± 7.4 vs 158.4 ± 12.1, P < .05) (Figure 6B).
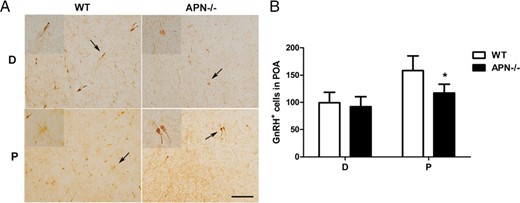
Influence of adiponectin deletion on GnRH immunoreactive neurons. A, Representative photographs of GnRH+ cells in POA of WT and APN−/− mice at 4 months of age. Scale bar, 100 μm (low magnification, ×200; high magnification, ×400). B, Bar graphs showed mean ± SEM of GnRH+ cells at D (diestrus) and P (proestrus); *, P < .05, vs WT mice; n = 7, significant differences were determined by t test.
Influence of adiponectin deficiency on LH surge
To further understand the impact of adiponectin deficiency on LH homeostasis, we evaluated whether the null mice could effectively mount an LH surge when facing an estrogen-initiated positive feedback. OVX and estrogen treatment were performed on the null and control mice (see Materials and Methods). A distinct peak of plasma LH was observed during 4–5 pm in all 3 genotype mice. Although the basal level of plasma LH was elevated at 2–3 pm in APN−/− mice (4.5 ± 0.4 vs 2.7 ± 0.4 ng/mL in WT and 5.0 ± 0.5 vs 3.0 ± 0.2 ng/mL in WT, P < .05) (Figure 7), the amplitude and the absolute concentration of LH were significantly decreased relative to the WT controls at peak (6.8 ± 0.4 vs 9.5 ± 0.9 ng/mL in WT, P < .05) (Figure 7).
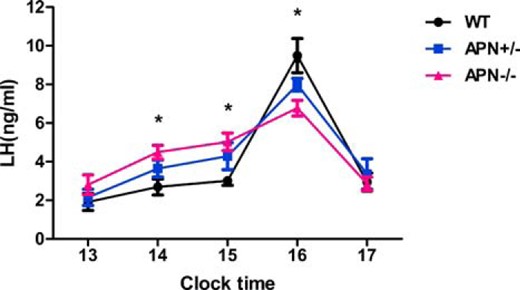
Influence of adiponectin deletion on LH surge. Hourly changes in plasma LH levels were measured at proestrus in the 3 genotypes at 4 months of age; *, P < .05, APN−/− vs WT mice. Data were expressed as mean ± SEM, n = 4. Significant differences were determined by one-way ANOVA, followed by Tukey’s post hoc test.
Reduction of LHR expression in the ovary of adiponectin null mice
To understand whether there was a functional change of LH in the event of adiponectin deficiency, we further investigated the expression of LHR in the ovarian tissues. As expected, strong LHR immunoreactivity was detected in mature GCs, theca-interstitial (TI) cells of the WT mice (Figure 8A). In the null mice, however, such expression was significantly decreased (P < .01) (Figure 8B). To further confirm this observation, we quantified the mRNA levels of LHR in the ovarian tissues and found its expression sharply lower in the heterozygotic and homozygotic mutant mice than in WT (0.17 ± 0.09 vs 0.97 ± 0.14 in WT, 0.05 ± 0.02 vs 0.97 ± 0.14 in WT, P < .001) (Figure 8C). In addition, Western blot analysis also showed that the expression of LHR in the homozygotic mutant mice was lower than in WT mice (P < .05) (Figure 8, D and E).
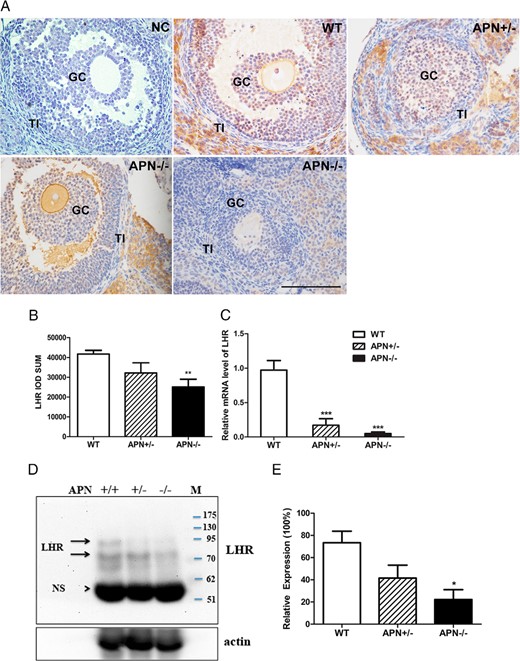
LHR expression is reduced in the adiponectin-deficient ovary. A, Immunohistochemical localization of LHR protein in the ovarian follicles of WT, APN+/−, and APN−/− mice at 4 months of age. Negative control (NC) is nonimmune serum control of LHR. LHR was detected in TI cells, GCs, of WT and APN+/− but was only weakly expressed or undetectable in TI cells and GCs of APN−/−. Original magnification, ×400; scale bar, 100 μm. B, Quantification of LHR-positive cells in the ovary; **, P < .01, APN−/− vs WT mice. C, Relative mRNA levels of LHR in ovarian tissue of the 3 genotypes; ***, P < .001, vs WT mice. Data were expressed as mean ± SEM, n = 7. Significant differences were determined by one-way ANOVA analysis, followed by Tukey’s post hoc test. D, Western blot analysis of LHR expression in the ovaries of WT, APN+/−, and APN−/−. E, Quantification of LHR expression in the 3 groups; *, P < .05, APN−/− vs WT mice. Owing to various glycosylation modifications, LHR appeared from 85–95 kDa (24), indicated by the 2 arrows’ NS, nonspecific band; M, marker.
Dysregulation of gene expression in adiponectin-deficient mice
To explore the potential molecular mechanisms underlying the fertility defect in female adiponectin null mice, several genes involved in ovarian oocyte maturation, ovulation and steroidogenesis were examined by quantitative PCR analysis (Figure 9, A–C). Accordingly, the expression of Bax was significantly elevated in the ovaries of the null mice relative to WT (2.6 ± 0.2 vs 1.1 ± 0.2 in WT, P < .001) (Figure 9A) and APN+/− mice (2.6 ± 0.2 vs 1.4 ± 0.2 in APN+/−, P < .01) (Figure 9A). Importantly, the expression level of IGFBP-4, often associated with atretic follicles, was markedly elevated relative to that in the WT mice (2.0 ± 0.1 vs 1.0 ± 0.1 in WT, P < .01) (Figure 9B). Furthermore, CYP11A1, a protein responsible for cholesterol side chain cleavage during the production of pregnenolone, was significantly lower in adiponectin null mice than in the WT littermates (0.5 ± 0.1 vs 1.1 ± 0.1 in WT, P < .01) (Figure 9C). Furthermore, TUNEL assay revealed that the number of apoptotic GCs in atretic follicles in adiponectin null mice was increased compared with that in WTs (P < .05) (Figure 9, D and E).
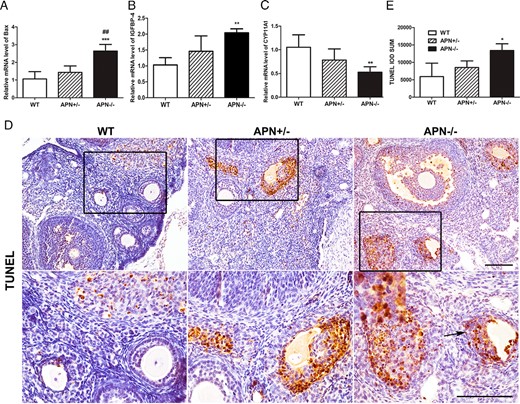
Gene expression profile is changed in the ovaries of adiponectin homozygotic mutation mice at 4 months of age. A–C, The expression of Bax (A), IGFBP-4 (B), and CYP11A1 (C) were measured by real-time PCR, normalized to GAPDH mRNA; **, P < .01; ***, P < .001, APN−/− vs WT mice; ##, P < .01, APN−/− vs APN+/− mice. Data were represented as mean ± SEM, n = 7 per genotype. D, Sections illustrated the typical low staining in healthy follicles in WT and APN+/− mice and high levels of TUNEL staining in the atretic follicles in APN−/− mice. Inset indicated TUNEL-positive cells. Scale bar, 100 μm (low magnification, ×200; high magnification, ×400). E, Quantification of TUNEL-positive cells in the ovary of the 3 genotype mice; *, P < .05, APN−/− vs WT mice. Data were represented as mean ± SEM, n = 4 per genotype. Significant differences were determined by one-way ANOVA, followed by Tukey’s post hoc test.
Discussion
Using adiponectin knockout mouse model, we have revealed that adiponectin deficiency could lead to decreased fertility in female mice with significantly reduced number of pups per female. In both the haploid (APN+/−) and null mutant (APN−/−) mice, the estrous cycle was disrupted, displaying significantly shortened proestrous/estrous phase and extended diestrous phase. The number of ovulated oocytes was down sharply in the null mice after PMSG/hCG treatment, suggesting that high basal adiponectin levels are associated with strong ovarian response to gonadotropin stimulation (25). The numbers of preantral and antral follicles in the null mutant mice were significantly reduced with a corresponding increase in atretic follicles. Adiponectin appears to play an important role in regulating the activity of hypothalamic-pituitary-gonadal axis, because its deficiency disrupts FSH and LH secretion as well as LH surge. Adiponectin mutation also causes significant reduction in GnRH immunoreactive neurons, which helps explain the disrupted estrous cyclicity and ovarian functions.
Adiponectin is an abundant circulating adipokine, playing pivotal roles in alleviating the pathogenesis of type 2 diabetes (26), cardiovascular disorders (27), inflammation (28), and potentially tumorigenesis (29). To our knowledge, the study described here represents the first genetic evidence to firmly establish the protective effect of adiponectin on folliculogenesis. Consistent with our results, previous studies have already reported the correlation between the reduction of all adiponectin isoforms with the subfertility of female pigs (11). Adiponectin incubation with cultured bovine GCs could stimulate the expression of genes critical for porcine ovulatory process (8) and IGF-1-induced proliferation (9). In addition, adiponectin has also been reported to enhance IGF-1-induced P4 secretion in hen preovulatory follicle GCs (7). Interestingly, such protective effect on reproduction may not be restricted to the females only. A recent study has implied that adiponectin could regulate sperm capacitation in the cultured sperms of Holstein bulls (30).
Several mechanisms may have contributed to the observed ovarian defects, including the dysregulation of gonadotropin hormones (and/or their receptors) as well as sex steroids. Follicular development is a highly organized and complex process, which is controlled by multiple hormones, such as E2, FSH, LH, and P4 (31). FSH controls follicular growth and E2 production, and LH stimulates ovulation and luteinization (32, 33). Follicle-Stimulating Hormone receptor-deficient females are sterile due to the arrest of folliculogenesis before antral follicle formation (34). Similarly, LHR-deficient mice are infertile due to arrested follicular development at late antral stage accompanied by increased number of atretic follicles (35). Already, adiponectin deficiency led to significant decrease in circulating levels of FSH and E2 in proestrous phase as well as P4 in diestorus phase. Although their baseline LH concentrations were modestly elevated, the estrogen-initiated LH surge was markedly attenuated in the adiponectin-deficient mice after oophorectomy. Furthermore, a reduction of ovarian expression of LHR was observed in the null mice. Together, these results indicate that the ovarian defects as a result of adiponectin deficiency is likely due to the decline of FSH and the totality of LH functions. These results are also in line with a recent report that adiponectin treatment in swine ovaries causes an elevation in GnRH-stimulated secretion of FSH (36). We did notice that although LHR expression was significantly reduced, there were still some dominant follicles ovulated in APN−/− mice (see Figure 2). We speculated that the residual LHR expression in the follicular cells might still exert some functions in follicle development. Alternatively, the development of antral follicles could also be boosted by FSH in the context of low LH activity, because FSH has been known to rescue antral follicles from atresia (37). Because pulsatile neuronal release of GnRH in the hypothalamus stimulates the secretion of LH and FSH from gonadotropes in the anterior pituitary (38), the sharply reduced GnRH immunoreactive cells might have also impaired the secretion of GnRH and, subsequently, the LH surge and FSH secretion in the context of adiponectin deficiency.
P4 production is a complex interplay of hormonal systems involving theca cells and GCs, as well as the actions of both LH and FSH (39). We found that the decrease of P4 levels was associated with the reduction of CYP11A1 expression. In GCs, FSH can stimulate the expression of CYP11A1 (40) that is best known for its catalysis of cholesterol to produce pregnenolone (41). Our result is also consistent with a recent report that the synthesis of P4 could be stimulated by adiponectin in rat ovary in vitro (42). The reduced FSH activity and the sharply reduced LHR expression in adiponectin null mice are also possible mechanisms underlying the drop of P4 concentrations. The combined effects of E2 and P4 reduction dictates the disruption of estrous cycle, which is exactly the phenotype observed in adiponectin-deficient mice.
Follicular atresia in the ovary is tightly regulated by proapoptotic factors, eg, Bax, BCL2 associated agonist of cell death, and BH3 interacting domain death agonist, as well as IGFBPs (IGFBP-2, IGFBP-4, and IGFBP-5) (43, 44). Adiponectin deficiency caused a significant increase of apoptosis in GCs, which was associated with a corresponding increase in the expression of apoptosis-related factors, Bax and IGFBP-4, in the atretic follicles, supporting a protective role of adiponectin in GCs from apoptosis through suppression of proapoptotic protein expression.
Collectively, our results revealed that adiponectin deficiency affected female reproduction, folliculogenesis, and steroidogenesis. Unlike humans, mice have a high reproductive capability by generating multiple babies upon delivery. Thus, we reason that maintaining a high circulating adiponectin concentration is likely even more critical for female fertility in humans than in rodents. Interestingly, the increase of LH and T and the decrease of FSH levels in adiponectin null mice are also reminiscent of the widely reported elevations of LH to FSH ratios and T in PCOS patients (45). Although obesity is a major risk factor for PCOS and impaired fecundity (46, 47), Mirza et al has also demonstrated that the reduction of adiponectin level was independently associated with the risk of PCOS (48). We have shown that insulin resistance and body weight change were unlikely the contributing factors for the observed ovarian dysfunction in the adiponectin null mice maintained on the regular diet. Therefore, we propose that the reduced adiponectin activity contributes independently to the lower ovarian response to gonadotropin stimulation for women undergoing in vitro fertilization and that the subfertility of PCOS patients and obese women is due at least in part to decreased adiponectin via the potential mechanisms described here. The diagnostic as well as therapeutic potential of adiponectin should be further evaluated for infertile women.
Appendix
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| m-Adiponectin antibody | SinoGen, SGp2018 | Mouse; polyclonal | 1:1000 | ||
| Mouse anti-GnRH monoclonal antibody | Millipore, MAB5456 | Mouse; monoclonal | 1:1000 | ||
| LHR antibody (H-50) | Santa Cruz Biotechnology, sc-25828 | Mouse; polyclonal | 1:200 | ||
| Antiadiponectin receptor 1 antibody [EPR6626] | Abcam, ab126611 | Mouse; monoclonal | 1:50 | ||
| Antiadiponectin receptor 2 antibody | Abcam, ab77612 | Mouse; polyclonal | 1:50 | ||
| Goat antimouse IgG, (H + L), peroxidase conjugated | Thermo, OI192080 | Goat | 1:1000 | ||
| Goat antirabbit IgG, (H + L), peroxidase conjugated | Thermo, OG188649 | Goat | 1:1000 | ||
| Biotin, goat antimouse IgG | Abbkine, A21210 | Goat | 1:1000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| m-Adiponectin antibody | SinoGen, SGp2018 | Mouse; polyclonal | 1:1000 | ||
| Mouse anti-GnRH monoclonal antibody | Millipore, MAB5456 | Mouse; monoclonal | 1:1000 | ||
| LHR antibody (H-50) | Santa Cruz Biotechnology, sc-25828 | Mouse; polyclonal | 1:200 | ||
| Antiadiponectin receptor 1 antibody [EPR6626] | Abcam, ab126611 | Mouse; monoclonal | 1:50 | ||
| Antiadiponectin receptor 2 antibody | Abcam, ab77612 | Mouse; polyclonal | 1:50 | ||
| Goat antimouse IgG, (H + L), peroxidase conjugated | Thermo, OI192080 | Goat | 1:1000 | ||
| Goat antirabbit IgG, (H + L), peroxidase conjugated | Thermo, OG188649 | Goat | 1:1000 | ||
| Biotin, goat antimouse IgG | Abbkine, A21210 | Goat | 1:1000 |
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| m-Adiponectin antibody | SinoGen, SGp2018 | Mouse; polyclonal | 1:1000 | ||
| Mouse anti-GnRH monoclonal antibody | Millipore, MAB5456 | Mouse; monoclonal | 1:1000 | ||
| LHR antibody (H-50) | Santa Cruz Biotechnology, sc-25828 | Mouse; polyclonal | 1:200 | ||
| Antiadiponectin receptor 1 antibody [EPR6626] | Abcam, ab126611 | Mouse; monoclonal | 1:50 | ||
| Antiadiponectin receptor 2 antibody | Abcam, ab77612 | Mouse; polyclonal | 1:50 | ||
| Goat antimouse IgG, (H + L), peroxidase conjugated | Thermo, OI192080 | Goat | 1:1000 | ||
| Goat antirabbit IgG, (H + L), peroxidase conjugated | Thermo, OG188649 | Goat | 1:1000 | ||
| Biotin, goat antimouse IgG | Abbkine, A21210 | Goat | 1:1000 |
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| m-Adiponectin antibody | SinoGen, SGp2018 | Mouse; polyclonal | 1:1000 | ||
| Mouse anti-GnRH monoclonal antibody | Millipore, MAB5456 | Mouse; monoclonal | 1:1000 | ||
| LHR antibody (H-50) | Santa Cruz Biotechnology, sc-25828 | Mouse; polyclonal | 1:200 | ||
| Antiadiponectin receptor 1 antibody [EPR6626] | Abcam, ab126611 | Mouse; monoclonal | 1:50 | ||
| Antiadiponectin receptor 2 antibody | Abcam, ab77612 | Mouse; polyclonal | 1:50 | ||
| Goat antimouse IgG, (H + L), peroxidase conjugated | Thermo, OI192080 | Goat | 1:1000 | ||
| Goat antirabbit IgG, (H + L), peroxidase conjugated | Thermo, OG188649 | Goat | 1:1000 | ||
| Biotin, goat antimouse IgG | Abbkine, A21210 | Goat | 1:1000 |
Acknowledgments
We thank Dr Philipp E. Scherer for kindly providing adiponectin knockout mice and Dr Jing Li and Dr Jie Wu (Nanjing Medical University) for their technical assistance and critical reading of this manuscript.
Author contributions: L.C., A.Z., and F.L. designed the experiments. L.C., H.S., Yan Jin, X.L., J.P., Y.La., Y.Li., YaJ., and G.R. conducted the experiments and analyzed the data. L.C., H.S., A.Z., and F.L. wrote the manuscript.
This work was supported by grants from the National Program on Key Basic Research Project of China (973 Program) (2013CB945202) (to A.Z. and F.L.), the National Natural Science Foundation of China (81630021 and 81170780 to A.Z., 81372798 to F.L., and 81200570 to X.L.), the PhD Programs Foundation of Ministry of Education of China (20113234110005) (to A.Z.), Scientific Support Program of Jiangsu Province (BE2012756) (to A.Z.), Natural Science foundation of Jiangsu Province of China (BK20130059 to A.Z. and 2011766 to X.L.), and Jiangsu University Scientific Research Projects (12kJB310006) (to X.L.).
Disclosure Summary: The authors have nothing to disclose.
Abbreviations
- Adipo R
adiponectin receptor
- Bax
BCL2 associated X, apoptosis regulator
- Ct
threshold cycle
- CYP11A1
Cytochrome P450 family 11 subfamily A member 1
- DAB
diaminobenzidine
- GC
granulosa cell
- E2
estradiol
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- hCG
human chorionic gonadotropin
- IGFBP-4
insulin-like growth factor binding protein 4
- LHR
LH receptor
- OVX
ovariectomy
- P4
progesterone
- PCOS
polycystic ovarian syndrome
- PMSG
pregnant mare serum gonadotropin
- POA
preoptic area
- T
testosterone
- TI
theca-interstitial
- TUNEL
terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphates nick end-labeling
- WT
wild type.
References
Author notes
L.C., H.S., Yan Jin, and X.L. contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}