

Abstract
The neurodegenerative tauopathies are a clinically diverse group of diseases typified by the pathological self-assembly of the microtubule-associated tau protein. Although tau nitration is believed to influence the pathogenesis of these diseases, the precise residues modified, and the resulting effects on tau function, remain enigmatic. Previously, we demonstrated that nitration at residue Tyr29 markedly inhibits the ability of tau to self-associate and stabilize the microtubule lattice (Reynolds et al., 2005b, 2006). Here, we report the first monoclonal antibody to detect nitration in a protein-specific and site-selective manner. This reagent, termed Tau-nY29, recognizes tau only when nitrated at residue Tyr29. It does not cross-react with wild-type tau, tau mutants singly nitrated at Tyr18, Tyr197, and Tyr394, or other proteins known to be nitrated in neurodegenerative diseases. By Western blot analysis, Tau-nY29 detects soluble tau and paired helical filament tau from severely affected Alzheimer's brain but fails to recognize tau from normal aged brain. This observation suggests that nitration at Tyr29 is a disease-related event that may alter the intrinsic ability of tau to self-polymerize. In Alzheimer's brain, Tau-nY29 labels the fibrillar triad of tau lesions, including neurofibrillary tangles, neuritic plaques, and, to a lesser extent, neuropil threads. Intriguingly, although Tau-nY29 stains both the neuronal and glial tau pathology of Pick disease, it detects only the neuronal pathology in corticobasal degeneration and progressive supranuclear palsy without labeling the predominant glial pathology. Collectively, our findings provide the first direct evidence that site-specific tau nitration is linked to the progression of the neurodegenerative tauopathies.
Introduction
Alzheimer's disease (AD) and other non-AD tauopathies (NADTs) are heralded by a remarkable degree of clinical and pathological heterogeneity. Clinically, AD manifests as an insidious deterioration of mental function, affecting memory and one or more cognitive domains (Morris et al., 1989). Pathologically, the signatures of postmortem AD brain include neurofibrillary tangles (NFTs), neuritic plaques, and neuropil threads. In early stage disease, these lesions heavily populate the entorhinal cortex and hippocampus (HP) (Braak and Braak, 1991). In advanced disease, tau pathology progresses into the temporal neocortex and later invades the frontal and parietal neocortices (Arnold et al., 1991). The NFT, the spatiotemporal distribution of which closely parallels the cognitive deficits in patients with AD (Arriagada et al., 1992), is primarily comprised of the tau protein assembled into paired helical filament tau (PHFtau) and straight filaments (Kidd, 1963; Kosik et al., 1986). The biochemical composition of these filaments includes all six CNS isoforms generated by alternative splicing of the tau message (Lee et al., 2001).
The NADTs differ from AD in several important ways. First, patients with NADTs exhibit a constellation of clinical features, including both cognitive and motor deficits (Feany and Dickson, 1996). Second, the pathological landmarks of disease consist of intracellular glial tau inclusions as well as neuronal pathology. This diverse spectrum of tau pathology not only extends throughout the frontotemporal neocortex but also involves the basal ganglia, deep cerebellar nuclei, and limbic system (Lee et al., 2001). Third, the tau aggregates in NADTs are biochemically distinct from those of AD. For example, in progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), filaments are comprised of tau isoforms harboring four microtubule-binding repeats (4R) (Chambers et al., 1999; Sergeant et al., 1999). In Pick disease (PiD), however, 3R tau isoforms dominate the pathology (Sergeant et al., 1997). Collectively, whereas the tauopathies manifest diverse pathological features, dysfunction of the tau protein remains a common denominator in each of these diseases.
Under normal conditions, tau is a highly soluble, natively unfolded protein (Schweers et al., 1994). During disease, however, specific regions of tau become highly ordered to assume a β-pleated sheet conformation (von Bergen et al., 2000). In fact, work from our laboratory has shown that, in AD, tau undergoes an ordered series of conformational changes that promote its pathological self-assembly (Garcia-Sierra et al., 2003). These conformational events are driven, in part, by abnormal tau modifications, including hyperphosphorylation (Grundke-Iqbal et al., 1986). Nitrative modification, an event known to influence protein function (Ischiropoulos, 2003), is also a salient feature of diverse tauopathies (Horiguchi et al., 2003). Recently, we showed that tau nitration occurs with site specificity toward residues Tyr18 and Tyr29, and, to a lesser extent, residues Tyr197 and Tyr394 (Reynolds et al., 2005a). Notably, the cumulative effect of nitration is to inhibit tau polymerization in vitro (Reynolds et al., 2005a).
In the present study, we generated a monoclonal antibody (mAb) that recognizes tau nitration specifically at position Tyr29. Intriguingly, whereas this antibody detects tau monomers and PHFtau from severely affected AD brains, it does not label tau proteins from normal aged brains. Furthermore, this reagent differentially stains the neuronal and glial tau pathology of diverse tauopathies. Together, our findings suggest that nitration at Tyr29 is a disease-related event that may alter the biochemical state of tau in the neurodegenerative tauopathies.
Materials and Methods
Tau-nY29 antibody production.
Mouse mAbs were raised against a synthetic tau peptide [25DQGGY(NO2)TMHQDQEC37] harboring the nitrated Tyr29 residue (Cell Essentials, Boston, MA). The peptide was coupled to keyhole limpet hemocyanin (Pierce, Rockford, IL) and diluted in Freund's adjuvant to a final concentration of 1 mg/ml. Female BALB/c mice were immunized, both subcutaneously and intraperitoneally, with 200 μg of peptide conjugate every 3 weeks over a 12 month period. For the final two immunizations, 200 μg of a tau mutant singly nitrated at Tyr29 (see below) was administered intraperitoneally. Once the desired serum titer was attained, immune spleens were removed, dissociated, and fused to SP2/o myeloma cells (Binder et al., 1985). Two weeks after hybridoma selection in HAT-containing media, positive clones were selected based on their ability to bind tau proteins singly nitrated at Tyr29, but not wild-type tau or tau nitrated at other Tyr residues. One cell line, Tau-nY29, was subcloned four times to ensure monoclonality and hybridoma stability. The clone was adapted to serum-free media, grown in a bioreactor, and purified to homogeneity before storage in borate-buffered saline, pH 7.4, containing 50% glycerol.
Recombinant tau proteins.
Wild-type and mutant tau proteins were expressed in Escherichia coli strain BL21 (DE3) cells using the pT7C-hτ40 plasmid. This plasmid drives high-level expression of full-length human tau (hτ40) fused to an N-terminal His6x affinity tag (Carmel et al., 1996). The hτ40 cDNA harbors the longest CNS isoform of tau (441 residues) containing two N-terminal insertions as well as alternatively spliced exons 2, 3, and 10 (Goedert et al., 1989). Genetic modifications of the pT7C-hτ40 plasmid were introduced using a QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). Four rounds of Tyr→Phe mutagenesis were used to generate quadruple hτ40 mutants that contain single Tyr residues at each position in the native hτ40 protein (Tyr18, Tyr29, Tyr197, Tyr310, and Tyr394). The identity and position of all mutations were confirmed by automated DNA sequencing. After passing each protein over a Ni-NTA metal affinity column (Qiagen, Valencia, CA) (Abraha et al., 2000), samples were treated with a 100-fold molar excess of peroxynitrite (see below). Size-exclusion chromatography was subsequently performed to purify nitrated, monomeric tau proteins to near-homogeneity. The nomenclature, description, and characterization of all Tyr→Phe hτ40 mutants have been described previously (Reynolds et al., 2005a,b, 2006). For example, the quadruple hτ40 mutant containing a single, nitrated Tyr residue at position Tyr18 (i.e., Y29F, Y197F, Y310F, and Y394F) is termed 18nY. It should be noted that a tau mutant singly nitrated at Tyr310 was not included in these analyses, because we have shown previously that nitration occurs infrequently at this position in vitro (Reynolds et al., 2005a).
Protein nitration.
Peroxynitrite was prepared from sodium nitrite and acidified H2O2 as described previously (Beckman et al., 1994). Residual H2O2 was removed by passing the peroxynitrite stock solution over a manganese dioxide column (Uppu et al., 1996). The peroxynitrite concentration was determined spectrophotometrically at 320 nm in 0.3 m NaOH (ε302 = 1670 M−1 cm−1) preceding each experiment (Souza et al., 1999). Proteins were exchanged into an inorganic phosphate buffer, pH 7.4, and then treated with a 100-fold molar excess of peroxynitrite. The final pH of the solution was measured and kept at 7.4 (Ischiropoulos and al-Mehdi, 1995). Recombinant protein concentrations were determined by the Lowry method using bovine serum albumin as a standard (Lowry et al., 1951).
Tau purification from human brain.
Biochemical enrichment of soluble tau and PHFtau from human brain was performed on three pathologically severe AD cases (Braak Stage V-VI) and three normal aged cases (Braak Stage III), as described previously (Ksiezak-Reding et al., 1992; Hanger et al., 1998). For each case, gray matter (10–20 g) from the temporal cortex was homogenized in ice-cold buffer (5 ml of buffer/g of tissue) and centrifuged at 27,000 × g for 30 min to remove nuclear and membrane-bound material. The resultant supernatant was centrifuged at 95,000 × g for 2 h to sediment PHFtau from the soluble tau fraction. To purify PHFtau to homogeneity, the pellet was solubilized in 4 m guanidine-HCl, dialyzed against bis-tris propane buffer, pH 7.0, heated at 100°C, cooled over ice, and centrifuged at 95,000 × g for 1 h to remove heat-labile material. To isolate soluble tau, the supernatant was boiled in 2% (v/v) β-mercaptoethanol, cooled over ice, and treated with ammonium sulfate. After centrifugation at 27,000 × g, the ammonium sulfate pellet was resuspended, dialyzed against homogenization buffer, and treated with perchloric acid to a final volume of 2.5% (v/v). A second centrifugation step was performed, and the supernatant was extensively dialyzed against homogenization buffer overnight. For both PHFtau and soluble tau, samples were concentrated using Centriprep YM-10 filter devices (Millipore, Bedford, MA). Protein concentrations were determined by the modified SDS-Lowry method using bovine serum albumin as a standard (Hartree, 1972).
Solid-phase assays.
For ELISAs, 100 ng of wild-type, nitrated wild-type, or nitrated mutant tau was diluted in borate-saline buffer, pH 7.4, and immobilized onto a 96-well microtitration plate (Corning, Corning, NY). Proteins were blocked for 1 h in 5% (w/v) nonfat dry milk and then incubated for 2 h at room temperature in a primary antibody solution. After a secondary incubation with either a horseradish peroxidase-conjugated anti-rabbit or anti-goat antibody (Jackson ImmunoResearch, West Grove, PA), the proteins were reacted with a 3,3′,5,5′-tetramethylbenzidine substrate solution for 10 min at room temperature. The reaction was stopped using a 3% (v/v) H2SO4 solution, and the absorbance of the immunoconjugate was read at 450 nm. Western blot and dot blot analyses were performed as described previously (Reynolds et al., 2005a).
Human brain tissue.
Postmortem brain tissue, fixed in 4% paraformaldehyde, was provided as 40-μm-thick sections by the Cognitive Neurology and Alzheimer's Disease Center Brain Bank at Northwestern University. These cases included eight pathologically confirmed AD (stages V-VI) and five normal aged (stage III) subjects staged according to the morphological criteria described by Braak and Braak (1991). In severe AD and normal aged cases, tissue sections from the inferior temporal gyrus (ITG), hippocampus (CA1), subiculum, and entorhinal/transentorhinal cortices were examined. In addition, pathological specimens from five confirmed cases of PSP, CBD, PiD, and the Lewy body variant of Alzheimer's disease (LBVAD) were also obtained from the Mayo Clinic Brain Bank (Jacksonville, FL). For non-AD tauopathy cases, sections from the hippocampus, frontal cortex, midbrain, and pons were examined.
Immunohistochemistry.
Immunohistochemistry was performed on free-floating, 40-μm-thick brain sections as described previously (Ghoshal et al., 2002). Briefly, sections were incubated overnight at 4°C in a Tau-nY29 (1 μg/ml), PHF-1 (33 ng/ml), or AD2 (0.02 μg/ml) antibody solution. All antibody titers were predetermined by testing serial dilutions on similar tissues. A biotinylated goat anti-mouse antibody (Jackson ImmunoResearch) was used as a secondary probe. After a 1 h incubation in avidin-biotin complex (Vectastain ABC Elite; Vector Laboratories, Burlingame, CA), tissue sections were developed using metal enhanced 3,3′-diaminobenzidine (Pierce), mounted onto glass slides, dehydrated through graded alcohols, cleared in xylene, and coverslipped with Permaslip (Alban Scientific, St. Louis, MO). Hematoxylin counterstaining was routinely used to demarcate cell nuclei. To assay for nonspecific binding of the secondary antibody, control sections were processed in a manner identical to that described above, except that the primary antibody was omitted.
Double-label immunofluorescence.
For single-label immunofluorescence studies, tissue sections were processed as described previously (Garcia-Sierra et al., 2003). Sections were incubated with the Tau-nY29 (1 μg/ml) primary antibody overnight at 4°C. After a secondary incubation with a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (Jackson ImmunoResearch), sections were counterstained with Thiazin Red (0.001%) and treated with Sudan Black to abolish lipofuscin autofluorescence. For double-label experiments, sections were stained with the Tau-nY29 (IgG subclass; 10 μg/ml) and Alz-50 (IgM subclass; 0.2 μg/ml) antibodies and visualized using γ- and μ-specific secondary antibodies linked to FITC and Texas Red fluorochromes, respectively (Jackson ImmunoResearch). Tissue sections were mounted on ionized slides, coverslipped with Vectashield media (Vector Laboratories), and visualized using a 510-Zeiss (Thornwood, NY) laser-scanning confocal microscope. All confocal images were acquired as z-stacks of single optical sections. Individual sections, two-dimensional, and three-dimensional rotations were analyzed using the software included with the microscope (Zeiss LSM Image Browser).
Results
Tau-nY29 detects tau nitration in a protein-specific and site-selective manner
A singular obstacle toward identifying nitrated proteins in neurological disease has been the lack of reagents that detect nitration in a protein-specific context. One recent development has been the generation of mAbs against nitrated α-synuclein that recognize both the α- and β-synuclein isoforms (Giasson et al., 2000). With one exception, the reactivity of these mAbs depends on multiple nitrated Tyr residues. Curiously, while screening these same antibodies for specificity, Horiguchi et al. (2003) discovered one mAb (termed n847) that reacts with nitrated α-synuclein, β-synuclein, and tau. In a later study, this group showed that n847 labels the fibrillar tau inclusions in several tauopathies (Horiguchi et al., 2003).
Since this report, work from our own laboratory has revealed that tau nitration is not a random event. In fact, after treatment with peroxynitrite, tau nitration occurs with site-selectivity toward the N-terminal residues Tyr18 and Tyr29 and, to a lesser degree, Tyr197 and Tyr394 (Reynolds et al., 2005a). These modifications not only inhibit the ability of tau to self-polymerize but also influence tau-microtubule interactions (Reynolds et al., 2005b, 2006). Therefore, to determine whether site-directed nitration influences the series of events leading to tau aggregation in situ, we raised mAbs against a synthetic tau peptide harboring the nitrated Tyr29 residue. These mAbs were screened by ELISA to measure their affinity toward full-length human tau (hτ40), nitrated tau (nτ40), and tau mutants singly nitrated at residues Tyr18, Tyr29, Tyr197, or Tyr394 (18nY, 29nY, 197nY, and 394nY, respectively).
One clone, an IgG1 termed Tau-nY29, showed remarkable specificity toward nitrated full-length tau and tau nitrated at residue Tyr29 (EC50 = 15 and 20 ng/ml, respectively) (Fig. 1A). While operating within the linear range of antibody-antigen binding, Tau-nY29 showed no reactivity toward wild-type tau and tau mutants singly nitrated at Tyr18, Tyr197, and Tyr394. In contrast, the n847 antibody, which recognizes nitrated α-synuclein, β-synuclein, and tau, reacts with all four 3-nitrotyrosine (3-NT) epitopes that span the length of the tau molecule (Fig. 1B). In fact, after comparing the n847 titering curve with that of a nonspecific 3-NT polyclonal antibody, it appears that n847 immunoreactivity is directly proportional to the degree of nitration of the individual tau proteins (Fig. 1C). Therefore, whereas the n847 mAb may recognize nitration within the context of the full-length tau protein, it fails to do so in a site-directed manner (see Discussion).

Epitope mapping of the Tau-nY29 and n847 mAbs. The relative affinity of the antibodies Tau-nY29 (A), n847 (B), and 3-NT (C) toward wild-type tau (hτ40), nitrated tau (nτ40), and tau mutants singly nitrated at residues Tyr18, Tyr29, Tyr197, and Tyr394 (18nY, 29nY, 197nY, and 394nY, respectively) was measured by ELISA. Only Tau-nY29 detects a single nitrated Tyr residue (Tyr29) within the linear range of antibody-antigen binding. Each titering curve is representative of five independent experiments.
As a control for antibody specificity, tau proteins singly nitrated at Tyr29 (29nY) were immobilized onto nitrocellulose membranes, treated with sodium hydrosulfite (dithionite) or vehicle alone, and then probed with the Tau-nY29 mAb. Dithionite is a strong reducing agent that converts 3-NT groups into nonreactive amines. Classically, the diminution of staining after dithionite reduction constitutes a chemical criterion of 3-NT-dependent probes (Ye et al., 1996). As shown by immunoblot, dithionite treatment of 29nY completely abolishes Tau-nY29 reactivity (Fig. 2A). The absence of Tau-nY29 staining is attributable to chemical reduction of the 3-NT moiety and not to loss of total tau protein, as evidenced by staining with the 3-NT and Tau-5 antibodies, respectively. In addition, preabsorption of Tau-nY29 with 5 μg of the nitrated, but not non-nitrated, Tyr29 peptide eliminates Tau-nY29 immunoreactivity (Fig. 2B). Together, these data strongly suggest that Tau-nY29 is a novel mAb, the epitope of which consists of both the nitrated Tyr29 residue as well as the unique tau sequence surrounding Tyr29.

Chemical reduction of 3-NT and preabsorption with a nitrated peptide abolish Tau-nY29 reactivity. A, As a control for antibody specificity, 50 ng of a tau mutant singly nitrated at Tyr29 (29nY) was absorbed onto nitrocellulose membranes and treated with either a strong reducing agent (dithionite) or vehicle alone. Membranes were then probed with the antibodies Tau-nY29 (1 μg/ml), 3-NT (1 μg/ml), and Tau-5 (20 ng/ml). B, The Tau-nY29 antibody was preabsorbed with 5 μg of either a nitrated (n-pep) or nonmodified (pep) peptide before membrane incubation. Results are representative of five independent experiments.
Because of the high degree of sequence homology between tau and other microtubule-associated proteins (MAPs), particularly MAP2, we assayed for Tau-nY29 cross-reactivity with other MAPs. To this end, a porcine MAP fraction, highly enriched for MAP2, MAP1A, MAP1B, and tau, was treated with either peroxynitrite or vehicle alone. The proteins were resolved by SDS-PAGE and probed with the antibodies 3-NT and Tau-nY29. Western blot analysis with the 3-NT antibody, which nonspecifically recognizes nitrated proteins, reveals a high-molecular mass smear in the peroxynitrite-treated samples (Fig. 3A). This higher-order smear likely represents nitrated MAPs. When the same samples were probed with Tau-nY29, however, no cross-reactivity was apparent in either the non-nitrated or nitrated MAP fraction. Importantly, the primary sequence of porcine tau contains an Asp residue, instead of Tyr, at position 29 (Himmler et al., 1989). Therefore, it was not surprising that Tau-nY29 showed no reactivity toward nitrated porcine tau proteins.
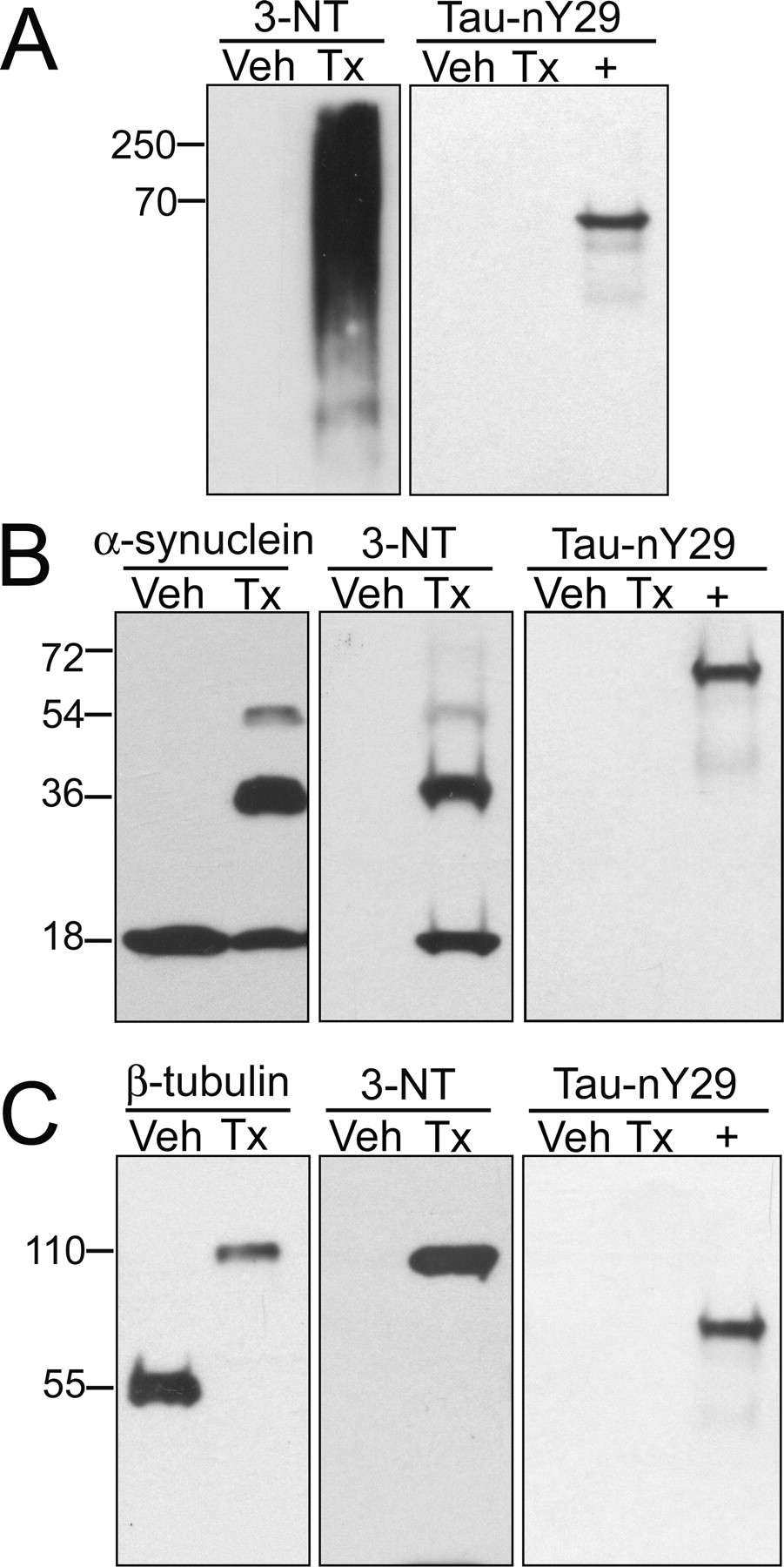
Tau-nY29 does not cross-react with other nitrated proteins. Western blot analysis was performed on proteins treated with either a 100-fold molar excess of ONOO− (Tx) or vehicle alone (Veh). A, A porcine MAP fraction highly enriched for MAP2, MAP1A, MAP1B, and tau was probed with 3-NT (1 μg/ml) and Tau-nY29 (1 μg/ml). B, Recombinant human α-synuclein was detected using the antibodies α-synuclein (0.5 μg/ml), 3-NT (1 μg/ml), and Tau-nY29 (1 μg/ml). C, Purified porcine tubulin was probed with antibodies reactive toward β-tubulin (0.2 μg/ml), 3-NT (1 μg/ml), and Tau-nY29 (1 μg/ml). In each panel, a 29nY mutant was included as a positive control (+) for Tau-nY29 reactivity. In all cases, 50 ng of protein was loaded per lane. Results are representative of five independent experiments.
Given that α-synuclein and tubulin are known to be nitrated in neurodegenerative diseases (Eiserich et al., 1999; Giasson et al., 2000), we also examined Tau-nY29 cross-reactivity with these proteins. After treatment with peroxynitrate, we observed both nitration and 3,3′-dityrosine cross-linking of the α-synuclein and tubulin proteins (Fig. 3B,C). Although the former modification was detectable using a 3-NT antibody, the latter event was manifest as SDS-insoluble oligomers after staining with antibodies against 3-NT or the native protein. Importantly, Tau-nY29 did not recognize either protein, regardless of their nitrative state. These findings indicate that the Tau-nY29 reagent reacts with nitrated tau in a biologically selective and site-directed manner.
Nitration at Tyr29 is a disease-related event
To examine whether tau nitration occurs at Tyr29 in human AD brain, Tau-nY29 was used to probe brain homogenates from the temporal and occipital cortex of three severe AD cases (Braak stage V-VI). Notably, the stereotyped progression of tau pathology in AD occurs first in the entorhinal cortex and hippocampus, followed later by involvement of the temporal neocortices (Braak and Braak, 1991). During this process, the occipital cortex remains relatively unaffected and may be used as a negative control for tau pathology. As a result of this predictable pattern of pathology, characterization of Tau-nY29 was first performed on AD-involved tissues.
Brain homogenates were fractionated by SDS-PAGE and probed with the antibodies Tau-46.1 (for total tau protein) and Tau-nY29 (for tau nitrated at Tyr29). As evidenced by Western blot analysis, Tau-46.1 recognizes many of the soluble tau isoforms (between 48 and 64 kDa) generated by alternative splicing of the tau message (Fig. 4A, top). In contrast, we could not detect these same tau proteins using the Tau-nY29 mAb, even after extended exposure times (Fig. 4A, bottom). Several studies have shown that only trace amounts of nitrated proteins are present within biological samples (Duncan, 2003; Nikov et al., 2003). For this reason, tau proteins were biochemically enriched from severely affected AD (Braak stage V-VI) and normal aged (Braak stage III) brains, resolved electrophoretically, and blotted with the Tau-46.1 and Tau-nY29 antibodies (see Materials and Methods). After detection with Tau-46.1, PHFtau proteins appeared as three major bands at ∼68, 64, and 60 kDa (Fig. 4B, top). These PHFtau bands exhibit reduced electrophoretic mobility, compared with soluble tau proteins, because of their hyperphosphorylated state (Lee et al., 2001). In contrast, soluble tau proteins isolated from both severely affected AD (τSEV) and normal aged (τMILD) brains display the characteristic profile of tau isoforms (Fig. 4B, top). Intriguingly, when these samples were probed with Tau-nY29, only the soluble tau and PHFtau proteins from severe AD cases were detected (Fig. 4B, bottom). Even after extended exposure times, soluble tau proteins from normal aged subjects were not visualized with the Tau-nY29 mAb. This observation suggests that nitration at Tyr29 is a disease-related event that may influence the pathological misfolding and deposition of the tau protein.

Tau nitration at Tyr29 is a disease-related event. Gray matter from the occipital cortex (Occ Ctx) and temporal cortex (Temp Ctx) of three severe AD cases (Braak V-VI) was homogenized and subjected to Western blot analysis. A, Immunodetection was performed using the antibodies Tau-46.1 (40 ng/ml) and Tau-nY29 (1 μg/ml), as indicated. A 29nY mutant was included as a positive control (+) for Tau-nY29 reactivity. In addition, soluble tau proteins from three aged control cases (Braak stage III) and three severe AD cases (τMILD and τSEV, respectively), as well as PHFtau from the same severe AD brains, were biochemically isolated. B, These highly enriched fractions were resolved via SDS-PAGE and probed with the antibodies Tau-46.1 (40 ng/ml) and Tau-nY29 (1 μg/ml). The purified tau proteins in each lane are representative of three independent preparations. C, Tau proteins from aged control brains (τMILD) were treated with ONOO− and immunoblotted with the Tau-nY29 antibody (1 μg/ml). In A and C, 50 μg and 1 ng of protein were loaded per lane, respectively. In the top and bottom sections of B, 1 and 10 μg of protein were loaded per lane, respectively. Results are representative of five independent experiments.
As an additional control experiment, we demonstrated that Tau-nY29 reactivity could be induced in soluble tau proteins from the brains of normal aged subjects by treatment with peroxynitrite (Fig. 4C). In fact, after peroxynitrite treatment, Tau-nY29 not only recognizes the soluble tau monomers at ∼50–70 kDa but also labels nitrated, 3,3′-dityrosine-linked oligomers at ∼120 kDa (Fig. 4C). These data indicate that other modifications at Tyr29, such as phosphorylation by Tyr-directed kinases, do not prevent nitration at Tyr29 under physiological conditions.
Tau-nY29 labels the fibrillar lesions of AD and other non-AD tauopathies
In previous studies, we showed that the N-terminal residue Tyr29 was a primary nitrative target in vitro (Reynolds et al., 2005a), and that nitration at this site markedly alters the ability of tau to self-associate and promote tubulin assembly (Reynolds et al., 2005b, 2006). Therefore, to examine whether tau nitration occurs at Tyr29 in situ, immunohistochemistry was performed on AD brain sections using the Tau-nY29 mAb. In pathologically advanced AD cases (Braak stage V-VI), we observed robust neurofibrillary staining of the large pyramidal neurons in the CA1 region and subiculum of the HP (Fig. 5A). Far fewer NFTs were visualized in the entorhinal and transentorhinal cortices (data not shown), regions known to be the first affected by NFT pathology in AD brain (Braak and Braak, 1991). This finding may be attributable to proteolytic events that remove the Tau-nY29 epitope in more mature NFTs (see Discussion). At higher magnification, we observed Tau-nY29 labeling of the signature tau pathologies, including NFTs (Fig. 5B, arrow), neuritic plaques (Fig. 5B, asterisk), and, to a far lesser degree, neuropil threads (Fig. 5B, arrowhead). It is worthy of note that, in severe AD cases, Tau-nY29 staining primarily occupies mature, compact fibrillar tangles.

Tau-nY29 labels the triad of fibrillar tau lesions in Alzheimer's brain. Immunohistochemical studies were performed using the Tau-nY29 antibody on sections of AD HP and ITG. A, Dense NFTs in the CA1 region and subiculum of a severe Alzheimer's HP (Braak stage V-VI). B, Higher magnification of A illustrating the compact NFTs (arrow), neuritic plaques (asterisk), and neuropil threads (arrowhead). Extensive NFT pathology in the ITG of a severe AD case labeled with PHF-1 (10 ng/ml) (C) and Tau-nY29 (1 μg/ml) (D). The arrows delineate Tau-nY29-positive neurons harboring intact apical and/or basal dendrites. Fewer tangles in the ITG of a pathologically mild, normal aged case (Braak stage III) stained with PHF-1 (E) and Tau-nY29 (F). Scale bars: A, 500 μm; B–F, 50 μm.
To delineate the timing and magnitude of Tau-nY29 reactivity as a function of disease severity, sections of ITG were stained using the antibodies Tau-nY29 and PHF-1. The PHF-1 antibody recognizes a phosphorylation-sensitive epitope that appears in the early stages of NFT pathology (Su et al., 1994). ITG sections were used for these experiments, because this region embraces a wide range of tau pathology: from few to no NFTs in normal aged cases (Braak stage III) to extensive NFT burden in severe cases (Braak stage V-VI). In severe AD cases, we found that PHF-1 labels numerous NFTs in cortical layers III-VI (Fig. 5C), whereas Tau-nY29 detects a much smaller number of these tangles (Fig. 5D). Intriguingly, when compared with staining with PHF-1, Tau-nY29 labels significantly fewer neuropil threads. We have shown previously that these neuritic lesions comprise the earliest manifestations of tau pathology in AD (Ghoshal et al., 2002). Similar to PHF-1, Tau-nY29 staining projects far into the apical and basal dendritic arbors (Fig. 5C,D, arrows). However, when PHF-1 was used to probe ITG sections from normal aged cases (Braak stage III), fewer tangles were observed compared with PHF-1 staining of severe AD cases (Fig. 5E). In these same cases, Tau-nY29 labeled few, if any, fibrillar tau structures (Fig. 5F). Collectively, in the cases examined, our qualitative observations indicate that the number of PHF-1- and Tau-nY29-positive NFTs may increase as a function of disease progression. Additionally, given that PHF-1-positive NFTs are greater in number than Tau-nY29-positive NFTs at each Braak stage, the appearance of the Tau-nY29 epitope likely occurs later than the appearance of the PHF-1 epitope.
To test the hypothesis that tau nitration at Tyr29 is a unifying feature among diverse tauopathies, Tau-nY29 was used to probe postmortem brain sections from PiD, PSP, and CBD cases. As shown by bright-field microscopy, Tau-nY29 labels numerous spherical tau inclusions, or Pick bodies, within the frontal cortex (Fig. 6A) and dentate gyrus (Fig. 6B) of PiD cases. We also observed intense Tau-nY29 staining in the perikarya and ramified processes of astroglia (Fig. 6C, arrow). For each case examined using Tau-nY29, adjacent sections were processed with the AD2 antibody as a positive control for tau pathology. This antibody shares a common epitope with PHF-1 and has been used to characterize tau pathology in CBD and PSP (Berry et al., 2004). Not surprisingly, AD2 detects both the Pick bodies (Fig. 6D, asterisk) and astroglial pathology (Fig. 6D, arrow) of PiD.

Tau-nY29 differentially labels the fibrillar tau inclusions of non-AD tauopathies. In PiD cases, Tau-nY29 stains numerous Pick body inclusions in the frontal cortex (A) and granular cell layer of the dentate gyrus (B). C, Ramified astroglia (arrow) also exhibit robust labeling with Tau-nY29. D, The AD2 antibody, which serves as a positive control for tau pathology in the non-AD tauopathies, labels both the ramified astroglia (arrow) and Pick bodies (asterisk). E, F, In PSP, Tau-nY29 detects globose NFTs in the frontal cortex. Notably, the glial pathology of PSP, including thorny astroglia (G), tufted astroglia (H, arrow), and coiled bodies (H, asterisk), lacks Tau-nY29 reactivity. Moreover, Tau-nY29 exhibits little-to-no staining of neuritic threads (G, arrow) in the gray matter of the frontal cortex. The perinuclear inclusions of CBD are highly reactive toward both the Tau-nY29 (I, asterisk) and AD2 (J, asterisk) antibodies. However, the neuritic thread-like processes (J, arrow) and astroglial plaques within the frontal cortex (K) lack significant Tau-nY29 staining. L, Dense Tau-nY29-positive threads (arrow) were observed in the pons of CBD cases. Scale bar, 25 μm.
In PSP cases, robust labeling of globose NFTs was demonstrated by both the Tau-nY29 (Fig. 6E) and AD2 (Fig. 6F) antibodies. However, unlike the staining of PiD cases, Tau-nY29 failed to detect any glial component of tau pathology in PSP. This glial pathology is extensive and includes thorny astrocytes within the subpial layer (Fig. 6G), coiled bodies in the gray and white matter (Fig. 6H, asterisk), and tufted astroglia (Fig. 6H, arrow). The latter manifestation constitutes a pathognomonic feature of PSP (Yamada et al., 1992) and underscores the significance of absent Tau-nY29 staining.
In cases of CBD, Tau-nY29 labeled arcuate inclusions directly adjacent to the neuronal nuclei (Fig. 6I,J, asterisks). These perinuclear inclusions have been described previously (Feany et al., 1996; Berry et al., 2004). However, similar to the Tau-nY29 staining of PSP cases, extensive glial pathology was not observed in CBD cases. For instance, astroglial plaques, a unique feature of CBD (Feany and Dickson, 1995; Berry et al., 2004), were not stained using the Tau-nY29 reagent. Moreover, the numerous thread-like processes coursing through the neuropil (Fig. 6J, arrow) were not readily detected with Tau-nY29. It is worthy of note, however, that Tau-nY29-positive neuritic threads were evident in the pons and midbrain of most CBD cases (Fig. 6L). Finally, to determine whether tau nitration occurs at Tyr29 in the hallmark inclusions of synucleinopathies, we stained sections of the LBVAD with Tau-nY29. In each case, we found no specific staining of cortical or nigral Lewy bodies (data not shown).
To examine the relationship between nitration at Tyr29 and β-pleated sheet structure in the tau lesions of severe AD brain, immunofluorescent labeling was performed using Tau-nY29 together with Thiazin Red (Mena et al., 1995). Laser scanning confocal microscopy demonstrates that Tau-nY29 recognizes only a subset of Thiazin Red-positive NFTs (Fig. 7A–C). For example, numerous NFTs that stain intensely with Thiazin Red show little to no staining with Tau-nY29 (Fig. 7A–C, arrowhead). In contrast, very few, if any, tangles stained with Tau-nY29 alone. Whereas Thiazin Red highlights the dystrophic neurites present within neuritic plaques, Tau-nY29 fails to strongly detect these structures (Fig. 7A–C, arrow).

Tau-nY29 staining cosegregates with markers of early stage tau pathology. A–C, Laser-scanning confocal microscopy was used to determine the degree of colocalization between Tau-nY29 (green) and Thiazin Red (red) in severe AD cases. The arrowhead depicts a Thiazin Red-positive, Tau-nY29-negative tangle. The arrow illustrates a Thiazin Red-positive neuritic plaque with absent Tau-nY29 reactivity. Double-label immunofluorescence was also performed using the Tau-nY29/Alz-50 antibody pairing on severe AD and PiD tissues. D–F, In AD cases, colocalization was concentrated in the proximal basal dendrites of NFTs (arrow). G–I, In contrast, Pick body inclusions exhibit a chimeric pattern whereby Tau-nY29 staining occupies a central core and Alz-50 decorates the periphery of the inclusion (arrow). Images represent projections of several optical sections. Scale bars: A, 50 μm; D–I, 10 μm.
To define whether the Tau-nY29 epitope colocalizes with markers of early stage tau pathology, we also performed double-label studies in which Tau-nY29 was paired with the antibody Alz-50. Alz-50 is a conformation-dependent probe that recognizes early filamentous changes in tau (Hyman et al., 1988; Carmel et al., 1996). Qualitatively, we found that the Tau-nY29 and Alz-50 antibodies colocalize to an appreciable extent in the NFTs of severe AD cases. In double-labeled, tangle-bearing neurons, intense colabeling was also observed in the proximal dendritic arborizations (Fig. 7F, arrow). Double-labeling experiments were also performed on sections of PiD brains. In these cases, Tau-nY29 and Alz-50 staining cosegregated in most, but not all, Pick body inclusions. Interestingly, the majority of double-labeled Pick bodies exhibit a chimeric pattern whereby Tau-nY29 staining occupies a central region, and Alz-50 decorates the periphery of the inclusion (Fig. 7G–I, arrow). Because of the marked cosegregation of Tau-nY29 and Alz-50 in these cases, our findings suggest that tau nitration at Tyr29 may occur in the early stages of diverse tauopathies (see Discussion).
Discussion
Tau-nY29 is a novel nitro-tau-specific mAb
Multiple lines of evidence suggest that tau nitration plays a mechanistic role in the pathogenesis of diverse tauopathies. Until recently, much of our understanding of tau nitration in vivo originated from studies using the n847 antibody (Horiguchi et al., 2003). It should be noted, however, that although n847 has been considered a reliable marker of tau nitration, it demonstrates specificity only for 3-nitrotyrosine. The n847 antibody is neither protein specific nor dependent on the amino acid sequence surrounding any given Tyr residue. In support of this, Horiguchi et al. (2003) acknowledge that n847 recognizes nitrated Tyr residues on α-synuclein, β-synuclein, and tau. Indeed, in the present study, we reveal that n847 detects each nitrated Tyr residue on tau in proportion to its degree of nitration.
By comparison, Tau-nY29 is a biologically selective, site-directed probe that detects tau nitration exclusively at the Tyr29 position. Using this reagent, we demonstrate that nitration at Tyr29 is a disease-related event that may influence the ability of tau to self-associate. In addition, whereas Tau-nY29 stains the fibrillar tau lesions in AD brain, it differentially labels glial and neuronal tau inclusions in the NADTs. This latter finding suggests that, during the progression of diverse tauopathies, differential posttranslational modifications may alter the biochemical properties of tau, leading to markedly dissimilar pathological and clinical phenotypes.
A primary motivation for generating the Tau-nY29 reagent was the lack of interspecies homology at this position. For example, whereas Tyr29 is highly conserved in humans and some mammals, most rodents harbor an Asp or Glu at residue 29. This observation is intriguing for several reasons. First, the Asp and Glu residues, both of which maintain a net negative charge, have been used extensively as phosphorylation mimetics in vitro (Abraha et al., 2000). Second, it has been suggested that nitration and phosphorylation not only compete for protein Tyr residues in vivo, but also exert disparate effects on protein function (Ischiropoulos, 2003). In fact, work from our laboratory reveals that although pseudophosphorylation at Tyr29 does not affect tau polymerization (Reynolds et al., 2005a), nitration at this same residue markedly delays tau assembly kinetics (Reynolds et al., 2005b). Therefore, in humans and higher mammals, it is entirely possible that tau nitration at Tyr29 may serve as a regulatory mechanism to modulate tau assembly. In other species, however, this system may be ineffective because of the persistent state of pseudophosphorylation (i.e., Asp or Glu) in place of residue Tyr29. The dynamic interplay between nitration and phosphorylation at Tyr29 may help to explain the species-specific manifestations of tau pathology in response to inciting stimuli (i.e., β-amyloid peptide deposition).
The timing of tau nitration at Tyr29
In both AD and PiD cases, Tau-nY29 staining cosegregates to a remarkable degree with the early stage marker of tau pathology, Alz-50. This observation suggests that nitration at Tyr29 may itself be an early stage event. In addition, the finding that Tau-nY29 labels the apical and basal dendritic tree of AD-affected neurons further supports the notion of tau nitration as an early stage event. These dendritic arbors, which remain extended in the early stages of tangle pathology, tend to retract in late-stage NFTs (Anderton et al., 1998). The preservation of these structures, even in the face of fibrillar tau pathology, suggests that the appearance of the Tau-nY29 epitope occurs early in the course of NFT evolution but likely after tau fibrils have begun to form.
In contrast, we observed that although Tau-nY29-positive NFTs were present in brain regions affected early in AD (i.e., entorhinal and transentorhinal cortices), staining was far more intense in regions involved later in the disease (i.e., CA1 and subiculum). Although the reason for this discrepancy is unclear, It has been established that, during tangle evolution, tau truncation occurs at both the N and C termini (Garcia-Sierra et al., 2003; Horowitz et al., 2004). In particular, proteolytic removal of the N terminus of tau signifies a progression to late-stage tau pathology (Ghoshal et al., 2002). Therefore, it is possible that N-terminal truncation events remove the Tau-nY29 epitope at later stages of tau pathology. This event could explain the paucity of Tau-nY29-positive NFTs in the entorhinal cortex of severe AD cases but is unlikely to be the only contributing factor given that Tau-nY29-positive tangles persist in the hippocampus. Additional studies on brains from normal aged individuals with “age-appropriate” NFTs in the entorhinal cortex and/or hippocampus may help clarify this issue.
Intriguingly, in sections of AD brain, Tau-nY29 strongly labels NFTs and neuritic plaques but stains far fewer neuropil threads. In our previous work, we demonstrated that neuropil threads are the earliest manifestations of fibrillar tau pathology in AD (Ghoshal et al., 2002). There are several possible explanations for this finding. First, multiple lines of evidence suggest that nitrative injury occurs more readily within NFTs and neuritic plaques than neuropil threads (Ii et al., 1996; Smith et al., 1997). As a result, neuropil threads may exhibit a paucity of tau proteins nitrated at the Tyr29 position. Another equally attractive explanation is that proteolytic events, such as N-terminal truncation, may remove the Tau-nY29 epitope from the early stage neuropil threads. In this event, the later-stage NFTs and neuritic plaques would retain the Tau-nY29 epitope and, therefore, react toward the Tau-nY29 antibody.
Still, other lines of evidence suggest that the Tau-nY29 epitope may be unmasked in the middle-to-late stages of tangle pathology. For example, soluble tau monomers purified from normal aged brains (Braak stage III) do not react with the Tau-nY29 antibody. Similarly, immunohistochemical characterization of Tau-nY29 demonstrates few-to-no tangles in Braak Stage III brains. It is also noteworthy that Tau-nY29 staining appears in fibrillar, compact tangles. These fibrillar NFTs likely represent more advanced stages of tau pathology. Therefore, although some of our data suggest that tau nitration at Tyr29 occurs early during the course of NFT evolution, other experiments intimate that this event occurs much later. At present, our data cannot exclude the possibility that tau nitration is a stochastic process that occurs at one or more stages in the disease process. To unambiguously assess the timing issue, a quantitative study using numerous well-staged brains will be required to map the spatiotemporal appearance of the Tau-nY29 epitope in relation to other known markers of tau pathology.
Tau-nY29 differentially stains tau pathology in diverse tauopathies
Perhaps most surprisingly, Tau-nY29 differentially labels the neuronal and glial tau inclusions in several tauopathies. In AD brain, Tau-nY29 recognizes the triad of fibrillar tau lesions. The tau filaments within these structures are comprised of all six CNS tau isoforms. In PiD sections, not only does Tau-nY29 label neuronal Pick body inclusions, but it also stains ramified astroglia in layers II-III of the frontal cortex. Similar to AD, PSP and CBD exhibit mostly neuronal tau pathology when stained with Tau-nY29. The predominant glial pathology that typifies CBD and PSP, however, does not stain with the Tau-nY29 reagent. Several possible mechanisms may explain this differential pattern of Tau-nY29 staining. First, peroxynitrite, the major in vivo nitrating agent, is generated from the reaction of superoxide and nitric oxide radicals (Beckman and Koppenol, 1996). Whereas superoxide is produced under a variety of physiological and pathological circumstances, nitric oxide is generated both constitutively and in high-output forms. The constitutive production of nitric oxide is governed by neuronal and endothelial nitric oxide synthase, whereas high-output nitric oxide is mediated by inducible nitric oxide synthase in activated neuroglia (Garthwaite and Boulton, 1995). Therefore, based on the levels of NOS isoform expression in the varying tauopathies, it is feasible that the Tau-nY29 epitope could differentially appear in the neuronal and glial pathology of these diseases. Second, given the isoform composition of tau filaments in PiD (3R) and PSP/CBD (4R), the Tau-nY29 staining pattern may be attributable to a differential susceptibility of certain tau isoforms toward nitration at Tyr29. For example, in the 3R tauopathies, tau monomers nitrated at Tyr29 may readily assemble into filaments. As a result, in PiD, both neuronal and glial tau pathology would exhibit Tau-nY29 positivity. In the 4R tauopathies, however, tau nitration at Tyr29 may reduce the ability of tau monomers to self-associate, thereby minimizing Tau-nY29-positive tau inclusions. In support of this contention, our immunohistochemical data reveal absent glial staining, and sparse neuronal labeling, of the tau lesions in PSP and CBD.
Footnotes
-
This work was supported by National Institutes of Health Grants AG 021661 (L.I.B.), AG 21184 (L.I.B.), and F30 NS051043 (M.R.R.). We thank Nichole E. LaPointe (Northwestern University, Chicago, IL) for helpful discussions and purification of recombinant α-synuclein. We acknowledge Virginia M.-Y. Lee (University of Pennsylvania, Philadelphia, PA) for providing the antibodies Tau46.1 and n847. We also thank Peter Davies (Albert Einstein College of Medicine, New York, NY) and Andre Delacourte (Institut National de la Santé et de la Recherche Médicale U422, Lille, France) for allowing us to use the antibodies PHF-1 and AD2, respectively.
- Correspondence should be addressed to Matthew R. Reynolds, Tarry Building 8-754, 303 East Chicago Avenue, Chicago, IL 60611. m-reynolds{at}md.northwestern.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}