Abstracts
Inherited epidermolysis bullosa (EB) is a heterogeneous group of genetic disorders that present with skin and, in some cases, mucosal fragility, predisposing patients to the development of blisters and/or erosions after minimal trauma or friction. Children with a recurrent history of these kinds of lesions or neonates that present them in the absence of another reasonable explanation should be investigated. Diagnosis must be based on clinical and histopathological findings. To date, management of inherited EB basically consists in avoiding traumas that trigger lesions, as well as preventing infection and facilitating healing of the wounds with the systematic use of bandages.
Epidermolysis bullosa; Epidermolysis bullosa dystrophica; Epidermolysis bullosa, junctional; Epidermolysis bullosa simplex
A epidermólise bolhosa hereditária (EBH) compreende um grupo heterogêneo de desordens genéticas que têm em comum a fragilidade cutânea e, em alguns casos mucosa, predispondo ao desenvolvimento de bolhas e/ou erosões após fricção ou trauma mínimo. Crianças com história recorrente deste tipo de lesão ou neonatos que as apresentem na ausência de outra explicação plausível devem ser investigados. O diagnóstico deve se basear em achados clínicos e histopatológicos. Até o presente momento, o manejo da EBH consiste basicamente em evitar os traumas desencadeadores das lesões, bem como evitar a infecção e facilitar a cicatrização das feridas com o uso sistemático de curativos.
Epidermólise bolhosa; Epidermólise bolhosa distrófica; Epidermólise bolhosa juncional; Epidermólise bolhosa simples
INTRODUCTION
Inherited epidermolysis bullosa (EB) is a group of genetically transmitted skin disorders characterized by spontaneous blistering or blistering caused by minor trauma.11. Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann N Y Acad Sci. 2010;1194:213-22. , 22. Pai S, Marinkovich MP. Epidermolysis bullosa: new and emerging trends. Am J Clin Dermatol. 2002;3:371-80. There are three classic types of inherited EB (simplex, junctional and dystrophic). They are differentiated by the level of blister cleavage and subdivided according to the pattern of genetic inheritance, morphology/topography of lesions and genetic mutation involved. After the Third International Consensus on Diagnosis and Classification of Inherited EB, there was the addition of a fourth entity to the group of inherited EB - Kindler syndrome (KS) previously considered a photosensitive poikiloderma.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 44. Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30:70-7. Currently, over 30 phenotypiccally and genetically distinct nosological entities have been described (Table 1).11. Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann N Y Acad Sci. 2010;1194:213-22. , 33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. Figure 1 illustrates the key molecules involved in the pathogenesis of EB.
Representation of the proteins affected in different types of inherited epidermolysis bullosa
Estimates of the incidence and prevalence of inherited EB were made using different techniques for various populations worldwide. More rigorous studies are derived from the National Epidermolysis Bullosa Registry in the United States, which estimated an incidence of 50 cases of the disease per 1,000,000 live births; 92% of these cases are the simplex variant, 5% dystrophic, 1% junctional, and 2% unclassified.55. Fine JD, Bauer EA, McGuire J, Moshell A. Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances and the findings of the National Epidermolysis Bullosa Registry. Baltimore: The Johns Hopkins University Press; 1999. Although there are no epidemiological data on the disease in Brazil, it is suggested that there is no geographical or racial interference, since studies in other countries have not shown significant variations.66. Risser J, Lewis K, Weinstock MA. Mortality of bullous skin disorders from 1979 through 2002 in the United States. Arch Dermatol. 2009;145:1005-8. Despite their rarity, inherited EB cause a huge impact on the lives of patients and their families, due to physical pain, emotional suffering or economic repercussions. 77. Margari F, Lecce PA, Santamato W, Ventura P, Sportelli N, Annicchiarico G, et al. Psychiatric Symptoms and Quality of Life in Patients Affected by Epidermolysis Bullosa. J Clin Psychol Med Settings. 2010;17:333-9. , 88. Sebaratnam DF, Frew JW, Davatchi F, Murrell DF. Quality-of-life measurement in blistering diseases. Dermatol Clin. 2010;30:301-7.
The clinical presentation of inherited EB varies according to the type of disease, and diagnosis can only be reached by skin biopsy and immunofluorescence or electron microscopy, the latter being considered the gold standard.99. Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203.
CLASSIFICATION
Epidermolysis bullosa simplex
Epidermolysis bullosa simplex (EBS) is characterized by a disorder of keratinocytes, intraepidermal blistering and little systemic involvement. Nail dystrophy, alopecia and mucosal lesions may occur in more severe forms of the disease. Skin lesions usually disappear without scarring. Blistering decreases with age. Inheritance is typically autosomal dominant, although rare cases of autosomal recessive inheritance have been documented.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 1010. Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32.
The intraepidermal cleavage observed in EBS is the result of mutations in the K5 and K14 genes, which encode the production of keratin and type I and II intermediate filament proteins, expressed in keratinocytes of the basal layer of the epidermis and epithelial-related complexes.1111. Mitsuhashi Y, Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa. Arch Dermatol R. 2003;295:S29-33. , 1212. Bolling MC, Lemmink HH, Jansen GH, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol. 2011;164:637-44.
EBS is subdivided as follows: localized EBS (Weber-Cockaine subtype), generalized EBS (Köebner subtype), and EBS herpetiformis (Dowling-Meara subtype). The milder forms of EBS present with blisters that are usually caused by an identifiable traumatic event. The Weber-Cockaine subtype (EBS-WC) is characterized by mild to severe blistering and palmoplantar topography, and patients may concomitantly show hyperhidrosis.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 1010. Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32. In its severe forms, hands, feet and limbs are also commonly involved, although in these cases blisters generally develop soon after birth. Palmoplantar hyperkeratosis and erosions occur mainly in the Köebner subtype. In the Dowling-Meara subtype (EBS-DM) there is involvement of the oral mucosa and formation of herpetiform blisters.1010. Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32.
Variable blistering, followed by muscular dystrophy in adulthood, can be seen in EBS with muscular dystrophy, a defect in the expression of plectin. Late myopathy is due to the fact that there is plectin in the composition of the cytoskeleton of skeletal muscles. The severity of skin lesions does not necessarily correlate with the degree of muscular dystrophy. Some patients may also present dental abnormalities.1313. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
As its name suggests, EBS with pyloric atresia presents with pyloric atresia at birth and, usually, blistering is widespread. In most patients, even with correction of pyloric atresia, prognosis is unfavorable, given the extent of systemic involvement. Although the disease is described as precociously fatal, some individuals with milder symptoms can survive during childhood.1010. Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32. , 1313. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa (JEB) is an autosomal recessive disorder characterized by separation of the lamina lucida in the dermo-epidermal junction. A mutation in the LAMB3 gene, which encodes laminin-5, occurs in more than half of patients with JEB. Mutations in the genes encoding collagen XVII and integrin α6β4 are also seen.1111. Mitsuhashi Y, Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa. Arch Dermatol R. 2003;295:S29-33.
Involvement of the oral mucosa, alopecia and anonychia are frequent.1414. Wright JT. Oral manifestations in epidermolysis bullosa spectrum. Dermatol Clin. 2010;28:159-64.
15. Tosti A, Duque-estrada B, Murrell DF. Alopecia in epidermolysis bullosa. Dermatol Clin. 2010;28:165-9.
-
1616. Tosti A, de Farias DC, Murrell DF. Nail involvement in epidermolysis bullosa. Dermatol Clin. 2010;28:153-7.
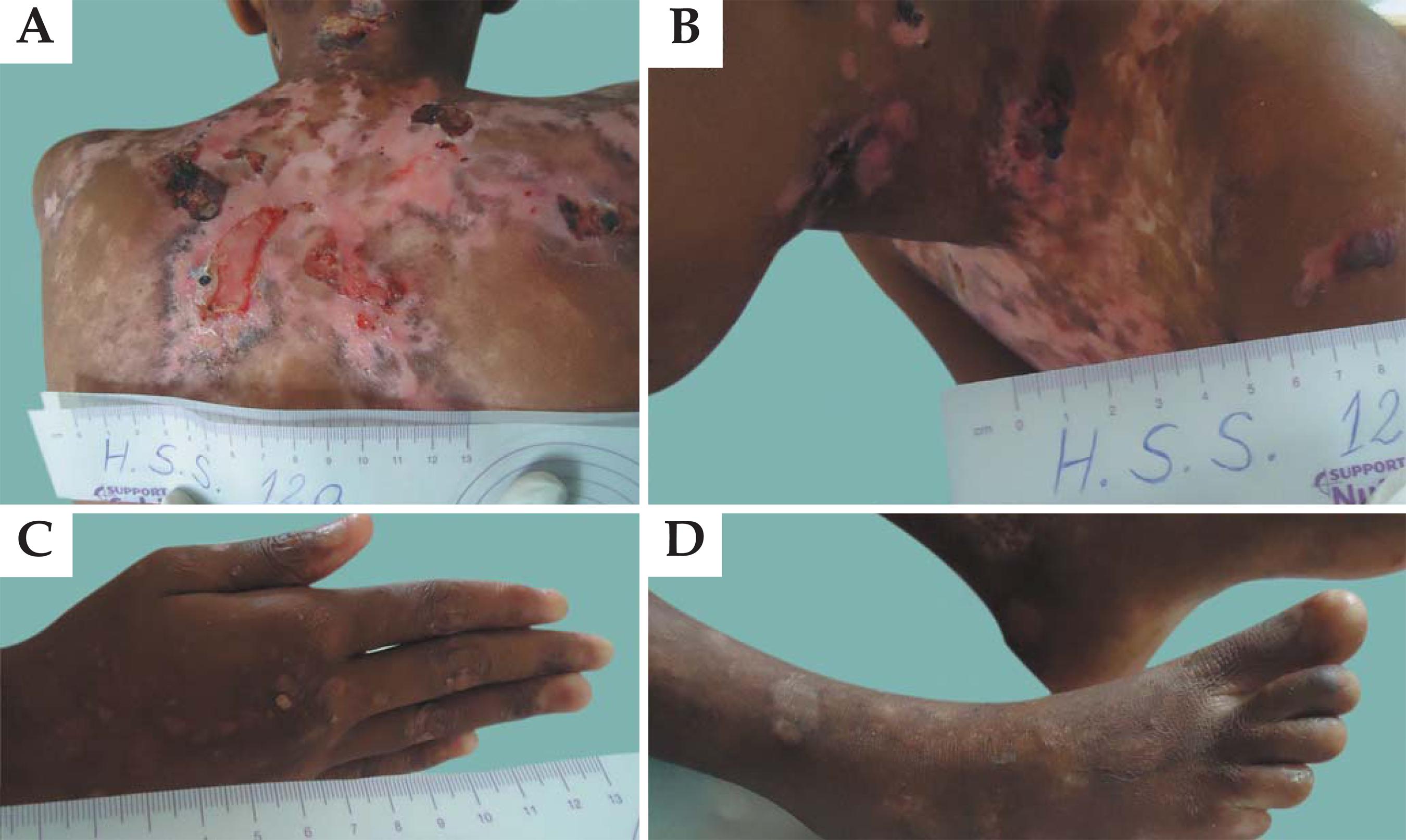
Figure 2 shows lesions observed in patients with JEB. Cases of JEB with congenital pyloric atresia and, more rarely, of other portions of the gastrointestinal tract, have been described in the literature. This disorder is associated with a significant risk of congenital abnormalities of the genitourinary tract and infant/neonatal death. Although patients present with a phenotype virtually identical to that of EBS with pyloric atresia, intra-lamina lucida cleavage of blisters characterizes these patients as having JEB.1010. Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32.
,
1313. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
,
1717. Puvabanditsin S, Garrow E, Kim DU, Tirakitsoontorn P, Luan J. Junctional epidermolysis bullosa associated with congenital localized absence of skin, and pyloric atresia in two newborn siblings. J Am Acad Dermatol. 2001;44:S330-5.
Herlitz JEB (lethal JEB) occurs due to an absence or significant deficiency in the expression of laminin-5. Patients present with erosions around the lips, eyes and nose, often accompanied by significant hypertrophy of the granulation tissue. There is corneal, conjunctival, tracheobronchial, oral, pharyngeal, esophageal, rectal, and genitourinary mucosal involvement. Hoarseness, coughing and other respiratory symptoms are frequent and exuberant. Patients with Herlitz JEB are at high risk of death from sepsis, often not surviving after childhood.1313. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
Among individuals with JEB, those who survive infancy may show clinical improvement with age and present what is called JEB mitis. Often, these patients' respiratory system is not as strongly involved as that of those with the Herlitz form of the disease. However, abnormalities in the scalp, nails and teeth become more apparent. Periorificial erosions and hypertrophy of the granulation tissue may be present. Mucous membranes are often affected by erosions, resulting in stenosis. Some patients with JEB mitis may present blisters in intertriginous areas.1313. Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12. , 1818. Yuen WY, Duipmans JC, Molenbuur B, Herpertz I, Mandema JM, Jonkman MF. Long-term follow-up of patients with Herlitz-type junctional Epidermolysis bullosa. Br J Dermatol. 2012;167:374-82.
The generalized atrophic benign HEB is a relatively mild subtype characterized by skin blisters present at birth. The activity of the lesions is compounded by increasing temperature, healing with a distinctive atrophic appearance. Extracutaneous involvement is rare, with the exception of enamel hypoplasia, which results in the development of cavities. Nail atrophy and alopecia are other common manifestations. Individuals with this type of JEB have a life expectancy similar to that of the general population.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50.
Dystrophic epidermolysis bullosa
Dystrophic epidermolysis bullosa (DEB) is due to mutations in the gene encoding type VII collagen, resulting in defective anchoring fibrils and consequent separation of the sub-basal lamina.1111. Mitsuhashi Y, Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa. Arch Dermatol R. 2003;295:S29-33. When healed, blisters give way to dystrophic lesions (Figure 3). Millium formation occurs due to damage in the hair follicles.1919. Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74.
DEB may be associated with autosomal recessive or dominant inheritance. In the dominant subtype (DDEB) clinical manifestations usually occur at birth or during childhood, with generalized blistering. With increasing age, blisters tend to be more localized.1919. Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74. , 2020. Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107-14. A common variable described as Cockayne-Touraine has acral distribution and minimal oral/dental involvement. In another variant, described by Pasini, there is also involvement of the oral mucosa and teeth, but blistering is more extensive and similar to papules on the trunk (albopapuloid lesions). Dystrophy or anonychia are common to both forms of DDEB.1919. Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74.
The recessive subtype (RDEB) may have a mild to severe clinical presentation. The mild/localized form is called RDEB mitis, usually with acral and nail involvement, but little involvement of the mucous membranes. It usually shows clinical manifestations similar to those of other inherited forms of dystrophic EB.2020. Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107-14. The severe form, described by Hallopeau and Siemens (RDEB-HS) usually shows generalized blistering, predominantly in acral surface, which can lead to pseudosyndactyly of the hands ("boxing glove hands") and feet. Flexural contractures of the extremities are common and intensify with age.1919. Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74. Nails and teeth are usually affected, and inner mucosal involvement can lead to esophageal obstruction, urethral and anal stenosis, phimosis, and corneal lesions. Malabsorption often leads to iron-deficiency anemia, and protein-calorie malnutrition causes deficit in global development. Patients with severe RDEB who survive childhood have a significant risk of developing aggressive squamous cell carcinoma in areas of chronic lesions.2020. Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107-14.
KINDLER SYNDROME
Kindler syndrome (KS) is an autosomal recessive genodermatosis that can clinically simulate all three classic types of inherited EB.2121. Wiebe CB, Larjava HS. Abnormal deposition of type VII collagen in Kindler syndrome. Arch Dermatol Res. 1999;291:6-13. It is a rare dermatosis characterized by acral blistering, fusion of fingers/toes, and generalized progressive poikiloderma. Other clinical findings include trauma-induced blistering (common to all inherited EB), dry and atrophic skin, lichenification and photosensitivity of proximal surfaces. Generally, KS is associated with disruption of the basement membrane and abnormal deposition of type VII collagen both in regions with active lesions and in lesion-free areas. Immunohistochemical examination shows that blistering occurs in the lamina lucida.2121. Wiebe CB, Larjava HS. Abnormal deposition of type VII collagen in Kindler syndrome. Arch Dermatol Res. 1999;291:6-13. Recently, it was shown that this entity results from mutation in the gene encoding Kindlin-1, a focal component of contact between basal keratinocytes. As opposed to other mechano-bullous diseases, there are multiple cleavage planes (intradermal, junctional or sub-lamina densa) and other dermatological findings such as poikiloderma and photosensitivity also differentiate KS from all other forms of inherited EB.11. Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann N Y Acad Sci. 2010;1194:213-22.
CLINICAL MANIFESTATIONS
Besides the typical blistering and erosions secondary to the mechanical fragility of the skin, inherited EB may lead to the formation of millium, nail dystrophy or anonychia. Exuberant granulation tissue (periorificial, in the axillary, occipital, lumbosacral, and periungual regions or at fingertips) and palmoplantar keratoderma (localized or confluent) may be present. Other less common and nonspecific findings include reduced or absent hair, recurrent albopapuloid lesions on the lower trunk, hypo-or hyperhidrosis.2222. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84.
Alopecia. Even in the recessive form of DEB, patients show no specific alopecia. In the presence of anemia, reversible telogen effluvium may occur. Complete absence of hair, eyelashes and eyebrows is a distinct finding that occurs in lethal acantholytic EB. Localized or diffuse alopecia can be observed in Herlitz JEB. In some patients, alopecia presents a typical androgenetic pattern. The degree of capillary involvement varies considerably between individuals with deficient type XVII collagen. There is gradual alopecia in areas of frictional trauma and blistering in patients with DEB. Kindler syndrome is not associated with alopecia.1515. Tosti A, Duque-estrada B, Murrell DF. Alopecia in epidermolysis bullosa. Dermatol Clin. 2010;28:165-9.
Gastrointestinal Tract. In theory, any portion of the gastrointestinal tract, except the gallbladder, pancreas and liver, may be affected in patients with Herlitz JEB, occurring more intensely in RDEB. The most severe complication is stenosis of the esophagus because it compromises swallowing. Malabsorptive syndrome may be secondary to denudation of the small bowel mucosa. As previously mentioned, patients with EBS and JEB may have pyloric atresia at birth.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402. , 2424. Fine JD, Johnson LB. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46:147-58.
Anemia. Herlitz JEB patients may present with severe anemia caused by multiple factors, especially those with JEB and generalized RDEB. Anemia can be partially improved with iron supplementation and blood transfusions.2222. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84. , 2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
Wound healing. This process is compromised by multiple factors including foreign bodies, bacteria, deficiency of nutritional factors and tissue hypoxia. Exogenous agents such as glucocorticoids and penicillamine contribute to impaired wound healing. Optimization of healing occurs with control of these factors. Patents with Herlitz JEB heal slowly, probably due to deficiency of laminin-5.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
Infections. Extensive areas of bare skin show loss of stratum corneum barrier and allow microbial penetration. The accumulation of lymph and moisture in the surface increases bacterial growth. Severe subtypes of Herlitz JEB correlate with immunological abnormalities, including reduced production of lymphocytes. Along with poor nutritional status, there is decreased resistance to infections. Staphylococcus aureus and Streptococcus pyogenes are often the etiological agents, although infections with gram-negative bacteria may also occur. Patients usually show greater susceptibility to develop sepsis, with a high risk of death in early childhood.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402. Prevention of infection is the preferred strategy. With extensive bare areas or areas of crusting, strict care must be taken. This regimen includes the use of topical antibiotics. Self-adhesive dressing is a good choice to keep the areas covered.2525. Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, et al. Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007;46:795-800.
Genitourinary Tract. The formation of recurring vesicles along the urethra, in the ureterovesical junction and ureters can generate obstructive processes culminating with hydronephrosis. There may be chronic renal failure secondary to hydronephrosis, streptococcal glomerulonephritis, mesangial IgA disease and amyloidosis. They are the most common complications in RDEB, causing the death of around 12% of these patients.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402. . 2424. Fine JD, Johnson LB. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46:147-58.
Eyes. They may be affected by recurring erosions or blisters, with greater frequency in JEB and RDEB. Both can occur in childhood, causing scarring and progressive visual impairment, if not treated.2222. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84. , 2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402. , 2626. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 19862006. J Am Acad Dermatol. 2009;60:203-11.
Ears, nose and throat. The most significant complication is partial or complete occlusion of the airways, usually resulting from stenosis of the vocal cords, which can quickly lead to death. It is seen almost exclusively in some subtypes of JEB and can occur in the first year of life.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
Dental manifestations. Tissues of the oral mucosa may be affected in RDEB and JEB. Enamel hypoplasia, which facilitates the formation of cavities and tooth loss, is a pathognomonic characteristic of all types of JEB, while microstomia and ankyloglossia are typical of RDEB. Aggressive dental intervention can increase functionality and contribute to increased nutrient uptake.2727. Feijoo JF, Bugallo J, Limeres J, Peñarrocha D, Peñarrocha M, Diz P. Inherited epidermolysis bullosa: an update and suggested dental care considerations. J Am Dent Assoc. 2011;142:1017-25.
Musculoskeletal system. Progressive contracture of the hands and feet (mitten deformities) may develop in the first year of life and is seen primarily in the Hallopeau-Siemenes subtype of RDEB. Surgical intervention may improve hand functionality, although repeated procedures are necessary to maintain this functionality. Osteopenia and osteoporosis are common in RDEB. Muscular dystrophy beginning in adulthood is typical of EBS with muscular dystrophy.2828. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, et al. Pseudosyndactyly and musculoskeletal contractures in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry, 1986-2002. J Hand Surg Br. 2005;30:14-22.
Cardiomyopathies. Although uncommon in patients with RDEB-HS, there may be dilated cardiomyopathy, possibly fatal, especially when there is associated renal failure. The cause may be multifactorial, including micronutrient deficiencies (selenium and carnitine), transfusion-related iron loss and viral myocarditis.2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
Skin tumors. Squamous cell carcinoma (SCC) usually occurs in multiple primary sites of chronic lesions, particularly in patients with DEB, especially in RDEB/RDEB-HS. In cases of DEB, there is no predilection for photoexposed areas. The peak of incidence of SCC increases dramatically in the second and third decades of life. These lesions may recur frequently even with aggressive surgical excision. Recent studies on the pathogenesis of SCC in patients with RDEB suggest that cancer occurs due to decreased expression of type VII collagen in the NC1 domain. Type VII collagen is required for Ras activation in epidermal tumorigenesis.2929. Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, et al. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773-6.
About 80-90% of patients with HS-RDEB between 45 and 55 years of age have or have already had SCC.2626. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 19862006. J Am Acad Dermatol. 2009;60:203-11. This high risk shows that early detection and treatment of SCC has great importance in the management of adults with RDEB, considering the development of severe and recurrent lesions or chronic skin ulcerations and erosions. However, the presence of scar tissue in patients with chronic Herlitz JEB cannot explain this phenomenon alone, because SCC that affect scar tissue are not usually as aggressive as those involving patients with RDEB, suggesting that other factors may be involved in its pathogenesis.2626. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 19862006. J Am Acad Dermatol. 2009;60:203-11.
EBS-DM patients have a substantial risk of developing basal cell carcinoma (BCC). Possibly, repeated injury to keratinocytes promotes tumorigenesis. The risk of BCC is low in other subtypes of EBS. The risk of melanoma and BCC in other subtypes is comparable to that of the general population.2222. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84. , 2929. Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, et al. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773-6.
QUALITY OF LIFE
Fifty-five percent of patients with Herlitz JEB show significant disease impact on quality of life.66. Risser J, Lewis K, Weinstock MA. Mortality of bullous skin disorders from 1979 through 2002 in the United States. Arch Dermatol. 2009;145:1005-8. , 3030. Fine JD, Johnson LB. Assessment of mobility, activities and pain in different subtypes of epidermolysis bullosa. Clin Exp Dermatol. 2004;29:122-7. There is also a correlation between the Dermatology Life Quality Index - DLQI -and intensity of the psychological disorders presented by these patients. Psychiatric symptoms are observed in all types of Herlitz JEB. Family is a major influence, with love and affection being important to improve the quality of life of Herlitz JEB patients, regardless of financial status, difficulties, emotional factors or time.3131. Pagliarello C, Tabolli S. Factors affecting quality of life in epidermolysis bullosa. Expert Rev Pharmacoecon Outcomes Res. 2010;10:329-38.
The coexistence of symptoms such as depression, anxiety and behavioral disorders should be taken into consideration, as they may compromise treatment strategies and worsen symptoms.66. Risser J, Lewis K, Weinstock MA. Mortality of bullous skin disorders from 1979 through 2002 in the United States. Arch Dermatol. 2009;145:1005-8.
DIAGNOSIS
The diagnosis of Herlitz JEB is based on clinical and laboratory findings. As genetic differentiation is not available in most Brazilian cities, subtypes are usually distinguished by immunological and ultra-structural analysis.3232. Hernández-Martín A, Torrelo A. Inherited epidermolysis bullosa: from diagnosis to reality. Actas Dermosifiliogr. 2010;101:495-505. , 3333. Oliveira ZNP, Perigo AM, Fukumori LMI, Aoki V. Immunological mapping in hereditary epidermolysis bullosa. An Bras Dermatol. 2010;85:856-61. Subclassification is important in determining prognosis (risk of mucosal involvement, development of malignancies and premature death), as well as in providing subsidies for genetic counseling.3232. Hernández-Martín A, Torrelo A. Inherited epidermolysis bullosa: from diagnosis to reality. Actas Dermosifiliogr. 2010;101:495-505.
Electron microscopy (EM) is still the gold standard in the diagnosis of Herlitz JEB, even though it has some limitations.99. Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203. Improper handling or problems in skin tissue sample fixation can result in misdiagnosis. These limitations can be overcome with the concomitant use of immunofluorescence to map the basement membrane in frozen tissue sections with a variety of antibodies, including laminin-1 and 5, collagen V, VII and XVII, bullous pemphigoid antigen, integrin α6β4 and plectin.99. Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203. , 3333. Oliveira ZNP, Perigo AM, Fukumori LMI, Aoki V. Immunological mapping in hereditary epidermolysis bullosa. An Bras Dermatol. 2010;85:856-61. EM is a relatively expensive method and one that is not yet routinely performed. Immunohistochemical study uses a limited number of antibodies and can be a useful alternative.99. Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203. Diagnostic confirmation is done by immunofluorescence, antigenic mapping, antigen-specific monoclonal study of Herlitz JEB and compatible electron microscopy.99. Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203. , 3232. Hernández-Martín A, Torrelo A. Inherited epidermolysis bullosa: from diagnosis to reality. Actas Dermosifiliogr. 2010;101:495-505. , 3333. Oliveira ZNP, Perigo AM, Fukumori LMI, Aoki V. Immunological mapping in hereditary epidermolysis bullosa. An Bras Dermatol. 2010;85:856-61.
Table 2 correlates the types of Herlitz JEB with level of cleavage and the genes/proteins involved in their pathogenesis.
THERAPEUTIC MANAGEMENT
There is no specific therapy for any form of Herlitz JEB. First, an inventory of the affected body surface area and the type of skin involvement (intact blisters, erosions and chronic lesions) should be made. The skin should be evaluated at least every six months, even though most patients are reluctant to this exposure.3434. Falabella AF, Schachner LA, Valencia IC, Eaglstein WH. The Use of Tissue-Engineered Skin (Apligraf) to Treat a Newborn with Epidermolysis Bullosa. Arch Dermatol. 1999;135:1219-22.
35. Shin KC, Park BY, Kim HK, Kim WS, Bae TH. The use of cultured allogenic keratinocyte grafting in a patient with epidermolysis bullosa simplex. Ann Dermatol. 2011;23:393-7.
-
3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
Infants require greater care and control of the environment around them to prevent trauma. This includes gentle manipulation techniques by their caregivers, use of foam to cover bony prominences and zinc oxide anti-adherent diapers. In older children, the use of special shoes and foam in the knee to prevent blistering is recommended. Older patients tend to have chronic ulcers and be colonized with antibiotic-resistant bacteria.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
With respect to the treatment of lesions, blister puncturing to prevent dissemination and use of sterile dressings are recommended. The skin should be left in place, functioning as a biological dressing and preventing bacterial colonization. Firm and easily torn crusts require debridement to prevent maintenance of the inflammatory process.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
The basic principle underlying the care of patients with Herlitz JEB is to prevent blistering with meticulous skin protection and prevention of infections through wound care. This is done with the use of non-adhesive synthetic hydrocolloid dressing.2525. Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, et al. Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007;46:795-800. , 3434. Falabella AF, Schachner LA, Valencia IC, Eaglstein WH. The Use of Tissue-Engineered Skin (Apligraf) to Treat a Newborn with Epidermolysis Bullosa. Arch Dermatol. 1999;135:1219-22. Treatment decision should consider the location of the lesions, need for extra cushioning and protection, use of special dressings and clothing. Lesions should be cleaned with solutions of low toxicity, such as saline solution and water.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
Curative options should be evaluated according to the type of lesion and Herlitz JEB subtype. Other characteristics such as extension and location of lesions, frequency of dressing changes, cost, and economic status of the patient should also be considered.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print] Although there is no single treatment protocol to be followed, the American Academy of Dermatology recently published a consensus recommending the ideal properties of dressings to assist in the various stages of wound healing.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50.
The antiadherent and absorbent characteristics of dressings were crucial in the choice of materials, for this is a disease that evolves with bullous lesions, extensive exulcerated wounds, high chances of infection and scarification. Dressings should also be protective - considering the development of lesions after minimal trauma - and durable enough for the exchanges to occur with the minimum possible frequency.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 3232. Hernández-Martín A, Torrelo A. Inherited epidermolysis bullosa: from diagnosis to reality. Actas Dermosifiliogr. 2010;101:495-505. , 3737. Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-5.
Silicone foam is the most complete and effective type of coverage in the healing process, offering protection, fluid absorption, hydration of the lesions, and antimicrobial properties when combined with additives such as silver.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 3737. Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-5. For drier wounds, foam continues to be the coverage with the broadest action. Hydrogel dressings, contact layers and biosynthetic cellulose can also be used. For scarified wounds, hydrogel dressings, biosynthetic cellulose and hydrocolloid are the best choices, given their debriding action.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50. , 3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
Debridement becomes less necessary when wounds are treated in their early stages with appropriate coverings; minimal debridement can be done during the patient's bath. These dressings are also effective to reduce pain and itching.33. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50.
Curative options for each type of lesion are shown in table 3; the properties of each category of dressings are shown in table 4.
Nutritional support plays a critical role in the resolution of wounds. Some patients require a gastrostomy tube for optimal nutritional status. It is also important to monitor and maintain hemoglobin levels above 8mg/dl. Iron supplementation (oral or intravenous) may be required in some patients. In symptomatic cases, even blood transfusion may be needed.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
Topical or systemic antibiotics may be used for short periods following established criteria to avoid bacterial resistance and sensitization.3434. Falabella AF, Schachner LA, Valencia IC, Eaglstein WH. The Use of Tissue-Engineered Skin (Apligraf) to Treat a Newborn with Epidermolysis Bullosa. Arch Dermatol. 1999;135:1219-22. , 3535. Shin KC, Park BY, Kim HK, Kim WS, Bae TH. The use of cultured allogenic keratinocyte grafting in a patient with epidermolysis bullosa simplex. Ann Dermatol. 2011;23:393-7. More than three of the following characteristics recommend the use of topical antibiotics: wound that does not heal, increased exudate, erythema, presence of friable tissue, presence of dead tissue and stench. Occurrence of at least three of the following characteristics indicates the use of systemic antibiotics: increased wound size, temperature difference greater than 5.4 º C in relation of degradation, erythema or swelling at the border of the lesion, excessive exudation and stench.3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
Avoiding trauma is essential to manage pain. Analgesic drugs should be prescribed according to pain severity, with the use of acetaminophen, NSAIDs, and even morphine in case of severe pain. 3636. Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
New local therapeutic strategies include the use of biological or skin-like dressings.3434. Falabella AF, Schachner LA, Valencia IC, Eaglstein WH. The Use of Tissue-Engineered Skin (Apligraf) to Treat a Newborn with Epidermolysis Bullosa. Arch Dermatol. 1999;135:1219-22. , 3535. Shin KC, Park BY, Kim HK, Kim WS, Bae TH. The use of cultured allogenic keratinocyte grafting in a patient with epidermolysis bullosa simplex. Ann Dermatol. 2011;23:393-7. Since heat and sweat are aggravating factors of EBS, the use of topical aluminum chloride to prevent sweating and improve or prevent deterioration of lesions was attempted, but still without a clear clinical significance. The use of botulinum toxin has also been tested to prevent plantar blistering.3737. Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-5. , 3838. Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34:20-5.
Patients with Herlitz JEB subtypes showing a well-known risk of extra-cutaneous complications require careful monitoring and appropriate intervention (medical, surgical, dental, nutritional and psychological) before the tissues involved become severely injured. Signs and symptoms suggestive of early disease activity in the cornea require a quick assessment by an ophthalmologist to avoid permanent scarring and visual impairment. Patients with esophageal stenosis must undergo dilation to maintain an adequate intake of nutrients orally. Children unable to ingest food orally should receive supplementation via gastrostomy.2424. Fine JD, Johnson LB. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46:147-58.
Hand deformities should be prevented with appropriate dressings involving all fingers at night. The "boxing glove hands" can be temporarily improved with surgical procedures.2828. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, et al. Pseudosyndactyly and musculoskeletal contractures in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry, 1986-2002. J Hand Surg Br. 2005;30:14-22. SCC, which may develop early in these patients, should be treated by wide surgical excision, and the patient should be monitored to prevent recurrences.3939. Venugopal SS, Murrell DF. Treatment of skin cancers in epidermolysis bullosa. Dermatol Clin. 2010;28:283-7. Patients with generalized forms of JEB and RDEB should be monitored for prevention and/or early detection of osteoporosis and osteopenia.2222. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84. , 2323. Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
PERSPECTIVES
New therapeutic strategies have been developed to treat RDEB, such as gene therapy, bone marrow stem cell transplantation and recombinant protein infusion. An example of a new gene therapy strategy was reported in a study conducted in Italy with a 36year-old patient with JEB. After retrovirus integration of the LAMB3 gene, which encodes LAM5-β3 (deficient in patients with JEB) to DNA epidermal stem cells from this patient, a reparative effect on the healing of lesions was shown.4040. Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nature Medicine. 2005;12:1397-9. A study involving mice with RDEB undergoing infusion of recombinant human type VII collagen demonstrated clinical improvement of these affected animals, suggesting possibilities for therapy in humans.4141. Remington J, Wang X, Hou Y, Zhou H, Burnett J, Muirhead T, et al. Injection of Recombinant Human Type VII Collagen Corrects the Disease Phenotype in a Murine Model of Dystrophic Epidermolysis Bullosa. Mol Ther. 2008;17:26-33. A recent study of allogeneic bone marrow transplantation in six children with RDEB showed, by immunofluorescence, production of collagen VII in the lamina densa, in the dermoepidermal junction, with consequent clinical benefit in the early healing of lesions. However, the mechanism and the risk and benefits of this aggressive treatment in the long term need to be better understood.4242. Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629-39.
In DDEB many studies are being conducted focusing on ways that negatively regulate the dominant negative gene, or, alternatively, that offset its presence with up regulation of other genes whose products may offer, even if partially, better skin structural stability, replacing the effect of the underlying mutation.4343. Fine JD. Inherited epidermolysis bullosa: recent basic and clinical advances. Current Opinion. 2010;22:453-8.
Herlitz JEB patients are usually unable to undergo grafting due to their age and disease characteristics. Recently, the development of skin through genetic engineering techniques has had a positive effect on wound healing in patients with Herlitz JEB. Studies of allogeneic keratinocyte grafting have shown improvement of unhealed wounds in patients with Herlitz JEB, reducing trauma and promoting reepithelialization.3838. Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34:20-5.
Although new topical and systemic therapeutic proposals have been suggested, such as the use of topical aluminum chloride 20%, 5% bufexamac cream, oral oxytetracycline 1-1.5 g daily to treat EBS, and the systemic use of phenytoin or trimethoprim to treat DEB, none of them were effective in the resolution or healing of lesions.3838. Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34:20-5. , 4444. Lara-Corrales I, Parkin PC, Stephens D, Hamilton J, Koren G, Weinstein M, et al. The efficacy of trimethoprim in wound healing of patients with epidermolysis bullosa: a feasibility trial. J Am Acad Dermatol. 2012;66:264-70. Recent publications have highlighted important limitations arising from the type of study design, small samples, short treatment period and loss of patient sample, which complicates the validation of many therapeutic attempts.3838. Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34:20-5. Currently, hope for these patients depends on ongoing studies on gene therapy. Meanwhile, local measures and prophylaxis of clinical complications, as well as multiprofessional contribution, are the only effective strategies to control the disease.
FINAL CONSIDERATIONS
Although Herlitz JEB is a quite rare dermatological disorder, its impact on the lives of patients and their families is immeasurable, bringing great physical and emotional suffering and several limitations to the individual. The absence of effective therapeutic measures to control this disease shows the need for monitoring aiming to provide greater physical and psychological comfort.
QUESTIONS
1. Which type of genetic inheritance is observed in EBS?a) Autosomal recessive; b) Autosomal dominant; c) X-linked recessive; d) X-linked dominant.
2. The intraepidermal cleavage observed in EBS is the result of mutation in which genes?a) K14; b) K5; c) LAMB3; d) A and B are correct.
3. In EBS, the late onset of muscular dystrophy results from defects in the expression of:a) Plectin; b) Collagen XVII; c) Laminin-5; d) α6β4 integrin.
4. Which type of inheritance is observed in JEB?a) Autosomal recessive; b) Autosomal dominant; c) X-linked recessive; d) X-linked dominant.
5. Which gene is mutated in more than half of patients with JEB?a) K5; b) LAMB3; c) Plectin; d) Collagen XVII.
6. Which type of inheritance is observed in DEB?a) Autosomal recessive; b) Autosomal dominant; c) X-linked recessive; d) A and B are correct.
7. DEB is due to mutation in the gene that encodes which molecule?a) Plectin; b) Laminin-5; c) Collagen VII; d) α6β4 integrin.
8. Which form of DEB is associated with generalized blistering, predominantly in acral surface, leading to pseudosyndactyly of the hands (boxing glove hands) and feet?a) Acral DDEB; b) RDEB-SH; c) DDEB of the nails; d) Disseminated DDEB.
9. Which kind of genetic inheritance is observed in KS?a) Autosomal recessive; b) Autosomal dominant; c) X-linked recessive; d) X-linked dominant;
10. Osteopenia and osteoporosis are common manifestations of which type of Herlitz JEB?a) DDEB; b) RDEB; c) JEB; d) EBS.
11. It is believed that the slow healing observed in patients with Herlitz JEB is due to a deficiency of:a) K14; b) Laminin-5; c) Collagen VII; d) α6β4 integrin.
12. Enamel hypoplasia and microstomia/ankyloglossia are typical manifestations of, respectively:a) DDEB and JEB; b) JEB and RDEB; c) JEB and DDEB; d) RDEB and JEB.
13. Which types of Herlitz JEB are associated with pyloric atresia?a) DEB and JEB; b) EBS and JEB; c) JEB and DEB; d) DEB and EBS.
14. Which subtype of EBS leads to a higher risk of developing SCC?a) Superficial EBS; b) Localized EBS; c) Lethal acantholytic EBS; d) EBS-DM.
15. It is correct to state the following about psychiatric comorbidities in Herlitz JEB patients:a) The disease appears not to interfere with quality of life when DLQI is considered; b) Psychiatric symptoms are proportional to disease severity; c) Regardless of the type of Herlitz JEB and its severity, there is a major impact on quality of life; d) Herlitz JEB treatment seems to influence quality of life. However, the converse is not true.
16. Which method is considered the gold standard in the diagnosis and classification of Herlitz JEB?a) Electron microscopy; b) Immunophenotyping; c) Immunohistochemistry; d) Simple clinical and histopathological examination.
17. Which characteristic below is taken into consideration in the choice of the curative for patients with Herlitz JEB?a) Anti-adherence; b) Durability; c) Ability to protect the area covered; d) All of the above.
18. It is correct to state the following about the usefulness of silicone foams as curatives for patients with Herlitz JEB:a) They are effective in healing, although they do not prevent new lesions due to minor protection; b) They are considered the best coverage for healing, protection, absorption of fluids and hydration of lesions; c) Their association with silver sulfadiazine has not shown benefits in the management of patients with Herlitz JEB; d) Because of their strong adherence to the wounds, their use is very limited.
19. Which criteria are taken into consideration prior to the use of topical antibiotic therapy in patients with Herlitz JEB?a) Erythematous wounds that do not heal; b) Presence of dead or friable tissue; c) Increased exudate and stench; d) All of the above.
20. It is correct to state the following about anemia in patients with Herlitz JEB:a) It is more common in patients with DDEB; b) JEB rarely evolves with anemia; c) Oral or intravenous iron supplementation is not effective in patients with Herlitz JEB; d) Hemoglobin levels should be kept at above 8mg/dL.
Answer keyParaneoplastic cutaneous manifestations: concepts and updates. An Bras Dermatol. 2013;88(1):9-22.
PapersInformation for all members: The EMC-D questionnaire is now available at the homepage of the Brazilian Annals of Dermatology: www.anaisdedermatologia.org.br. The deadline for completing the questionnaire is 30 days from the date of online publication.
REFERENCES
-
1Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann N Y Acad Sci. 2010;1194:213-22.
-
2Pai S, Marinkovich MP. Epidermolysis bullosa: new and emerging trends. Am J Clin Dermatol. 2002;3:371-80.
-
3Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatology. 2008;58:931-50.
-
4Intong LR, Murrell DF. Inherited epidermolysis bullosa: new diagnostic criteria and classification. Clin Dermatol. 2012;30:70-7.
-
5Fine JD, Bauer EA, McGuire J, Moshell A. Epidermolysis bullosa: clinical, epidemiologic, and laboratory advances and the findings of the National Epidermolysis Bullosa Registry. Baltimore: The Johns Hopkins University Press; 1999.
-
6Risser J, Lewis K, Weinstock MA. Mortality of bullous skin disorders from 1979 through 2002 in the United States. Arch Dermatol. 2009;145:1005-8.
-
7Margari F, Lecce PA, Santamato W, Ventura P, Sportelli N, Annicchiarico G, et al. Psychiatric Symptoms and Quality of Life in Patients Affected by Epidermolysis Bullosa. J Clin Psychol Med Settings. 2010;17:333-9.
-
8Sebaratnam DF, Frew JW, Davatchi F, Murrell DF. Quality-of-life measurement in blistering diseases. Dermatol Clin. 2010;30:301-7.
-
9Petronius D, Bergman R, Ben Izhak O, Leiba R, Sprecher E. A Comparative Study of Immunohistochemistry and Electron Microscopy Used in the Diagnosis of Epidermolysis Bullosa. Am Jour Dermatopathol. 2003;25:198-203.
-
10Sprecher E. Epidermolysis Bullosa Simplex. Dermatol Clin. 2010;28:23-32.
-
11Mitsuhashi Y, Hashimoto I. Genetic abnormalities and clinical classification of epidermolysis bullosa. Arch Dermatol R. 2003;295:S29-33.
-
12Bolling MC, Lemmink HH, Jansen GH, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol. 2011;164:637-44.
-
13Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
-
14Wright JT. Oral manifestations in epidermolysis bullosa spectrum. Dermatol Clin. 2010;28:159-64.
-
15Tosti A, Duque-estrada B, Murrell DF. Alopecia in epidermolysis bullosa. Dermatol Clin. 2010;28:165-9.
-
16Tosti A, de Farias DC, Murrell DF. Nail involvement in epidermolysis bullosa. Dermatol Clin. 2010;28:153-7.
-
17Puvabanditsin S, Garrow E, Kim DU, Tirakitsoontorn P, Luan J. Junctional epidermolysis bullosa associated with congenital localized absence of skin, and pyloric atresia in two newborn siblings. J Am Acad Dermatol. 2001;44:S330-5.
-
18Yuen WY, Duipmans JC, Molenbuur B, Herpertz I, Mandema JM, Jonkman MF. Long-term follow-up of patients with Herlitz-type junctional Epidermolysis bullosa. Br J Dermatol. 2012;167:374-82.
-
19Horn HM, Tidman MJ. The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol. 2002;146:267-74.
-
20Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107-14.
-
21Wiebe CB, Larjava HS. Abnormal deposition of type VII collagen in Kindler syndrome. Arch Dermatol Res. 1999;291:6-13.
-
22Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61:367-84.
-
23Fine JD, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61:387-402.
-
24Fine JD, Johnson LB. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the National Epidermolysis Bullosa Registry. J Pediatr Gastroenterol Nutr. 2008;46:147-58.
-
25Mellerio JE, Weiner M, Denyer JE, Pillay EI, Lucky AW, Bruckner A, et al. Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007;46:795-800.
-
26Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 19862006. J Am Acad Dermatol. 2009;60:203-11.
-
27Feijoo JF, Bugallo J, Limeres J, Peñarrocha D, Peñarrocha M, Diz P. Inherited epidermolysis bullosa: an update and suggested dental care considerations. J Am Dent Assoc. 2011;142:1017-25.
-
28Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, et al. Pseudosyndactyly and musculoskeletal contractures in inherited epidermolysis bullosa: experience of the National Epidermolysis Bullosa Registry, 1986-2002. J Hand Surg Br. 2005;30:14-22.
-
29Ortiz-Urda S, Garcia J, Green CL, Chen L, Lin Q, Veitch DP, et al. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science. 2005;307:1773-6.
-
30Fine JD, Johnson LB. Assessment of mobility, activities and pain in different subtypes of epidermolysis bullosa. Clin Exp Dermatol. 2004;29:122-7.
-
31Pagliarello C, Tabolli S. Factors affecting quality of life in epidermolysis bullosa. Expert Rev Pharmacoecon Outcomes Res. 2010;10:329-38.
-
32Hernández-Martín A, Torrelo A. Inherited epidermolysis bullosa: from diagnosis to reality. Actas Dermosifiliogr. 2010;101:495-505.
-
33Oliveira ZNP, Perigo AM, Fukumori LMI, Aoki V. Immunological mapping in hereditary epidermolysis bullosa. An Bras Dermatol. 2010;85:856-61.
-
34Falabella AF, Schachner LA, Valencia IC, Eaglstein WH. The Use of Tissue-Engineered Skin (Apligraf) to Treat a Newborn with Epidermolysis Bullosa. Arch Dermatol. 1999;135:1219-22.
-
35Shin KC, Park BY, Kim HK, Kim WS, Bae TH. The use of cultured allogenic keratinocyte grafting in a patient with epidermolysis bullosa simplex. Ann Dermatol. 2011;23:393-7.
-
36Garcia-Doval I, Davila-Seijo P, Langan SM. Updated systematic review of randomized controlled trials of treatments for inherited forms of Epidermolysis bullosa. Clin Exp Dermatol. 2012 [ Epub ahead of print]
-
37Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-5.
-
38Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2009;34:20-5.
-
39Venugopal SS, Murrell DF. Treatment of skin cancers in epidermolysis bullosa. Dermatol Clin. 2010;28:283-7.
-
40Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nature Medicine. 2005;12:1397-9.
-
41Remington J, Wang X, Hou Y, Zhou H, Burnett J, Muirhead T, et al. Injection of Recombinant Human Type VII Collagen Corrects the Disease Phenotype in a Murine Model of Dystrophic Epidermolysis Bullosa. Mol Ther. 2008;17:26-33.
-
42Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629-39.
-
43Fine JD. Inherited epidermolysis bullosa: recent basic and clinical advances. Current Opinion. 2010;22:453-8.
-
44Lara-Corrales I, Parkin PC, Stephens D, Hamilton J, Koren G, Weinstein M, et al. The efficacy of trimethoprim in wound healing of patients with epidermolysis bullosa: a feasibility trial. J Am Acad Dermatol. 2012;66:264-70.
-
*
Work conducted at the Service of Dermatology, Complexo Hospitalar Universitário Prof. Edgard Santos, Federal University of Bahia (C-HUPES-UFBA) - Salvador (BA), Brazil.
Publication Dates
-
Publication in this collection
Apr 2013
History
-
Received
26 Aug 2012 -
Accepted
24 Oct 2012