
Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database
Abstract
Background:
Recent short-term clinical trials in patients with Duchenne Muscular Dystrophy (DMD) have indicated greater disease variability in terms of progression than expected. In addition, as average life-expectancy increases, reliable data is required on clinical progression in the older DMD population.
Objective:
To determine the effects of corticosteroids on major clinical outcomes of DMD in a large multinational cohort of genetically confirmed DMD patients.
Methods:
In this cross-sectional study we analysed clinical data from 5345 genetically confirmed DMD patients from 31 countries held within the TREAT-NMD global DMD database. For analysis patients were categorised by corticosteroid background and further stratified by age.
Results:
Loss of ambulation in non-steroid treated patients was 10 years and in corticosteroid treated patients 13 years old (p = 0.0001). Corticosteroid treated patients were less likely to need scoliosis surgery (p < 0.001) or ventilatory support (p < 0.001) and there was a mild cardioprotective effect of corticosteroids in the patient population aged 20 years and older (p = 0.0035). Patients with a single deletion of exon 45 showed an increased survival in contrast to other single exon deletions.
Conclusions:
This study provides data on clinical outcomes of DMD across many healthcare settings and including a sizeable cohort of older patients. Our data confirm the benefits of corticosteroid treatment on ambulation, need for scoliosis surgery, ventilation and, to a lesser extent, cardiomyopathy. This study underlines the importance of data collection via patient registries and the critical role of multi-centre collaboration in the rare disease field.
LIST OF ABBREVIATIONS
DMD | Duchenne Muscular Dystrophy |
ENMC | European Neuromuscular Centre |
GCP | Good clinical practice |
TGDOC | TREAT-NMD Global Database Oversight Committee |
TREAT-NMD | Translational Research in Europe – Assessment & Treatment of Neuromuscular Diseases |
INTRODUCTION
TREAT-NMD, an “EU funded network of excellence” founded in 2007, has pioneered the formation of national and global patient registries in the field of rare neuromuscular diseases. In Duchenne muscular dystrophy (DMD), TREAT-NMD has facilitated the establishment of standardised national patient registries in multiple countries worldwide via the use of a standardised mandatory data set collected by each registry [1]. These national registries have combined data in an effort to create the TREAT-NMD global database for DMD [2], a unique global resource containing mutational information for over 7000 DMD patients and clinical data from over 5000 DMD patients from 31 countries. Data derived from registries and natural history studies are complementary. Registries involve much wider population cohorts and may demonstrate country-specific differences usually not captured by natural history (observational) clinical studies. At the same time, natural history studies provide high quality longitudinal data, including clinical and multi-disciplinary assessment not captured by registries. Collectively, they lead to an increased understanding of the complexity, variability and progression of DMD [3, 4].
DMD is a progressive, muscle-wasting disease with an X-linked mode of inheritance, affecting between 1 in 3500 to 1 in 5000 live male births globally [5]. Affected boys become symptomatic at 3–5 years of age due to proximal muscle weakness. Untreated DMD boys lose independent ambulation at an average age of 9.5 years of age [6]. The use of corticosteroids in controlled clinical trials has been shown to delay this loss of ambulation by 1-2 years [7]. Life-expectancy is typically limited to the second or third decade due to respiratory failure or cardiomyopathy. However, the application of ventilatory support in addition to a multidisciplinary care approach has lead to a growing adult DMD population, with several patients surviving as far as the fourth or fifth decades [8, 9]. Currently there is no cure for DMD. The implementation of care recommendations (including corticosteroids, cardiac medications and assisted ventilation) improve outcomes and quality of life, but their effects on the underlying disease mechanisms are as yet unknown [10–12]. Many experimental therapies designed to repair the primary genetic defect are being pursued, including the mutation specific approach of ‘exon-skipping’ and stop codon read through therapies (e.g. Altaluren, TranslarnaTM). However, a lack of comprehensive information on the natural history of DMD poses significant barriers to applying meaningful outcome measures to clinical trials, and subsequently to gaining regulatory approval. In order to gain a comprehensive understanding of clinical outcomes and the evolving heterogeneity of the disease, data from large numbers of patients from a variety of countries (where healthcare often differs) is required. In this study we determined clinical outcomes in relation to genotype and corticosteroid treatment in a large, multinational registry cohort of DMD patients.
MATERIALS AND METHODS
Patients and study design
In this cross-sectional study, we analysed clinical data from 5345 genetically confirmed DMD (identification of a pathogenic dystrophin mutation leading to lack of expression of function dystrophin protein) patients from 31 countries, held within the TREAT-NMD global DMD database. Standardised specific data based on the TREAT-NMD mandatory and highly encouraged items were collected via the national TREAT-NMD patients registries. Anonymised data from these registries [13] were transferred to the global DMD-database via a secure File Transfer Protocol in November 2013 in order to provide a single anonymised cohort of DMD phenotypes, clinical data and outcomes.
Patient organisations and patient advocacy groups were involved and consulted when the study was designed. Patient groups were represented in the European Neuromuscular Centre (ENMC) workshops, and TREAT-NMD Global Database Oversight Committee (TGDOC) meetings. In some cases, national DMD registries are run by patient organisations, so these directly contributed to the data collection. Patient organisations were also involved in publicising information about the registry and assisting with patient recruitment. The results are disseminated to the patients via registry newsletters and TREAT-NMD newsletters.
For analysis patients were categorised by corticosteroid background (yes, no, past and unknown use) and then further stratified by age (under the age of 20 and 20 years and older). Age stratification was carefully chosen to reduce selection bias in the analysis of the group of patients under the age of 20 years. Above the age of 20, study participation declined likely due to loss to clinical follow-up or death. Analysis of patients aged 20 years and older should therefore be interpreted with caution.
Statistical analysis
In the group of patients under the age of 20 years we used Turnbull estimator to analyse the time to reach disease milestones (wheelchair dependence, the need for scoliosis surgery, the need for ventilation and cardiomyopathy). Turnbull analysis was chosen because actual event time was not collected (interval-censored). Turnbull analysis is a generalization of the Kaplan-Meier estimator and allows the analysis of interval-censored data [14]. Turnbull estimates were computed in the software R (www.r-project.org). In the group of patients aged 20 years and older the number of deceased patients was considered significantly and increasing with time, but an exact number was not known. Therefore Turnbull or Kaplan Meier estimator could not be used. Instead we used chi-square analysis to analyse the effect of corticosteroids on disease milestones above the age of 20 years. Chi-square analysis was carried out in Graph pad Prism (GraphPad Software, La Jolla, CA 92037 USA).
RESULTS
Patients
The global TREAT-NMD DMD registry contains 7149 DMD patients. For one quarter of the records (25.2%: n = 1804) incomplete clinical information was provided, leaving 5345 patients suitable for in-depth clinical analysis. 84.4% (n = 4509) of the patients were under the age of 20 years and 15.6% (n = 836) were 20 years and older. (Table S1. in the Supplementary Appendix shows an overview of the number of patients in the database per age group and the number of patients analysed per clinical milestone).
Country specific differences in patient populations were observed in the dataset. Generally, long-standing registries (in existence for more than 5 years) had higher numbers of older patients than newer registries. The oldest patient populations in the database come from Japan and the Netherlands, with as high as 7% of their registry population being older than age 30.
Corticosteroid use
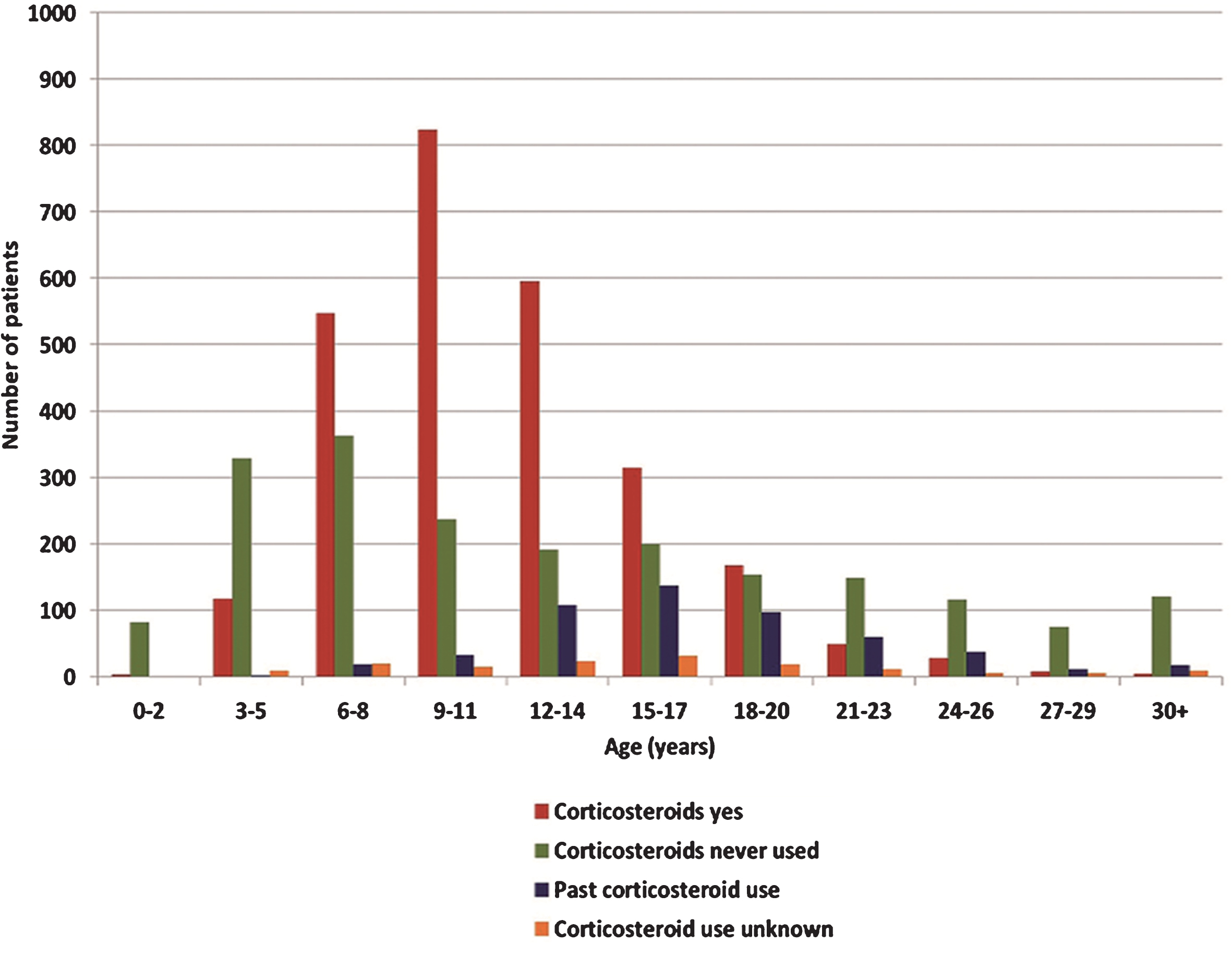
Corticosteroid use within the registry was recorded in four main categories: “corticosteroids yes” (reflecting current corticosteroid use, dose unspecified), “corticosteroids no” (reflecting never used corticosteroids) and “past corticosteroid use”(dose, duration of past use and stopping reasons not specified). Almost half of the study population (49.7%: n = 2658) were currently using steroids, 37.7% (n = 2015) had never used corticosteroids and 9.8% (n = 522) reported past steroids used. For the remaining 2.8% (n = 150) patients the use of corticosteroids was unknown. Corticosteroid use varied with age; younger patients reported more corticosteroid use than older patients. Current care standards recommend commencing corticosteroid use at around 4–6 years of age [15]; and this was reflected within the database. Until the age of 14 years corticosteroid use within the database increased in an age dependent manner, with patients aged 6–8, 9–11 and 12–14 reporting the greatest corticosteroid use (57%, 74% and 64% respectively) (Fig. 1). Corticosteroid use notably decreased after the age of 14 as ambulation decreased. In the database, population 20 years and older, only 15.2% (n = 127) of patients reported current corticosteroid use. This was further reduced in the population aged 30 years and older where only 4.4% (n = 6 of 135) reported current corticosteroid use. The early use of corticosteroids has only been common practice since 2004, and DMD patients born before 1998 are less likely to have ever used corticosteroids. (Table S2. in the supplementary index provides an overview of corticosteroid use per age group within the database).
Fig.1
Overview of corticosteroid use within the global TREAT-NMD DMD database. Corticosteroid was reported as “corticosteroids yes” (current corticosteroid users/red bars), “corticosteroids never used ”(corticosteroids never used/green bars), “past corticosteroid use” (used corticosteroids in the past/blue bars) or “corticosteroid use unknown” (corticosteroid use unknown/orange bars”).
Corticosteroids and clinical milestones in patients under the age of 20 years
Ambulation
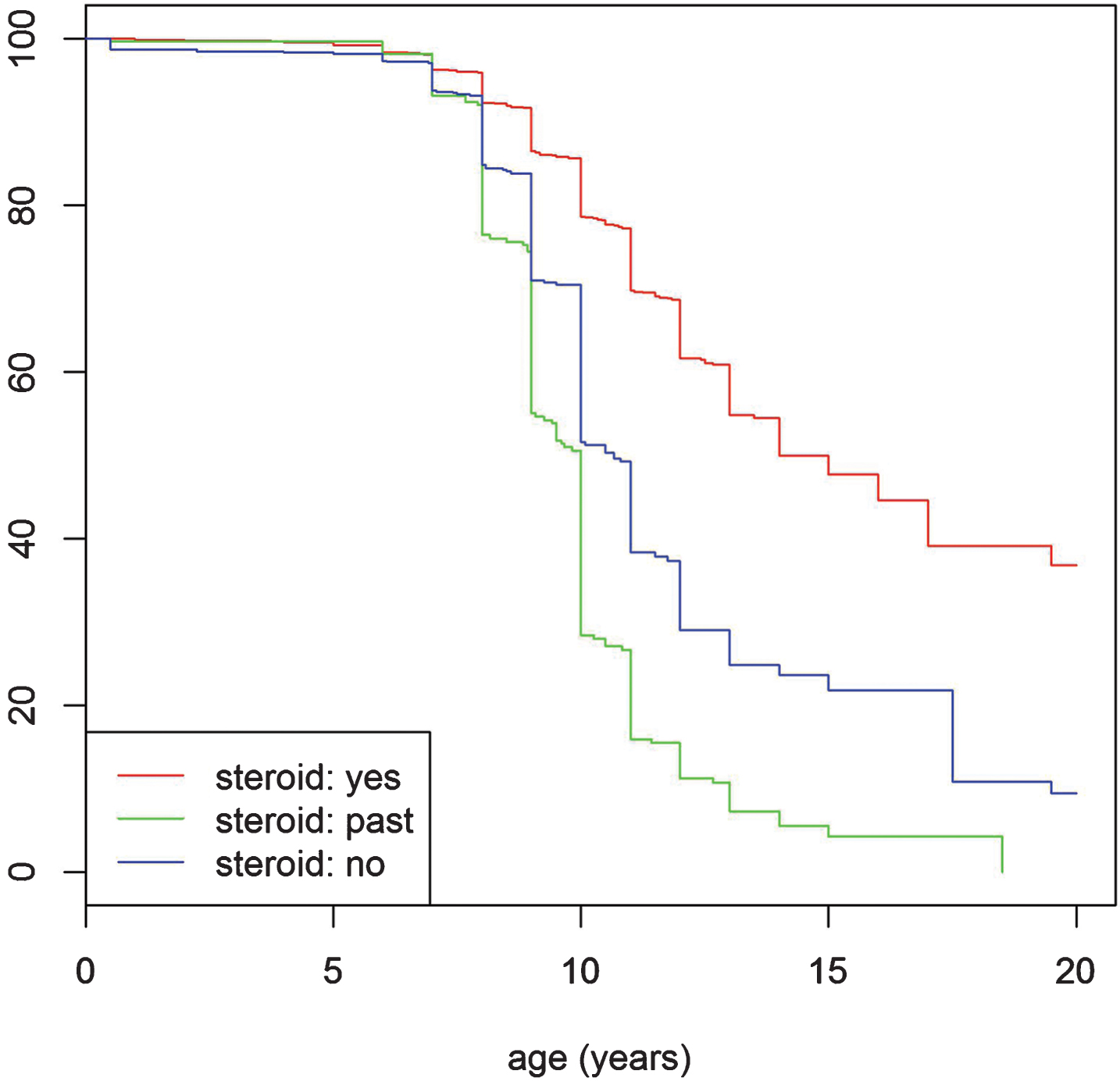
The proportion of ambulant and non-ambulant patients correlated with age and also independently with the use of corticosteroids. In the non-steroid treated population, 71% (n = 633) of patients under the age of 10 years remained ambulant (Fig. 2 and the corresponding Table S3. in the supplementary index). Beyond this age range, ambulation decreased significantly, with 52% (n = 464) of the non-steroid treated patients remaining ambulant at the age of 10 years. By comparison 79% (n = 1375) of patients currently taking steroids, remained ambulant at the age of 10 years. Median age at loss of ambulation in non-steroid treated patients was 10 years old, compared with 13 years in the steroid treated patients (p < 0.001).
Fig.2
Turnbull analysis for loss of ambulation. The red line indicates the “corticosteroid yes” group, the green line the “corticosteroid no (never)” group and the blue line the “past corticosteroid use” group. P-value indicates the difference between the steroid: yes and steroid: no groups.
Scoliosis surgery
A total of 9% (n = 488) of the registry patients have had scoliosis surgery. Forty two percent (n = 205) of these patients were under the age of 20 years. Turnbull analysis showed that from the age where patients begin to require scoliosis surgery (14 years and onwards), the use of corticosteroids significantly reduced the number of patients having scoliosis surgery (p < 0.001) (Figure S1A. in the Supplementary Appendix).
Ventilation
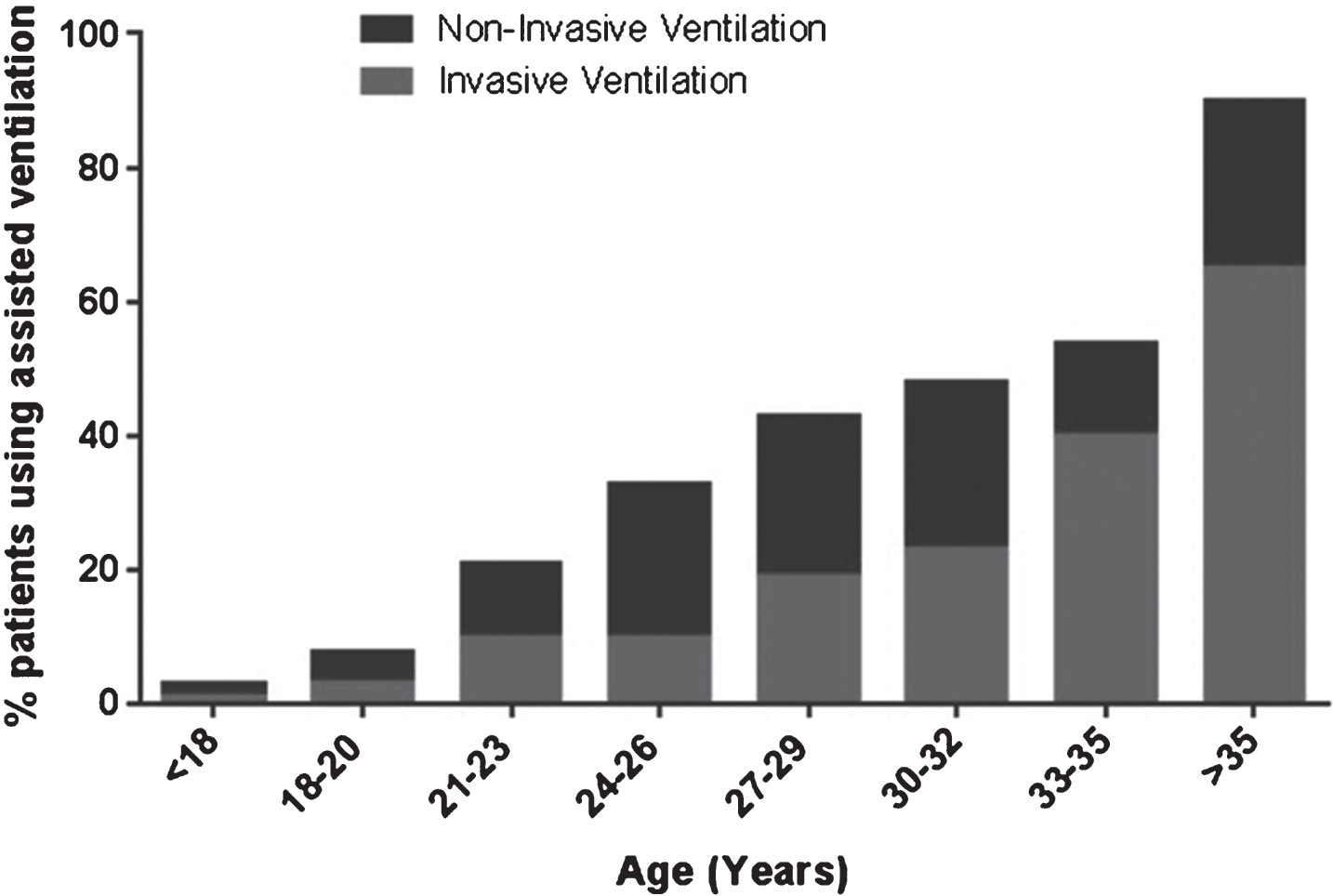
Overall 10.3% (n = 549) of registry patients used assisted ventilation. Assisted ventilation fell into two categories, non-invasive (nasal/mouth mask) or invasive (tracheostomy). Non-invasive ventilation accounted for 74.0% (n = 406) of the ventilated population (average age 25.5 years) and invasive ventilation for 25.0% (n = 137) (average age 28.1 years). For 1.1% (n = 6) of the patients type of ventilation was unknown. The use of assisted ventilation increased with age (Fig. 3) with 2.9% (n = 129) of the patients under the age of 20 years reporting the use of assisted ventilation, compared with 50.4% (n = 420) of patients aged 20 years and older. As shown by Turnbull analysis corticosteroids had a positive effect on ventilation (p < 0.001) (Figure S1B. in the Supplementary Appendix).
Fig.3
Assisted ventilation within the global TREAT-NMD DMD database.
Cardiomyopathy
Cardiomyopathy was reported in 12.0% (n = 639) of registry patients. In 66.5% (n = 425) diagnosis was based on a reduced ejection fraction on echocardiogram. In the remaining 34.5% (n = 214) the diagnostic methods were not available. Prevalence of cardiomyopathy in the database increased with age with 6.7% (n = 302) of patients aged 20 and under reporting cardiomyopathy and 40.4% (n = 337) of patients aged 20 and above having cardiomyopathy. Until the age of 20 no significant effect of corticosteroids on the development of cardiomyopathy was observed (p = 0.94). Importantly, no trend for a negative effect of the use of corticosteroids was seen (Figure S1C. in the Supplementary Appendix).
Corticosteroids and clinical milestones in patients aged 20 years and older
Whilst the majority of patients in the DMD global database were less than 20 years old, there was a sizable cohort of patients aged 20 and over 15.6% (n = 836).
33.9% (n = 283) of patients aged 20 years and older underwent scoliosis surgery. As observed in the younger patients group, the frequency of scoliosis surgery was lower in the corticosteroid treated population (χ2 4.4 p < 0.0001) (Fig. 4A). Assisted ventilation was reported in 50.4% (n = 420) of patients over the age of 20 years. 4.8% (n = 20) of these patients reported using corticosteroids, 16.9% (n = 70) reported past use, 75.5% (n = 318) reported never having used corticosteroids, and for 2.6% (n = 11) corticosteroid use was unknown. Chi-square testing of this group showed that corticosteroid use is associated with significantly less use of assisted ventilation (χ2 7.2 p < 0.0001) (Fig. 4B). Generally, non-invasive ventilation was utilised more than invasive ventilation except for patients over the age of 35. Patients aged 30 or older (n = 152) showed 75.5% (n = 115) dependence on assisted ventilation (36.5%: n = 42 invasive, 50%: n = 76 non-invasive).
Fig.4
Chi-square testing for scoliosis surgery (A), ventilation (B) and cardiomyopathy (C) in the patient group aged 20 years and older.
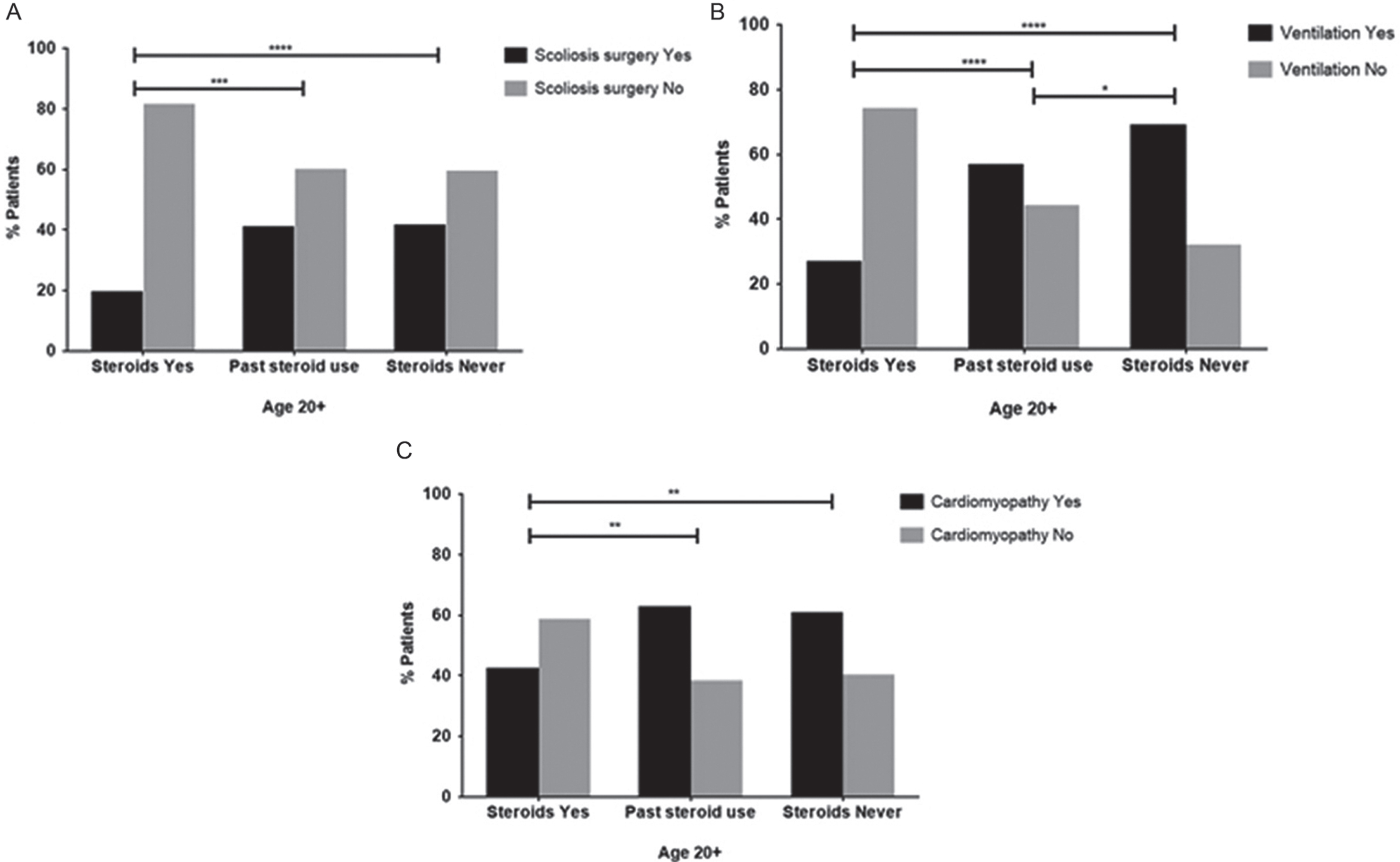
In contrast to the younger age group, further analysis of patients aged 20 years and older showed a cardioprotective effect of corticosteroids. Chi-square testing showed that in this age group cardiomyopathy was less frequent in the cohort using steroids (42%: n = 31) than in the past treated (62%: n = 71) or never steroid treated (60%:n = 229) cohorts (χ2 2.9, p = 0.0035) (Fig. 4C).
Genotype analysis in older age groups
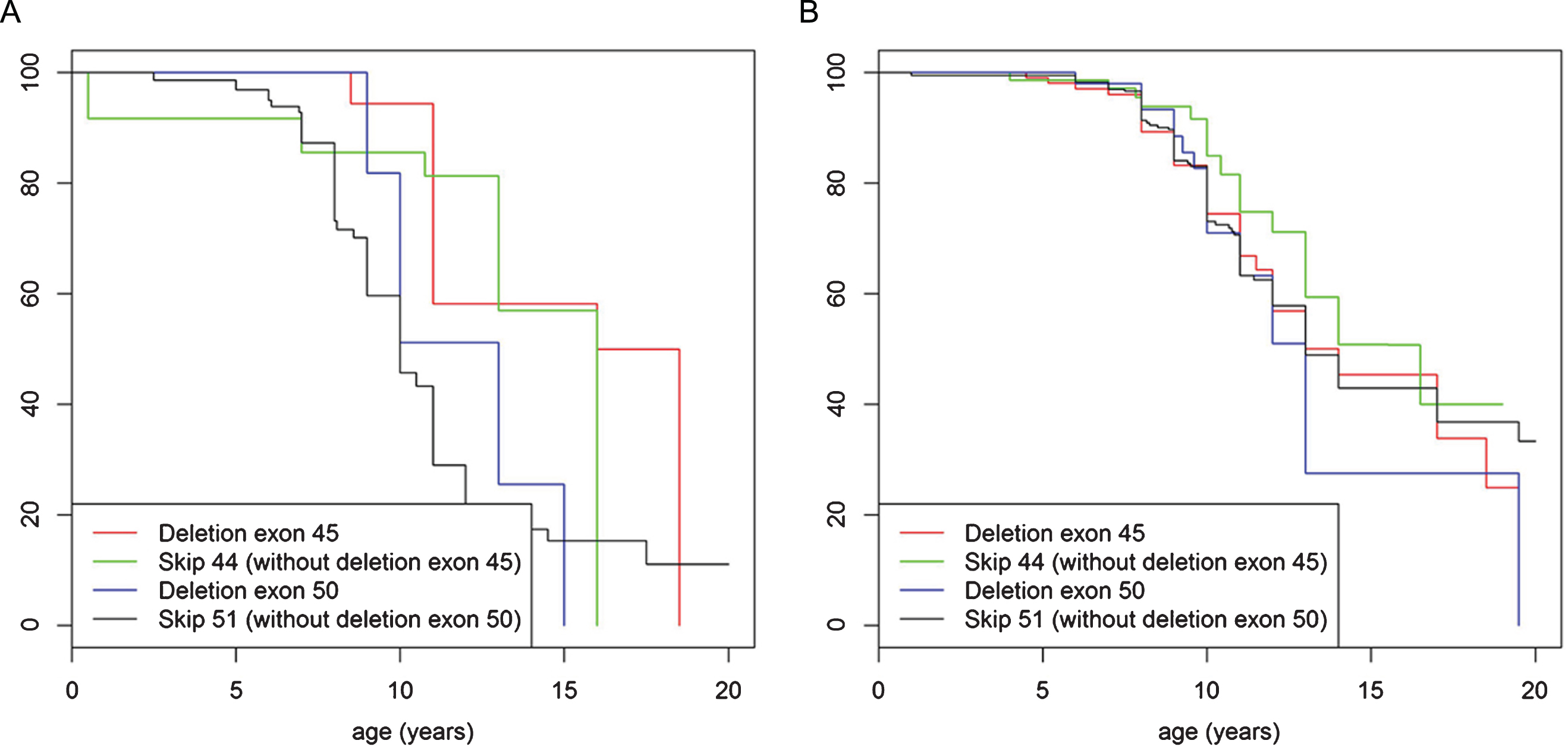
We examined different age ranges of the older cohort of patients for most frequent mutations to assess whether any mutations in particular predispose to more severe or less severe course of disease. Whilst distribution of mutation type within the population aged 20 years and older was similar to the genotype distribution in the patient group under the age of 20 years (with deletions being most frequent followed by small rearrangements then duplications), detailed analysis showed that deletion of exon 45 was the most frequent mutation in the cohort aged 20 years and older accounting for 6.7% (n = 56) of the population. In contrast, this mutation was present in only 4.3% (n = 196) of patients under the age of 20 years. (Fig. S2A. and B. in the Supplementary Appendix). In addition, patients under the age of 20 years, never treated with corticosteroids and with a single exon deletion of exon 45 lost ambulation significantly later (mean age 15 years old) than patients in this age group with other most common single exon deletions (deletion exon 51, 44, 52 and 50) (mean age 11 years) (p = 0.027) (Fig. 5A). When treated with corticosteroids no significant difference in loss of ambulation was observed (p = 0.52) (Fig. 5B). The data was then further stratified to compare the abovementioned group of patients with single exon deletion 45 with mutations that would be rescued by current exon skipping strategies, specifically “skip exon 44” (excluding single exon deletion 45) and “skip exon 51” (excluding single exon deletion 50). An advantage was observed in terms of loss of ambulation for both single exon deletion 45 and the skip exon 44 group (Fig. 6A), but not for deletions including exon 45 that cannot be brought back into frame by exon 44 skipping. In the corticosteroid treated population median age of loss of ambulation for single exon deletion 45, skip exon 44, as well as the single exon deletion 50 and the skip exon 51, were in the same median age range (14 to15 years), (Fig. 6B).
Fig.5
Turnbull analysis of loss of ambulation in patients under the age of 20 years, never (A) and ever (B) treated with corticosteroids and with a single exon deletion of exon 45 (red line), exon 51 (green line), exon 44 (purple line), exon 52 (black line) and exon 50 (blue line).
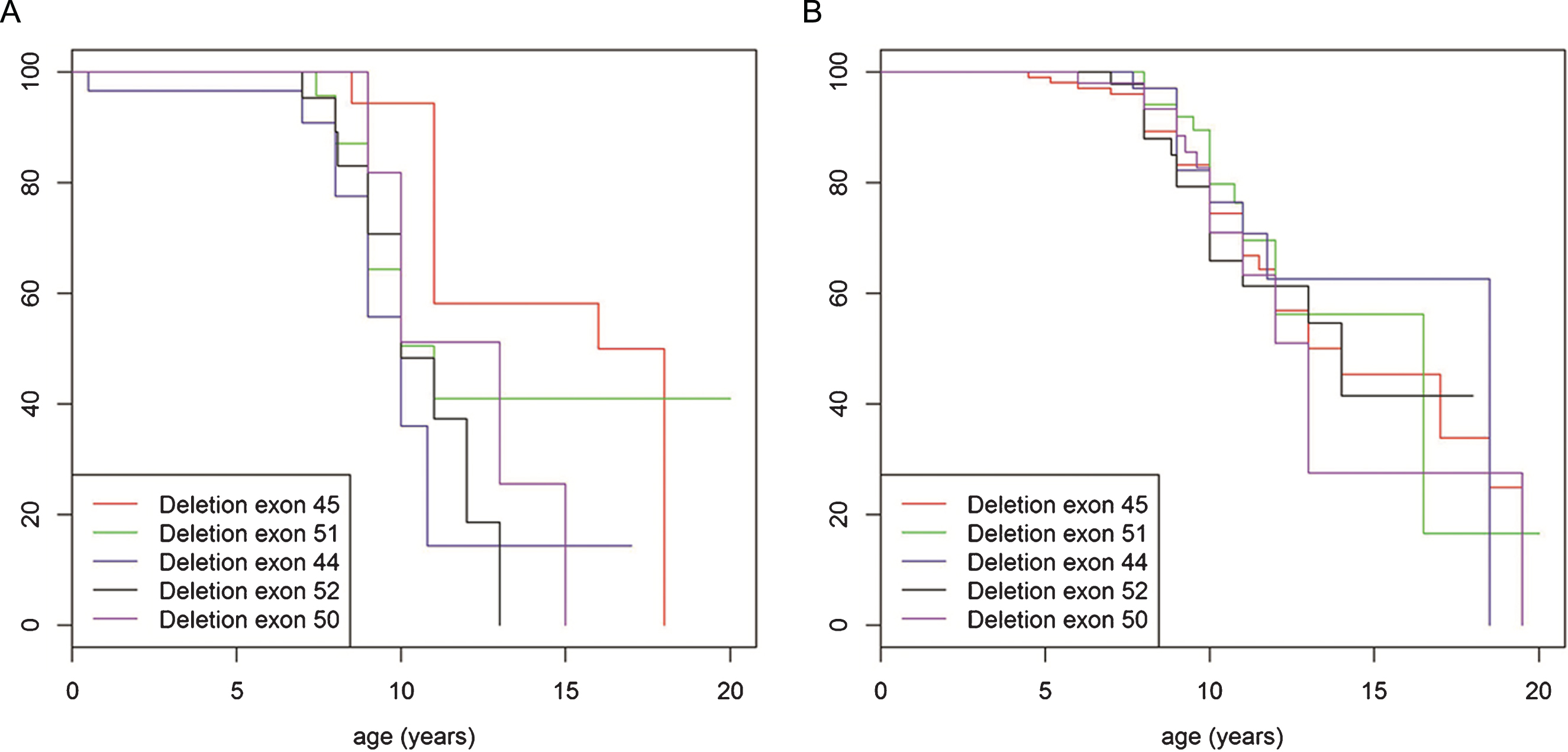
Fig.6
Turnbull analysis of loss of ambulation in patients under the age of 20 years in mutations rescued by exon skip 44 and 51. (A) represents patients never treated with corticosteroids and (B) represents patients ever treated with corticosteroids. In both figures the red line indicates patients with deletion of exon 45 mutation, the green line indicates patients with mutations amenable to exon skipping with exon 44 (but not deletion of exon 45 mutations), the blue line indicates patients with deletion of exon 50 mutation and the black line indicates patients with mutations amenable to exon skipping with exon 51 (but not deletion of exon 50 mutations).
DISCUSSION
DMD is a severe, progressive condition affecting 1 in 3600–6000 live male births [15–16]. A range of therapeutic approaches targeted at causative mutations of the dystrophin gene have been proposed including exon-skipping, which aims to restore the correct reading frame and result in expression of at least partly functional dystrophin protein [17–21], and techniques to enable readthrough of premature stop codons. Despite promising results in early stage studies, failure of recent phase 3 trials to meet primary endpoints [22] suggests the clinical variability of DMD is greater than anticipated and that this may mask treatment benefit. Variability may partly be due to the disease-modifying effect of long-term corticosteroid use, increasingly common amongst younger patients, as recommended in international consensus guidelines [15, 16, 23]. Data from smaller natural history studies and national registries has also suggested that changes in care and particular mutations in the dystrophin gene may be correlated with clinical outcomes [24, 25].
The current evidence for corticosteroid treatment in DMD is largely from short-term data from randomised control trials; long-term data, particularly on prolongation of ambulation, are limited [26]. Our study shows that corticosteroids are advantageous for all major clinical outcomes. Patients taking corticosteroids retain ambulation status for longer, have scoliosis surgery less frequently, require ventilatory support later in life, and have a lower incidence of cardiomyopathy (in patients ≥20 years). While the mechanism underlying this advantage is still not completely understood, it is thought that corticosteroids may act to reduce the rate of muscle breakdown [27], and may reduce muscle necrosis and inflammation [28]. However, not all immunosuppressive or anti-inflammatory drugs have an effect on DMD, suggesting immunosuppression is not the only function of corticosteroids [29]. Alternative possible mechanisms of action are through increasing the number of muscle precursor cells [30, 31].
While advantages of corticosteroids are clear in terms of ambulation, their impact on skeletal defects, ventilation or cardiomyopathy has been controversial so far. Retrospective studies in Japanese DMD patients using the Remudy patient registry have shown patients using prednisolone lose ambulation on average 11 months later than their non-steroid treated counterparts [32]. Our results confirm the profound effect of corticosteroid treatment on ambulation, scoliosis surgery, ventilatory support and, to a lesser extent, cardiomyopathy [33]. Patients not treated with corticosteroids have a 90% probability of developing a clinically significant progressive scoliosis [34]. Daily corticosteroid use has been shown to reduce the incidence of scoliosis [35, 36]. Our data show that patients currently taking corticosteroids are less likely to have had scoliosis surgery. Furthermore, our data show that patients currently taking corticosteroids are less likely to require ventilatory support.
There have been previous concerns that, due to preserved skeletal muscle function and thus increased stress on a vulnerable myocardium, corticosteroids may worsen cardiomyopathy in DMD [37]. In our cohort, no negative effects of the use of corticosteroids on the heart were observed. Moreover, in the cohort of patients aged ≥20 years a cardioprotective effect was observed.
Corticosteroids tend to be initially prescribed in patients around age 4–6 years and discontinued at or shortly after permanent wheelchair dependence. We acknowledge this as a limitation of our study analysis since some patients will have stopped taking corticosteroids once they have permanently lost ambulation and were therefore considered “past steroid users” in our study. In this group, patients with unfavourable genotypes, lack of response to corticosteroids or intolerable side effects may be overrepresented. In general, our analysis in the older DMD population may be biased towards favourable outcomes, as –based on voluntary registration of living individuals – deceased patients are not included. This may be reflected in a lower than expected percentage of scoliosis surgery, cardiomyopathy and ventilation in patients aged ≥20 years.
Historically the clinical progression of DMD was considered relatively homogeneous with predictable outcomes for muscle function and survival. We show that there is greater variability in clinical progression and outcomes than previously reported, with increasing evidence for the role of genetic modifiers [24, 38, 39]. Our analysis revealed that specific exons were associated with either positive or negative effects on disease severity. Such mutation specific responses have been suggested in case reports and smaller clinical studies [25, 40]. Our data highlights that single deletions of exon 45 may infer and advantage to survival when compared to other common single exon deletions. This could be due to exon 45 deletions having a higher frequency of spontaneous exon 44 skipping, giving rise to small amounts of functional dystrophin protein and resulting in long-term clinical benefit, as suggested by data from a smaller cohort [41]. This supports the use of exon-skipping antisense oligonucleotides to restore the reading frame of dystrophin mRNA and the further exploration of its potential therapeutic applications in DMD [17–21].
The global DMD platform continues its development by recruiting new members and national registries under the TREAT-NMD framework. Active research in the treatment of DMD introduces the need for post-marketing surveillance in the long-term assessment of safety and efficacy of therapeutic approaches. The global DMD registry is a good starting point in disease specific long-term data collection, which will allow the collection of specific post-marketing data and comparison of different therapeutic approaches. This work requires multicentre, multinational collaboration, an agreed additional standardized data set (e.g. adverse events forms) and agreed user roles (e.g. patient reported/clinician reported data) inbuilt in national registries to allow comprehensive, Good Clinical Practice (GCP) compliant data collection. Data collected in this way can contribute to better understanding of the course of the disease, disease management, and safety and efficacy profile of new therapies, and ultimately may lead to new policies and disease management protocols.
The infrastructure provided by TREAT-NMD and the standardized, common data set collected by the TREAT-NMD national registries has ultimately facilitated the formation of the TREAT-NMD global database for DMD [2]. This is a collection of more than 7000 genetically confirmed DMD patients worldwide of which we have clinical data, which passed quality control for 5345 patients. To our knowledge this is the single largest cohort of genetically confirmed DMD patients to date and has allowed us to highlight important DMD clinical milestones and outcomes. To facilitate the inclusion of patients, a flexible reporting methodology has been applied that makes use of both patient self-report and professional report depending on differences in medical and cultural conditions as well as practical considerations such as cost effectiveness. Great care has been taken in training, standardization, and curation, but some variability in reporting between different sites and countries is inherent to the registry approach. The trends uncovered will need to be taken into consideration for novel DMD treatments like exon-skipping, but will also have impact on DMD prognosis, management and care standards.
In addition to allowing access to large amounts of data, registries provide a mechanism for collecting longitudinal data at relatively low cost and often include information on patients who would be excluded from natural history studies or clinical trials. Unlike in randomized clinical trials, causality cannot be firmly established from registry data and we accept this as a limitation of our study. However, our data may help to power future clinical trials by highlighting the need for trial design to take into account the heterogeneity of the DMD population including patient age, mutation type and corticosteroid background. The data collected via registries contribute to better understanding of the cost-effectiveness of new therapies, allows assessment of the burden of illness, and helps in planning health economic actions and model healthcare system for a specific rare condition in the most effective way. It is also very reassuring that the association of corticosteroid treatment with improved clinical outcomes can be observed in the “real world” across many different countries with different healthcare settings and ethnic backgrounds and regardless of a specific corticosteroid regime.
CONCLUSIONS
Clinical trials in rare disorders such as DMD are challenging, expensive and liable to low recruitment numbers, lack of validation of outcomes in larger cohorts, and variability of assessment across multiple centres or countries. Power calculations are often based on assumptions and on data on a limited number of participants in single centres, and recent trials to prove the efficacy of novel drugs in DMD have not reached their primary endpoint in measures like the 6-minute-walk test [22]. While well-controlled clinical studies including natural history studies are expected to improve this knowledge and to de-risk future trials, complementary and supporting information can be obtained from pooling a limited, but well standardized set of data across multiple registries and countries. This cross-sectional approach allows correcting for country- or site-specific differences and for long-term changes in “real-world” standards of care and treatment. We suggest that through this approach, better assumptions of the variability in outcomes can be obtained, and power calculations and stratification strategies improved. In addition, with the development of new genetic therapies, comes the need for post-marketing surveillance to assess the long-term safety and efficacy of novel therapies. The global DMD registry will serve as an important initial source this information, allowing the generation of disease specific long-term data and facilitating the comparison of different therapeutic approaches, using standardized methods of data collection.
DECLARATIONS
Ethics approval and consent to participate
Ethics approval was obtained by national registries from local REC committees. Patients consented for participation and by national registries in accordance with national standards.
Consent for publication
Participants gave informed consent for data sharing.
Availability of data and material
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Conflict of interest
All authors declare: no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work. Professor Hanns Lochmüller was elected chair of the TREAT-NMD Alliance in April 2012. He and Jan Verschuuren both served as chairs of the oversight committee.
Funding
This work was supported by TREAT-NMD operating grants, FP6 LSHM-CT-2006-036825, 20123307 UNEW_FY2013 and AFM 16104. Further support came from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 305444 (RD-Connect) and 305121 (Neuromics) and Medical Research Council UK (reference G1002274, grant ID 98482). Original data collection was facilitated by national registries, which have received public and patient organisation funding. No private company funding was received for any aspect of this work.
Table 1
| AUTHORS’ CONTRIBUTIONS | |||
| Initials | Name | Role | Contribution |
| CLB | Catherine L Bladen | Senior Process Development Scientist | Was a major contributor in writing the manuscript. Performed data analysis and interpretation |
| ZK | Zaïda Koeks | 3rd year PhD student and neurologist in training | Was a major contributor in writing the manuscript. Performed data analysis and interpretation |
| DS | David Salgado | Research Engineer | Analysed and interpreted the patient data |
| EvZ | Erik van Zwet | Associate Professor | Analysed and interpreted the patient data |
| OP | Oksana Pogoryelova | Senior Research Associate | Analysed and interpreted the patient data, contributed to manuscript writing |
| GM | Grace McMacken | Clinical Research Associate | Analysed and interpreted the patient data, contributed to manuscript writing |
| SM | Soledad Monges | Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| MEF | Maria E Foncuberta | Genetic Epidemiology, Pediatrics, Otolaryngology | Analysed and interpreted the patient data, Contributed to data acquisition |
| KK | Kuriaki Kekou | Biologist PhD | Analysed and interpreted the patient data, Contributed to data acquisition, Contributed to data acquisition |
| KK | Konstantina Kosma | Paediatrician | Analysed and interpreted the patient data, Contributed to data acquisition |
| HD | Hugh Dawkins | Director, Office of Population Health Genomics | Analysed and interpreted the patient data, Contributed to the study design and concept |
| LL | Leanne Lamont | Office of Population Health Genomics | Analysed and interpreted the patient data, Contributed to data acquisition |
| MIB | Matthew I Bellgard | Director, Centre for Comparative Genomics (CCG) | Analysed and interpreted the patient data, Contributed to data acquisition |
| AJR | Anna J Roy | Research Scientist | Analysed and interpreted the patient data, Contributed to data acquisition |
| TC | Teodora Chamova | Medical doctor at Clinic of neurology | Analysed and interpreted the patient data, Contributed to data acquisition |
| VG | Velina Guergueltcheva | Child neurologist at EduSeeker | Analysed and interpreted the patient data, Contributed to data acquisition |
| SC | Sophelia Chan | Associate Consultant | Analysed and interpreted the patient data, Contributed to data acquisition |
| LK | Lawrence Korngut | Director, Calgary ALS and Motor Neuron Disease Clinic; Neurologist, Calgary Neuromuscular Clinic | Analysed and interpreted the patient data, Contributed to data acquisition |
| CC | Craig Campbell | Paediatric Neurology | Analysed and interpreted the patient data, Contributed to data acquisition |
| YD | Yi Dai | Gynaecology | Analysed and interpreted the patient data, Contributed to data acquisition |
| JW | Jen Wang | Joint founder of the China DMD Care and Support Association | Analysed and interpreted the patient data, Contributed to data acquisition |
| NB | Nina Barišić | Paediatrician | Analysed and interpreted the patient data, Contributed to data acquisition |
| PB | Petr Brabec | Vice-director for quality and management | Analysed and interpreted the patient data, Contributed to data acquisition |
| JL | Jaana Lahdetie | Senior Consultant in Child Neurology | Analysed and interpreted the patient data, Contributed to data acquisition |
| MCW | Maggie C Walter | Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| OLK | Olivia Schreiber-Katz | Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| VK | Veronika Karcagi | Head of Department of Molecular Genetics and Diagnostics | Analysed and interpreted the patient data, Contributed to data acquisition |
| MG | Marta Garami | TREAT-NMD database manager for Eastern-European Countries | Analysed and interpreted the patient data, Contributed to data acquisition |
| AH | Agnes Herczegfalvi | Leader of Neuromuscular Diseases Centre | Analysed and interpreted the patient data, Contributed to data acquisition |
| VV | Venkatarman Viswanathan | Faculty Associate | Analysed and interpreted the patient data, Contributed to data acquisition |
| FB | Farhad Bayat | Head of Laboratory at the Pasteur Institute of Iran | Analysed and interpreted the patient data, Contributed to data acquisition |
| FB | Filippo Buccellla | Italian DMD Registry Curator | Analysed and interpreted the patient data, Contributed to data acquisition |
| AF | Alessandra Ferlini | Associate Professor in Medical Genetics, Director of the section of Medical Genetics | Analysed and interpreted the patient data, Contributed to data acquisition |
| EK | En Kimura | Chief of Early Stage Exploratory Clinical Trial Unit | Analysed and interpreted the patient data, Contributed to data acquisition |
| JCvdB | Janneke C van den Bergen | MD – Neurology | Analysed and interpreted the patient data, Contributed to data acquisition |
| MR | Miriam Rodrigues | New Zealand NMD Registry Curator | Analysed and interpreted the patient data, Contributed to data acquisition |
| RR | Richard Roxburgh | Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| AL | Anna Lusakowska | Clinical expert; Principal investigator of clinical trial | Analysed and interpreted the patient data, Contributed to data acquisition |
| AKP | Anna Kostera-Pruszczyk | Clinical expert, Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| RS | Rosário Santos | Manager of registry; Director of laboratory | Analysed and interpreted the patient data, Contributed to data acquisition |
| EN | Elena Neagu | Paediatric Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| SA | Svetlana Artemieva | Paediatric Neurologist | Analysed and interpreted the patient data, Contributed to data acquisition |
| VMR | Vedrana Milic Rasic | Curator of DMD and SMA Registry in Serbia | Analysed and interpreted the patient data, Contributed to data acquisition |
| DV | Dina Vojinovic | PhD candidate at Erasmus MC | Analysed and interpreted the patient data, Contributed to data acquisition |
| MP | Manuel Posada | Director, Institute of Rare Diseases Research | Analysed and interpreted the patient data, Contributed to data acquisition |
| CB | Clemens Bloetzer | Clinical expert | Analysed and interpreted the patient data, Contributed to data acquisition |
| AK | Andrea Klein | Dr; Contact person of registry | Analysed and interpreted the patient data, Contributed to data acquisition |
| JDM | Jordi Díaz-Manera | Clinical expert; Investigator of research project | Analysed and interpreted the patient data, Contributed to data acquisition |
| EG | Eduard Gallardo | Biologist; Senior Researcher | Analysed and interpreted the patient data, Contributed to data acquisition |
| AAK | A. Ayşe Karaduman | Turkish DMD and SMA Registry Curator (Ankara) | Analysed and interpreted the patient data, Contributed to data acquisition |
| TOY | Tunca Oznur Yilmaz | Professor of Neonatal and Paediatrics Hereditary Muscle Disorders | Analysed and interpreted the patient data, Contributed to data acquisition |
| HT | Haluk Topaloğlu | Professor of Paediatrics and Neurology | Analysed and interpreted the patient data, Contributed to data acquisition |
| EeS | Rasha El Sherif | Consultant of Neurology and Head of Neurogenetics Unit – Air Hospital, Egypt | Analysed and interpreted the patient data, Contributed to data acquisition |
| AS | Angela Stringer | DMD Registry Curator | Analysed and interpreted the patient data, Contributed to data acquisition |
| AVS | Andriy V Shatillo | Neurologist, DMD registry curator | Analysed and interpreted the patient data, Contributed to data acquisition |
| ASM | Ann S Martin | Genetic Counselor &DuchenneConnect Coordinator | Analysed and interpreted the patient data, Contributed to data acquisition |
| HLP | Holly L Peay | Assistant Professor | Analysed and interpreted the patient data, Contributed to data acquisition |
| JK | Jan Kirschner | Consultant Pediatric Neurologist, Deputy Head of Division | Contributed to the study design and concept |
| KMF | Kevin M Flanigan | Attending Neurologist | Contributed to the study design and concept |
| VS | Volker Straub | Harold Macmillan Professor of Medicine | Contributed to the study design and concept |
| KB | Kate Bushby | Act. Res.Chair of Neuromuscular Genetics | Contributed to the study design and concept |
| CB | Christophe Béroud | Assistant Professor of Genetics, head of the bioinformatics team at the UMR_S 910 research unit | Contributed to the study design and concept |
| JV | Jan Verschuuren | Head of Neuromuscular Section, Department of Neurology | Contributed to the study design and concept |
| HL | Hanns Lochmüller | Professor of Experimental Myology | Contributed to the study design and concept |
All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by TREAT-NMD operating grants, FP6 LSHM-CT-2006-036825, 20123307 UNEW_FY2013 and AFM 16104. Further support came from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 305444 (RD-Connect) and 305121 (Neuromics) and Medical Research Council UK (reference G1002274, grant ID 98482).
The authors would like to acknowledge the families of those living with Duchenne muscular dystrophy who have been instrumental in the formation of the DMD national registries. We also acknowledge former and current members of the TREAT-NMD office at the Institute of Genetic Medicine in Newcastle including Stephen Lynn, Emma Heslop and Rachel Thompson. We also extend our thanks to the members of the TREAT-NMD executive committee: Hanns Lochmüller, Annemieke Aartsma-Rus, Anna Ambrosini, Filippo Buccella, Kevin Flanigan, Eric Hoffman, Janbernd Kirschner, Eugenio Mercuri, Ichizo Nishino, Kathy North, Jes Rahbek and Thomas Sejersen. We also acknowledge the members of the current TGDOC: Nathalie Goemans (Chair), Craig Campbell (Vice Chair), Hugh Dawkins (Outgoing Chair), Anna Ambrosini, Svetlana Artemieva, Alexander N. Baranov, Farhad Bayat, Christophe Béroud, Ria Broekgaarden, Filippo Buccella, Craig Campbell, Nick Catlin (former TGDOC member), Monica Ensini, Pat Furlong, Kevin Flanigan, Ole Gredal, Lauren P. Morgenroth, Serap İnal, Jacqueline Jackson, Pierre-Yves Jeannet, Anna Kaminska, A. Ayse Karaduman, Veronika Karcagi, En Kimura, Janbernd Kirschner, Jaana Lähdetie, Hanns Lochmüller, Vitaliy Matyushenko, Vedrana Milic Rasic, Violeta Mihaylova, Marie-Christine Ouillade, Ian Murphy, Miriam Rodrigues, Rosario dos Santos, Pascale Saugier-Veber, Inge Schwersenz, Thomas Sejersen, Rasha El Sherif, Eduardo Tizzano, Isabela Tudorache, Sylvie Tuffery-Giraud, Jen Wang, Simon Woods, W. Ludo van der Pol, Peter Van den Bergh, Petr Vondráček, Jan Verschuuren.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-170280.
REFERENCES
[1] | Bladen CL , Rafferty K , Straub V , Monges S , Moresco A , Dawkins H , et al. The TREAT-NMD duchenne muscular dystrophy registries: Conception, design, and utilization by industry and academia. Human Mutation. (2013) ;34: (11):1449–57. |
[2] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Human Mutation. (2015) ;36: (4):395–402. |
[3] | Pane M , Mazzone ES , Sivo S , Sormani MP , Messina S , D′Amico A , et al. Long term natural history data in ambulant boys with duchenne muscular dystrophy: 36-month changes. PLoS One. (2014) ;9: (10):e108205. |
[4] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A , et al. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle & Nerve. (2013) ;48: (1):55–67. |
[5] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. |
[6] | Gowers WR , Taylor J . A manual of diseases of the nervous system. London: J. & A. Chuchill; (1892) . |
[7] | Humbertclaude V , Hamroun D , Bezzou K , Bérard C , Boespflug-Tanguy O , Bommelaer C , et al. Motor and respiratory heterogeneity in Duchenne patients: Implication forclinical trials. European Journal of Paediatric Neurology. (2012) ;16: (2):149–60. |
[8] | Baxter P . Treatment of the heart in Duchenne muscular dystrophy. Developmental Medicine & Child Neurology. (2006) ;48: (03):163. |
[9] | Emery AEH . The muscular dystrophies. The Lancet. (2002) ;359: (9307):687–95. |
[10] | Hoffman EP , Reeves E , Damsker J , Nagaraju K , McCall JM , Connor EM , et al. Novel Approaches to Corticosteroid Treatment in Duchenne Muscular Dystrophy. Physical Medicine and Rehabilitation Clinics of North America. (2012) ;23: (4):821–8. |
[11] | Sejerson T , Bushby K . Standards of Care for Duchenne Muscular Dystrophy: Brief Treat-NMD Recommendations. In: Espinós C , Felipo V , Palau F , editors. Inherited Neuromuscular Diseases. Springer Netherlands; (2009) . pp. 13–21. |
[12] | Landfeldt E , Lindgren P , Bell CF , Schmitt C , Guglieri M , Straub V , et al. Compliance to care guidelines for duchenne muscular dystrophy. Journal of Neuromuscular Diseases. (2015) ;2: (1):63–72. |
[13] | Bladen CL , Rafferty K , Straub V , Monges S , Moresco A , Dawkins H , et al. The TREAT-NMD Duchenne muscular dystrophy registries: Conception, design, and utilization by industry and academia. Hum Mutat. (2013) ;34: (11):1449–57. |
[14] | Turnbull BW . The empirical distribution function with arbitrarily grouped, censored and truncated data. Journal of the Royal Statistical Society. Series B (Methodological). (1976) ;290–95. |
[15] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Diagnosis, and pharmacological and psychosocial management. The Lancet Neurology. (2010) ;9: (1):77–93. |
[16] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Implementation of multidisciplinary care. The Lancet Neurology. (2010) ;9: (2):177–89. |
[17] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen G-J , et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation. (2009) ;30: (3):293–9. |
[18] | Aartsma-Rus A , Janson AAM , Kaman WE , Bremmer-Bout M , den Dunnen JT , Baas F , et al. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Human Molecular Genetics. (2003) ;12: (8):907–14. |
[19] | Aartsma-Rus A , Janson AAM , Kaman WE , Bremmer-Bout M , van Ommen G-JB , den Dunnen JT , et al. Antisense-induced multiexon skipping for duchenne muscular dystrophy makes more sense. The American Journal of Human Genetics. (2004) ;74: (1):83–92. |
[20] | van Ommen G-J , van Deutekom J , Aartsma-Rus A . The therapeutic potential of antisense-mediated exon skipping. Curr Opin Mol Ther. (2008) ;10: (2):140–9. |
[21] | van Ommen G-JB , Aartsma-Rus A . Advances in therapeutic RNA-targeting. New Biotechnology. (2013) ;30: (3):299–301. |
[22] | Bushby K , Finkel R , Wong B , Barohn R , Campbell C , Comi GP , et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle & Nerve. (2014) ;50: (4):477–87. |
[23] | Gloss D , Moxley RT 3rd , Ashwal S , Oskoui M . Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. (2016) ;86: (5):465–72. |
[24] | Bello L , Piva L , Barp A , Taglia A , Picillo E , Vasco G , et al. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology. (2012) ;79: (2):159–62. |
[25] | van den Bergen JC , Ginjaar HB , Niks EH , Aartsma-Rus A , Verschuuren JJGM . Prolonged ambulation in duchenne patients with a mutation amenable to exon 44 skipping. Journal of Neuromuscular Diseases. (2014) ;1: (1):91–4. |
[26] | Matthews E , Brassington R , Kuntzer T , Jichi F , Manzur AY . Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. (2016) ;(5):CD003725. |
[27] | Rifai Z , Welle S , Moxley RT , Lorenson M , Griggs RC . Effect of prednisone on protein metabolism in Duchenne dystrophy. American Journal of Physiology - Endocrinology and Metabolism. (1995) ;268: (1):E67–E74. |
[28] | Spuler S , Engel AG . Unexpected sarcolemmal complement membrane attack complex deposits on nonnecrotic muscle fibers in muscular dystrophies. Neurology. (1998) ;50: (1):41–6. |
[29] | Griggs RC , Moxley RT 3rd , Mendell JR , Fenichel GM , Brooke MH , Pestronk A , et al. Duchenne dystrophy: Randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology. (1993) ;43: (3 Pt 1):520–7. |
[30] | Pasquini F , Guerin C , Blake D , Davies K , Karpati G , Holland P . The effect of glucocorticoids on the accumulation of utrophin by cultured normal and dystrophic human skeletal muscle satellite cells. Neuromuscular Disorders. (1995) ;5: (2):105–14. |
[31] | Sklar RM , Brown RH Jr . Methylprednisolone increases dystrophin levels by inhibiting myotube death during myogenesis of normal human muscle in vitro. Journal of the Neurological Sciences. (1991) ;101: (1):73–81. |
[32] | Takeuchi F , Yonemoto N , Nakamura H , Shimizu R , Komaki H , Mori-Yoshimura M , et al. Prednisolone improves walking in Japanese Duchenne muscular dystrophy patients. J Neurol. (2013) ;260: (12):3023–9. |
[33] | Barber BJ , Andrews JG , Lu Z , West NA , Meaney FJ , Price ET , et al. Oral Corticosteroids and Onset of Cardiomyopathy in Duchenne Muscular Dystrophy. The Journal of Pediatrics. (2013) ;163: (4):1080–4.e1. |
[34] | Smith AD , Koreska J , Moseley CF . Progression of scoliosis in Duchenne muscular dystrophy. The Journal of Bone & Joint Surgery. (1989) ;71: (7):1066–74. |
[35] | Alman BA , Raza SN , Biggar WD . Steroid treatment and the development of scoliosis in males with duchenne muscular dystrophy. The Journal of Bone & Joint Surgery. (2004) ;86: (3):519–24. |
[36] | Yilmaz Ö , Karaduman A , Topaloğlu H . Prednisolone therapy in Duchenne muscular dystrophy prolongs ambulation and prevents scoliosis. European Journal of Neurology. (2004) ;11: (8):541–4. |
[37] | Townsend D , Yasuda S , Chamberlain J , Metzger JM . Cardiac consequences to skeletal muscle-centric therapeutics for duchenne muscular dystrophy. Trends in Cardiovascular Medicine. (2009) ;19: (2):49–54. |
[38] | van den Bergen JC , Hiller M , Bohringer S , Vijfhuizen L , Ginjaar HB , Chaouch A , et al. Validation of genetic modifiers for Duchenne muscular dystrophy: A multicentre study assessing SPP1 and LTBP4 variants. Journal of Neurology, Neurosurgery, and Psychiatry. (2015) ;86: (10):1060–5. |
[39] | Flanigan KM , Ceco E , Lamar KM , Kaminoh Y , Dunn DM , Mendell JR , et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Annals of Neurology. (2013) ;73: (4):481–8. |
[40] | Pane M , Mazzone ES , Sormani MP , Messina S , Vita GL , Fanelli L , et al. 6 Minute walk test in duchenne MD patients with different mutations: 12 month changes. PLoS One. (2014) ;9: (1):e83400. |
[41] | Findlay AR , Wein N , Kaminoh Y , Taylor LE , Dunn DM , Mendell JR , et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Annals of Neurology. (2015) ;77: (4):668–74. |

