Abstracts
Iron imbalance/accumulation has been implicated in oxidative injury associated with many degenerative diseases such as hereditary hemochromatosis, beta-thalassemia, and Friedreich's ataxia. Mitochondria are particularly sensitive to iron-induced oxidative stress - high loads of iron cause extensive lipid peroxidation and membrane permeabilization in isolated mitochondria. Here we detected and characterized mitochondrial DNA damage in isolated rat liver mitochondria exposed to a Fe2+-citrate complex, a small molecular weight complex. Intense DNA fragmentation was induced after the incubation of mitochondria with the iron complex. The detection of 3' phosphoglycolate ends at the mtDNA strand breaks by a 32P-postlabeling assay, suggested the involvement of hydroxyl radical in the DNA fragmentation induced by Fe2+-citrate. Increased levels of 8-oxo-7,8-dihydro-2'-deoxyguanosine also suggested that Fe2+-citrate-induced oxidative stress causes mitochondrial DNA damage. In conclusion, our results show that iron-mediated lipid peroxidation was associated with intense mtDNA damage derived from the direct attack of reactive oxygen species.
iron; lipid peroxidation; mitochondria; mtDNA; oxidative stress
Desequilíbrio/acúmulo de ferro tem sido implicado em injúria oxidativa associada a diversas doenças degenerativas tais como, hemocromatose hereditária, beta-talassemia e ataxia de Friedreich. As mitocôndrias são particularmente sensíveis a estresse oxidativo induzido por ferro - um carregamento alto de ferro em mitocôndrias isoladas pode causar uma extensiva peroxidação lipídica e a permeabilização de membrana. Nesse estudo, nós detectamos e caracterizamos danos do DNA mitocondrial em mitocôndrias isoladas de fígado de rato, expostas ao complexo Fe2+-citrato, um dos complexos de baixo peso molecular. A intensa fragmentação do DNA foi induzida após a incubação das mitocôndrias com o complexo de ferro. A detecção de finais 3' de fosfoglicolato nas quebras de fitas de DNA mitocondrial pelo ensaio 32P-postlabeling sugere um envolvimento de radicais hidroxila na fragmentação do DNA induzido por complexo Fe2+-citrato. Os níveis elevados de 8-oxo-7,8-diidro-2'-desoxiguanosina também sugerem que o estresse oxidativo induzido por Fe2+-citrato causa danos no DNA mitocondrial. Em conclusão, nossos resultados mostram que a peroxidação lipídica mediada por ferro esteve associada com severos danos do DNA mitocondrial derivados de ataque direto das espécies reativas de oxigênio.
ferro; peroxidação lipídica; mitocôndrias; DNAmt; estresse oxidativo
BIOMEDICAL AND MEDICAL SCIENCES
Mitochondrial DNA damage associated with lipid peroxidation of the mitochondrial membrane induced by Fe2+-citrate
Andréa M. AlmeidaI,III; Clélia R.A. BertonciniII; Jiri BoreckýI; Nadja C. Souza-PintoI,IV; Aníbal E. VercesiI
IDepartamento de Patologia Clínica, Faculdade de Ciências Médicas (NCME), Universidade Estadual de Campinas, Cx. Postal 6111, 13083-970 Campinas, SP, Brasil
IILaboratório de Controle Genético, CEDEME, Universidade Federal de São Paulo, 04023-900 São Paulo, SP, Brasil
IIIDepartamento de Biologia Vegetal, Universidade Federal de Viçosa, 36570-000 Viçosa, MG, Brasil
IVLaboratory of Molecular Gerontology, National Institute on Aging, Baltimore, 21224-6825 Maryland, USA
ABSTRACT
Iron imbalance/accumulation has been implicated in oxidative injury associated with many degenerative diseases such as hereditary hemochromatosis, b-thalassemia, and Friedreich's ataxia. Mitochondria are particularly sensitive to iron-induced oxidative stress - high loads of iron cause extensive lipid peroxidation and membrane permeabilization in isolated mitochondria. Here we detected and characterized mitochondrial DNA damage in isolated rat liver mitochondria exposed to a Fe2+-citrate complex, a small molecular weight complex. Intense DNA fragmentation was induced after the incubation of mitochondria with the iron complex. The detection of 3' phosphoglycolate ends at the mtDNA strand breaks by a 32P-postlabeling assay, suggested the involvement of hydroxyl radical in the DNA fragmentation induced by Fe2+-citrate. Increased levels of 8-oxo-7,8-dihydro-2'-deoxyguanosine also suggested that Fe2+-citrate-induced oxidative stress causes mitochondrial DNA damage. In conclusion, our results show that iron-mediated lipid peroxidation was associated with intense mtDNA damage derived from the direct attack of reactive oxygen species.
Key words: iron, lipid peroxidation, mitochondria, mtDNA, oxidative stress.
RESUMO
Desequilíbrio/acúmulo de ferro tem sido implicado em injúria oxidativa associada a diversas doenças degenerativas tais como, hemocromatose hereditária, b-talassemia e ataxia de Friedreich. As mitocôndrias são particularmente sensíveis a estresse oxidativo induzido por ferro - um carregamento alto de ferro em mitocôndrias isoladas pode causar uma extensiva peroxidação lipídica e a permeabilização de membrana. Nesse estudo, nós detectamos e caracterizamos danos do DNA mitocondrial em mitocôndrias isoladas de fígado de rato, expostas ao complexo Fe2+-citrato, um dos complexos de baixo peso molecular. A intensa fragmentação do DNA foi induzida após a incubação das mitocôndrias com o complexo de ferro. A detecção de finais 3' de fosfoglicolato nas quebras de fitas de DNA mitocondrial pelo ensaio 32P-postlabeling sugere um envolvimento de radicais hidroxila na fragmentação do DNA induzido por complexo Fe2+-citrato. Os níveis elevados de 8-oxo-7,8-diidro-2'-desoxiguanosina também sugerem que o estresse oxidativo induzido por Fe2+-citrato causa danos no DNA mitocondrial. Em conclusão, nossos resultados mostram que a peroxidação lipídica mediada por ferro esteve associada com severos danos do DNA mitocondrial derivados de ataque direto das espécies reativas de oxigênio.
Palavras-chave: ferro, peroxidação lipídica, mitocôndrias, DNAmt, estresse oxidativo.
INTRODUCTION
Iron is an essential element, necessary for the catalytic activity of a large number of enzymes. Mitochondria play a crucial role in iron homeostasis (Foury and Talibi 2001) and synthesis of iron-sulfur clusters (Beinert et al. 1997) and heme (Ryter and Tyrrell 2000). Unbalanced iron homeostasis can have severe consequences. Iron overload originates usually from excessive dietary intake, multiple blood transfusions, or genetic diseases associated with alterations of expression and/or mutations in genes encoding ferroportin (hemochromatosis, Adams et al. 2003) and globin (b-thalassemia, May and Sadelain 2001). Iron deficiency caused by altered expression of frataxin, a mitochondrial iron storage protein, leads to Friedreich's ataxia (Adamec et al. 2000). The studies on iron pathology in several cellular and animal models suggested that oxidative damage to membranes should be a crucial event in iron toxicity. Evidences from studies in vitro with isolated organelles (Burkitt and Gilbert 1989), cells (Poli et al. 1987), and tissue homogenates (Arthur et al. 1988) implicated the hydroxyl radical species as the central mediator of most cellular damage.
Hydroxyl radical and other reactive oxygen species (ROS) are constantly generated in every cell during its normal life under aerobic conditions. It is well recognized that several physiological and pathological processes are linked to oxidative stress, defined as an imbalance between the rates of ROS formation and detoxification (Halliwell and Gutteridge 1999). In this regard, mitochondria are important cellular sites of ROS production and oxidative damage caused by these species (reviewed by Kowaltowski et al. 2001).
Lipid peroxidation is generally thought to be the major mechanism of biomembrane injury promoted by ROS and is strongly affected by transition metals that catalyze many of the reactions involved in this process (Halliwell and Gutteridge 1999). Iron complexes induce extensive lipid peroxidation of the mitochondrial membrane, initiated by ROS generated through Fenton chemistry (Liochev 1999). Lipid peroxidation is considered to be the main form of irreversible permeabilization of the mitochondrial membrane caused by Ca2+ in the presence of Fe2+-citrate (Castilho et al. 1994), Fe2+-ATP (Hermes-Lima et al. 1995), or high phosphate concentrations (Kowaltowski et al. 1996). Under these conditions, a series of alterations, including a drop in mitochondrial membrane potential, an increase in inner mitochondrial membrane permeability, mitochondrial swelling, and the loss of matrix components, occur (Castilho et al. 1994, 1999, Hermes-Lima et al. 1995, Kowaltowski et al. 1996, 2001). These observations have suggested a critical role for membrane lipid peroxidation in mitochondrial dysfunction under oxidative stress.
In this study, we investigated whether exposure of isolated rat liver mitochondria (RLM) to iron complexes can also induce damage to mitochondrial DNA (mtDNA). It has been proposed that accumulation of oxidative damage and mutations in mtDNA play a pivotal role in degenerative diseases and in the aging process (Hudson et al. 1998, Souza-Pinto and Bohr 2001). Furthermore, mtDNA seems to be more susceptible to attack by ROS than nuclear DNA because it lacks protective structural proteins and it is attached transiently to the inner mitochondrial membrane (Clayton 1982) close to the site of ROS generation. Our results show that the exposure of RLM to Fe2+-citrate, under conditions in which the mitochondrial inner membrane is permeabilized, induces extensive oxidative damage to mtDNA. We detected DNA fragmentation, increased levels of the oxidized base 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxo-dG) and 3'-phosphoglycolate termini (3'-PG) at the strand breaks. Both 8-oxo-dG and 3'-phosphoglycolate termini are indicative of hydroxyl radical attack.
MATERIALS AND METHODS
ISOLATION OF RAT LIVER MITOCHONDRIA
Mitochondria were isolated from the livers of adult male Wistar rats fasted overnight by conventional differential centrifugation as described by Kowaltowski et al. (1996). Protein concentrations were determined by the biuret assay (Gornall et al. 1949), using bovine serum albumin as standard.
STANDARD INCUBATION PROCEDURE
All incubations were carried out at 25ºC, with continuous stirring, in a standard reaction medium containing 125 mM sucrose, 65 mM KCl, and 10 mM HEPES, pH 7.2. For the treatments with Fe-citrate, mitochondria were pre-incubated in reaction medium containing 2 mM citrate for 2 min before the addition of 50 µM Fe2+. Other additions are indicated in the figure legends. The results shown are representatives of at least three independent experiments.
PURIFICATION OF MITOCHONDRIAL DNA
mtDNA was isolated by a modified method described previously by Palva and Palva (1985).Mitochondria (80 mg protein) were incubated under desired conditions and then pelleted by centrifugation (15,000 × g, 15 min). The pellets were resuspended in a final volume of 3.0 ml in ice-cold buffer containing 50 mM glucose, 1.5 mM EDTA, and 25 mM Tris-HCl (pH 7.4). Mitochondria were lysed and contaminating nuclear DNA denatured by addition of 500 µl of 10% SDS. Nuclear DNA was precipitated by 2 ml of 3.0 M potassium acetate and removed by centrifugation (10,000 × g, 10 min). Samples were further incubated with proteinase K (7.5 mg/ml) at 37ºC for 1 h and mtDNA was extracted twice with Tris-buffered phenol:chloroform:isoamyl alcohol (25:24:1) and once with chloroform:isoamyl alcohol (24:1). mtDNA was precipitated from the aqueous phase by the addition of two volumes of 100% ethanol and resuspended in 200 µl of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The samples were treated with ribonuclease A (100 µg/ml) for 2 h and once more extracted with chloroform:isoamyl alcohol (24:1). mtDNA was precipitated, washed, suspended in 10 mM Tris-HCl (pH 7.0), and quantified by spectrophotometric analysis. mtDNA samples were separated in 0.8% agarose gels, stained with ethidium bromide, and visualized by a UV transilluminator.
DETERMINATION OF MITOCHONDRIAL SWELLING
Mitochondrial swelling was monitored as a decrease of sample turbidity at 520 nm using a SLM Aminco DW 2000 spectrophotometer, as described by Beavis and Vercesi (1992).
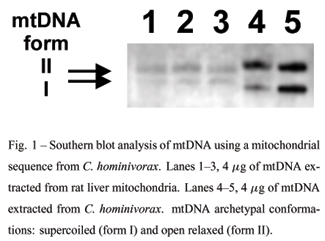
SOUTHERN BLOTTING
DNA samples (4 µg) were separated by electrophoresis and transferred to Hybond-N membranes (Amersham), as described by Vargas and Azeredo-Espin (1995). Membranes were hybridized with probes generated by random-primed DNA labeling with digoxigenin-dUTP (DIG DNA labeling and detection kit, Boehringer Mannheim). The probe raised against Cochliomyia hominivorax mtDNA (generously supplied by Dr. A.M.L. Azeredo-Espin, Universidade Estadual de Campinas, Brazil) has been described previously (Vargas and Azeredo-Espin 1995).
32P-POSTLABELING ASSAY
Detection of 3'-PG termini was described previously (Bertoncini and Meneghini 1995). mtDNA (10 µg) was digested with 0.08 U DNase I, 0.08 U phosphodiesterase I, and 0.8 U alkaline phosphatase in 100 µl of a digestion buffer (4 mM MgCl2, 10 mM Tris-HCl, pH 7.5) at 37ºC overnight. Subsequently, 0.04 U phosphodiesterase I and 0.2 U alkaline phosphatase were added and incubation continued for another 6 h. The enzymes were precipitated by the addition of three volumes of cold ethanol and removed by centrifugation (10,000 x g, 15 min). The supernatant was dried under vacuum and digested DNA resuspended in 100 µl of bi-distilled water, heated at 100ºC for 10 min to inactivate residual nucleases and phosphatases, and stored at -20ºC. For 5' 32P-labeling, 1 µg of digested DNA was incubated at 37ºC with 2 pmol [g-32P]ATP and 5 U polynucleotide kinase, in a 20-µl reaction containing 10 mM MgCl2, 5 mM dithiothreitol, 0.1 mM spermidine, 0.1 mM EDTA, and 50 mM Tris-HCl, pH 7.5, for 40-60 min. The excess of [g-32P]ATP was scavenged by sample incubation with 2 µg of oligonucleotide, (dN)21 or (dN)38, and 2.5 U polynucleotide kinase for 30 min. The samples were diluted with 1 volume of formamide buffer (90% formamide, 0.02% bromophenol blue, and 0.02% xylene cyanol in 1x TBE), heated at 97ºC for 3 min, cooled on ice, and loaded onto a 15% polyacrylamide/7 M urea sequencing gels. The samples were run until the bromophenol blue had migrated 15 cm. The gel was dried under vacuum and the radiolabeled products were visualized by autoradiography. To quantify the 3'-PG termini, the radioactive bands were cut out and their Cerenkov radiation counted. The resulting value was divided by the total counts in the lane and multiplied by the original amount of [g-32P]ATP in the phosphorylation reaction mixture loaded onto the gel (2 pmol).
HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY AND ELECTROCHEMICAL DETECTION
mtDNA samples (200 µg) were digested with nuclease P1 (10 µg/200 µg of DNA) in 200 µl of 50 mM Tris-HCl, 20 mM sodium acetate, pH 7.4, at 70ºC for 10 min. The samples were dephosphorylated with 1 U alkaline phosphatase in 100 mM Tris-HCl buffer (pH 7.4) for 2 h. The resulting hydrolysate was filtered in microcentrifuge filters (0.22 µm pore size) and stored at -20ºC prior to analysis. Complete mtDNA hydrolysis was monitored by electrophoresis in agarose gels stained with ethidium bromide. Nucleosides obtained by mtDNA hydrolysis were analyzed by HPLC as described by Shigenaga et al. (1990) with some modifications. The HPLC system used (Shimadzu) consisted of a LC 10AD pump, a model 7161 Rheodyne injector and an L-ECD-6A electrochemical detector. Chromatography was carried out on a C-18 Sephadex reverse phase column (5 µm, 4.6 × 250 mm) at flow rate of 1 ml/min. The mobile phase consisted of deaerated 50 mM KH2PO4 buffer, pH 5.5, containing 10% methanol and 2.5 mM EDTA. The elution profile was monitored by UV detection at 285 nm in series with electrochemical detection at +600 mV. The analyses were performed using specific software (EZChromTM Chromatography data system, version 6.2).
CHEMICALS
[g-32P]ATP (10 mCi/ml, 5 Ci/mol) was from Amersham. Oligonucleotides were synthesized by National Biosciences, Inc. All other reagents were ACS grade from Sigma. Phenol (molecular biology grade, Sigma, cat no. P1037) was buffered with Tris-base to pH > 8.
RESULTS
EFFECT OF FE2+-CITRATE ON MTDNA INTEGRITY
To avoid the contamination of the mitochondrial DNA preparation with the significantly more abundant nuclear DNA, mtDNA used here was extracted directly from isolated rat liver mitochondria (RLM). Southern hybridization of isolated liver mtDNA with a HindIII fragment of mtDNA from C. hominivorax (Vargas and Azeredo-Espin 1995) revealed two bands in the lanes containing the samples extracted from RLM (Fig. 1, lanes 1-3), corresponding to the supercoiled and closed circular forms of mtDNA, comparable to bands of mtDNA isolated from C. hominivorax (lanes 4-5). No hybridization with a probe for a conserved nuclear sequence was observed (not shown), indicating that the samples were free of nuclear DNA contamination.
Incubation of 1 mg/ml RLM with the Fe2+-citrate complex resulted in extensive mitochondrial swelling (Fig. 2A, lane b) that was not observed in the absence of the complex (Fig. 2A, lane a). This swelling was completely prevented by the addition of 20 µM butylhydroxytoluene (BHT, Fig. 2A, lane c), a lipophilic antioxidant that efficiently inhibits free-radical propagation during lipid peroxidation (Castilho et al. 1994). These results confirmed that the Fe2+-citrate-induced mitochondrial swelling under our experimental conditions is associated with mitochondrial membrane disruption due to lipidperoxidation. The integrity of mtDNA obtained from RLM incubated under the same conditions was analyzed by agarose gel electrophoresis. In samples obtained from control RLM, incubated with EGTA alone (Fig. 2B, lane 2), mtDNA was detected in its three archetypal conformations: supercoiled (form I), open relaxed (form II) and linear (form III). In contrast, a drastic decrease in the 16-kb band amount and the presence of smear were observed in the samples obtained from RLM incubated with the Fe2+-citrate complex (Fig. 2B, lane 3), indicating extensive fragmentation of the mtDNA. The presence of 20 µM BHT in incubation medium completely prevented the mtDNA damage, as shown by the reappearance of the major 16 kb band (compare lane 3 with the control mtDNA in lane 1).
INVOLVEMENT OF OXIDATIVE STRESS IN THE MECHANISM OF MTDNA DAMAGE INDUCED BY IRON COMPLEX
We investigated whether oxidative damage occurred in mtDNA from RLM treated with the Fe2+-citrate complex. ROS attack to DNA can lead to a wide variety of modifications. DNA strand breaks and base oxidations are the most abundant alterations. Single strand breaks can be generated in DNA by the direct attack of hydroxyl radical (Henner et al. 1983), resulting in 3'-phosphoglycolate termini, non-ligatable ends that are per-se a type of DNA damage. To investigate whether iron overload generates 3'-PG termini in mtDNA, DNA was extracted from control and Fe2+-citrate-treated RLM and 3'-PG termini levels determined by 32P-postlabeling. A typical autoradiogram obtained from mtDNA samples from control and treated RLM is shown in Fig. 3A. The indicated bands were assigned to deoxynucleoside 5'-phosphate-3'-phosphoglycolate (d-pNpg) and 32P-ortho-phosphate (bottom of the gel). The oligodeoxynucleotides added at the end of the post-labeling assay to scavenge the excess [g-32P]ATP are seen at the top of the autoradiogram. Lane 1 shows g-irradiated (150 Gy) calf thymus DNA as a positive control of 3'-PG termini production. Mitochondrial DNA from RLM incubated in the presence of 50 µM Fe and 2 mM citrate had higher levels of d-pNpgs than control mitochondria (compare lanes 4 and 6 with lanes 3 and 5). Quantification of the bands (panel B), indicated a relative intensity of 67.84±7.65 fmol of d-pNpg/µg DNA in the Fe2+-citrate-treated samples compared to the control samples (42.19±3.00 fmol of d-pNpg/µg DNA). This represents an increase of about 60% in 3'-PG termini, indicating that iron-induced mtDNA damage occurs via hydroxyl radical generation.
Furthermore, the levels of 8-oxo-dG were quantified by HPLC in mtDNA from RLM incubated under the conditions described above (Fig. 3, Panel C). This modified base is one of the main products detected in cellular DNA after oxidative stress; it is highly mutagenic and leads to G to T tranversions (Grollman and Moriya 1993). The quantification of 8-oxo-dG by the relative area of the peaks showed that mtDNA from the Fe2+-citrate treated mitochondria had almost three times more 8-oxo-dG (40.96±3.76 residues/106 dG) than mtDNA from control mitochondria (14.59 ±0.05 residues/106 dG). Together with 3'-PG termini analysis, these results suggest that iron-induced oxidative stress leads to the accumulation of oxidative damage in mtDNA.
DISCUSSION
The experimental model of isolated mitochondria used in this study simulates situations of either iron overload in the cell or deficiency of ferritin, where increasing non-protein-bound iron is chelated preferably by intracellular citrate or ADP. Fe2+-citrate and Fe2+-ADP complexes are particularly interesting for studies with mitochondrial membranes, because of their high probability of being present in the mitochondrial matrix under physiological and pathological conditions (Bacon and Britton 1990, Castilho et al. 1994, Minotti and Aust 1987, Halliwell and Gutteridge 1999). Chelation of iron by citrate was found to induce the auto oxidation of Fe2+ that results in the peroxidation of lipids in mitochondria and oxidative damage (Chen et al.2002). Much of the tissue damage in ischemia-reperfusion injury is also due to increased levels of cytosolic iron and can be further potentiated by a concomitant 4- to 6-fold increase in intracellular citrate concentration (Hassel et al. 1998).
In this study, we evaluated the effect of a metal-catalyzed oxidation system, the Fe2+-citrate complex (Pierre and Gautier-Luneau 2000), on mtDNA damage and determined the extent and nature of this damage, employing the experimental system used previously to demonstrate that Fe2+-citrate induced lipid peroxidation in isolated RLM (Castilho et al. 1994). This effect was associated with extensive mitochondrial swelling and membrane potential collapse, both sensitive to BHT, an antioxidant that stops the free-radical propagation of lipid peroxidation reaction among other processes (Castilho et al. 1994). Our results show that the exposure of isolated mitochondria to oxidative stress generated by the Fe2+-citrate complex also induced extensive mtDNA damage. This was demonstrated by the extensive DNA fragmentation and the increase in the levels of 3'-PG termini and of 8-oxo-dG in Fe2+-citrate-treated mitochondria.
Our previous study (Castilho et al. 1994) demonstrated that Fe2+-citrate induces extensive mitochondrial inner membrane permeabilization through membrane lipid peroxidation. BHT, a potent lipid chain-breaking peroxidation inhibitor, completely prevented these effects. Note that the ratio between BHT/mitochondrial protein (20 nmol/mg protein) was below the range of BHT-induced swelling (60-120 nmol/mg protein, Sokolove and Haley 1996) and thus BHT itself did not contribute to the swelling rates. In the present study, BHT also prevented DNA fragmentation under the same experimental conditions, suggesting that membrane permeabilization and DNA breakage are related phenomena. a-tocopherol, another lipid peroxidation inhibitor, also prevented both lipid peroxidation and mtDNA fragmentation induced by FeCl2 (Hruszkewycz 1988).
There is some debate as to which oxidant radical is involved in Fe2+-citrate-mediated lipid peroxidation-superoxide anion (Minotti and Aust 1987) and/or hydroxyl radical (Gutteridge 1991). The increased levels of 3'-PG termini (Fig. 3) and 8-oxo-dG in mtDNA from Fe2+-citrate-treated mitochondria presented in this study favor the involvement of hydroxyl radical in the mechanism of mtDNA damage. The 3'-PG termini are the end product of hydroxyl radical attack on the deoxyribose ring (Henner et al. 1983), which leads to a break in the phosphate backbone of mtDNA. Thus, the higher levels of 3'-PG in mtDNA obtained from Fe2+-citrate-treated mitochondria indicate that, at least to some degree, DNA fragmentation arose directly from hydroxyl radical attack or from an oxidant species with similar reactivity. Note that the low amounts of d-pNpg visible in DNA from control mitochondria may arise either during DNA extraction or may be present in DNA under normal cellular metabolic conditions (Meniel and Waters 1999).
8-Oxo-dG formation is also a result of hydroxyl radical hydrogen abstraction. Indeed, the hydroxylation of guanine is considered to be a biomarker of radical damage (Helbock et al. 1999), and 8-oxo-dG was reported to accumulate in mtDNA from animals exposed to oxidative stress and from aged animals (Hamilton et al. 2001). Although the absolute values of 8-oxo-dG detected in mtDNA from various sources are still very controversial due to the introduction of artificial oxidation, there is evidence that accumulation of 8-oxo-dG is associated with situations of oxidative stress (for review, see Souza-Pinto and Bohr 2001).
Since the hydroxyl radical is a very short-lived species (Pryor 1986), its formation has to occur near to the subsequent reaction site (reviewed by Meneghini 1997). mtDNA is transiently attached to the mitochondrial inner membrane (Clayton 1982) and transition metals bound to mtDNA can act as Fenton reaction centers, catalyzing hydroxyl radical formation from H2O2in situ (Lloyd and Phillips 1999). Iron may be involved in the initiation of mitochondrial peroxidation through the formation of the transition state complex Fe3+-O2- (perferryl), complexes of oxygen with both ferric and ferrous ion, or of hydroxyl radical-like species (Kachur et al. 1998, Pierre and Gautier-Luneau 2000) following the oxidation of Fe2+-citrate to Fe3+-citrate. We have shown (Hermes-Lima et al. 1995) that the formation of lipid peroxidation initiator species from an analogous iron complex (Fe2+-ATP) is H2O2-dependent and superoxide-independent, suggesting that iron-catalyzed Fenton reactions in situ may play a significant role in the mechanism of mtDNA damage.
In conclusion, our results provide strong evidence that mtDNA represents a critical target of iron-promoted free radical formation and lipid peroxidation in conditions related to abnormal intracellular iron distribution and/or iron overload, reported to occur in hereditary hemochromatosis, b-thalassemia, and Friedreich's ataxia (Britton et al. 1994, Gordon 2000). The accumulation of mtDNA mutations has been reported to lead to mitochondrial dysfunction and has been recently associated with a variety of cancers (Fliss et al. 2000) and aging (Trifunovic et al. 2004). Although mammalian mitochondria can remove various kinds of DNA damage from their genomes by the base excision repair pathway (Hashiguchi et al. 2004), the capacity of this mtDNA repair is limited. In this context, it is possible that extensive iron-catalyzed mitochondrial membrane peroxidation can contribute to the accumulation of mtDNA mutations and/or deletions.
ACKNOWLEDGMENTS
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grants No. 98/13012-5 and 97/11841-1),Conselho Nacional de Desenvolvimento Científico e Tecnológico and Programa de Apoio a Núcleos de Excelência (CNPq/PRONEX, grants No. 66.1192/ 1997-2 and 305381/02-4 NV). We thank Dr. Janice Onuki and Dr. Paolo Di Mascio for help with the HPLC analysis, Dr. Ana Cláudia Lessinger for help with the Southern analysis and Dr. Rogerio Meneghini for the use of his lab for the postlabeling assay. We also thank Mrs. Luis H.G. Ribeiro and Matheus P.C. Vercesi for excellent technical assistance.
Manuscript received on October 30, 2005; accepted for publication on November 24, 2005; contributed by ANíBAL E. VERCESI*
* Member Academia Brasileira de Ciências
Correspondence to: Jiri Borecký
E-mail: jiri@unicamp.br
- ADAMEC J, RUSNAK F, OWEN WG, NAYLOR S, BENSON LM, GACY AM AND ISAYA G. 2000. Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia. Am J Human Genet 67: 549-562.
- ADAMS PC, BARBIN YP, KHAN ZA AND CHAKRABARTI S. 2003. Expression of ferroportin in hemochromatosis liver. Blood Cells Mol Dis 31: 256-261.
- ARTHUR JR, MCPHAIL DB AND GOODMAN BA. 1988. Spin trapping of free radicals in homogenates of heart from selenium and vitamin E deficient rats. Free Rad Res Commun 4: 311-315.
- BACON BR AND BRITTON RS. 1990.The pathology of hepatic iron overload: a free radical-mediated process? Hepatology 11:127-137.
- BEAVIS AD AND VERCESI AE. 1992. Anion uniport in plant mitochondria is mediated by a Mg(2+)-insensitive inner membrane anion channel. J Biol Chem 267: 3079-3087.
- BEINERT H, HOLM RH AND MUNCK E. 1997. Iron-sulfur clusters: Nature's modular, multipurposestructures. Science 277: 653-659.
- BERTONCINI CRA AND MENEGHINI R. 1995. DNA strand breaks produced by oxidative stress in mammalian cells exhibit 3'-phosphoglycolate termini.Nucl Acid Res 23: 2995-3002.
- BRITTON RS, BACON BR AND TAVILL AS. 1994. Mechanisms of iron toxicity. In: BROCK JH ET AL. (Eds), Iron metabolism in health and disease, London: W.B. Saunders, p. 311-351.
- BURKITT MJ AND GILBERT BC. 1989. The control of iron-induced oxidative damage in isolated rat-liver mitochondria by respiration state and ascorbate. Free Rad Res Commun 5: 333-344.
- CASTILHO RF, MEINICKE AR, ALMEIDA AM, HERMES-LIMA M AND VERCESI AE. 1994. Oxidative damage of mitochondria induced by Fe(II)citrate is potentiated by Ca2+ and includes lipid peroxidation and alterations in membrane proteins. Arch Biochem Biophys 308: 158-164.
- CASTILHO RF, MEINICKE AR, VERCESI AE AND HERMES-LIMA M. 1999. The role of Fe(III) in Fe(II)-citrate mediated peroxidation of mitochondrial membrane lipids. Mol Cell Biochem 196: 163-168.
- CHEN OS, HEMENWAY S AND KAPLAN J. 2002. Genetic analysis of iron citrate toxicity in yeast: implications for mammalian iron homeostasis. Proc Natl Acad Sci USA 99: 16922-16927.
- CLAYTON PA. 1982. Replication of animal mitochondrial DNA. Cell 28: 693-705.
- FLISS MS, USADEL H, CABALLERO OL, WU L, BUTA MR, ELEFF SM, JEN J AND SIDRANSKY D. 2000. Facile detection of mitochondrial DNA mutationsin tumors and body fluids. Science 287: 2017-2019.
- FOURY F AND TALIBI D. 2001. Mitochondrial control of iron homeostasis. A genome wide analysis of gene expression in a yeast frataxin-deficient strain. J Biol Chem 276: 7762-7768.
- GORDON N. 2000. Friedreich's ataxia and iron metabolism. Brain Develop 22: 465-468.
- GORNALL AG, BARDAWILL CJ AND DAVID MM. 1949. Determination of serum proteins by meansof biuret reaction. J Biol Chem 177: 751-766.
- GROLLMAN AP AND MORIYA M. 1993. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet 9: 246-249.
- GUTTERIDGE JM. 1991. Hydroxyl radical formation from the auto-reduction of a ferric citrate complex. Free Rad Biol Med 11: 401-406.
- HALLIWELL B AND GUTTERIDGE JMC. 1999. Free radicals in biology and medicine, Oxford University Press Inc, New York, USA, 246 p.
- HAMILTON ML, VAN REMMEN H, DRAKE JA, YANG H, GUO ZM, KEWITT K, WALTER CA AND RICHARDSON A. 2001. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci USA 98: 10469-10474.
- HASHIGUCHI K, BOHR VA AND DE SOUZA-PINTO NC. 2004. Oxidative stress and mitochondrial DNA repair: implications for NRTIs induced DNA damage. Mitochondrion 4: 215-222.
- HASSEL B, ILEBEKK A AND TONNESSEN T. 1998. Cardiac accumulation of citrate during brief myocardial ischaemia and reperfusion in the pig in vivo. Acta Physiol Scand 164: 53-59.
- HELBOCK HJ, BECKMAN KB AND AMES BC. 1999. 8-Hydroxydeoxyguanosine as 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol 300: 156-166.
- HENNER WD, GRUNBERG SM AND HASELTINE WA. 1983. Enzyme action at 3'termini of ionizing radiation-induced DNA strand breaks. J Biol Chem 258: 15198-15205.
- HERMES-LIMA M, CASTILHO RF, MEINICKE AR AND VERCESI AE. 1995. Characteristics of Fe(II)ATP complex-induced damage to the rat liver mitochondria. Mol Cell Biochem 145: 53-60.
- HRUSZKEWYCZ AM. 1988. Evidence for mitochondrial DNA damage by lipid peroxidation. Biochem Biophys Res Commun 153: 191-197.
- HUDSON EK, HOGUE BA, SOUZA-PINTO NC, CROTEAU DL AND ANSON RM. 1998. Age-associated change in mitochondrial DNA damage. Free Rad Res Commun 29: 573-579.
- KACHUR AV, TUTTLE SW AND BIAGLOW JE. 1998. Auto-oxidation of ferrous ion complexes: a method for the generation of hydroxyl radicals. Rad Res 50: 475-482.
- KOWALTOWSKI AJ, CASTILHO RF, GRIJALBA MT, BECHARA EJH AND VERCESI AE. 1996. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J Biol Chem 271:2929-2934.
- KOWALTOWSKI AJ, CASTILHO RF AND VERCESI AE. 2001. Mitochondrial permeability transition and oxidative stress. FEBS Lett 495: 12-15.
- LIOCHEV SI. 1999. The mechanism of 'Fenton-like' reactions and their importance for biological systems. A biologist's view. Met Ions Biol Syst 36: 1-39.
- LLOYD DR AND PHILLIPS DH. 1999. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links. Mutat Res 424: 23-36.
- MAY C AND SADELAIN M. 2001. A promising genetic approach to the treatment of b-thalassemia. Trends Cardiovasc Med 11: 276-280.
- MENEGHINI R. 1997. Iron homeostasis, oxidative stress, and DNA damage. Free Rad Biol Med 23: 783-792.
- MENIEL V AND WATERS R. 1999. Spontaneous and photosensitiser-induced DNA single-strand breaksand formamidopyrimidine-DNA glycosylase sensitive sites at nucleotide resolution in the nuclear and mitochondrial DNA of Saccharomyces cerevisiaeNucl Acids Res 27: 822-830.
- MINOTTI G AND AUST SD. 1987. An investigation into the mechanism of citrate-Fe2+-dependent lipid peroxidation. Free Rad Biol Med 3: 379-387.
- PALVA TK AND PALVA ET. 1985. Rapid Isolation of animal mitochondrial DNA by alkaline extraction. FEBS Lett 192: 267-270.
- PIERRE JL AND GAUTIER-LUNEAU I. 2000. Iron and citric acid: a fuzzy chemistry of ubiquitous biological relevance. Biometals 13: 91-96.
- POLI G, ALBANO E AND DIANZANI MU. 1987. The Role of Lipid-Peroxidation in Liver-Damage. Chem Phys Lipids 45: 117-142.
- PRYOR WA. 1986. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol 48: 657-667.
- RYTER SW AND TYRRELL RM. 2000. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Rad Biol Med 28: 289-309.
- SHIGENAGA MK, PARK J, CUNDY KC, GIMENO CJ AND AMES B. 1990. In vivo oxidative DNA damage: measurement of 8-hydroxy-2'-deoxyguanosine in DNA and urine by high-performance liquid chromatography with electrochemical detection. Methods Enzymol 186: 521-530.
- SOKOLOVE PM AND HALEY LM. 1996. Butylated hydroxytoluene and inorganic phosphate plus Ca2+ increase mitochondrial permeability via mutually exclusive mechanisms. J Bioenerg Biomembr 28: 199-206.
- SOUZA-PINTO NC AND BOHR VA. 2001. DNA repair in mammalian mitochondria. In: RODRIGUES H AND CUTLER RG (Eds), Oxidative stress andaging: advances in basic science, diagnostics, andintervention. World Scientific Publishing Co., Chapter 43.
- TRIFUNOVIC A ET AL. 2004. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417-423.
- VARGAS MEI AND AZEREDO-ESPIN AML. 1995. Genetic variability in mitochondrial DNA of the screw worm, Cochliomyia hominivorax (diptera: calliphoridae), from Brazil. Biochem Genet 33: 237-256.
Publication Dates
-
Publication in this collection
18 Aug 2006 -
Date of issue
Sept 2006
History
-
Accepted
24 Nov 2005 -
Received
30 Oct 2005