

Abstract
Methamphetamine (METH) is an illicit drug that causes neuronal apoptosis in the mouse striatum, in a manner similar to the neuronal loss observed in neurodegenerative diseases. In the present study, injections of METH to mice were found to cause the death of enkephalin-positive projection neurons but not the death of neuropeptide Y (NPY)/nitric oxide synthase-positive striatal interneurons. In addition, these METH injections were associated with increased expression of neuropeptide Y mRNA and changes in the expression of the NPY receptors Y1 and Y2. Administration of NPY in the cerebral ventricles blocked METH-induced apoptosis, an effect that was mediated mainly by stimulation of NPY Y2 receptors and, to a lesser extent, of NPY Y1 receptors. Finally, we also found that neuropeptide Y knock-out mice were more sensitive than wild-type mice to METH-induced neuronal apoptosis of both enkephalin- and nitric oxide synthase-containing neurons, suggesting that NPY plays a general neuroprotective role within the striatum. Together, our results demonstrate that neuropeptide Y belongs to the class of factors that maintain neuronal integrity during cellular stresses. Given the similarity between the cell death patterns induced by METH and by disorders such as Huntington's disease, our results suggest that NPY analogs might be useful therapeutic agents against some neurodegenerative processes.
Introduction
Methamphetamine (METH) is a psychostimulant for which use has spread throughout the United States (Freese et al., 2002). Repeated exposures to this drug lead to tolerance to its psychotropic effects with resulting increases in the consumed doses via compulsive binges to maintain its euphoric and stimulatory effects (Davidson et al., 2001). Recurrent use of high doses of METH can cause neurotoxic abnormalities in humans (Wilson et al., 1996; Volkow et al., 2001). Neurodegenerative manifestations of METH have been demonstrated also in several mammalian species by using various drug administration schedules (Zalis et al., 1967; Escalante and Ellinwood, 1970; Ellison and Switzer, 1993; Davidson et al., 2001; Cadet et al., 2003). Importantly, METH can also induce cell death in various brain regions, including the striatum (Deng et al., 1999, 2002; Cadet et al., 2003).
The striatum contains various classes of neuronal cell bodies, which include projection neurons and interneurons. Projection neurons are GABAergic spiny neurons, subclasses of which also synthesize the neuropeptides substance P, dynorphin, or enkephalin (ENK) (Kawaguchi, 1997). Four types of interneurons have been described: (1) cholinergic neurons; (2) GABAergic cells that coexpress neuropeptide Y (NPY), somatostatin (SS), and neuronal nitric oxide synthase (nNOS); (3) GABAergic cells that express parvalbumin; and (4) GABAergic neurons that express calretinin (Kawaguchi et al., 1995; Kawaguchi, 1997). These classes of neurons are known to show differential sensitivity to certain neurodegenerative agents (Reiner et al., 1988). For example, ENK-positive neurons are sensitive to excitotoxic injuries, whereas nNOS- and NPY-containing neurons are resistant to these insults (Koh et al., 1986; Beal et al., 1991; Mitchell et al., 1999). Similar observations have been reported in previous in vivo models of strokes (Ferriero et al., 1988; Gonzales et al., 1992; Behrens et al., 1996). NPY interneurons are also spared in Huntington's disease (Ferrante et al., 1985; Reiner et al., 1988; Mitchell et al., 1999), which is a genetic neurodegenerative disorder that affects the human striatum (de la Monte et al., 1988).
NPY participates in several physiological functions, including control of food intake, anxiety, and memory (Grundemar and Hakanson, 1994). Its actions are mediated by five G-protein-coupled receptors (Y1, Y2, Y4, Y5, and Y6) (Pedrazzini et al., 2003). Activation of all NPY receptor subtypes inhibits adenylyl cyclase and regulates calcium-signaling pathways (Pedrazzini et al., 2003). Y1, Y2, and Y5 receptors are expressed in several areas of the mouse brain, whereas Y4 and Y6 are located in peripheral organs (Naveilhan et al., 1998). NPY-containing neurons express only Y2 receptors (Caberlotto et al., 2000), whereas Y1 and Y5 are colocalized in neurons that do not contain NPY (Wettstein et al., 1995; Naveilhan et al., 1998), suggesting that only Y2 receptors function as autoreceptors. The Y2 receptor also was found in non-NPY-expressing neurons (Caberlotto et al., 2000). We now report that NPY exerts protective effects against METH-induced neurotoxicity in the striatum, and that these effects are mediated by both Y1 and Y2 NPY receptors, with Y2 receptors playing the most significant role.
Materials and Methods
Animals. We used 10- to 12-week-old (30-40 g) male CD-1 (Charles River, Raleigh, NC), 129S6/SvEv, and NPY knock-out (KO) mice on a 129S6/SvEv background. NPY KO mice were generated as described previously (Erickson et al., 1996). Animals were housed three or two (if implanted with chronic cannula) per cage. Mice were maintained at a temperature-controlled (22 ± 2°C) and light-controlled environment; food and water were available ad libitum. Animal use procedures were used according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local Animal Care Committee.
METH and NPY treatment. Mice received intraperitoneal injections of saline (0.9%) or METH (four times with 10 mg/kg each at 2 h intervals). Animals showed no evidence of seizures after METH treatment. Mice were killed at various time points (12 h, 1 d, 3 d, or 7 d) after METH injections. NPY (H-6375; Bachem, King of Prussia, PA), the NPY Y1 agonist Leu31-Pro34 NPY (H-8575; Bachem), and the NPY Y2 agonist NPY (3-36) (H-8570; Bachem) were administered intracerebroventricularly through injecting needles that were anchored to chronically implanted cannulas. For cannula implantation, animals were anesthetized with a mixture of ketamine, xylazine, and acepromazine (100, 20, and 3 mg/kg, respectively), and guide cannulas (28GA; Plastics One, Roanoke, VA) were implanted unilaterally in one of the lateral ventricles [coordinates with respect to bregma: anterior (A), -0.1; lateral (L), 0.7; ventral (V), 1.2 from the skull]. The intracerebroventricular (icv) injection studies were performed 3-5 d after surgery. Obturators were used to prevent dust from entering the cannulas when they were not in use. For microinjections, the obturators were removed, and an injection needle (31GA; Plastics One) was inserted, which extended 2 mm below the guide cannula. Mice received an icv injection of 2.0 μl of artificial CSF (aCSF) or solution of NPY or the NPY analogs in aCSF delivered over a period of 120 s via a 10 μl glass syringe. The injector was left in place for an additional 60 s, and subsequently the obturators were replaced. Mice received three icv injections: the first injection was given 1 h before they received systemic injections of saline or METH; the second and third icv injections were given 24 and 48 h after the first injection. Mice were killed 24 h after the last icv injection, corresponding to 3 d after METH administration.
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling histochemistry. The animal was killed 3 d after METH administration. Subsequently, the brains were removed and cut with a cryostat. Section preparation was done according to methods reported previously (Deng et al., 1999, 2002). Slide-mounted sections were fixed for ∼30 min in 4% paraformaldehyde. After washing three times in 0.01 m PBS, the sections were rinsed in 0.5% Triton X-100 for 20 min at 80°C. To label damaged nuclei, 50 μl of the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) reaction mixture was added to each sample followed by a 60 min incubation at 37°C in a humidified chamber. For statistical analysis, TUNEL-positive cells in the striatum (one side only) at the bregma level 1.00 ± 0.30 were counted using a Zeiss (Thornwood, NY) microscope by an individual blinded to the experimental treatment. For nonoperated animals, the left or right striatum was chosen randomly for quantification. For operated animals, only the controlateral striatum was chosen for counting to avoid the additional cell death induced by cannula implantation. Three to four sections were counted for each animal. Three to seven animals were used in each group.
Double-labeling: immunocytochemistry and TUNEL staining. To identify the striatal cells undergoing apoptosis, we used dual staining of sections with specific immunochemistry followed by TUNEL histochemistry as described previously (Deng et al., 2002). We used antibodies generated against NPY, nNOS, and ENK (Chemicon, Temecula, CA) to identify the different neuronal classes. Briefly, 3 d after the last METH injection, the animals were perfused transcardially under deep pentobarbital anesthesia, first with saline followed by 40 ml of 4% paraformaldehyde in 0.1 m phosphate buffer at 4°C. The brains were removed, post-fixed overnight in 4% paraformaldehyde, and then allowed to equilibrate in 30% sucrose for 24 h. Coronal sections (30 μm) were first exposed to 1% hydrogen peroxide for 20 min and then to 1% bovine serum albumin/0.3% Triton X-100 for 30 min. This was followed by incubation with primary antibodies (1 h at 37°C and then overnight at 4°C) and subsequently with biotinylated secondary antibody and Texas Red-avidin-D of cell-sorter grade (Vector Laboratories, Burlingame, CA). After mounting the sections on slides, we stained the sections with TUNEL histochemistry as described above. For statistical analysis, three sections were taken from three to five animals. These sections were taken between the coordinates 0.7 and 1.3 anterior to bregma. In each section, double-positive cells were counted in striatal area using a Zeiss laser scanning confocal system with Axiovert 135-inverted microscopy by an individual blinded to the experimental treatment.
Quantitative real-time-PCR. We used the real-time (RT)-PCR approach (LightCycler; Roche Molecular Biochemicals, Mannheim, Germany) to investigate the levels of NPY and Y1, Y2, and Y5 receptor mRNAs in mice striata after the METH injections using methods described previously (Thiriet et al., 2002). We extracted RNA from six to eight animals per time points and performed reverse transcription reactions with oligodT primers using the Advantage RT for PCR kit (Clontech, Palo Alto, CA). We then performed PCR experiments using Light Cycler-FastStart DNA Master SYBR Green I kit (Roche Molecular Biochemicals). We generated the sequences of the primers used for each gene using Light Cycler Probe Design software. For the quantitative RT-PCR amplification, the sequences of the primers used to study the change in gene expression are as follows: NPY, 5′-GTG TTT GGG CAT TCT G-3′/5′-TTC TGT GCT TTC CTT CAT-3′; Y1 receptor, 5′-CAA GAT ATA CAT TCG CTT GA-3′/5′-AGA TTG TGG TTG CAG G-3′; Y2 receptor, 5′-CCA TCT TCC GGG AAT AC-3′/5′-TGA CGT GGT TCC TCA G-3′; Y5 receptor, 5′-TCA AGC GTT CCC TCA C-3′/5′-ACA ACA GGA CAT CAT GC-3′; clathrin, 5′-AAG TAT CCG TAA GTG GAG-3′/5′-GGG GTT AAA GTC ACA CAG-3′. We incubated DNA Master SYBR Green I mix (containing TaqDNA polymerase, deoxynucleotide triphosphate, MgCl2, and SYBR Green I dye) with primers and cDNA template. Fluorescence data collection was performed at the end of each extension phase. A negative control without cDNA template was run with every assay to assess the overall specificity and to verify that no primer-dimer was generated. The mean concentration of clathrin light chain was used to normalize the RNA input. The relative change in gene expression was recorded as the ratio of normalized data over saline.
Temperature measurements. Mice were implanted with cannulas as described. Mice were transported from the animal care to the experimental room and habituated for 30 min before temperature measurements began. Temperature was determined by the use of a rectal probe (YSI telethermometer; Yellow Springs Instruments, Yellow Spring, OH). Temperature was recorded 30 min before the icv injections and at 30 min intervals until the administration of METH or saline. Subsequently, temperature measurements were obtained at 1 h intervals until 1 h after the last drug or saline injection. Mice were killed the day after the experiment for histological verification of cannula placement.
Statistics. For statistical analysis, we used one-way ANOVA followed by Fisher's protected least significant difference (PLSD) test for determining differences between the different time points and the saline-treated animals. For temperature experiments, we used a two-way ANOVA for repeated measures followed by a PLSD test. All analyses were done using Statview 4.02 (SAS Institute, Cary, NC). The null hypothesis was rejected at p < 0.05.
Results
METH kills ENK-positive neurons but not NPY-containing neurons
We performed double-label experiments using immunohistochemistry (Fig. 1, left column) and TUNEL histochemistry (Fig. 1, middle column) in an attempt to identify which striatal cells undergo METH-induced apoptosis in the striata of mice killed on the third day after drug administration. No TUNEL-positive signal was detected in saline-treated animals (Fig. 1Ab,Bb). METH killed several ENK-positive cell bodies (Fig. 1Af). Nevertheless, many of the TUNEL-positive cells were not labeled with the ENK antibody. We used NPY immunohistochemistry to test the possibility that the NPY neurons might undergo METH-induced apoptosis (Fig. 1B). NPY-containing neurons showed complete resistance to METH toxicity (Fig. 1Bf). These results led us to further evaluate possible interactions of METH with the NPY system of the mouse striatum.

METH causes selective neuronal death in the mouse striatum. Results of the immunohistochemical experiments are shown in red, and TUNEL-positive labels are shown in green. Immunopositive cells undergoing cell death are double labeled and appear in orange. A, ENK-positive cells are sensitive to METH-induced apoptosis. B, NPY-positive interneurons are spared from METH-induced apoptosis. No TUNEL-positive cells were observed in the saline-treated animals (Ab or Bb). The inset in Af shows an enlargement of an ENK-positive cell that was also TUNEL positive. Scale bar, 50 μm.

METH administration significantly changes NPY and NPY receptor expression. Left, METH increased NPY expression significantly at 3 and 7 d. Middle, Y1 receptor level was progressively decreased at all time points after METH treatment. Right, Y2 receptor mRNA level was significantly decreased 12 h after METH injections and increased 1 d after METH injections. Statistical analysis was done by ANOVA, followed by Fisher's PLSD. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the saline-treated group. Rec., Receptor.
METH alters NPY and NPY receptor mRNA levels
Because the immunocytochemical results using the NPY antibody suggested that there might be some increase in the level of the peptide after METH administration (Fig. 1B), we used quantitative PCR techniques to measure any possible changes in NPY mRNA in the mouse striatum after METH administration. We also measured transcript levels for Y1, Y2, and Y5, because these receptors are expressed in the brain (Naveilhan et al., 1998). METH caused significant increases in NPY mRNA levels at 3 and 7 d after drug administration (Fig. 2, left). Y1 mRNA levels showed progressive decreases in expression (Fig. 2, middle), whereas Y2 mRNA levels decreased at 12 h but then increased at 1 d after drug administration (Fig. 2, right). Y2 expression returned to control levels at later time points. Y5 mRNA levels were not affected by the drug (data not shown).
NPY administration protects against METH-induced neurotoxicity
We also tested the possibility that infusion of exogenous NPY could provide protection against the neurotoxic effects of METH. Two doses of NPY were administered (1 and 10 μg, i.c.v.). NPY given alone did not cause any cell death in the mouse brain. At the dose of 1 μg, NPY did not attenuate the number of cells undergoing cell death after METH administration (Fig. 3, left). In contrast, when given at the dose of 10 μg, NPY significantly reduced the number of TUNEL-labeled cells in comparison with METH-treated animals, and the number of TUNEL-labeled cells was comparable with the number observed in saline-treated animals (Fig. 3, left).
To determine which NPY receptor subtype might be involved in the protective effects of NPY, we used selective agonists for Y1 and Y2 receptors (Fig. 3, right). The Y1 agonist, Leu31-Pro34 NPY (10 μg, i.c.v.), reduced METH-induced cell death to 44% compared with METH alone, and the number of apoptotic cells remained significantly higher than control (Fig. 3, right). In contrast, the Y2 agonist, NPY (3-36) (10 μg, i.c.v.), reproduced NPY (10 μg) effects and completely blocked METH-induced cell death (Fig. 3, right).
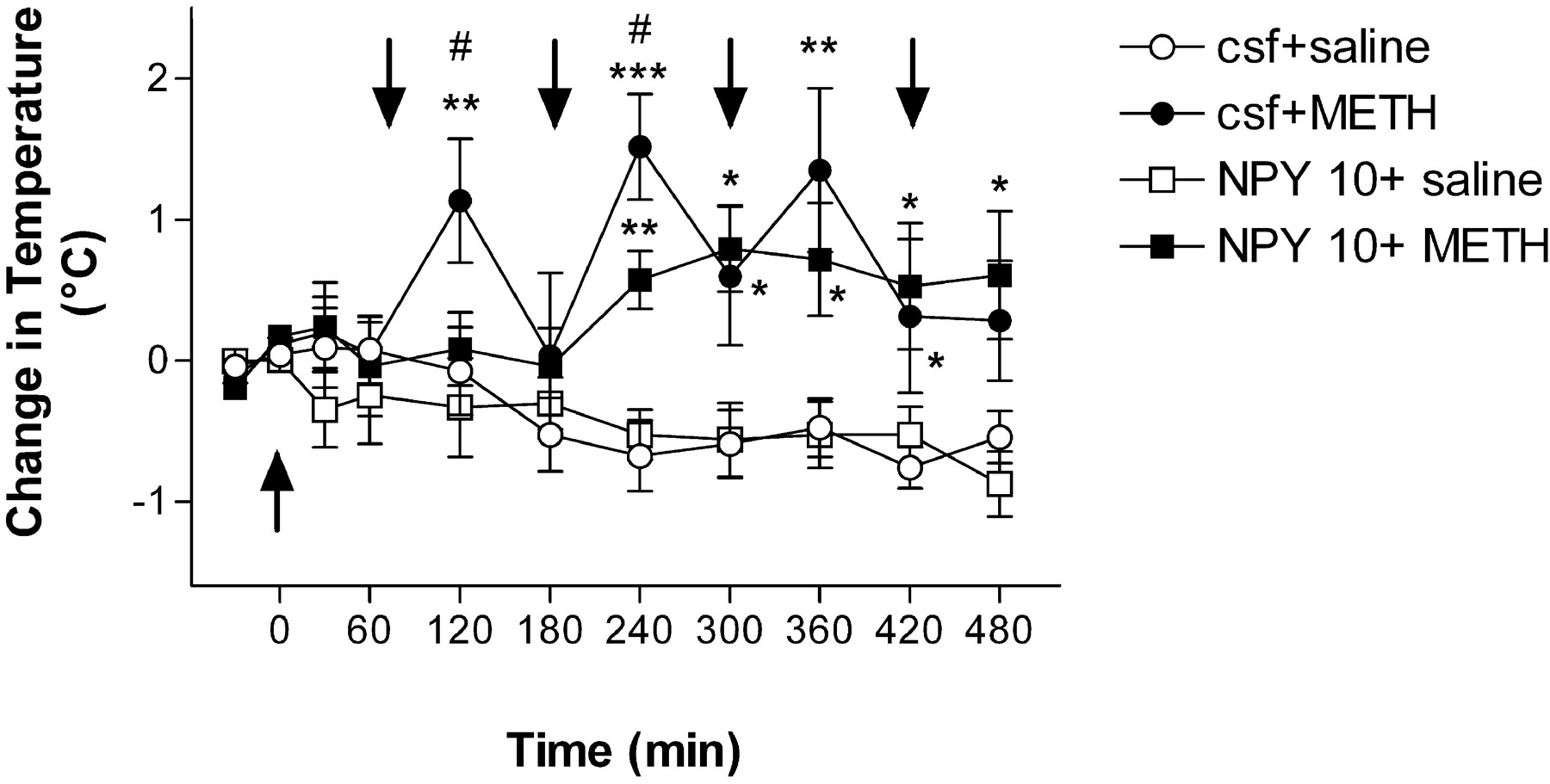
Administration of NPY reduces METH-induced hyperthermia
METH administration is associated with increases in body temperature (Brown et al., 2003; Jayanthi et al., 2005), which is thought to participate in METH-induced toxicity (for review, see Cadet et al., 2003). Because NPY is involved in body thermoregulation (Bouali et al., 1995; Richard, 1995), we sought to determine whether the protective effects of NPY might be associated with NPY-mediated prevention of METH-induced hyperthermia. Initial basal temperature was not significantly different across experimental groups. In the control (CSF plus saline) and NPY alone (NPY plus saline) groups, temperature tended to decrease over the duration of the experiments (Fig. 4). Administration of METH caused significant increases in body temperature with peak increases of 1.5°C occurring 60 min after the second drug injection (Fig. 4). Intracerebral injection of NPY (10 μg) significantly blocked the initial increases in temperature induced by METH at 60 min after the first METH injection (Fig. 4). The injection of NPY attenuated the METH-induced increases after the second METH injections but not during the late phases of the experiment (Fig. 4).

NPY and NPY agonists inhibit METH-induced neuronal death in the mouse striatum. Quantitative data comparing the number of TUNEL-positive cells after METH administration alone or in combination with NPY at two dosages (1 or 10 μg) or with Y1 and Y2 receptor agonists are shown. The number of TUNEL-labeled cells induced by METH was reduced by NPY when given at 10 μg. Few cells were labeled after saline (Sal) or NPY administration (left). The Y1 agonist (Leu31-Pro34 NPY; 10 μg) partially blocked METH-induced apoptosis, whereas the Y2 receptor agonist [NPY (3-36); 10 μg] completely blocked the apoptotic effects of METH (right). Statistical analysis was done by ANOVA followed by Fisher's PLSD. **p < 0.01, *** p < 0.001, compared with saline-treated animals; ∘∘∘p < 0.001, compared with METH-treated animals.

Effects of NPY on METH-induced hyperthermia. Quantitative data represent the changes in temperature (in °C) after METH administration alone or in combination with 10 μg of NPY over 9.5 h of experimental session. Arrows indicate the times of injections: the first arrow (arrowhead up) indicates the intracerebroventricular injection of CSF or NPY (10 μg); the other arrows (arrowhead down) indicate intraperitoneal injections of either saline or METH. METH caused significant increases in body temperature that were attenuated by administration of NPY at early time points. Statistical analyses were done using ANOVA followed by Fisher's PLSD. *p < 0.05, **p < 0.01, *** p < 0.001, compared with saline-treated (csf+saline) animals; #p < 0.05, compared with CSF plus METH (csf+METH) animals.

METH-induced neuronal cell death is potentiated in NPY KO mice. A, Representative photomicrographs illustrating the effects of METH in the striatum of WT and NPY KO mice. No TUNEL-positive cells (green) were observed in the saline-treated animals for WT (Ab) or NPY KO (Ae) mice. After METH treatment, WT mice (Ah) show less TUNEL-positive cells than NPY KO animals (Ak). The number of ENK-positive neurons sensitive to METH-induced apoptosis (orange) is increased in the NPY KO animals (compare Ai with Al). The insets in Ai and Al show an enlargement of an ENK- and TUNEL-positive cell (arrowheads). Scale bar, 50 μm. B, Quantitative data comparing the two genotypes and their responses to METH. Significantly more neurons were killed by METH in NPY KO mice compared with WT animals (ANOVA, followed by Fisher's PLSD; *p < 0.05). C, Quantitative data comparing the response of ENK neurons to METH in the two genotypes. Significantly more ENK-positive neurons were killed by METH in NPY KO mice compared with WT animals (ANOVA, followed by Fisher's PLSD; *p < 0.05).
NPY KO mice are more sensitive to the apoptotic effects of METH
The observations described above suggested that NPY might be exerting neuroprotective effects on striatal neurons. To test that idea, we used KO mice that were engineered not to express NPY (Erickson et al., 1996). No TUNEL-positive signal was detected in the saline-treated animals (Fig. 5Ab,Ae). Injections of METH caused the appearance of TUNEL-positive cells in both wild-type (WT) and NPY KO mice (Fig. 5, Ah and Ak, respectively). The toxic effects of METH were potentiated in the NPY KO mice (Fig. 5, compare Ah, Ak; see B for quantitative data).
To determine whether the ENK-positive cells became more sensitive to the actions of METH in the absence of NPY, we examined sections labeled with both ENK and TUNEL histochemistry. The number of ENK-positive cells in the striatum of untreated WT and NPY KO mice was similar (Fig. 5Aa,Ad). In agreement with what is observed in Figure 1A, many of the cells undergoing apoptosis after METH treatment were ENK-positive cells (Fig. 5Ai,Al). Statistical analysis revealed that significantly more ENK-positive cells were affected by METH in the NPY KO mice than in the WT mice (Fig. 5C).

nNOS neurons in NPY KO mice become sensitive to METH. nNOS-positive cells are shown in red, whereas TUNEL-positive cells are shown in green. The merged results, which show nNOS-positive cells that are also TUNEL positive, are shown in orange. No TUNEL-positive cells were observed in the saline-treated WT (b) or NPY KO (e) mice. After METH treatment, no nNOS-positive cells were TUNEL positive in the WT mice (i), whereas some nNOS neurons were also TUNEL positive in the NPY KO animals (l). The inset in l shows an enlargement of a nNOS- and TUNEL-positive cell (arrowheads). Scale bar, 50 μm.
To determine whether NPY also exerts an autoprotective effect on the NPY/nNOS cells, we tested the possibility that the absence of NPY might render these cells sensitive to METH-induced death. We examined sections that were processed for nNOS immunochemistry and TUNEL histochemistry. Injections of METH caused the appearance of a greater number of cells labeled with the nNOS antibody in both genotypes (Fig. 6g,j). This is consistent with our previous report (Deng and Cadet, 1999). Injections of METH caused the appearance of many TUNEL-positive cells in the striatum of both genotypes (Fig. 6h,k). None of the TUNEL-positive cells were colabeled with the nNOS antibody in the WT mice (Fig. 6i), whereas a mean of 4.3 ± 0.37 nNOS-positive cells per striatal area per section were also TUNEL positive in the NPY KO mice (Fig. 6l).
Discussion
In the present study, we investigated the possible protective role of NPY against METH-induced neurotoxicity in the striatum. Our findings are as follows: (1) striatal ENK-positive neurons are sensitive to METH-induced cell death, whereas NPY-expressing neurons are spared, in WT mice; (2) METH administration causes upregulation of NPY mRNA, downregulation of Y1 receptor mRNA, and biphasic changes in the expression of the Y2 receptor gene; (3) NPY icv administration protects against METH-induced neurotoxicity; (4) both Y1 and Y2 receptors are involved in the protective effects of NPY; (5) NPY KO mice are more sensitive to METH toxicity; and (6) striatal nNOS-positive neurons from NPY KO mice, but not from control mice, are vulnerable to METH-induced cell death. These results indicate that NPY acts as a neuroprotective factor in the mouse brain.
Resistance of NPY neurons to neurotoxic processes
METH is a neurotoxin that causes neuronal apoptosis in the mouse striatum (Deng et al., 1999; Cadet et al., 2003). METH-induced apoptosis does not affect NPY/nNOS interneurons, because these neurons are spared, whereas some ENK-positive projection neurons are killed by a toxic METH regimen (Fig. 1). Similar sparing of nNOS/NPY neurons has been reported in models that cause striatal injury. For example, Gonzales et al. (1992) reported that NPY interneurons were spared in the striata of gerbils that had suffered ischemic lesions. Exposure of striatal neurons to the excitotoxin quinolinic acid, both in vitro (Koh et al., 1986) and in vivo (Beal et al., 1991), also killed ENK-positive neurons but spared NPY/nNOS-containing striatal interneurons. Of broader clinical interest, sparing of NPY-expressing neurons has been reported in the brains of Huntington's disease patients (Ferrante et al., 1985; Reiner et al., 1988; Mitchell et al., 1999).
NPY acts as a neuroprotective agent in the striatum
Based on our present results and on these previous reports (Beal et al., 1991; Gonzales et al., 1992; Mitchell et al., 1999), we hypothesized that the NPY/nNOS neurons possess properties that render them resistant to excitotoxic insults. We propose that NPY and its receptors are potential mediators of this resistance to injuries. Our observations that NPY KO mice were more sensitive to the apoptotic effects of METH (Fig. 5) support the view that NPY is neuroprotective. We also observed that nNOS-containing neurons, which are resistant to METH toxicity in wild-type animals (Deng and Cadet, 1999) (Fig. 6i), were actually sensitive to METH-induced apoptosis in the absence of NPY (Fig. 6l), suggesting that NPY itself might be responsible for this protection. These suggestions are supported by our demonstration that administration of NPY or NPY receptor agonists blocks METH-induced cell death. Several recent observations are in agreement with such a potential protective role for NPY in the brain. For example, NPY protects hippocampal cells against glutamate excitotoxicity (Silva et al., 2003). In addition to these neuroprotective effects, NPY stimulates cell proliferation as demonstrated by the fact that NPY increases the number of olfactory neuronal precursor cells in culture and that the number of these cells are reduced in the olfactory bulb of NPY-deficient mice (Hansel et al., 2001).
The idea that NPY acts as a neuroprotective factor in our model is consistent with the delayed increases in NPY mRNA levels and the changes in the transcripts for its Y1 and Y2 receptors observed after administration of the toxic METH regimen. Specifically, the delayed increases in NPY mRNA levels, which occurred 3-7 d after METH administration, might represent a feedback regulatory response that serves to replenish NPY after METH-induced release of the neuropeptide from its intracellular compartments. This interpretation is consistent with the reduced levels of NPY-like immunoreactivity observed by Westwood and Hanson (1999) in the striata of rats killed 18 h after injections of toxic doses of METH. Finally, it is intriguing to suggest that the massive release of NPY associated with injections of toxic doses of METH might represent a protective response of the organism against extensive METH-induced apoptotic changes in the striatum.
Involvement of Y1 and Y2 receptors in the protective effects of NPY
Administration of either Y1 or Y2 agonists provided protection against METH-induced toxicity, with the Y2 agonist more effective than the Y1 agonist. The differential effects of the two agonists might be attributable to the number and/or distribution of these receptors, with Y2 receptors more numerous and widespread than Y1 receptors. Unfortunately, the lack of commercially available antibodies against NPY receptor subtypes prevented us from performing immunohistochemical studies to determine the protein levels of these receptors in subpopulations of neurons in saline-treated and METH-treated animals. It is also possible that the difference in potency observed between the two NPY agonists might be attributable to more efficient coupling of the Y2 receptors to intracellular signaling cascades. This interpretation is in agreement with the findings that Y2 receptors are more efficient than Y1 and Y5 receptors at decreasing glutamate release and in inhibiting calcium channels in the hippocampus (Silva et al., 2003).
Hypothetical mechanisms for NPY neuroprotective actions
METH-induced apoptosis is the result of a complex cascade of events triggered by the entrance of METH into the terminals of dopamine (DA) neurons (Davidson et al., 2001; Cadet et al., 2003). The influx of METH in the terminals results in a massive release of DA in the synaptic cleft. METH toxicity is thought to be related to the oxidation of DA (Cadet et al., 2003), which causes the formation of quinones and reactive species, such as superoxide radicals and hydrogen peroxide, with resulting neurodegeneration of dopaminergic terminals and neuronal apoptosis in the rodent striatum (Davidson et al., 2001; Cadet et al., 2003). These ideas are supported by the observations that METH-induced cell death and the destruction of striatal DA terminals are attenuated in mice that overexpress CuZn superoxide dismutase (SOD) (Cadet et al., 1994; Jayanthi et al., 1998; Deng and Cadet, 2000). Mice that overexpress manganese (Mn) SOD are also protected against METH effects on DA terminals (Maragos et al., 2000). Because NPY/nNOS neurons in the cortex and striatum show higher expression of MnSOD than other interneurons (Inagaki et al., 1991a,b; Gonzalez-Zulueta et al., 1998), it might be suggested that MnSOD is responsible for the sparing of these neurons from METH toxicity. However, because nNOS neurons from NPY knock-out mice are susceptible to METH-induced apoptosis (Fig. 5), it appears that the high-basal expression of MnSOD in striatal nNOS/NPY neurons, by itself, is not enough to rescue these neurons. Thus, NPY itself appears to be the main factor responsible for the observed protection against METH toxicity.
METH-induced formation of reactive oxygen species causes perturbations at levels of the mitochondria and the endoplasmic reticulum (ER) (Cadet et al., 2003). The ER is an organelle that is involved in the control of intracellular calcium homeostasis and that plays an important role in the induction of cell death (Ferri and Kroemer, 2001; Jayanthi et al., 2004). Because METH-induced cell death is, in part, related to dysregulations of intracellular calcium homeostasis (Uramura et al., 2000; Cadet et al., 2003) and NPY receptors activate signaling pathways that regulate intracellular calcium (Silva et al., 2002), it is possible that NPY might exert its protective effects via its action on calcium influx. This idea is consistent with the demonstration that activation of both Y1 and Y2 receptors in hippocampal cells blunts the increases in intracellular calcium concentration observed after KCl depolarization (Silva et al., 2001) and provides protection against kainate excitotoxicity (Silva et al., 2001, 2003).
Another possible mechanism for NPY-induced protection against METH toxicity might involve the activation of mitogen-activated protein kinase pathways, because both Y1 and Y2 receptors activate extracellular signal-regulated kinase 1 (ERK1) and ERK2 (Mannon and Raymond, 1998; Keffel et al., 1999; Mullins et al., 2002). Activation of the ERKs themselves by fructose-1,6-bisphosphate has also been shown to protect against neuronal death caused by hypoxia (Fahlman et al., 2002). These protective effects of the ERKs appear to occur via the maintenance of mitochondrial function (Lee et al., 2004).
Increases in body temperature are thought to be involved in METH toxicity on monoaminergic systems (for review, see Cadet et al., 2003). For example, environmental manipulations and some drugs that protect against METH toxicity also have been shown to prevent or attenuate METH-induced hyperthermia (Ali et al., 1994; Bowyer et al., 1994). In contrast, other reports have indicated that METH-induced hyperthermia and monoaminergic toxicity also can be dissociated (Battaglia et al., 2002; Halladay et al., 2003). The latter possibility is supported by reports that METH-induced hyperthermia was not affected by genetic manipulations that either exacerbate (Deng and Cadet, 1999) or protect against (Deng et al., 2002) neuronal apoptosis. Because NPY can regulate body temperature (Bouali et al., 1995; Richard, 1995), we tested the hypothesis that NPY causes protection against METH-induced neuronal apoptosis through reduction of METH-induced hyperthermia. The observations that NPY blocked early METH-induced increases in temperature suggest that the effects of NPY on temperature might be involved, in part, in its protective effects against METH-induced neuronal apoptosis.
In conclusion, when taken together with the previous reports on NPY neuroprotective effects, the present observations that NPY can protect against METH-induced neuronal apoptosis suggest a more general role for NPY as a potential neuroprotective agent that could be used against neurodegenerative processes that occur in the human brain.
Footnotes
We thank Dr. S. Jayanthi for help with preparation of this manuscript and Prof. M. Jaber for providing materials for the temperature measurements.
Correspondence should be addressed to Dr. Jean Lud Cadet, Molecular Neuropsychiatry Branch, Department of Health and Human Services, National Institutes of Health-National Institute on Drug Abuse, Intramural Research Program, 5500 Nathan Shock Drive, Baltimore, MD 21224. E-mail: jcadet{at}intra.nida.nih.gov.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255273-07$15.00/0
↵* N.T. and X.D. contributed equally to this work.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}