Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases
Abstract
It is well-established that inflammation plays an important role in Alzheimer’s disease (AD) and frontotemporal lobar dementia (FTLD). Inflammation and synapse loss occur in disease prior to the formation of larger aggregates, but the contribution of tau to inflammation has not yet been thoroughly investigated. Tau pathologically aggregates to form large fibrillar structures known as tangles. However, evidence suggests that smaller soluble aggregates, called oligomers, are the most toxic species and form prior to tangles. Furthermore, tau oligomers can spread to neighboring cells and between anatomically connected brain regions. In addition, recent evidence suggests that inspecting the retina may be a window to brain pathology. We hypothesized that there is a relationship between tau oligomers and inflammation, which are hallmarks of early disease. We conducted immunofluorescence and biochemical analyses on tauopathy mice, FTLD, and AD subjects. We showed that oligomers co-localize with astrocytes, microglia, and HMGB1, a pro-inflammatory cytokine. Additionally, we show that tau oligomers are present in the retina and are associated with inflammatory cells suggesting that the retina may be a valid non-invasive biomarker for brain pathology. These results suggest that there may be a toxic relationship between tau oligomers and inflammation. Therefore, the ability of tau oligomers to spread may initiate a feed-forward cycle in which tau oligomers induce inflammation, leading to neuronal damage, and thus more inflammation. Further mechanistic studies are warranted in order to understand this relationship, which may have critical implications for improving the treatment of tauopathies.
INTRODUCTION
In Alzheimer’s disease (AD), the increased production of amyloid-β (Aβ), and the altered processing of the protein tau, are associated with synaptic dysfunction and neuronal death that leads to cognitive and behavioral deficits. It is becoming increasingly apparent that another prominent feature of AD brains is inflammation, which has a significant influence on disease progression [1]. The majority of transgenic mouse models of AD exhibit substantial reactive gliosis with the accumulation of activated astrocytes and microglia in affected brain regions, often prior to the appearance of plaque and tangle pathology [2]. The severity of inflammation in many of these animals correlates with neuronal death [2], and moreover, the local inflammatory environment directly influences the rate of disease progression and the extent of neuronal loss [3]. Recent data shows that astrocyte-mediated inflammatory responses play a significant role in mediating neurotoxic responses to Aβ [4]. Importantly, these results suggest that disrupting inflammatory signaling, in combination with other therapies such as anti-oligomer therapies, could provide a novel neuroprotective intervention for AD and related diseases.
The pathological aggregation of the microtubule-associated protein, tau, culminates in large insoluble aggregates known as neurofibrillary tangles (NFTs) and other inclusions containing hyperphosphorylated tau. These aggregates are the defining histopathological features of AD and other tauopathies, including frontotemporal lobar dementia with tau (FTLD-tau), Pick’s disease, and corticobasal degeneration [5]. FTLD encompasses a broad spectrum of distinct pathologies and protein aggregations that manifest with similar symptoms, ranging from primary tauopathy to transactive response DNA/RNA binding protein-43 (TDP-43) or Fus positive inclusions [6, 7]. In FTLD-TDP there are TDP-43 positive, α-synuclein negative, and tau negative inclusions. They typically do not have extensive tangle pathology [8], but little to no research has been conducted on smaller tau oligomers in FTLD-TDP which we have investigated here. Recent research has shown that the NFTs themselves are not the most toxic form of tau, but rather the smaller aggregates called tau oligomers likely initiate neurodegeneration in tauopathies [9–23].
Inspection of the retina has been suggested as a potential biomarker for the diagnosis of neurodegeneration [24–26]. There is a higher incidence of age-related macular degeneration in patients with AD [27]. While the link remains unclear between retinal degeneration and neurodegeneration, there is increasing evidence that they may be linked. Tau oligomers were recently found in the retina of a rat model for glaucoma and suppression of tau reduced the occurrence of retinal degeneration [28]. Also, there is evidence of large protein deposits, such as Aβ and tau, being present in the retina of AD patients and in tauopathy mice as well as the presence of inflammation [24–26, 29, 30]. Early detection of AD, possibly even prodromal detection, would allow for the early intervention and prevention of neurodegeneration prior to major synapse loss and cognitive decline. Using the retina for the detection of both retinal damage and AD would also be non-invasive, inexpensive, and could be translated into the clinic as part of a normal screening done at patientcheckups.
Inflammation is a critical marker of neurodegenerative diseases, in addition to protein aggregates. Inflammation and cell death occur prior to, or independently, from the formation of NFTs in AD and in transgenic mouse models [9, 12, 13, 15, 31]. Therefore, it is likely that tau oligomers, which form prior to NFTs, may be causing the inflammation [23]. This inflammatory response may induce a self-perpetuating cycle, whereby tau oligomers increase inflammation, causing more damage to the neurons, which in turn may increase the rate of oligomer formation. Furthermore, a majority of transgenic mouse models of AD exhibit astrogliosis and microgliosis in affected brain regions, often prior to the appearance of plaque and tangle pathology [2]. The severity of inflammation in many of these animals correlates with neuronal death [2]. Moreover, the local inflammatory environment directly influences the rate of disease progression and the extent of neuronal loss [3, 32], suggesting that disrupting inflammatory signaling, in addition to removing tau oligomers, could provide a novel neuroprotective intervention for neurodegenerative diseases.
Neuroinflammation is often thought to be a response induced by Aβ in AD [3, 32], given this, there are few reports investigating the role of tau oligomers in neuroinflammation. The studies that have investigated tau indicate tau may play a role in neuroinflammation and may be an early contributor to neurodegeneration [33, 34]. These results warrant further investigation. Thus, we used tauopathy mice and postmortem FTLD-TDP, and AD brain samples to study tau oligomers and inflammation. The tauopathy mice that we used contained the P301L mutation, which is implicated in some genetic tauopathies. In addition, this mutation, along with the related P301S mutation, has been associated with increased inflammation in the brain as well as retinal deficits [24, 33]. However, these events were investigated in isolation and were not investigated thoroughly to determine if they are linked. Also, FTLD-TDP samples were used because they do not have Aβ plaques or tau tangles, thus removing these conditions as potential confounding factors for inflammation. Additionally, tau oligomers have never been examined in FTLD-TDP and may play a role in the late stages of the disease. Thus, we used this model to determine the role tau oligomers play in inflammation in the absence of Aβ and NFTs. We hypothesized that tau oligomers play a role in the feed forward cycle of neuroinflammation in neurodegenerative tauopathies. We performed systematic immunohistochemical and biochemical analyses of brain tissue from transgenic tau mice and FTLD patients. Analyses were performed using a tau oligomer-specific antibody in combination with antibodies for inflammatory markers, including astrocytes and microglia. Our data shows that tau oligomers are associated with markers for neuroinflammation suggesting that tau oligomers may play a role in a feed forward mechanisms resulting in chronic neuroinflammation. These descriptive results call attention to the important and novel role tau oligomers may play in inflammation and in accelerating neurodegeneration. Furthermore, these results validate the potential of the retina to be used as biomarker of neurodegeneration. This study demonstrates a need to identify and characterize the relationship and mechanism behind the association of tau oligomers and inflammation. Perhaps, disrupting this association through anti-oligomer and anti-inflammatory combination therapies can slow the progression, or even halt, neurodegenerative tauopathies.
MATERIALS AND METHODS
Tissue harvesting and preparation
Animals were handled in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch. Animals were euthanized with CO2 and perfused transcardially with ice-cold phosphate buffered saline (PBS). Brains and eyes were collected and snap frozen. They were stored at –80°C. The right eye and hemisphere were homogenized, while the left hemisphere and eye were sectioned. Briefly, frozen tissues were diced and homogenized in PBS with a protease inhibitor mixture (Roche, Indianapolis, IN) and 0.02% NaN3 using a 1:3 mass: volume dilution ratio. Samples were then centrifuged at 10,000 rpm for 10 min at 4°C. The supernatants were divided into aliquots, snap-frozen, and stored at –80°C until use. Postmortem human brain tissues were homogenized by the same protocol. Human samples were obtained from the Oregon Brain Bank and were collected by that institution following approved protocols and laws regarding human tissue.
Immunofluorescence
Immunofluorescent labeling was conducted on frozen cortical brain sections of Htau (Jackson Laboratory, stock #004808, n = 3), and wildtype (WT) (Taconic, BALB/c, n = 2) mice as well as brain samples from FTLD (n = 4), AD (n = 4), and age-matched controls (n = 4) (Oregon Brain Bank). P301L (Taconic, stock #2508, n = 3) mouse eye sections were also used. Immunofluorescence was conducted as previously published [35]. Briefly, the sections were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton-X 100 (Sigma-Aldrich, St. Louis, MO), and the human sections were treated with an autofluorescence eliminator as per manufacturer instructions (Millipore, Darmstadt, Germany). Mouse and human sections were blocked in 3% bovine serum albumin (Fisher Scientific, Pittsburgh, PA), and were then incubated with tau oligomer-specific polycolonal antibody T22 (mouse-1:250, human-1:200, [22]) in 4°C overnight. The following day, the sections were incubated with goat anti-rabbit IgG Alexa-488 or Alexa-568 (1:500, Invitrogen, Waltham, MA) for 1 h. Sections were then incubated overnight with glial fibrillary acidic protein (GFAP) (mouse and human-1:150, BioLegend, San Diego, CA), Iba1 (mouse and human- 1:100, Abcam, Cambridge, MA), or high motility group box 1 (HMGB1) (Mouse-1:50, Abcam, human-1:500, Sigma-Aldrich) at 4°C. The sections were incubated with goat anti-mouse IgG Alexa-568 (1:500, Invitrogen) or goat anti-rabbit 568 for 1 h. The sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and mounted in Fluoromount-G (SouthernBiotech, Birmingham, AL). The retinal slides were imaged with a confocal LSM510 Zeiss laser scanning microscope (Carl Zeiss, Thornwood, NY, USA) as a single digital image. The brain sections were imaged on a Nikon A1R MP laser scanning microscope (Nikon, Melville, NY) at 60X magnification under oil submersion. The objective numerical aperture was 1.4. Using ImageJ, the corrected cell fluorescence method was used to quantify the images as previously described [35, 36]. Briefly, there were four separate images from each section analyzed using this method where five cells were measured as well as five background regions. In order to account for cell size and background, the background was multiplied by the area of the cell and subtracted from the total fluorescence. One-way ANOVA, student’s t-tests, or Pearson’s correlation were used to analyze the corrected cell fluorescence on Graphpad Prism 6 (Graphpad Software, La Jolla, CA). In addition, Pearson’s correlation coefficient was measured to quantify the strength of co-localization between T22 and GFAP using ImageJ software.
ELISA
An enzyme linked immunosorbent assay (ELISA) was conducted to detect the astrocyte marker, GFAP, and total tau. All measurements were performed in triplicate. Plates were coated with 30 μg/well using 0.05 M sodium bicarbonate, pH 9.6, as coating buffer, followed by overnight incubation at 4°C. Age-matched control (n = 4), FTLD (n = 4), and AD (n = 4) brains obtained from the Oregon Brain bank, were used in the ELISA. Negative and positive controls were conducted. The positive control was Tg2576 insoluble brain homogenate with 0.05% Triton-X in PBS. Plates were washed 1 time with Tris-buffered saline with low Tween (0.01%) (TBST), then blocked for 2 h at room temperature with 10% nonfat dry milk in TBST. The plates were then washed with TBST 1 time. Anti-GFAP (1:2000, Covance, Princeton, NJ) was diluted in 5% nonfat milk in TBST and added to one plate. Anti-Tau5 (1:1000, Millipore), diluted in 5% nonfat milk in TBST, was added to another plate and all were allowed to react for 1 h at room temperature. The plates were then washed 3 times with TBST and 100 μl of horseradish peroxidase-conjugated anti-mouse IgG (1:3000, BioRad, Hercules, CA) was added. The plates were incubated for 1 h at room temperature. Finally, plates were washed 3 times with TBST and developed with 3,3’,5,5’,-tetramethylbenzidine substrate from DAKO (Carpentaria, CA). The reaction was stopped with 100 μl 1 M HCl, and samples were read at 450 nm using a POLARstar OMEGA plate reader (BMG Labtechnologies, Ortenberg, Germany). One-way ANOVA with a multiple comparisons test was used to analyze results on GraphPad Prism 6 (GraphPad Software).
Statistics
Images were quantified using corrected cell fluorescence, as described previously [35, 36]. Four images per subject were obtained and five cells were outlined and measured in each image. To account for both background and the size of the cell, five background measurements were also taken and the background fluorescence was multiplied by the area. This was subtracted from the total intensity for the cell in order to obtain a corrected cell fluorescence. The corrected cell fluorescence for five cells per image were averaged and a One-Way ANOVA with a multiple comparisons test was conducted to compare control (n = 4, 20 cells quantified), FTLD (n = 4, 20 cells quantified), and AD (n = 4, 20 cells quantified). A student’s unpaired t-test was used for the mouse quantifications with two groups while a One-Way ANOVA was used for the mouse samples with more than two groups. For the ELISA, One-Way ANOVA was used with multiple comparisons test. All statistics were performed on GraphPad Prism6 (GraphPad Software) and p < 0.05 was considered significant.
RESULTS
P301L mouse retina exhibits inflammation in association with tau oligomers
Tau abnormalities in the retina of transgenic animals have been investigated extensively, but the association of tau oligomers with inflammation remains unknown. We investigated the co-localization of tau oligomers with inflammatory markers in the retina of an aggressive tauopathy mouse model, P301L. T22 is a novel conformational polyclonal antibody developed by our laboratory. It recognizes tau oligomers but does not recognize tau monomers or fibrils [22, 37]. We discovered the presence of activated astrocytes in the retina, associated with oligomeric tau aggregates. We also found that tau oligomers are present in the outer nuclear layer of the eye, localized in close proximity to activated astrocyte processes in the outer plexiform layer (Fig. 1).
In order to further characterize the pattern of inflammation and tau oligomer pathology in the eye of transgenic tau mice, we investigated the presence of activated microglia and tau oligomers in the rod outer segment (ROS) of the eye. Labeling the P301L mouse retina with T22, Iba1, and DAPI revealed microglial cells co-localizing with tau oligomers in the ROS (Fig. 1). There was also evidence of hyper-activation and proliferation of microglial cells at the ROS and retinal pigment epithelial (RPE) cell layers. Activation of microglia was seen throughout the retina of P301L transgenic mice. Of note, are small cell bodies with irregular, short, cell processes that are present near the deposits of T22 positive tau oligomers. Co-localization of the microglia with tau oligomers may indicate the engulfment of the oligomers by the activated microglia. Additionally, we detected glial cell processes of Muller cells extending to the outer nuclear cells, co-localizing with mutant tau oligomers, and concurring with the phagocytic nature of the Muller cells in a pathological event (Fig. 1). Both thick and thin ramifications of the Muller cell processes were noted throughout the outer nuclear layer, as well as in close apposition to the outer limiting membrane. Also noted were multiple small cell bodies of Muller cells in the inner nuclear layer, with some dendritic spurs towards the ganglion cell layers. These findings help support the idea that eyes can be used as a means to study some of the pathological changes in the central nervous system.
FTLD, AD subjects, and Htau mice exhibit an association between inflammation and tau oligomers
Frozen frontal cortical brain sections from FTLD, AD, and age-matched controls were analyzed with immunofluorescent staining. We found that tau oligomers were surrounded by the astrocyte marker, GFAP. Additionally, there was a significant increase in GFAP protein levels as compared to the control (Fig. 2). It is important to note that GFAP did not appear to co-localize with tau oligomers directly, yet GFAP was in close proximity suggesting that tau oligomers are interacting with astrocytes but not engulfing the tau oligomers. The astrocytes appear to be surrounding the tau oligomer containing cells but not fully internalizing the tau oligomers (Fig. 2F, I). To more accurately quantify GFAP protein levels and total tau protein, we conducted a direct ELISA. We found that in FTLD and AD, astrocyte levels were significantly higher than in the age-matched controls (Fig. 2J). However, total tau was not significantly different between age-matched controls, FTLD, and AD suggesting a gain of function toxicity for the tau oligomers rather than increased toxicity due to the upregulation of tau protein.
In order to confirm and further validate the association between astrocytes and tau oligomers we used the Htau mouse model. The Htau mouse model was used here because it expresses all six human tau isoforms without expressing mouse tau making it a better model for sporadic tauopathy. In addition, Htau mice have been shown to have neuroinflammation previously allowing us to investigate if this inflammation can be linked to tau oligomers [34]. Using immunofluorescence of cortical regions with GFAP and T22 tau oligomer-specific antibodies, we found that the number of astrocytes in Htau mice changes with age and in comparison to WT mice (Fig. 3). Seven-month-old WT mice exhibited low levels of reactive astrocytes and negligible tau oligomer staining. We showed that at 3 months of age, Htau mice have only limited astrocyte activation, but by 11 months, Htau mice begin to show evidence of fragmented astrocytes, as well as some reactive morphology. Astrocyte activation is dramatically increased when tau oligomers are present at higher levels in the brains of 24-month-old Htau mice. GFAP corrected cell fluorescence was significantly increased in the brains of Htau mice, increasing with the age of the mice as well as in comparison to the WT mice. Additionally, there was a positive correlation between GFAP and T22 corrected cell fluorescence regardless of age or genotype (r = 0.662, r2 = 0.439, p < 0.01). These data suggest a possible interaction between tau oligomers and reactive astrocytes in inflammation.
The FTLD, AD, and control subject frontal cortical sections were stained for tau oligomers and microglia. Tau oligomers co-localized with microglia in FTLD and AD subjects, suggesting that tau oligomers may play a role in stimulating inflammation (Fig. 4). In contrast to the astrocytes, co-localization suggests internalization of the tau oligomers. Microglial quantification by corrected cell fluorescence also showed a significant increase in Iba1 in FTLD and AD, which is common in inflammation and neurodegenerative diseases. Next, we examined the relationship of microglia and tau oligomers in Htau cortices. We used Iba1 and T22 to immunolabel brains from WT and Htau mice. We found little evidence of microglial activation in WT mice and negligible labeling for tau oligomers, as expected for a WT animal. However, in Htau mice at 24 months of age, a significant increase in activated microglia was detected, largely co-localizing with tau oligomers (Fig. 5). This was consistent with the results seen in the human cases and indicates a potential interplay between toxic oligomeric tau and microglia. In addition, this co-localization could indicate that tau oligomers are being internalized by the microglia, which may increase the spread of pathology [38].
To further characterize inflammation, we investigated the association of tau oligomers with the chromatin protein, high motility group box 1 (HMGB1). HMGB1 is a protein normally found within the nucleus of cells that are not undergoing inflammatory processes. During inflammation, however, HMGB1 can translocate from the nucleus to the cytosol and can then be secreted as a pro-inflammatory cytokine. We conducted T22 and HMGB1 immunofluorescence on tissues from the frontal cortex of FTLD, AD, and age-matched controls. HMGB1 localized to the cytoplasm rather than the nucleus, as was seen in the control subjects indicating inflammatory signaling in the FTLD and AD subjects (Fig. 6). HMGB1 showed significantly increased corrected cell fluorescence in FTLD and AD samples compared to controls. The increase in HMGB1 and its translocation to the cytoplasm indicate it has been activated as a pro-inflammatory signal in FTLD and AD brains. The co-localization of tau oligomers with HMGB1 suggests there may be an interaction between tau oligomers and inflammatory signaling. To further characterize the relationship of HMGB1 and tau oligomers in the cortex we conducted immunofluorescence in WT and Htau mice. We found that HMGB1 was detected at low levels that were, primarily, associated with the nucleus in WT mice (Fig. 7). HMGB1 was transported out of the nuclei in 24-month-old Htau mice, a condition that was representative of the activation of HMGB1 as an inflammatory cytokine. These results were consistent with those seen in the brains of human subjects. However, tau oligomers co-localized with HMGB1 to a much lesser extent in mice as compared to the human samples, suggesting the relationship between tau oligomers and the pro-inflammatory cytokine HMGB1 may be different between species or that the relationship is more complex. These results warrant further research to determine more specifically the mechanisms in human subjects.
DISCUSSION
A growing body of evidence suggests that large meta-stable tau aggregates, including NFTs, are not causally linked to tauopathy phenotypes and appear after neuronal toxicity [9–23], suggesting that soluble tau oligomers may underlie early disease stages. We recently demonstrated that tau oligomers form prior to NFTs, are present intra- and extracellularly, are highly toxic, induce cognitive deficits in vivo, and play a crucial role in neuronal cytopathology [11, 22, 39, 40]. We also established the specificity and efficacy of one tau oligomer-specific monoclonal antibody that recognizes a conformationally distinct subset of tau oligomers in the same tauopathy animal models used in the present study: aged P301L mice [41, 42] and Htau mice [43, 44]. Furthermore, we published evidence that tau oligomer transformation into pore-like structures is cell-specific and disease-specific [45]. In addition we were able to isolate, characterize, and propagate tau oligomers from postmortem brain tissue of patients with various neurodegenerative diseases, as well as, patients with traumatic brain injury [35, 36, 44].
Our results demonstrate that tau oligomers may induce inflammation in FTLD, AD, and potentially, other neurodegenerative diseases, thus linking tau oligomer deposition to inflammatory signaling in the brain. It is interesting that our results suggest that astrocytes interact with tau oligomers but do not appear to engulf them, while microglia showed extensive co-localization. This is consistent with data in current literature that indicates that microglia can engulf tau oligomers and secrete them in exosomes, facilitating their propagation to surrounding cells [38]. Tau oligomers and inflammation may be self-reinforcing stimuli leading to chronic inflammation, physiological impairments, cellular dysfunction, and ultimately, neurodegeneration. Additionally, the spread of tau oligomers from one brain region to another [37, 46, 47] may initiate inflammation, inducing a toxic cycle of tau oligomers, inflammation, and cell death in larger anatomical areas (Fig. 8). This suggests that the propagation of tau oligomers may also propagate inflammation and increase the rate of degeneration. Future studies characterizing the inflammation phenotypes specific to tau oligomers in FTLD, AD, and other neurodegenerative diseases should elucidate the mechanism and confirm that tau oligomers are inducing the inflammation. Our results are novel in that they associate tau oligomers specifically with inflammation, a process that occurs early in disease pathogenesis, and they lay the foundation for future studies to explore this relationship. Further studies should be done to investigate whether inflammation can induce oligomer production, which would contribute to this vicious cycle. Our results confirm inflammation as a contributing factor to the neurodegenerative disease cascade, and they have implications for therapeutic approaches [32].
Moreover, the findings reported here, and by other investigators, regarding retinal damage in tau transgenic mice provide a basis for the under-reported visual deficits in these animals [30]. These data also provide evidence that detecting neurodegeneration may be possible by detecting retinal damage. Development of non-invasive techniques to detect early neurodegeneration is essential for the advancement of therapies. Our descriptive data shown here call for the characterization of eye pathology in neurodegenerative diseases such as FTLD and AD in order to determine how the retina changes throughout disease progression. Perhaps, non-invasive eye examinations could be used as a diagnostic for the detection of these diseases.
Another exciting result from our research is the discovery of the association between tau oligomers and HMGB1, which is an important mediator for inflammation in the brain, as well as peripherally. HMGB1 has been well-established as a pro-inflammatory cytokine when it is released into the extracellular space and is produced by neurons, astrocytes, and microglia [48–50]. In addition, astrocytes and microglia can respond to HMGB1 by producing pro-inflammatory conditions, which is suggested to play an important role in neuroinflammation in neurodegenerative diseases [51–53]. HMGB1 is an important pro-inflammatory cytokine, but it has also been implicated in a necrotic cell death response [54, 55]. Following acute injuries, such as ischemic brain insults, HMGB1 is translocated out of the nucleus and appears to play a role in necrosis or necroptosis (programed cell death in response to injury) [55]. However, it is unlikely that the HMGB1 response seen here is necrosis related due to that fact that neurodegeneration is a process lasting many years to many decades. Rather, it is much more likely that the HMGB1 translocation out of the nucleus is stimulating the pro-inflammatory signaling pathways leading to the chronic state of inflammation that is a known characteristic of most neurodegenerative diseases.
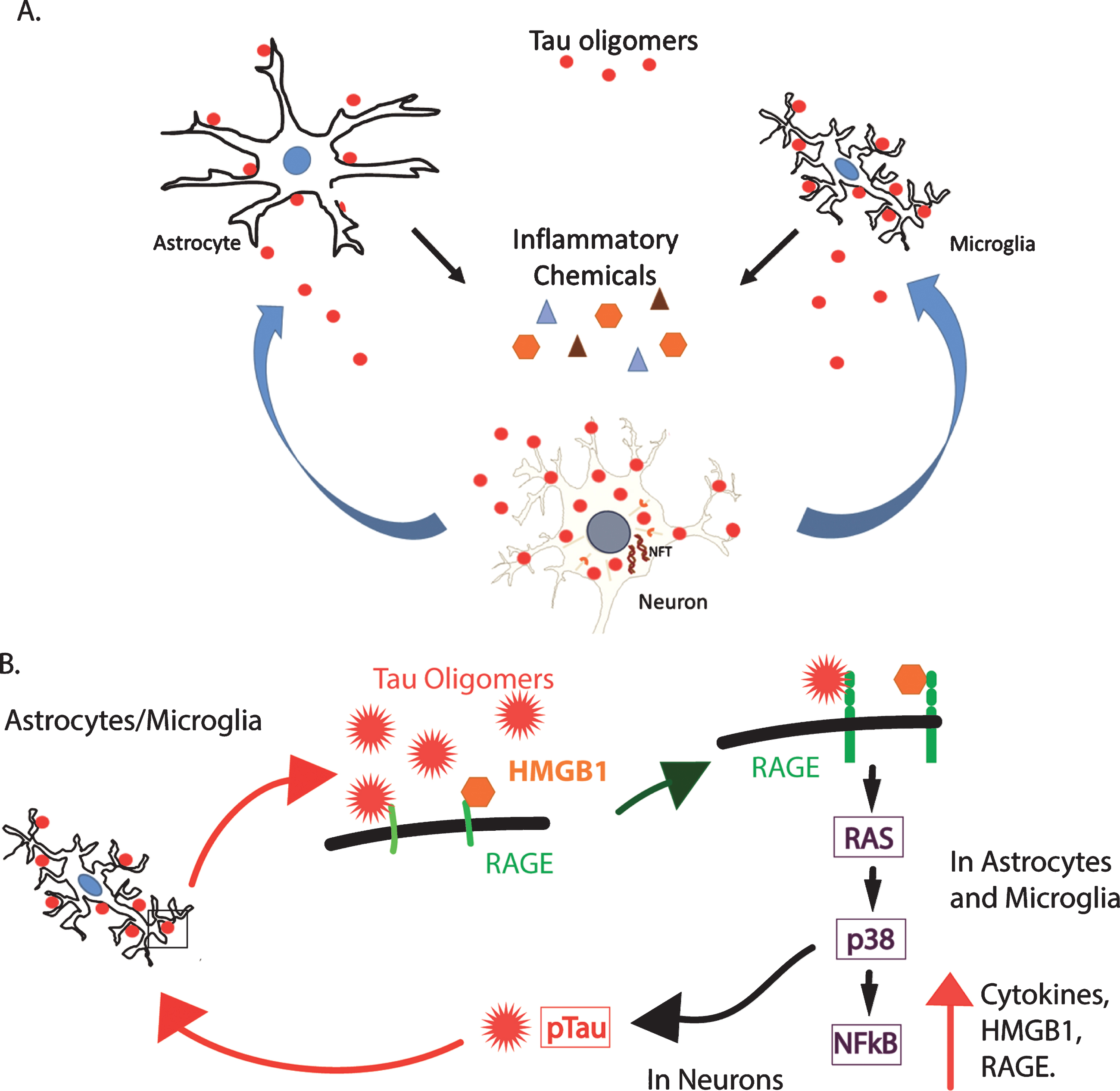
One of several possible pathways by which HMGB1 may be acting as a pro-inflammatory cytokine is through the receptor for advanced glycation end products (RAGE). RAGE is a multi-ligand receptor that binds HMGB1, Aβ, and advanced glycation end products [56]. It is possible that RAGE may also bind tau oligomers and initiate pro-inflammatory signaling through the RAS, p38, and nuclear factor-κB (NFκB) pathway [56]. The NFκB transcription factor increases the expression of HMGB1 and its receptor RAGE, increasing pro-inflammatory RAGE signaling by a feed-forward mechanism [57, 58]. HMGB1 is well established as a pro-inflammatory cytokine, which can induce the production of other cytokines through the NFκB pathway [59]. Figure 8 illustrates a hypothetical model for these interactions. Tau oligomers may interact with astrocytes and microglia to induce inflammation, potentially through RAGE signaling. This inflammation, in turn, damages neurons resulting in more inflammation. In addition, signaling via the RAGE receptor on the surface of neurons can activate p38, which is known to phosphorylate tau, and may increase the presence of hyperphosphorylated tau. Hyperphosphorylated tau is more prone to aggregate, leading to the production of more oligomers that can produce their own toxicity and induce further inflammation. Our results implicate HMGB1 as a possible key player in tau-induced inflammation and toxicity, and therefore, further investigation of its role in tauopathies and brain injuries are warranted. Thus, the association of tau oligomers with cells producing and secreting HMGB1 could have important implications for inflammation associated with tauopathies.
Finally, the existence of tau oligomers in FTLD-TDP is a novel finding since FTLD-TDP is associated with TDP-43 inclusions rather than tau inclusions. These results may be relevant to disease progression, and it would be interesting to investigate the temporal appearance of tau oligomers and disease progression. It is likely that the appearance of tau oligomers is a late occurrence in FTLD-TDP due to the lack of larger insoluble aggregates of tau in this disease. However, it is also possible that the oligomers are more stable in FTLD-TDP, thus prolonging their toxic longevity and inhibiting the formation of the less-toxic fibrils. The specificity of the T22 antibody has been established in previous studies. We found that it reliably and specifically detects tau oligomers[22, 35–37, 39].
In summary, our results demonstrate increased deposition of tau oligomers and neuroinflammation in the retina and brain tissue from tauopathy mice and from patients with FTLD-TDP and AD. The tau oligomer pathology and inflammation in the animal models showed a clear association. Furthermore, there is promise in the development of non-invasive eye examinations for the early detection of neurodegenerative diseases. Additionally, these results can be translated into FTLD and AD, and they indicate that tau oligomers are associated with inflammation. The descriptive results shown here demonstrate a novel association between tau oligomers and inflammation. However, the mechanism and pathways that associate tau oligomers and inflammation need to be elucidated, and this should be a priority for neuroinflammation and neurodegeneration researchers. Moreover, these results suggests that the degeneration of neurons due to chronic inflammation induced by tau oligomers may be inhibited through the combination of anti-tau oligomer and anti-inflammatory therapeutics for the treatment of AD and other tauopathies. This combination would inhibit the proposed feed forward cycle shown in Fig. 8 from two different angles, tau oligomers and inflammatory signaling.
ACKNOWLEDGMENTS
This work was supported in part by the Mitchell Center for Neurodegenerative Disease, the University of Texas System Neuroscience and Neurotechnology Research Institute and Retina Research Foundation (R.K. and W.Z.), NIH grants AG054025 (R.K.) and EY022694 (W.Z). We thank the Kayed lab members for their support and suggestions in preparing the manuscript.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0912).
REFERENCES
[1] | McGeer EG , McGeer PL ((2010) ) Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J Alzheimers Dis 19: , 355–361. |
[2] | Schwab C , Klegeris A , McGeer PL ((2010) ) Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta 1802: , 889–902. |
[3] | Janelsins MC , Mastrangelo MA , Park KM , Sudol KL , Narrow WC , Oddo S , LaFerla FM , Callahan LM , Federoff HJ , Bowers WJ ((2008) ) Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol 173: , 1768–1782. |
[4] | Garwood CJ , Pooler AM , Atherton J , Hanger DP , Noble W ((2011) ) Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis 2: , e167. |
[5] | Ballatore C , Lee VM , Trojanowski JQ ((2007) ) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8: , 663–672. |
[6] | Lashley T , Rohrer JD , Mead S , Revesz T ((2015) ) Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol 41: , 858–881. |
[7] | Irwin DJ , Cairns NJ , Grossman M , McMillan CT , Lee EB , Van Deerlin VM , Lee VM , Trojanowski JQ ((2015) ) Frontotemporal lobar degeneration: Defining phenotypic diversity through personalized medicine. Acta Neuropathol 129: , 469–491. |
[8] | Geser F , Lee VM , Trojanowski JQ ((2010) ) Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: A spectrum of TDP-43 proteinopathies. Neuropathology 30: , 103–112. |
[9] | Berger Z , Roder H , Hanna A , Carlson A , Rangachari V , Yue M , Wszolek Z , Ashe K , Knight J , Dickson D , Andorfer C , Rosenberry TL , Lewis J , Hutton M , Janus C ((2007) ) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci 27: , 3650–3662. |
[10] | Polydoro M , Acker CM , Duff K , Castillo PE , Davies P ((2009) ) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29: , 10741–10749. |
[11] | Lasagna-Reeves CA , Castillo-Carranza DL , Jackson GR , Kayed R ((2011) ) Tau oligomers as potential targets for immunotherapy for Alzheimer’s disease and tauopathies. Curr Alzheimer Res 8: , 659–665. |
[12] | Spires-Jones TL , Kopeikina KJ , Koffie RM , de Calignon A , Hyman BT ((2011) ) Are tangles as toxic as they look? J Mol Neurosci 45: , 438–444. |
[13] | Yoshiyama Y , Higuchi M , Zhang B , Huang SM , Iwata N , Saido TC , Maeda J , Suhara T , Trojanowski JQ , Lee VM ((2007) ) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53: , 337–351. |
[14] | Cowan CM , Quraishe S , Mudher A ((2012) ) What is the pathological significance of tau oligomers? Biochem Soc Trans 40: , 693–697. |
[15] | Andorfer C , Acker CM , Kress Y , Hof PR , Duff K , Davies P ((2005) ) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25: , 5446–5454. |
[16] | Morsch R , Simon W , Coleman PD ((1999) ) Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol 58: , 188–197. |
[17] | de Calignon A , Fox LM , Pitstick R , Carlson GA , Bacskai BJ , Spires-Jones TL , Hyman BT ((2010) ) Caspase activation precedes and leads to tangles. Nature 464: , 1201–1204. |
[18] | Gomez-Isla T , Hollister R , West H , Mui S , Growdon JH , Petersen RC , Parisi JE , Hyman BT ((1997) ) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41: , 17–24. |
[19] | Terry RD ((2000) ) Do neuronal inclusions kill the cell? J Neural Transm Suppl 59: , 91–93. |
[20] | van de Nes JA , Nafe R , Schlote W ((2008) ) Non-tau based neuronal degeneration in Alzheimer’s disease – an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res 1213: , 152–165. |
[21] | Maeda S , Sahara N , Saito Y , Murayama S , Ikai A , Takashima A ((2006) ) Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci Res 54: , 197–201. |
[22] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Sarmiento J , Troncoso J , Jackson GR , Kayed R ((2012) ) Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 5: , 1946–1959. |
[23] | Patterson KR , Remmers C , Fu Y , Brooker S , Kanaan NM , Vana L , Ward S , Reyes JF , Philibert K , Glucksman MJ , Binder LI ((2011) ) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286: , 23063–23076. |
[24] | Mazzaro N , Barini E , Spillantini MG , Goedert M , Medini P , Gasparini L ((2016) ) Tau-driven neuronal and neurotrophic dysfunction in a mouse model of early tauopathy. J Neurosci 36: , 2086–2100. |
[25] | Koronyo-Hamaoui M , Koronyo Y , Ljubimov AV , Miller CA , Ko MK , Black KL , Schwartz M , Farkas DL ((2011) ) Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54: (Suppl 1), S204–S217. |
[26] | Guo L , Duggan J , Cordeiro MF ((2010) ) Alzheimer’s disease and retinal neurodegeneration. Curr Alzheimer Res 7: , 3–14. |
[27] | Frost S , Guymer R , Aung KZ , Macaulay SL , Sohrabi HR , Bourgeat P , Salvado O , Rowe CC , Ames D , Masters CL , Martins RN , Kanagasingam Y , Group AT ((2016) ) Alzheimer‘s disease and the early signs of age-related macular degeneration. Curr Alzheimer Res. doi: 10.2174-1567205013666160603003800 |
[28] | Chiasseu M , Cueva Vargas JL , Destroismaisons L , Vande Velde C , Leclerc N , Di Polo A ((2016) ) Tau accumulation, altered phosphorylation, and missorting promote neurodegeneration in glaucoma. J Neurosci 36: , 5785–5798. |
[29] | Gupta V , Gupta VB , Chitranshi N , Gangoda S , Vander Wall R , Abbasi M , Golzan M , Dheer Y , Shah T , Avolio A , Chung R , Martins R , Graham S ((2016) ) One protein, multiple pathologies: Multifaceted involvement of amyloid beta in neurodegenerative disorders of the brain and retina. Cell Mol Life Sci. doi: 10.1007/s00018-016-2295-x |
[30] | Liu B , Rasool S , Yang Z , Glabe CG , Schreiber SS , Ge J , Tan Z ((2009) ) Amyloid-peptide vaccinations reduce beta-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer’s transgenic mice. Am J Pathol 175: , 2099–2110. |
[31] | Cook C , Kang SS , Carlomagno Y , Lin WL , Yue M , Kurti A , Shinohara M , Jansen-West K , Perkerson E , Castanedes-Casey M , Rousseau L , Phillips V , Bu G , Dickson DW , Petrucelli L , Fryer JD ((2015) ) Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum Mol Genet 24: , 6198–6212. |
[32] | Golde TE ((2009) ) The therapeutic importance of understanding mechanisms of neuronal cell death in neurodegenerative disease. Mol Neurodegener 4: , 8. |
[33] | Jaworski T , Lechat B , Demedts D , Gielis L , Devijver H , Borghgraef P , Duimel H , Verheyen F , Kugler S , Van Leuven F ((2011) ) Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol 179: , 2001–2015. |
[34] | Garwood CJ , Cooper JD , Hanger DP , Noble W ((2010) ) Anti-Inflammatory Impact of Minocycline in a Mouse Model of Tauopathy. Front Psychiatry 1: , 136. |
[35] | Gerson JE , Castillo-Carranza DL , Sengupta U , Bodani R , Prough DS , DeWitt D , Hawkins BE , Kayed R ((2016) ) Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in Htau mice. J Neurotrauma. doi: 10.1089/neu.2015.4262 |
[36] | Gerson JE , Sengupta U , Lasagna-Reeves CA , Guerrero-Munoz MJ , Troncoso J , Kayed R ((2014) ) Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun 2: , 73. |
[37] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Guerrero-Munoz MJ , Kiritoshi T , Neugebauer V , Jackson GR , Kayed R ((2012) ) Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2: , 700. |
[38] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kugler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[39] | Lasagna-Reeves CA , Castillo-Carranza DL , Guerrero-Muñoz MJ , Jackson GR , Kayed R ((2010) ) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49: , 10039–10041. |
[40] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Clos AL , Jackson GR , Kayed R ((2011) ) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6: , 39. |
[41] | Guerrero-Munoz MJ , Castillo-Carranza DL , Krishnamurthy S , Paulucci-Holthauzen AA , Sengupta U , Lasagna-Reeves CA , Ahmad Y , Jackson GR , Kayed R ((2014) ) Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol Dis 71: , 14–23. |
[42] | Castillo-Carranza DL , Sengupta U , Guerrero-Munoz MJ , Lasagna-Reeves CA , Gerson JE , Singh G , Estes DM , Barrett AD , Dineley KT , Jackson GR , Kayed R ((2014) ) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34: , 4260–4272. |
[43] | Gerson JE , Castillo-Carranza DL , Kayed R ((2014) ) Advan-ces in therapeutics for neurodegenerative tauopathies: Moving toward the specific targeting of the most toxic tau species. ACS Chem Neurosci 5: , 752–769. |
[44] | Castillo-Carranza DL , Gerson JE , Sengupta U , Guerrero-Munoz MJ , Lasagna-Reeves CA , Kayed R ((2014) ) Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis 40: (Suppl 1), S97–S111. |
[45] | Lasagna-Reeves CA , Sengupta U , Castillo-Carranza D , Gerson JE , Guerrero-Munoz M , Troncoso JC , Jackson GR , Kayed R ((2014) ) The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol Commun 2: , 56. |
[46] | Gerson JE , Kayed R ((2013) ) Formation and propagation of tau oligomeric seeds. Front Neurol 4: , 93. |
[47] | Wu JW , Herman M , Liu L , Simoes S , Acker CM , Figueroa H , Steinberg JI , Margittai M , Kayed R , Zurzolo C , Di Paolo G , Duff KE ((2013) ) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288: , 1856–1870. |
[48] | Fonken LK , Frank MG , Kitt MM , D’Angelo HM , Norden DM , Weber MD , Barrientos RM , Godbout JP , Watkins LR , Maier SF ((2016) ) The alarmin HMGB1 mediates age-induced neuroinflammatory priming. J Neurosci 36: , 7946–7956. |
[49] | Passalacqua M , Patrone M , Picotti GB , Del Rio M , Sparatore B , Melloni E , Pontremoli S ((1998) ) Stimulated astrocytes release high-mobility group 1 protein, an inducer of LAN-5 neuroblastoma cell differentiation. Neuroscience 82: , 1021–1028. |
[50] | Piras S , Furfaro AL , Domenicotti C , Traverso N , Marinari UM , Pronzato MA , Nitti M ((2016) ) RAGE expression and ROS generation in neurons: Differentiation versus damage. Oxid Med Cell Longev 2016: , 9348651. |
[51] | Gonzalez-Reyes RE , Graciela Rubiano M ((2016) ) Astrocyte’s RAGE: More than just a question of mood. Cent Nerv Syst Agents Med Chem. doi: 10.2174-1871524916999160505105121 |
[52] | Frank MG , Weber MD , Fonken LK , Hershman SA , Watkins LR , Maier SF ((2016) ) The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav Immun 55: , 215–224. |
[53] | Pedrazzi M , Patrone M , Passalacqua M , Ranzato E , Colamassaro D , Sparatore B , Pontremoli S , Melloni E ((2007) ) Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J Immunol 179: , 8525–8532. |
[54] | Hyun HW , Ko AR , Kang TC ((2016) ) Mitochondrial translocation of high mobility group box 1 facilitates LIM kinase 2-mediated programmed necrotic neuronal death. Front Cell Neurosci 10: , 99. |
[55] | Xu M , Zhou GM , Wang LH , Zhu L , Liu JM , Wang XD , Li HT , Chen L ((2016) ) Inhibiting High-Mobility Group Box 1 (HMGB1) Attenuates inflammatory cytokine expression and neurological deficit in ischemic brain injury following cardiac arrest in rats. Inflammation 39: , 1594–1602. |
[56] | Ray R , Juranek JK , Rai V ((2016) ) RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev 62: , 48–55. |
[57] | Walker D , Lue LF , Paul G , Patel A , Sabbagh MN ((2015) ) Receptor for advanced glycation endproduct modulators: A new therapeutic target in Alzheimer’s disease. Expert Opin Investig Drugs 24: , 393–399. |
[58] | Matrone C , Djelloul M , Taglialatela G , Perrone L ((2015) ) Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: Is RAGE the key? Histol Histopathol 30: , 125–139. |
[59] | Tian J , Dai H , Deng Y , Zhang J , Li Y , Zhou J , Zhao M , Zhao M , Zhang C , Zhang Y , Wang P , Bing G , Zhao L ((2015) ) The effect of HMGB1 on sub-toxic chlorpyrifos exposure-induced neuroinflammation in amygdala of neonatal rats. Toxicology 338: , 95–103. |
Figures and Tables
Fig.1
Analysis of the P301L tau transgenic mouse model retina demonstrates the presence of tau oligomers and an association with inflammatory cells. A, B) The retina was stained for tau oligomers (T22, red), astrocytes (GFAP, green), and the nucleus (DAPI). A) The glial cell processes of Muller cells are shown extending to the outer nuclear cells. The co-localizing of mutant tau oligomers with the Muller cell processes concurs with the phagocytic nature of the Muller cells in a pathologic event. Both thick and thin ramifications of the Muller cell processes are noted throughout the outer nuclear layer (ONL) and as well as in close apposition to the outer limiting membrane. Also noted, were multiple small cell bodies of the Muller cells in the inner nuclear layer (INL), with some dendritic spurs towards the ganglion cell layers. The arrowhead in (A) indicates where tau oligomers were co-localizing with GFAP positive Muller cell processes near the rod outer segment (ROS). B) Mutant tau oligomers were observed in the ONL near glial cell processes in the outer plexiform layer (OPL). Arrows indicate the position of the tau oligomers in the ONL. C, D) P301L mouse model retina stained for tau oligomers (T22, red), microglia (Iba1, green), and DAPI. C) Staining revealed microglia cells co-localizing with mutant tau oligomers in the ROS. It is clear that there is hyper-activation and proliferation of microglia cells at the ROS and retinal pigment epithelial (RPE) cell layer. Activation of microglial cells were seen throughout the retina of the P301L transgenic mouse model. Of note, were the small cell bodies with irregular short cell processes that were present in increased numbers near the deposits of tau oligomers. Also, co-localization of the microglia with tau oligomers were observed, suggesting the engulfment of the oligomers by the activated microglial cells. D) Microglia cells co-localized with mutant tau oligomers in the ONL of the retina. GCL- ganglion cell layer.
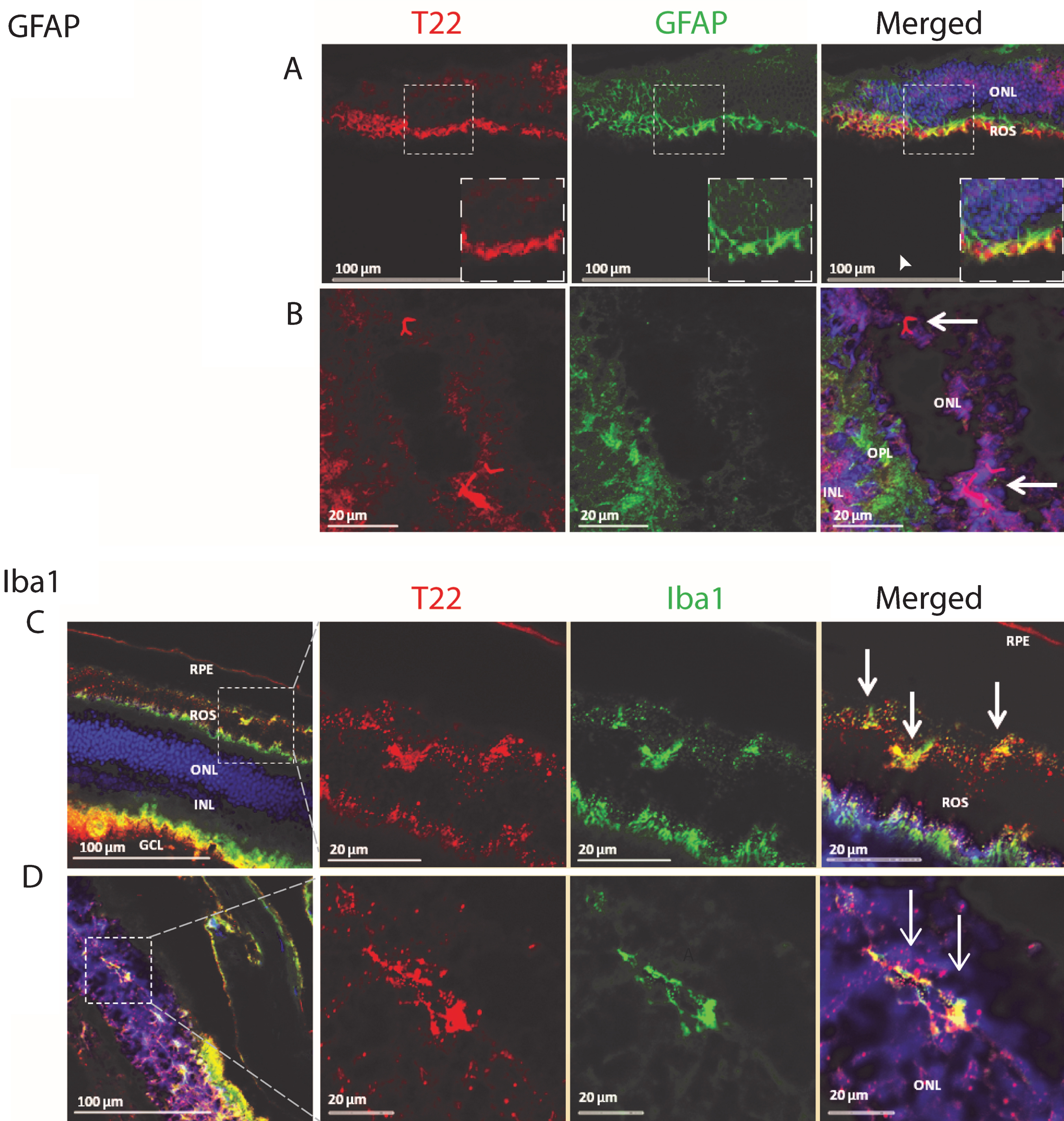
Fig.2
Astrocytes are associated with tau oligomers in the frontal cortex of FTLD and AD. A-I) There was increased staining of the astrocyte marker GFAP (red) in FTLD and AD sections as compared to control. F, I) Inset images are enlarged to show how astrocytes surround the T22 positive tau oligomer (green) containing cell. J) Control, FTLD, and AD homogenates were analyzed by direct ELISA with GFAP. GFAP levels increased significantly in FTLD and AD brains compared to control. K) The co-localization of T22 positive tau oligomers and GFAP was measured using Pearson’s correlation coefficient. Pearson’s correlation coefficient was significantly greater in FTLD and AD images when compared to controls. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar 20 μm.
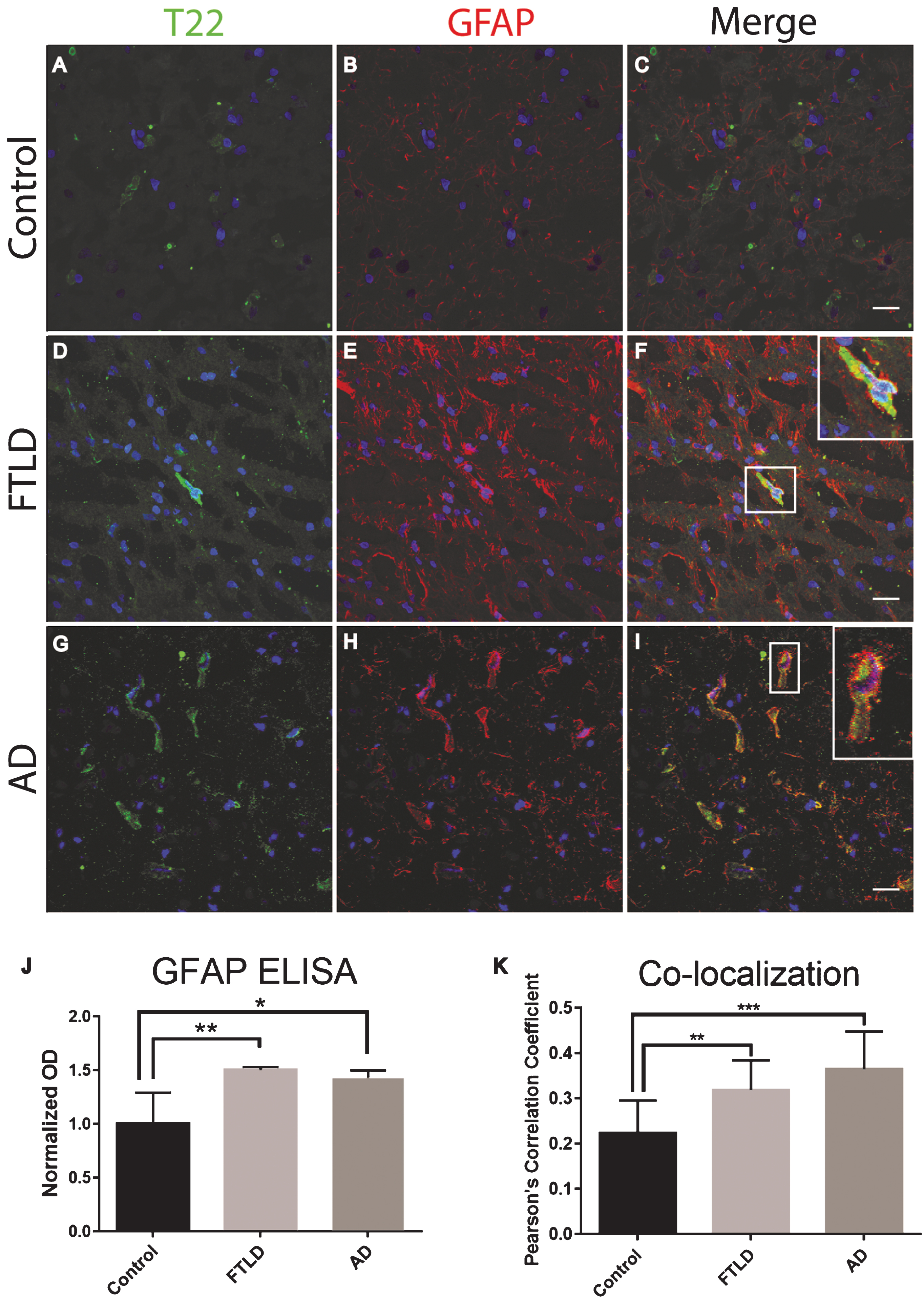
Fig.3
Immunofluorescence analysis of the human tau transgenic mouse model demonstrates an age-dependent increase in tau oligomers and astrocytes in cortical tissues. A-C) WT mice exhibit low astrocyte levels, low levels of reactive astrocytes, and negligible T22 positive tau oligomer (green) staining. D-L) There was an age dependent increase in astrocyte marker GFAP (red) and T22 positive tau oligomers at 11 and 24 months of age. D-F) At 3 months, Htau mice have limited astrocyte activation. G-I) At 11 months of age, Htau mice begin to show evidence of fragmented astrocytes, as well as an increased abundance of both astrocytes and tau oligomers in comparison to WT and 3-month-old Htau mice. J-L) Astrocyte activation is dramatically increased with T22 positive tau oligomers in the brain tissue of 24-month-old Htau mice. M) Quantified GFAP intensity using corrected cell fluorescence. ANOVA with multiple comparisons showed a significant difference between WT and 24-month-old Htau mice. 24-month-old Htau mice were also significantly higher than 3-month-old Htau mice. There was a steady increase in GFAP as the Htau mice aged, consistent with astrogliosis and disease progression. N) As GFAP increased, T22 positive tau oligomer staining also increased with WT and 3-month-old Htau mice in the lower left portion of the correlation and increasing with the 24-month-old mice as shown in the upper right. There was a moderate correlation between GFAP and T22 positive tau oligomer staining (r = 0.662, r2 = 0.439, p < 0.01). ****p < 0.0001, ***p < 0.001. Scale bar 20 μm.
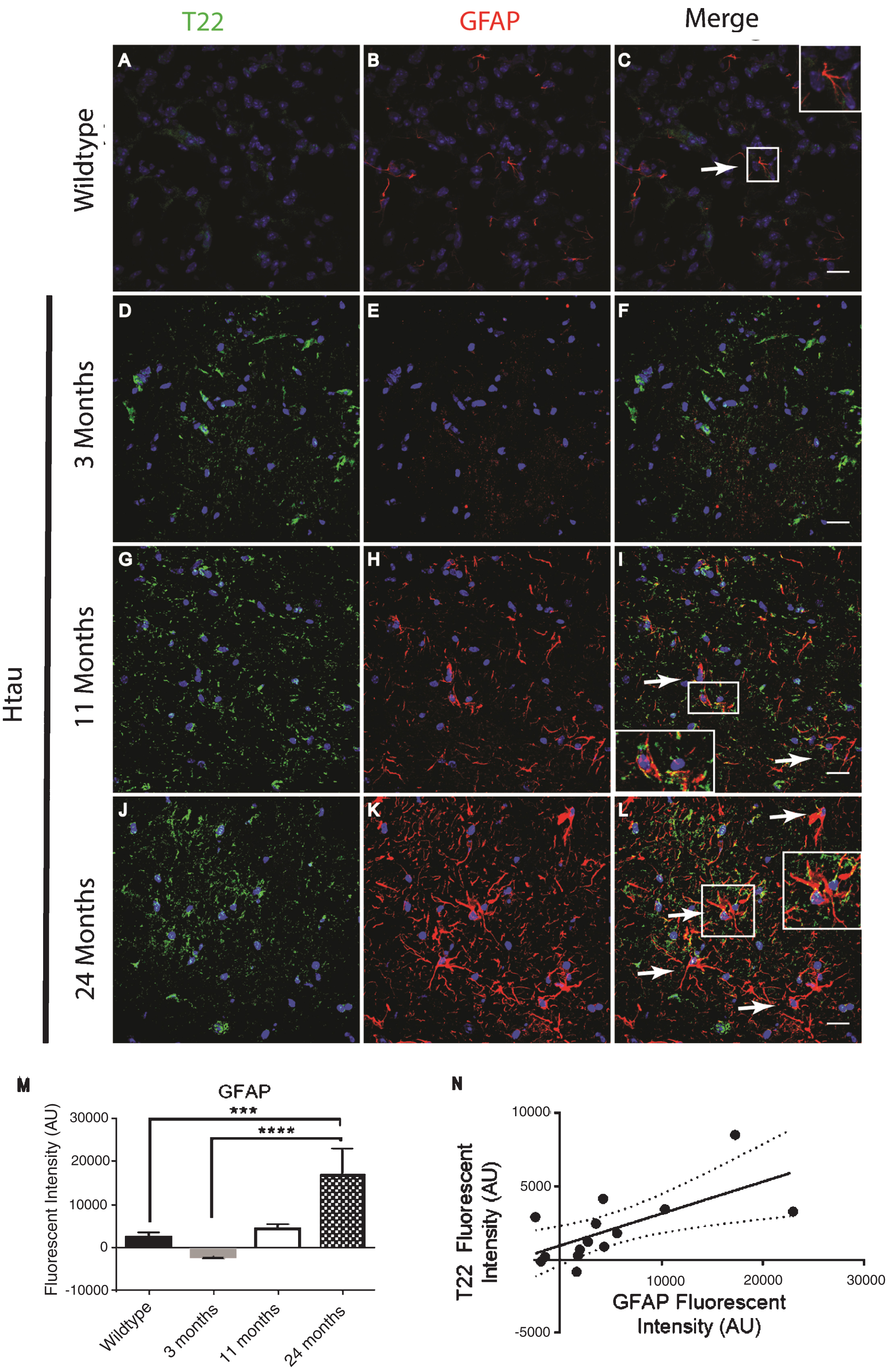
Fig.4
Microglia co-localized with tau oligomers in the frontal cortex of FTLD and AD. A-I) Microglia Iba1 (red) staining increased in FTLD and AD compared to control and co-localized with T22 positive tau oligomers (green). D-F) Representative FTLD sections. Enlargement in (F) shows microglia co-localizing with T22 positive tau oligomers suggesting engulfment of oligomers by microglia. G-I) Representative AD images also showed co-localization between T22 positive tau oligomers and microglia. J) Iba1 corrected cell fluorescence showed significantly elevated levels of Iba1 in FTLD and AD compared to controls, indicating microgliosis. **p < 0.01. Scale bar 20 μm.
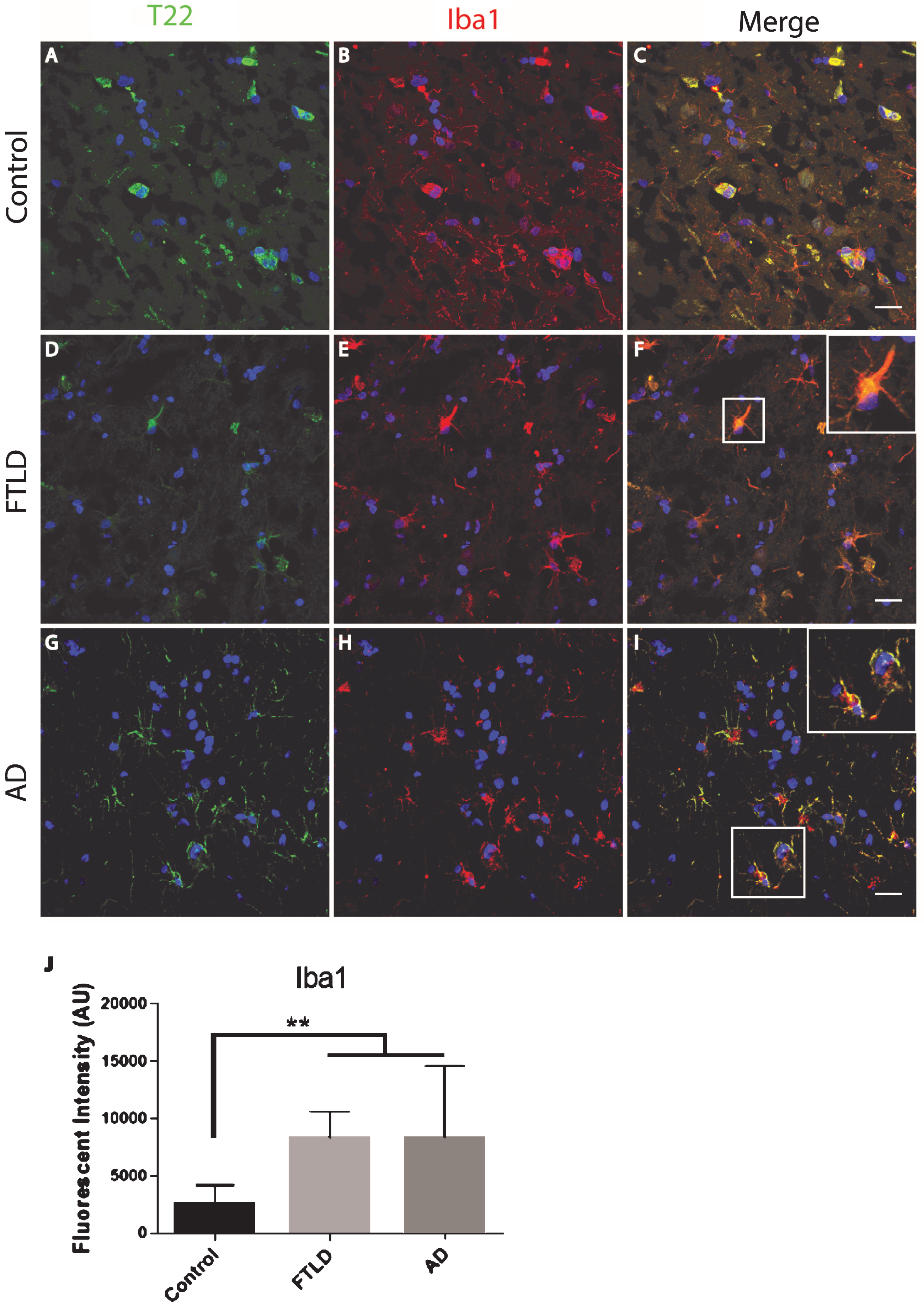
Fig.5
Microglia co-localized with tau oligomers in an aged human tau transgenic mouse model but not in wildtype mouse cortical brain regions. A-C) Minimal microglia Iba1 (red) activation was observed in WT mice. D-F) An increase in activated microglia detected by Iba1 was seen co-localizing with T22 positive tau oligomers (green) in 24-month-old Htau mice. A few examples of microglia co-localizing with T22 positive tau oligomers are indicated by the arrows. G) 24-month-old Htau mice had significantly higher Iba1 compared to WT according to corrected cell fluorescence. **p < 0.01. Scale bar 20 μm.
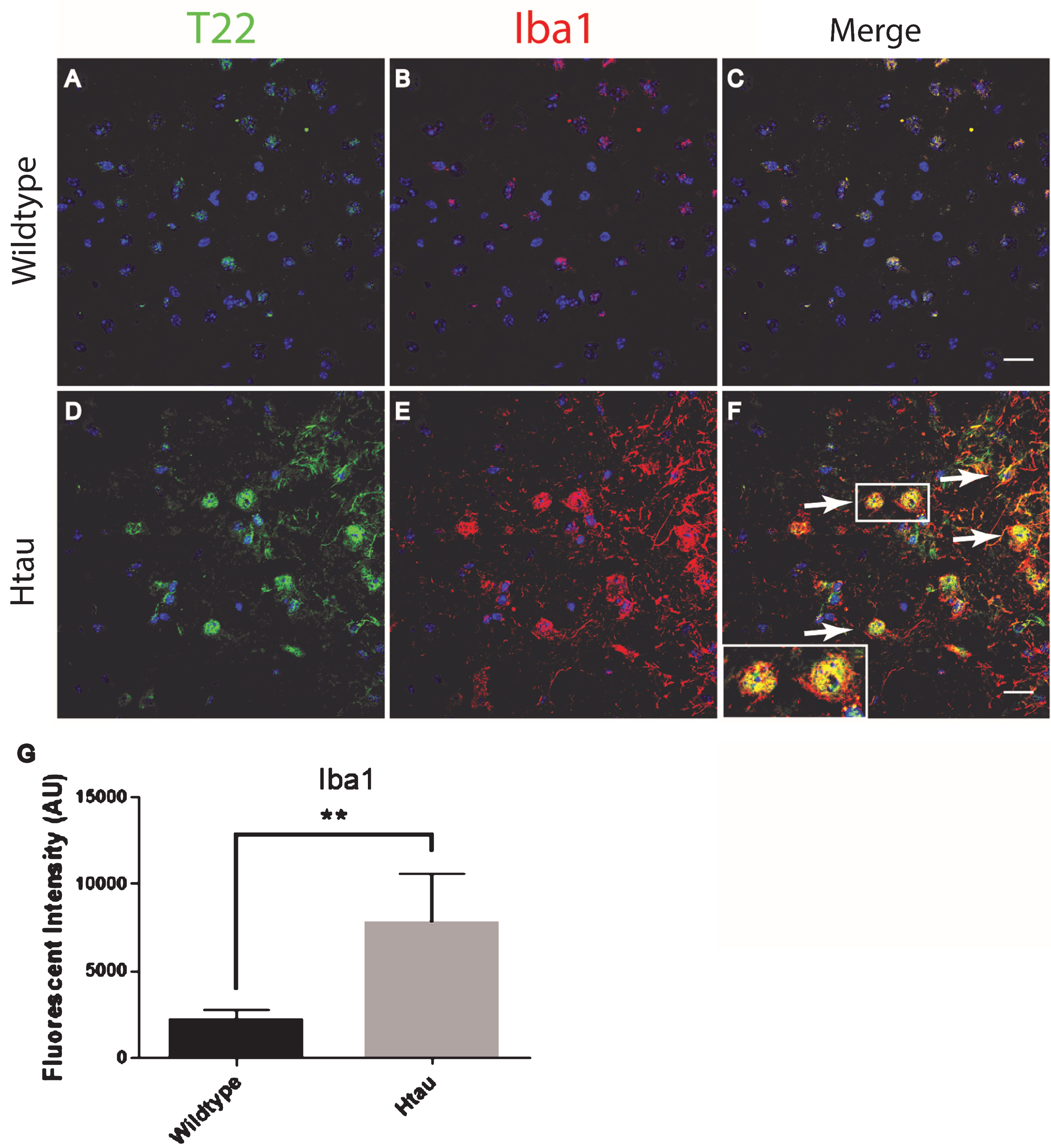
Fig.6
Tau oligomers co-localized with HMGB1 in the frontal cortex of FTLD and AD subjects. A-C) Control subjects showed HMGB1 (red) in proximity to the nucleus (enlargement in C). D-F) FTLD samples displayed HMGB1 translocated out of the nucleus (enlargement in F). T22 positive tau oligomers (green) were co-localized with HMGB1 in FTLD. G-I) AD frontal cortex showed a consistent co-localization between HMGB1 and T22 positive tau oligomers as seen in FTLD. J) There were significantly elevated levels of HMGB1 in FTLD and AD brains compared to controls that were quantified using corrected cell fluorescence. *p < 0.05, ****p < 0.0001. Scale bar 20 μm.
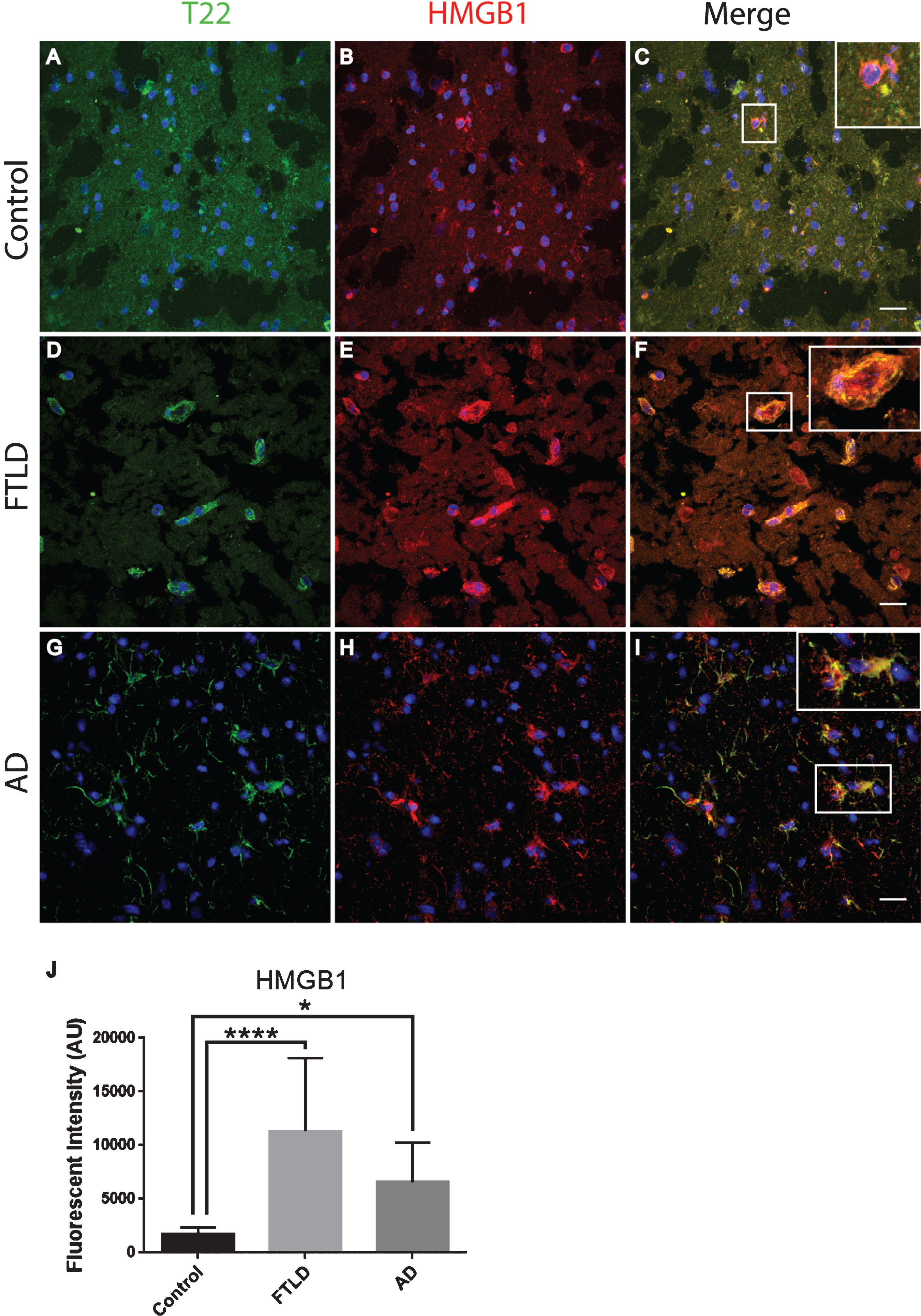
Fig.7
Altered localization of HMGB1in aged human tau transgenic mouse cortical tissue compared to wildtype. HMGB1 co-localized with tau oligomers. A-C) HMGB1 (red) was detected primarily co-localizing with the nucleus (arrow) in WT mice with negligible T22 positive tau oligomers (green). D-F) HMGB1 translocated out of the nuclei (arrowhead) in 24-month-old Htau mice, representative of the activation of HMGB1 as a pro-inflammatory cytokine. Enlargement in (D-F) illustrates the translocation of HMGB1 out of the nucleus and co-localization with T22 positive tau oligomers. G) The HMGB1 corrected cell fluorescence showed a significant increase in aged Htau compared to WT. This is consistent with HMGB1 translocating to the cytoplasm during inflammatory signaling. *p < 0.05. Scale bar 20 μm.
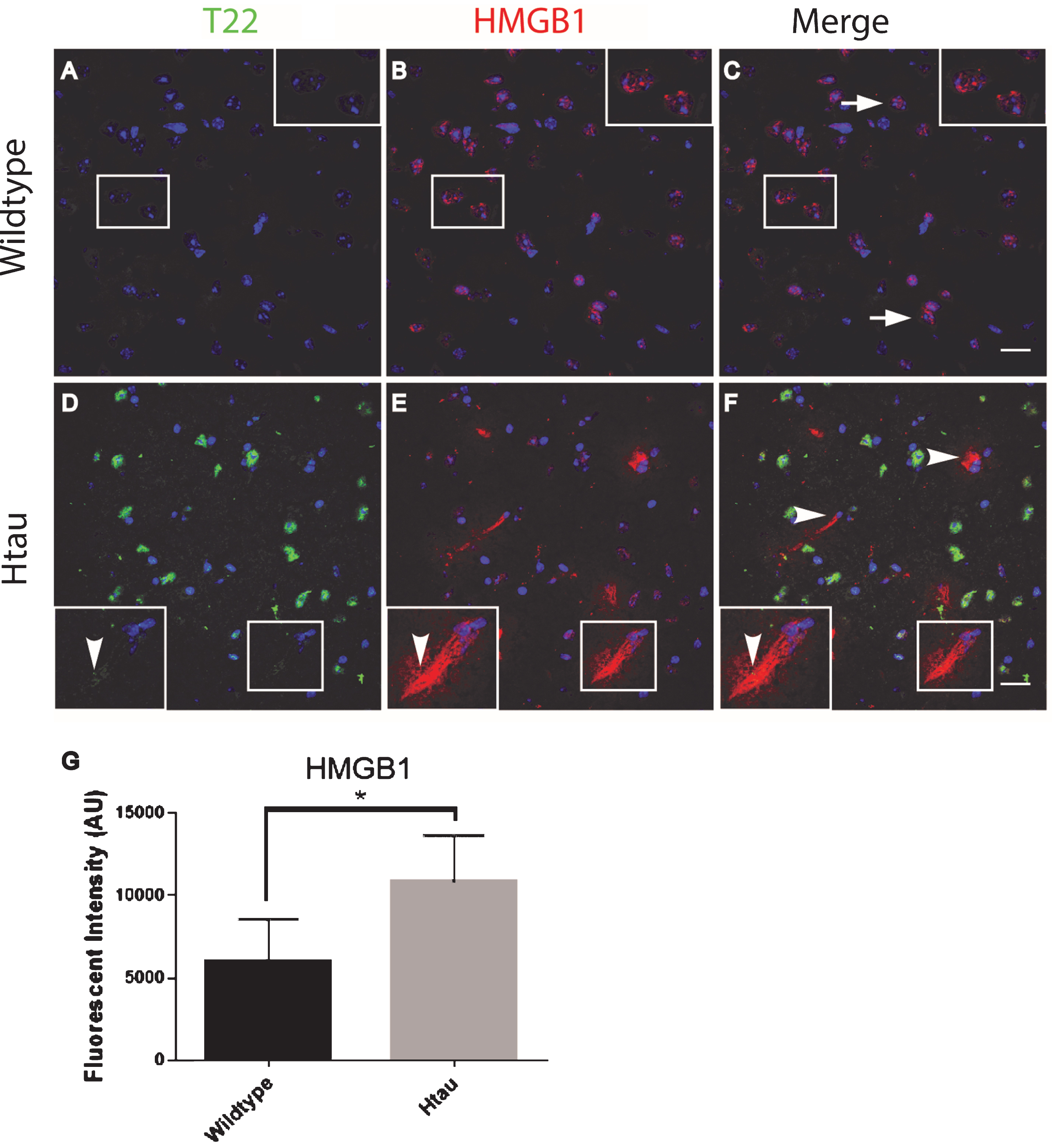
Fig.8
A hypothetical model of the toxic relationship between tau oligomers and inflammation. A) Tau oligomers can spread from brain region to brain region, potentially triggering inflammation and the secretion of pro-inflammatory cytokines by interacting with astrocytes and microglia. The inflammation may become chronic, resulting in further damage to the neurons and in turn, increased inflammation and eventual cell death. B) RAGE, a potential receptor for tau oligomers, signals NFκB through several pathways including through RAS and p38. This increases the production of both RAGE and its ligand, HMGB1, initiating a feed-forward mechanism producing a state of chronic inflammation. Additionally, p38 is known to phosphorylate tau and may increase the abundance of hyperphosphorylated tau, which is more prone to aggregate into oligomers. This may, in turn, induce more inflammation through the production of more oligomers.