Identification of a Potentially Functional microRNA–mRNA Regulatory Network in Lung Adenocarcinoma Using a Bioinformatics Analysis
 Xiao-Jun Wang
Xiao-Jun Wang Jing Gao
Jing Gao Zhuo Wang
Zhuo Wang Qin Yu
Qin Yu- 1The First School of Clinical Medicine, Lanzhou University, Lanzhou, China
- 2Department of Respiratory Medicine, Gansu Provincial Hospital, Lanzhou, China
- 3Respiratory Medicine Unit, Department of Medicine, Karolinska Institute, Stockholm, Sweden
- 4Department of Pulmonary Medicine, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 5Department of Pathology Medicine, Gansu Provincial Hospital, Lanzhou, China
- 6Department of Respiratory Medicine, The First Hospital of Lanzhou University, Lanzhou, China
Background: Lung adenocarcinoma (LUAD) is a common lung cancer with a high mortality, for which microRNAs (miRNAs) play a vital role in its regulation. Multiple messenger RNAs (mRNAs) may be regulated by miRNAs, involved in LUAD tumorigenesis and progression. However, the miRNA–mRNA regulatory network involved in LUAD has not been fully elucidated.
Methods: Differentially expressed miRNAs and mRNA were derived from the Cancer Genome Atlas (TCGA) dataset in tissue samples and from our microarray data in plasma (GSE151963). Then, common differentially expressed (Co-DE) miRNAs were obtained through intersected analyses between the above two datasets. An overlap was applied to confirm the Co-DEmRNAs identified both in targeted mRNAs and DEmRNAs in TCGA. A miRNA–mRNA regulatory network was constructed using Cytoscape. The top five miRNA were identified as hub miRNA by degrees in the network. The functions and signaling pathways associated with the hub miRNA-targeted genes were revealed through Gene Ontology (GO) analysis and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. The key mRNAs in the protein–protein interaction (PPI) network were identified using the STRING database and CytoHubba. Survival analyses were performed using Gene Expression Profiling Interactive Analysis (GEPIA).
Results: The miRNA–mRNA regulatory network consists of 19 Co-DEmiRNAs and 760 Co-DEmRNAs. The five miRNAs (miR-539-5p, miR-656-3p, miR-2110, let-7b-5p, and miR-92b-3p) in the network were identified as hub miRNAs by degrees (>100). The 677 Co-DEmRNAs were targeted mRNAs from the five hub miRNAs, showing the roles in the functional analyses of the GO analysis and KEGG pathways (inclusion criteria: 836 and 48, respectively). The PPI network and Cytoscape analyses revealed that the top ten key mRNAs were NOTCH1, MMP2, IGF1, KDR, SPP1, FLT1, HGF, TEK, ANGPT1, and PDGFB. SPP1 and HGF emerged as hub genes through survival analysis. A high SPP1 expression indicated a poor survival, whereas HGF positively associated with survival outcomes in LUAD.
Conclusion: This study investigated a miRNA–mRNA regulatory network associated with LUAD, exploring the hub miRNAs and potential functions of mRNA in the network. These findings contribute to identify new prognostic markers and therapeutic targets for LUAD patients in clinical settings.
Introduction
Lung adenocarcinoma (LUAD) is the most common histological subtype of lung cancer, accounting for approximately 40% of all cases of lung cancer in China and other countries (Bray et al., 2018; Cao and Chen, 2019; Siegel et al., 2020). LUAD is characterized by rapid progression and early development of metastases and a high recurrence rate, with the highest morbidity and mortality in both genders (Bray et al., 2018; Cao and Chen, 2019; Siegel et al., 2020). The overall 5-year survival rates for LUAD only reach approximately 20% (Bray et al., 2018; Miller et al., 2019) given the lack of early detection and limited effective therapies at earlier stages of disease (Gan et al., 2017). LUAD patients have an 80% chance of surviving 5 years if diagnosed at an early stage (Henschke et al., 2006). Thus, there is a growing need to identify and characterize the molecular pathogenesis of LUAD in order to better understand the underlying disease mechanisms and to improve treatment outcomes of this malignancy.
MicroRNAs (miRNAs) are a family of small non-coding RNAs that downregulate gene expression by repressing or degrading messenger RNA (mRNA) targets, thereby controlling the genes involved in cellular processes (Di Leva et al., 2014; Du et al., 2018). miRNAs regulate the expression of approximately 30% of all human genes, playing important regulatory roles in many human diseases (Lewis et al., 2005; Di Leva et al., 2014; Ramassone et al., 2018; Wu K.L. et al., 2019; Wu S.G. et al., 2019; Condrat et al., 2020). miRNAs could be readily detected in circulation and carry information regarding the origin of a neoplasm, thus serving as diagnostic and prognostic biomarkers in the development of tumors (Hummel et al., 2010; Blondal et al., 2013; Qi et al., 2013; Zhang et al., 2016; Liu et al., 2017; Świtlik et al., 2019; Cojocneanu et al., 2020; Wang et al., 2020). An increasing number of studies have reported various expression levels of miRNAs in LUAD (Cazzoli et al., 2013; Zhong et al., 2018), although findings remain inconsistent. Extensive genomic studies have verified that abnormal expressions of multiple mRNAs of genes involved in LUAD tumorigenesis and progression play vital roles (Herbst et al., 2018). The miRNA–mRNA regulatory network is characterized in such a way that individual miRNA could regulate a wealth of different mRNAs of genes, and the individual mRNA of a target gene could be correspondingly suppressed by multiple different miRNAs (Cui et al., 2020; Li et al., 2020; Liu et al., 2020). Thus, it is necessary to examine the miRNA–mRNA regulatory network in LUAD to advance our understanding of its molecular mechanisms.
Microarray analysis has been widely used in the evaluation of miRNA in cancers to understand the complexity and heterogeneity of malignant disease (Jurj et al., 2017; Cojocneanu et al., 2020), but has not been fully elucidated in LUAD. A public dataset from the Cancer Genome Atlas (TCGA) is widely used in LUAD analyses, although cases primarily originate from a non-Asian population, yet include primary tissue samples. Hence, the TCGA dataset and actual clinical patient data from multiple sample types contribute to determining the biomarkers of LUAD and exploring the underlying mechanisms (Rheinbay et al., 2020).
Here, we constructed a miRNA–mRNA regulatory network of LUAD using TCGA data and our microarray analysis data, including multiple sample types and ethnicities, to subsequently identify hub miRNAs in the network. The functional enrichment analysis of the targeted mRNAs of hub miRNAs were investigated using the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. In addition, we analyzed the protein–protein interaction (PPI) network to understand the potential mechanism of mRNAs in LUAD occurrence and development. In doing so, we identified the top ten key mRNAs using the PPI network and CytoHubba. The Gene Expression Profiling Interactive Analysis (GEPIA) allowed us to evaluate these key mRNAs and identify the hub genes in LUAD using survival analysis.
Materials and Methods
Figure 1 illustrates the workflow. This study received ethical approval from the Ethics Committee of the Gansu Provincial Hospital (14 April 2020, No. 2020-117). Informed consent was obtained from all participants in the microarray experiment, and the research adhered to the principles of the Declaration of Helsinki.
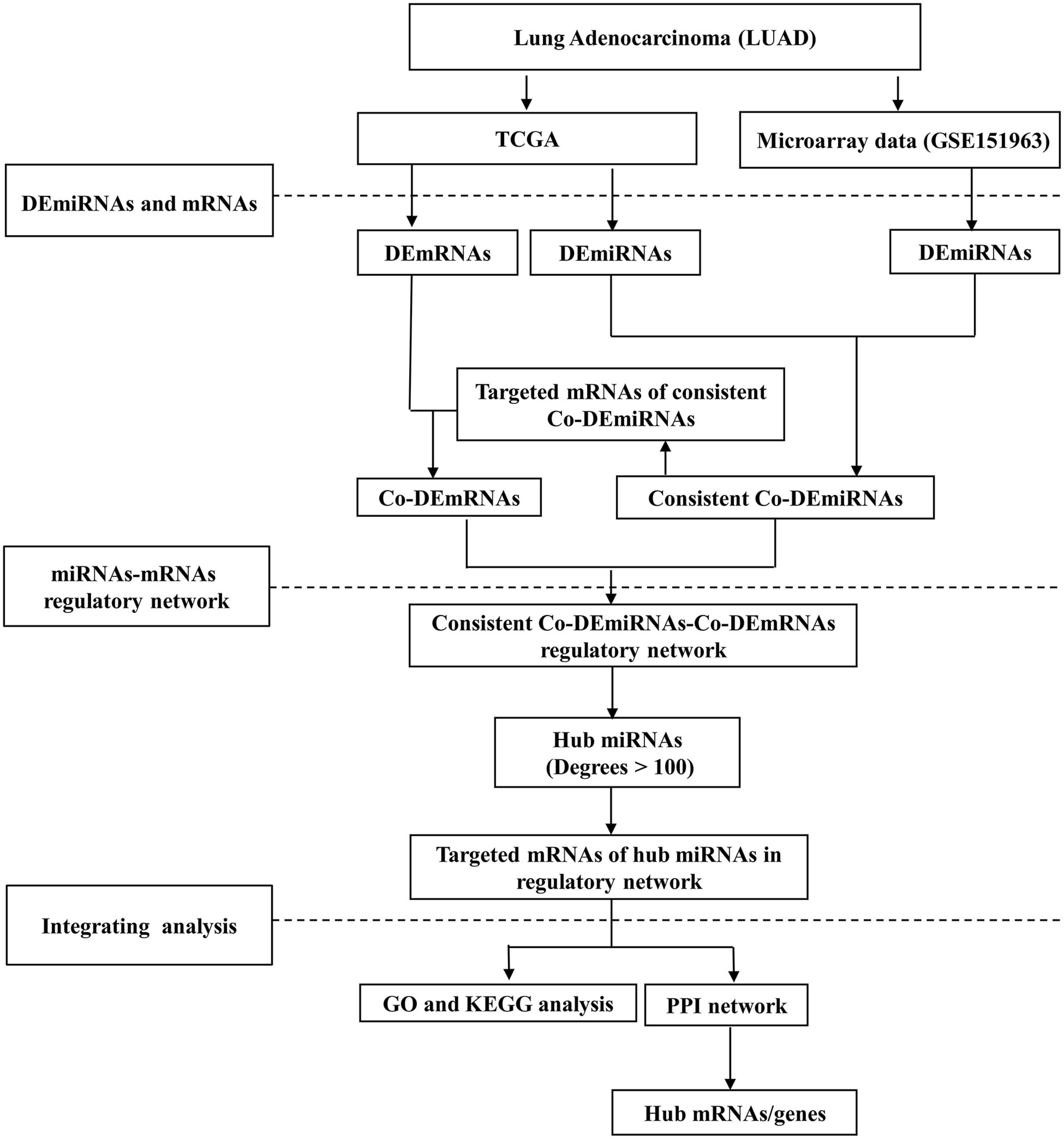
Figure 1. The study workflow. LUAD, lung adenocarcinoma; DE, differential expression; Co-DE, common differentially expressed; Co-mRNAs, common mRNAs; TCGA, the Cancer Genome Atlas; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein–protein interaction.
The TCGA dataset from tissue samples and the microarray data from plasma in LUAD were included in this study. Supplementary Tables 1,2 provide the clinical characteristics in LUAD from the TCGA and microarray data. The TCGA data included miRNA (400 LUAD and 15 normal lung tissue) and mRNA (515 LUAD and 20 normal lung tissue) samples, all downloaded using Firehose1 on 16 April 2020 (Izadi et al., 2017). The mRNA samples were obtained by filtering the samples with low-quality reads as expressions <0.8. The miRNA data were obtained from intersecting the mRNA-sequence data and filtering the samples with low-quality reads as expressions <0.5. The microarray data (n = 10) were collected between October 2018 and March 2019 at Gansu Provincial Hospital (China), and included initially diagnosed LUAD patients (n = 6) and controls (n = 4). We excluded patients who (a) previously received chemotherapy, radiotherapy, molecular-targeted therapy, immunotherapy or surgery before blood samples were collected; (b) had other combined cancers; (c) were pregnant or lactating; or (d) presented with cardiopulmonary insufficiency, serious cardiovascular disease and a serious infection as well as severe malnutrition (Wang et al., 2014; Li et al., 2019).
RNA degradation and contamination, especially DNA contamination, was monitored on 1.5% agarose gels. RNA concentration and purity were measured using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). RNA integrity was assessed using the RNANano 6000 Assay Kit of the Agilent Bioanalyzer 2100 System (Agilent Technologies, CA, United States). A total amount of 2.5 ng RNA per sample was used as the input material for the RNA sample preparations. Sequencing libraries were generated using NEBNextR UltraTM small RNA Sample Library Prep Kit for IlluminaR (NEB, United States) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Raw data (raw reads) of fastq format were first processed through in-house perl scripts. The clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low-quality reads from raw data. The reads were trimmed and cleaned by removing sequences <15 nt or >35 nt. At the same time, Q20, Q30, and GC-content of the clean data were calculated. All downstream analyses were based on clean data with a high quality. Then, using the Bowtie soft tools, clean reads were compared to the Silva database, GtRNAdb database, Rfam database, and Repbase database, respectively, to identify the sequence alignment, and filtered for ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and other ncRNA and repeats. The remaining reads were used to detect known miRNA and novel miRNA predicted through comparison with known miRNAs from miRBase. Detailed information on the microarray appears elsewhere (Zhong et al., 2018). The sequencing reads were uploaded to the gene expression omnibus (GEO) dataset (access number GSE151963).
Construction of the miRNA–mRNA Regulatory Network
The differentially expressed miRNAs (DEmiRNAs) and mRNA (DEmRNAs) were analyzed comparing the LUAD and normal samples using the R package limma. The DEmiRNAs consistent with the expression of TCGA were further analyzed. The isoform expressions of the DEmiRNAs were identified using the R package miRNAmeConverter. The common consistently and differentially expressed (Co-DE) miRNAs were obtained using intersected analyses comparing TCGA and microarray datasets (Cojocneanu et al., 2020). The targeted mRNAs of the Co-DEmiRNAs were predicted using three online analytical software tools including miRDB2, TargetScanHuman (version 7.2)3, and miRWalk (Agarwal et al., 2015; Dweep and Gretz, 2015; Chen and Wang, 2020)4. An overlap was applied to confirm the Co-DEmRNAs identified both in targeted mRNAs and DEmRNAs using FunRich tools (Pathan et al., 2015). Adjusted p-values (adj. p) were used to correct false-positive results (Patten et al., 2016). DEmiRNAs and DEmRNAs were identified applying p < 0.05 and | Log 2 fold-change (log2FC) | >1. The volcano plots were drawn using R package ggplot2. Based on the results of the Co-DEmiRNAs and Co-DEmRNAs, we constructed the miRNA–mRNA regulatory network using the Cytoscape software version 3.7.0 (Shannon et al., 2003). The top five miRNAs (degree >100) were selected as hub miRNAs in the network.
Functional Enrichment Analysis and Confirmation of Hub Genes
The biological functions of the targeted mRNAs of these hub miRNAs were evaluated using the GO and KEGG pathway enrichment analyses with the R package ClusterProfiler (Yu et al., 2012). We considered p < 0.05 statistically significant. Using the STRING database,5 we downloaded data from the PPI network for targeted mRNAs for the hub miRNAs applying the following criteria: (a) homo sapiens; and (b) medium confidence 0.400 (Zhu et al., 2020). Then, the PPI network was further adjusted using the Cytoscape software version 3.7.0, and we identified the top ten key mRNAs of the PPI network using cytoHubba’s Maximal Clique Centrality (MCC) ranking (Yang et al., 2019). The hub mRNAs were further verified using survival analysis using the Gene Expression Profiling Interactive Analysis (GEPIA) tools6. Significant survival was identified through survival analysis (log-rank p < 0.05). The key mRNAs with significant survival outcomes were identified as hub genes in LUAD.
Results
Identification of Differentially Expressed miRNAs and mRNAs
Through small RNA sequence and data standardization, we harvested a total of 3,166 miRNAs for subsequent analysis of the microarray data. In total, we identified 6 patients (50% male, median age of 62.5 years, 100% Asian, and 100% stage III–IV) with LUAD using plasma samples from the microarray data. In total, 319 DEmiRNAs (145 upregulated and 174 downregulated miRNAs) were identified from the plasma comparing LUAD and controls in the microarray data (Figure 2A). In addition, we identified 515 LUAD patients (46% male, median age of 66 years, 2% Asian) from TCGA tissue samples, including stage I (53.4%), stage II (23.7%), and stage III–IV (25.2%). Furthermore, 4,206 DEmRNAs (1,105 upregulated and 3,101 downregulated) and 197 DEmiRNAs (126 upregulated and 71 downregulated) were identified using TCGA [(| log2 FC) | > 1 and p < 0.05, respectively, Figures 2B,C]. In addition, 147 of 197 DEmiRNAs (95 upregulated and 52 downregulated) exhibited isoform expressions.
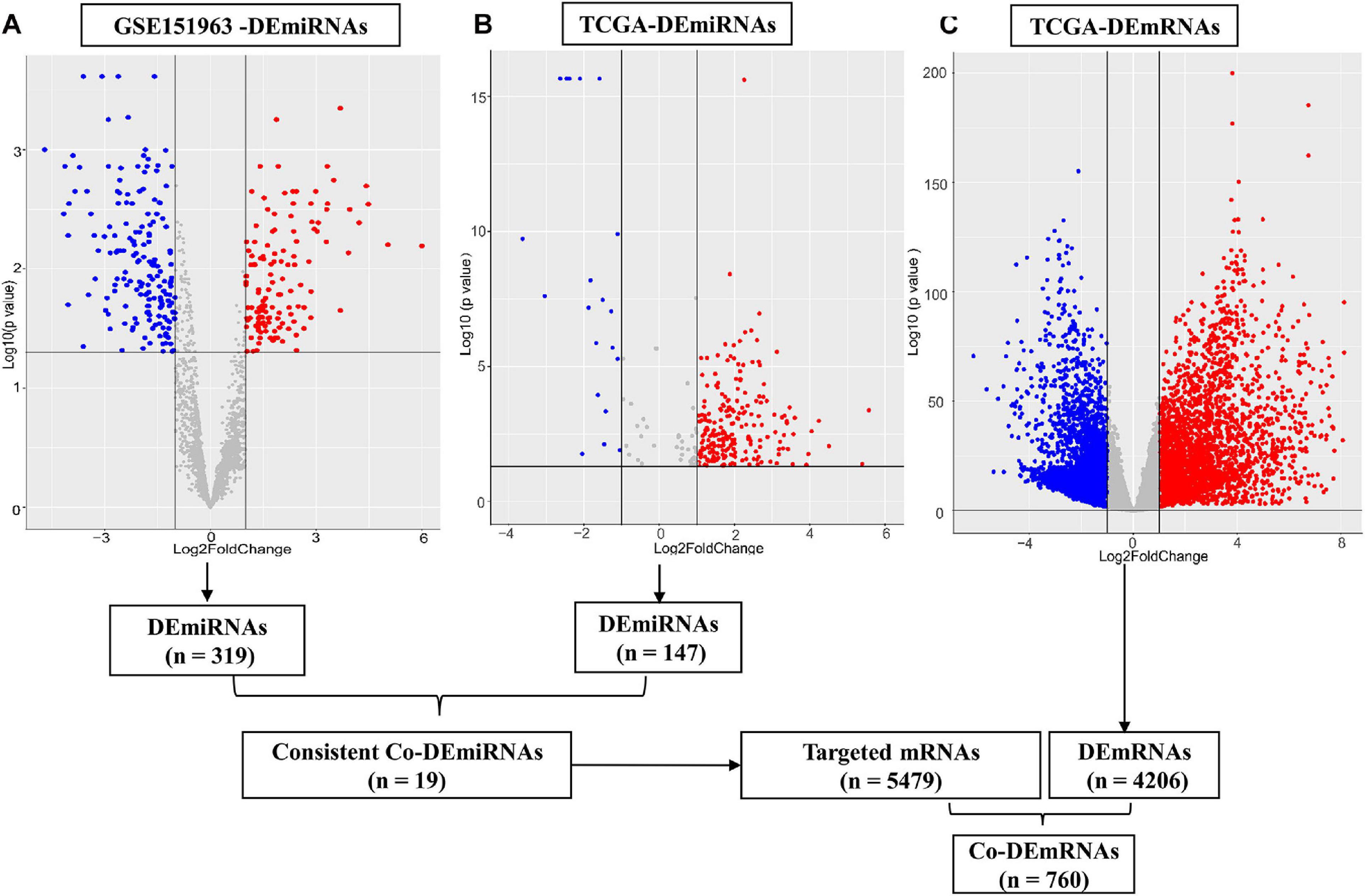
Figure 2. Differential expression of miRNAs and mRNAs. (A) Differential expression (DE) miRNAs in patient cohort (GSE151963), (B) DEmiRNAs in TCGA and (C) DEmRNAs in the TCGA dataset. The red plots show the upregulated expression of miRNA or mRNA; the blue plots show the downregulated expression of miRNA or mRNA; the gray plots show the normal expression of miRNA or mRNA. DE, differential expression; Co-DE, common differentially expressed; miRNA, microRNA; mRNA, messenger RNA.
Constructing the miRNA–mRNA Regulatory Network
The 30 common DEmiRNAs (Co-DEmiRNAs), including 19 consistent DEmiRNAs (6 upregulated and 13 downregulated miRNAs) and 11 inconsistent DEmiRNAs (2 upregulated and 9 downregulated miRNAs from the microarray data and 9 upregulated and 2 downregulated miRNAs from TCGA), were identified through an intersection analysis between the TCGA and microarray datasets (Table 1). In total, 5,479 target mRNAs from 19 consistently Co-DEmiRNAs were identified. Next, 760 Co-DEmRNAs (152 upregulated and 608 downregulated) were identified through the intersection analysis comparing the DEmRNAs in the TCGA and microarray datasets. The miRNA–mRNA regulatory network consists of 19 Co-DEmiRNAs and 760 Co-DEmRNAs (Figure 3). Overall, 19 consistently Co-DEmiRNAs identified 985 targeted relationships with 760 Co-DEmRNAs. Furthermore, the top five miRNAs were identified as hub miRNAs in the network, including miR-539-5p (216 degrees), miR-656-3p (209 degrees), let-7b-5p (135 degrees), miR-2110 (125 degrees), and miR-92b-3p (117 degrees). Finally, 677 Co-DEmRNAs consisted of targeted mRNAs from five hub miRNAs.
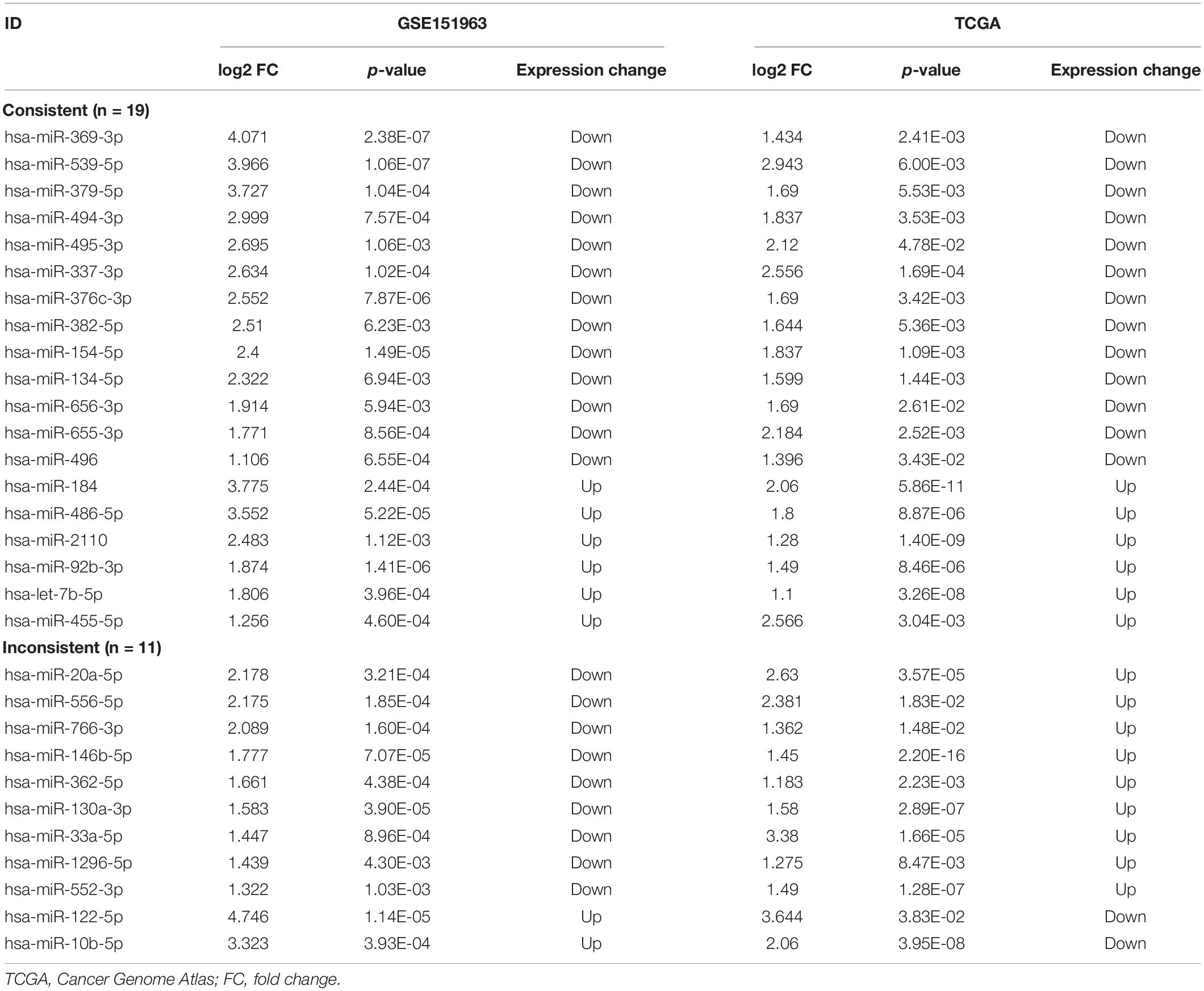
Table 1. Common differentially expressed miRNAs (n = 30) in the GSE151963 and TCGA datasets.
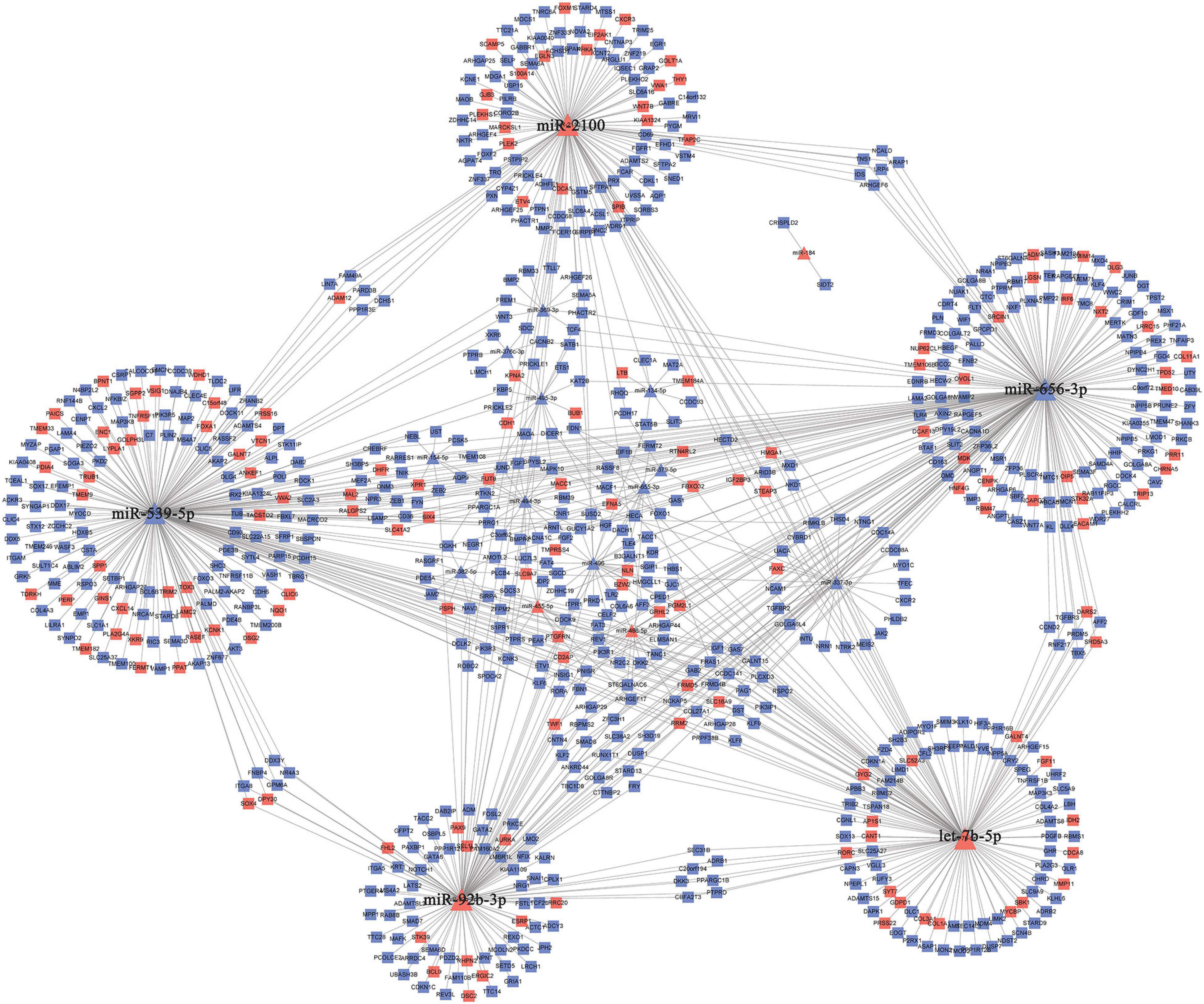
Figure 3. The miRNA–mRNA regulatory network. The rectangles and triangles represent mRNAs and miRNAs, respectively. Red represents the upregulated expression of miRNA or mRNA; blue represents the downregulated expression of miRNA or mRNA. miRNA, microRNA; mRNA, messenger RNA.
GO and KEGG Analyses of Targeted mRNAs for Hub miRNAs
In total, we used 677 Co-DEmRNAs to identify the potential functions of hub miRNAs using GO and KEGG pathway enrichment analyses. We obtained 836 results in the GO analysis, in which Figure 4A illustrates the top 10 results from the biological process, cellular component and molecular function. Some results associated with the occurrence and progression of a tumor, including the regulation of the cellular response to the growth factor stimulus, the collagen-containing extracellular matrix and the basement membrane among others. In addition, 48 signaling pathways emerged from the KEGG pathways analysis (Table 2), with the top ten results shown in Figure 4B. Most of these pathways associated with the occurrence and progression of a tumor, including focal adhesion, proteoglycans in cancer, extracellular matrix–receptor interaction and the Wnt signaling pathway (Table 2). The focal adhesion pathway contained the largest number of mRNAs and the smallest adj. p-value.
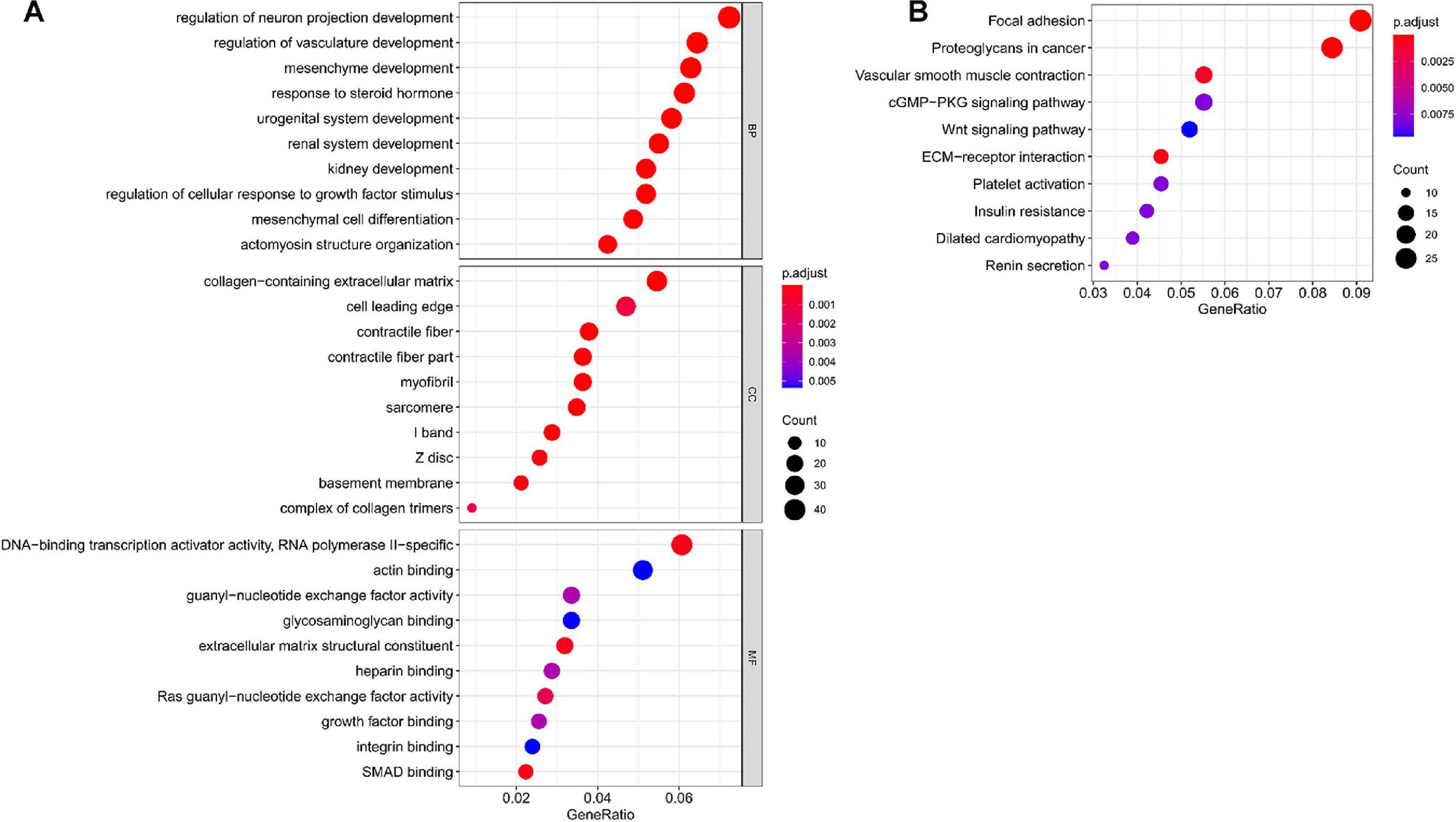
Figure 4. Functional enrichment analysis of targeted mRNAs from the hub miRNAs. (A) Dotplot of the top ten results from the GO analysis in terms of BP, CC and MF with the adjusted p-value from smallest to largest, respectively, and (B) dotplot of the top ten KEGG enrichment pathways. GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes; miRNA, microRNA; mRNA, messenger RNA.
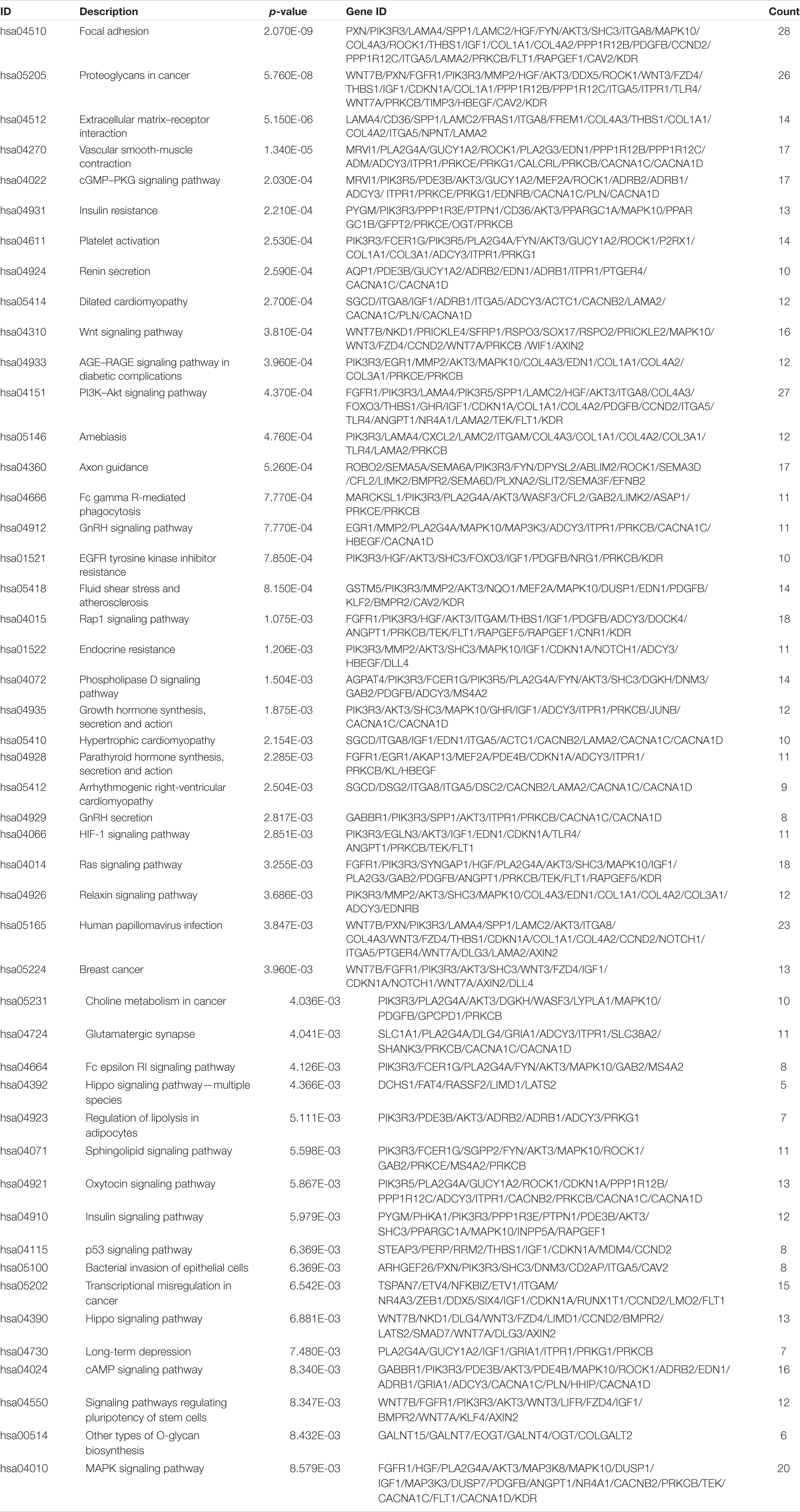
Table 2. Results from the Kyoto Encyclopedia of Genes and Genomes enrichment pathway analysis.
Identification of Hub Genes
The PPI networks identified 597 mRNAs from 677 Co-DEmRNAs with 2,246 edges related to each other (Figure 5). The top ten key mRNAs were identified using the cytoHubba plugin MCC ranking, including neurogenic locus notch homolog protein 1 (NOTCH1), matrix metalloproteinase 2 (MMP2), insulin-like growth factor 1 (IGF1), kinase-insert domain-containing receptor (KDR), secreted phosphoprotein 1(SPP1), vascular endothelial growth factor receptor 1 (FLT1), hepatocyte growth factor (HGF), angiopoietin-1 receptor (TEK), angiopoietin-1 gene (ANGPT1) and platelet-derived growth factor-beta (PDGFB; Figure 6). These key mRNAs were further verified using GEPIA through the calculation of the survival analysis. Here, only two (SPP1 and HGF) were identified as hub genes and associated with survival (Figure 7). The survival analysis revealed that the expression of SPP1 was negatively correlated with survival, while HGF positively correlated with survival in LUAD (Figure 7). In addition, SPP1 and HGF were both enriched in the focal adhesion signaling pathway (Table 2).
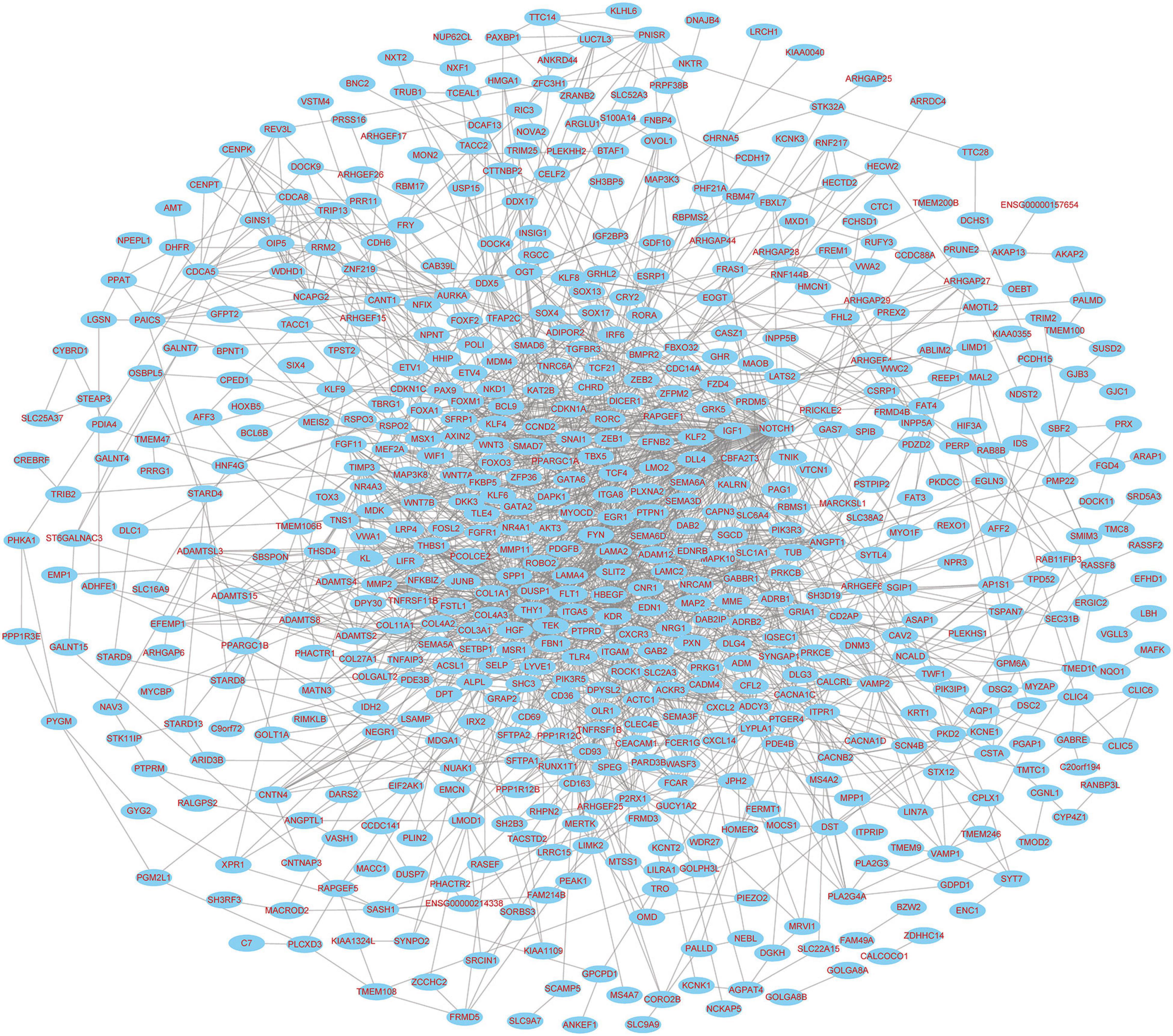
Figure 5. PPI network diagram of targeted mRNAs from the hub miRNAs. PPI, protein–protein interaction; miRNA, microRNA; mRNA, messenger RNA.
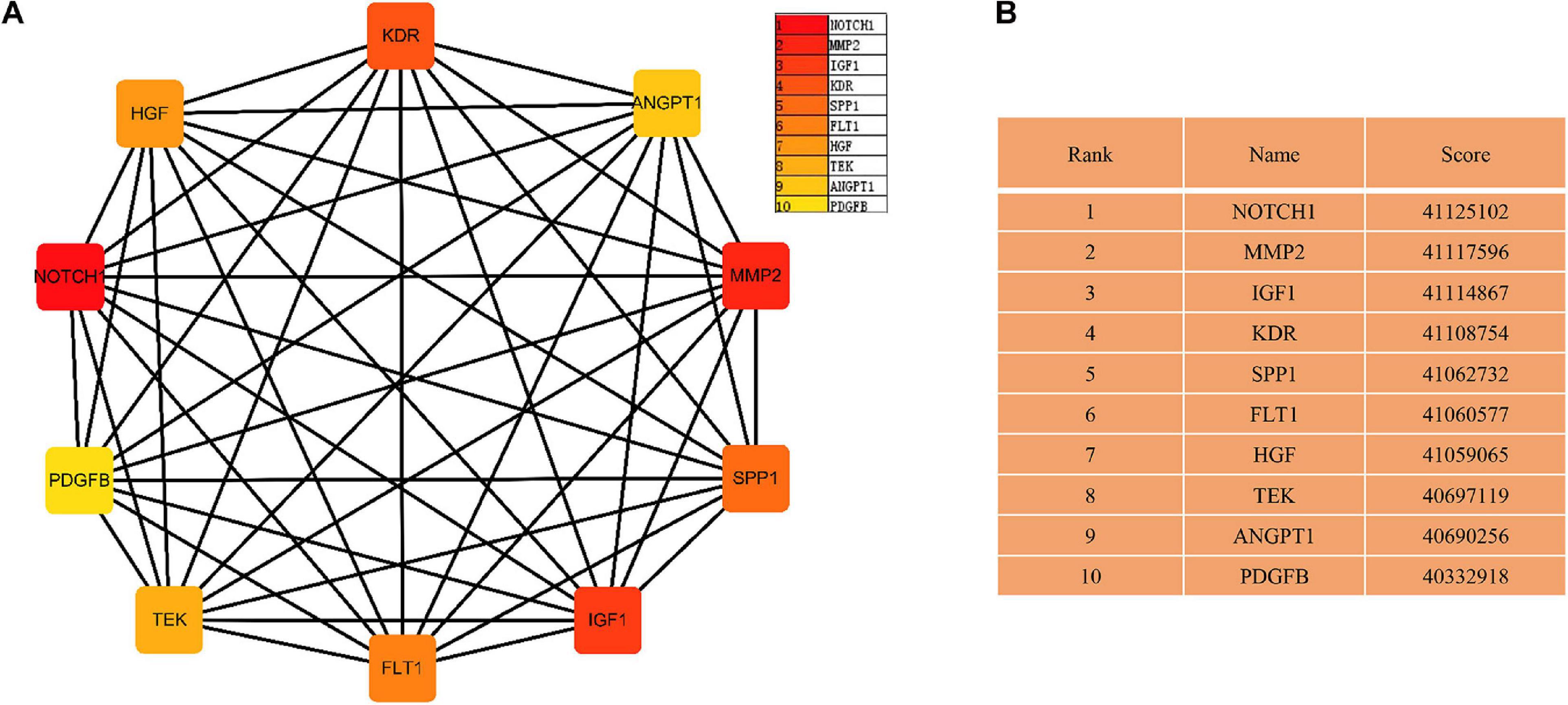
Figure 6. The network of key mRNAs. (A) The network diagram of the top ten hub miRNAs and (B) the rank and score of the top ten hub mRNAs in the PPI network. PPI, protein–protein interaction; miRNA, microRNA; mRNA, messenger RNA; NOTCH1, neurogenic locus notch homolog protein 1; MMP2, matrix metalloproteinase 2; IGF1, insulin-like growth factor 1; KDR, kinase-insert domain-containing receptor; SPP1, secreted phosphoprotein 1; FLT-1, vascular endothelial growth factor receptor 1; HGF, hepatocyte growth factor; TEK, angiopoietin-1 receptor; ANGPT1, angiopoietin-1 gene; PDGFB, platelet-derived growth factor-beta.
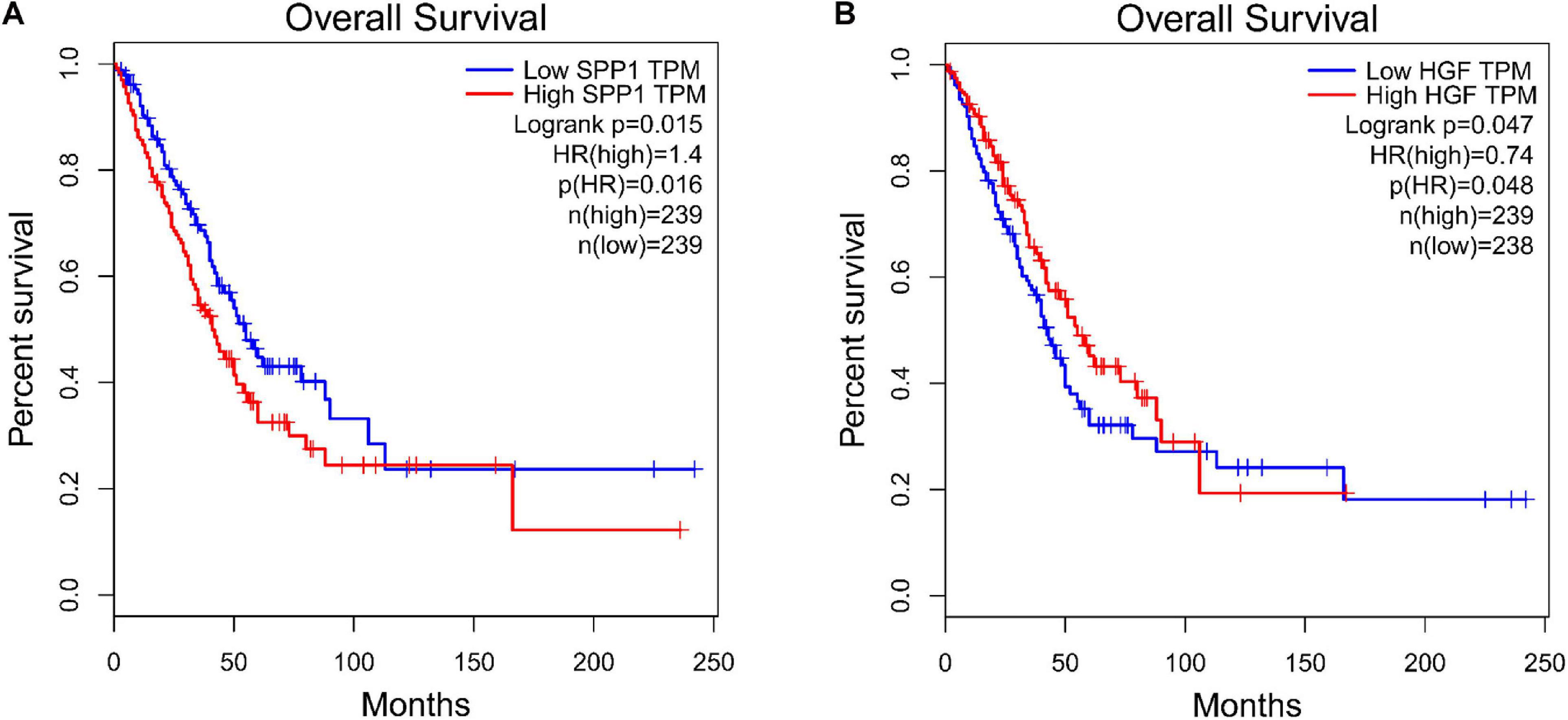
Figure 7. Survival analysis for the hub genes. (A) SPP1, secreted phosphoprotein 1 and (B) HGF, hepatocyte growth factor. TPF, transcripts per kilobase of exon model per million mapped reads; HR, hazard ratio.
Discussion
LAUD is a high-risk disease with a high mortality, and its potential occurrence and development mechanisms have not been fully identified. Previously, several studies reported the role of miRNAs in LAUD, although those findings lacked agreement (Cazzoli et al., 2013; Zhong et al., 2018). Here, we identified several hub miRNAs and hub mRNAs in LUAD using the miRNA–mRNA regulatory network using multiple sample types and ethnicities. In total, five hub miRNAs—namely, miR-539-5p, miR-656-3p, let-7b-5p, miR-2110, and miR-92b-3p—played important roles in LUAD tumorigenesis and progression. Two hub mRNAs, namely, SPP1, and HGF, associated with LUAD patient survival. These findings suggest that the specific miRNA and mRNA expression patterns and functional analyses contribute to identifying new predictive markers and therapeutic targets for LUAD patients in clinical settings.
Five hub miRNAs in the network played the most important roles in LUAD tumorigenesis and progression. Specifically, miR-539-5p emerged with the highest node degrees compared to other nodes in the network. Several studies found that miR-539-5p expression levels associated with a poor prognosis and negatively correlated with hypoxia and the stem index among LUAD patients (González-Vallinas et al., 2018; Guo et al., 2020). Yet, further experiments are needed to verify these results given the limited evidence documenting the direct mechanisms of miR-539-5p in LUAD. For example, Chen et al. showed that miR-656-3p could reduce AKT serine/threonine kinase 1 (AKT1) expression and suppress the occurrence and development of non-small cell lung cancer (NSCLC) (Chen et al., 2019). Additionally, upregulating miR-656-3p might improve the chemotherapeutic efficacy in NSCLC by target-regulating sex-determining region Y-related high-mobility group box 4 (SOX4) (Wang et al., 2020). The expression of let-7b-5p was upregulated and might play a vital role in NSCLC based on the GEO dataset using bioinformatics analysis (Zhou et al., 2020), and participate in lymph node micro-metastases of LUAD stage IA (Zhu et al., 2019). miR-2110 and miR-92b-3p express at different levels in different tumors (Long et al., 2017; Gong et al., 2018; Neerincx et al., 2018; Zhao et al., 2018; Ma et al., 2019). To the best of our knowledge, very few studies have attempted to clarify the effect of these two miRNAs in LUAD. In our study, we found that miR-2110 and miR-92b-3p were upregulated in LUAD. Thus, these studies confirmed that miR-539-5p, miR-656-3p, and let-7b-5p are indeed involved in the occurrence and progression of LUAD. Furthermore, miR-92b-3p and miR-2110 may represent new potential therapeutic targets in LUAD.
SPP1 and HGF were identified as hub mRNAs in PPI and significantly predicted survival outcomes in LUAD. These two mRNAs appear enriched in the focal adhesion signaling pathway, containing the largest number of mRNAs with the smallest significant adj. p-value. The focal adhesion signaling pathway could involve the tumor microenvironment (TME), resulting in tumor progression and indicative of a poor outcome in LUAD (Yue et al., 2019; Wei et al., 2020). Accumulating evidence has revealed that the TME could influence malignant behavior and the progression of tumors. Previous studies found that SPP1 played a role in lung cancer escape and mediating macrophage polarization, while inhibiting SPP1 expression might overcome resistance to second-generation epidermal growth factor receptor gene (EGFR)–tyrosine kinase inhibitors (TKIs) (Zhang et al., 2017; Wang et al., 2019). HGF is a pleiotropic cytokine composed of an α-chain and a β-chain, which mediates malignant biological behaviors in LUAD, including growth, invasion, metastasis and the epithelial-to-mesenchymal transition (EMT), as well as increases resistance to EGFR–TKIs in LUAD (Tarhini et al., 2017; Suzuki et al., 2019). Importantly, it is worth noting that SPP1 presented with the targets of miR-539-5p, correlated with a poor survival in LUAD and appeared involved in the regulation of focal adhesion and the extracellular matrix–receptor interaction pathways. Thus, we found a significant miR-539-5p–SPP1 axis, which played important roles in LUAD occurrence and progression by regulating the TME through the aforementioned pathways in our regulatory network. Although the other eight key mRNA played some important roles in LUAD or lung cancer (Ding et al., 2008; Roybal et al., 2011; Licciulli et al., 2013; Li et al., 2017; Neri et al., 2017; Tian et al., 2018; Yao et al., 2019), these were not the significant predictors of survival in LUAD.
Aside from the strengths of our study, we should point out several limitations. Firstly, the small sample size in our cohort mirrors the limitations of similar previous studies (Patnaik et al., 2012; Xu et al., 2020). Yet, all of these miRNA expression profiling studies offer opportunities to develop new therapeutic targets. Although increasing numbers of miRNAs have been identified, a meta-analysis approach and a larger cohort are needed in future in order to minimize the drawbacks resulting from small sample sizes and different technological platforms. Secondly, given our findings related to the target genes from the Co-DEmiRNAs and hub miRNAs identified primarily based on bioinformatics analyses, further mechanistic studies from cells as well as animal models and clinical validation studies may be necessary in the future.
In this study, we constructed a miRNA–mRNA regulatory network for LUAD, identifying five hub miRNAs (namely, miR-539-5p, miR-656-3p, let-7b-5p, miR-2110, and miR-92b-3p) and two hub mRNAs (SPP1and HGF) through our microarray data and TCGA dataset. We also performed a functional enrichment analysis on these final target genes to understand the potential functional mechanisms in LUAD. These findings provide new molecular markers for the prediction, prognosis and therapeutic targets in clinical settings, as well as emphasize new mechanistic insights into LUAD.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Gansu Provincial Hospital, China. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
QY, X-JW, and JG contributed to the design of the study and revised the manuscript. X-JW and ZW performed the sample collection, analysis and downloaded the data. X-JW and JG contributed to the data analysis and writing the manuscript. All authors approved the final version of the manuscript.
Funding
This study was supported by the Science-Technology Foundation for Young Scientist of the Gansu Province of China (Grant No. 18JR3RA059) and the Young Scientists Fund of the Gansu Provincial Hospital of China (Grant No. 17GSS7-1). JG was supported by the Sigrid Jusélius Foundation, the Finnish Anti-Tuberculosis Association Foundation and the Jalmari and Rauha Ahokas Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We extend our deepest gratitude to all subjects who volunteered to participate in our study. In addition, we also thank the Biomarker Technologies Corporation (Beijing, China) for the sequence technology and support. We are also grateful to Vanessa L. Fuller, from Language Services at the University of Helsinki, Finland, for assistance in the English-language revision of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.641840/full#supplementary-material
Supplementary Table S1 | Clinical characteristics of LUAD patients in the microarray data (GSE151963).
Supplementary Table S2 | Clinical characteristics of LUAD patients in the TCGA data.
Footnotes
- ^ http://gdac.broadinstitute.org/
- ^ http://www.mirdb.org/
- ^ http://www.targetscan.org/
- ^ zmf.umm.uni-heidelberg.de/mirwalk2
- ^ http://string-db.org
- ^ http://gepia.cancer-pku.cn
References
Agarwal, V., Bell, G. W., Nam, J. W., and Bartel, D. P. (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4:e05005. doi: 10.7554/eLife.05005
Blondal, T., Jensby, N. S., Baker, A., Andreasen, D., Mouritzen, P., Wrang, T. M., et al. (2013). Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods 59, S1–S6. doi: 10.1016/j.ymeth.2012.09.015
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Cao, M., and Chen, W. (2019). Epidemiology of lung cancer in China. Thorac. Cancer 10, 3–7. doi: 10.1111/1759-7714.12916
Cazzoli, R., Buttitta, F., Di Nicola, M., Malatesta, S., Marchetti, A., Rom, W. N., et al. (2013). microRNAs derived from circulating exosomes as noninvasive biomarkers for screening and diagnosing lung cancer. J. Thorac. Oncol. 8, 1156–1162. doi: 10.1097/JTO.0b013e318299ac32
Chen, T., Qin, S., Gu, Y., Pan, H., and Bian, D. (2019). Long non-coding RNA NORAD promotes the occurrence and development of non-small cell lung cancer by adsorbing MiR-656-3p. Mol. Genet. Genomic Med. 7, e757–e794. doi: 10.1002/mgg3.757
Chen, Y., and Wang, X. (2020). miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, D127–D131. doi: 10.1093/nar/gkz757
Cojocneanu, R., Braicu, C., Raduly, L., Jurj, A., Zanoaga, O., Magdo, L., et al. (2020). Plasma and tissue specific miRNA expression pattern and functional analysis associated to colorectal cancer patients. Cancers 12, 843–863. doi: 10.3390/cancers12040843
Condrat, C. E., Thompson, D. C., Barbu, M. G., Bugnar, O. L., Boboc, A., Cretoiu, D., et al. (2020). miRNAs as biomarkers in disease: latest findings regarding their role in diagnosis and prognosis. Cells 9, 276–307. doi: 10.3390/cells9020276
Cui, X., Liu, Y., Sun, W., Ding, J., Bo, X., and Wang, H. (2020). Comprehensive analysis of miRNA-gene regulatory network with clinical significance in human cancers. Sci. China Life Sci. 63, 1201–1212. doi: 10.1007/s11427-019-9667-0
Di Leva, G., Garofalo, M., and Croce, C. M. (2014). MicroRNAs in cancer. Annu. Rev. Pathol. 9, 287–314. doi: 10.1146/annurev-pathol-012513-104715
Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. doi: 10.1038/nature07423
Du, X., Zhang, J., Wang, J., Lin, X., and Ding, F. (2018). Role of miRNA in lung cancer-potential biomarkers and therapies. Curr. Pharm. Des. 23, 5997–6010. doi: 10.2174/1381612823666170714150118
Dweep, H., and Gretz, N. (2015). miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods 12:697. doi: 10.1038/nmeth.3485
Gan, T. Q., Chen, W. J., Qin, H., Huang, S. N., Yang, L. H., Fang, Y. Y., et al. (2017). Clinical value and prospective pathway signaling of MicroRNA-375 in lung adenocarcinoma: a study based on the cancer genome Atlas (TCGA), gene expression omnibus (GEO) and bioinformatics analysis. Med. Sci. Monit. 23, 2453–2464. doi: 10.12659/msm.901460
Gong, L., Ren, M., Lv, Z., Yang, Y., and Wang, Z. (2018). miR-92b-3p promotes colorectal carcinoma cell proliferation, invasion, and migration by inhibiting FBXW7 in vitro and in vivo. DNA Cell Biol. 37, 501–511. doi: 10.1089/dna.2017.4080
González-Vallinas, M., Rodriguez-Paredes, M., Albrecht, M., Sticht, C., Stichel, D., Gutekunst, J., et al. (2018). Epigenetically regulated chromosome 14q32 miRNA cluster induces metastasis and predicts poor prognosis in lung adenocarcinoma patients. Mol. Cancer Res. 16, 390–402. doi: 10.1158/1541-7786.MCR-17-0334
Guo, L., Li, H., Li, W., and Tang, J. (2020). Construction and investigation of a combined hypoxia and stemness index lncRNA-associated ceRNA regulatory network in lung adenocarcinoma. BMC Med. Genomics 13:166. doi: 10.1186/s12920-020-00816-8
Guo, Z., Huang, J., Wang, Y., Liu, X. P., Li, W., Yao, J., et al. (2020). Analysis of expression and its clinical significance of the secreted phosphoprotein 1 in lung Adenocarcinoma. Front. Genet. 11:547. doi: 10.3389/fgene.2020.00547
Henschke, C. I., Yankelevitz, D. F., Libby, D. M., Pasmantier, M. W., Smith, J. P., and Miettinen, O. S. (2006). Survival of patients with stage I lung cancer detected on CT screening. N. Engl. J. Med. 355, 1763–1771. doi: 10.1056/NEJMoa060476
Herbst, R. S., Morgensztern, D., and Boshoff, C. (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. doi: 10.1038/nature25183
Hummel, R., Hussey, D. J., and Haier, J. (2010). MicroRNAs: predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur. J. Cancer 46, 298–311. doi: 10.1016/j.ejca.2009.10.027
Izadi, F., Zamanian-Azodi, M., Mansouri, V., Khodadoostan, M., and Naderi, N. (2017). Exploring conserved mRNA-miRNA interactions in colon and lung cancers. Gastroenterol. Hepatol. Bed Bench 10, 184–193. doi: 10.22037/ghfbb.v0i0.1198
Jurj, A., Braicu, C., Pop, L. A., Tomuleasa, C., Gherman, C. D., and Berindan-Neagoe, I. (2017). The new era of nanotechnology, an alternative to change cancer treatment. Drug Des. Devel. Ther. 11, 2871–2890. doi: 10.2147/DDDT.S142337
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Li, J., Yin, X., Zhang, B., Li, C., and Lu, P. (2020). Bioinformatical analysis of miRNA-mRNA interaction network underlying macrophage aging and cholesterol-responsive difference between young and aged macrophages. Biomed. Res. Int. 2020:9267475. doi: 10.1155/2020/9267475
Li, L., Zhang, L., Tian, Y., Zhang, T., Duan, G., Liu, Y., et al. (2019). Serum chemokine CXCL7 as a diagnostic biomarker for colorectal cancer. Front. Oncol. 9:921. doi: 10.3389/fonc.2019.00921
Li, S., Huang, L., Gu, J., Wu, J., Ou, W., Feng, J., et al. (2017). Restoration of KLF4 inhibits invasion and metastases of lung adenocarcinoma through suppressing MMP2. J. Cancer 8, 3480–3489. doi: 10.7150/jca.21241
Licciulli, S., Avila, J. L., Hanlon, L., Troutman, S., Cesaroni, M., Kota, S., et al. (2013). Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 73, 5974–5984. doi: 10.1158/0008-5472.CAN-13-1384
Liu, P., Zhang, R., Yu, W., Ye, Y., Cheng, Y., Han, L., et al. (2017). FGF1 and IGF1-conditioned 3D culture system promoted the amplification and cancer stemness of lung cancer cells. Biomaterials 149, 63–76. doi: 10.1016/j.biomaterials.2017.09.030
Liu, Q., Yu, Z., Yuan, S., Xie, W., Li, C., Hu, Z., et al. (2017). Circulating exosomal microRNAs as prognostic biomarkers for non-small-cell lung cancer. Oncotarget 8, 13048–13058. doi: 10.18632/oncotarget.14369
Liu, Y., Zhang, M., Shan, Y., Ji, G., Ju, X., Tu, Y., et al. (2020). miRNA-mRNA network regulation in the skeletal muscle fiber phenotype of chickens revealed by integrated analysis of miRNAome and transcriptome. Sci. Rep. 10:10619. doi: 10.1038/s41598-020-67482-9
Long, M., Zhan, M., Xu, S., Yang, R., Chen, W., Zhang, S., et al. (2017). miR-92b-3p acts as a tumor suppressor by targeting Gabra3 in pancreatic cancer. Mol. Cancer 16:167. doi: 10.1186/s12943-017-0723-7
Ma, G., Song, G., Zou, X., Shan, X., Liu, Q., Xia, T., et al. (2019). Circulating plasma microRNA signature for the diagnosis of cervical cancer. Cancer Biomark. 26, 491–500. doi: 10.3233/CBM-190256
Miller, K. D., Nogueira, L., Mariotto, A. B., Rowland, J. H., Yabroff, K. R., Alfano, C. M., et al. (2019). Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 69, 363–385. doi: 10.3322/caac.21565
Neerincx, M., Poel, D., Sie, D., van Grieken, N., Shankaraiah, R. C., van der Wolf-de, L. F., et al. (2018). Combination of a six microRNA expression profile with four clinicopathological factors for response prediction of systemic treatment in patients with advanced colorectal cancer. PLoS One 13:e201809. doi: 10.1371/journal.pone.0201809
Neri, S., Miyashita, T., Hashimoto, H., Suda, Y., Ishibashi, M., Kii, H., et al. (2017). Fibroblast-led cancer cell invasion is activated by epithelial-mesenchymal transition through platelet-derived growth factor BB secretion of lung adenocarcinoma. Cancer Lett. 395, 20–30. doi: 10.1016/j.canlet.2017.02.026
Pathan, M., Keerthikumar, S., Ang, C. S., Gangoda, L., Quek, C. Y., Williamson, N. A., et al. (2015). FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15, 2597–2601. doi: 10.1002/pmic.201400515
Patnaik, S. K., Yendamuri, S., Kannisto, E., Kucharczuk, J. C., Singhal, S., and Vachani, A. (2012). MicroRNA expression profiles of whole blood in lung adenocarcinoma. PLoS One 7:e46045. doi: 10.1371/journal.pone.0046045
Patten, S. B., Wilkes, T. C., Williams, J. V., Lavorato, D. H., El-Guebaly, N., Wild, T. C., et al. (2016). Childhood adversity and subsequent mental health status in adulthood: screening for associations using two linked surveys. Epidemiol. Psychiatr. Sci. 25, 160–170. doi: 10.1017/S2045796015000104
Qi, J., Wang, J., Katayama, H., Sen, S., and Liu, S. M. (2013). Circulating microRNAs (cmiRNAs) as novel potential biomarkers for hepatocellular carcinoma. Neoplasma 60, 135–142. doi: 10.4149/neo_2013_018
Ramassone, A., Pagotto, S., Veronese, A., and Visone, R. (2018). Epigenetics and MicroRNAs in cancer. Int. J. Mol. Sci. 19:459. doi: 10.3390/ijms19020459
Rheinbay, E., Nielsen, M. M., Abascal, F., Wala, J. A., Shapira, O., Tiao, G., et al. (2020). Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111. doi: 10.1038/s41586-020-1965-x
Roybal, J. D., Zang, Y., Ahn, Y. H., Yang, Y., Gibbons, D. L., Baird, B. N., et al. (2011). miR-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol. Cancer Res. 9, 25–35. doi: 10.1158/1541-7786.MCR-10-0497
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Siegel, R. L., Miller, K. D., and Jemal, A. (2020). Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30. doi: 10.3322/caac.21590
Suzuki, E., Yamazaki, S., Naito, T., Hashimoto, H., Okubo, S., Udagawa, H., et al. (2019). Secretion of high amounts of hepatocyte growth factor is a characteristic feature of cancer-associated fibroblasts with EGFR-TKI resistance-promoting phenotype: a study of 18 cases of cancer-associated fibroblasts. Pathol. Int. 69, 472–480. doi: 10.1111/pin.12838
Świtlik, W. Z., Karbownik, M. S., Suwalski, M., Kozak, J., and Szemraj, J. (2019). Serum miR-210-3p as a potential noninvasive biomarker of lung adenocarcinoma: a preliminary study. Genet. Test. Mol. Biomark. 23, 353–358. doi: 10.1089/gtmb.2018.0275
Tarhini, A. A., Rafique, I., Floros, T., Tran, P., Gooding, W. E., Villaruz, L. C., et al. (2017). Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancer 123, 2936–2944. doi: 10.1002/cncr.30717
Tian, W., Li, Y. S., Zhang, J. H., Li, J. J., and Gao, J. H. (2018). Comprehensive analysis of DNA methylation and gene expression datasets identified MMP9 and TWIST1 as important pathogenic genes of lung adenocarcinoma. DNA Cell Biol. 37, 336–346. doi: 10.1089/dna.2017.4085
Wang, J., Zhang, C., Peng, X., Liu, K., Zhao, L., Chen, X., et al. (2020). A combination of four serum miRNAs for screening of lung adenocarcinoma. Hum. Cell 33, 830–838. doi: 10.1007/s13577-020-00346-6
Wang, L., Meng, L., Wang, X. W., Ma, G. Y., and Chen, J. H. (2014). Expression of RRM1 and RRM2 as a novel prognostic marker in advanced non-small cell lung cancer receiving chemotherapy. Tumour Biol. 35, 1899–1906. doi: 10.1007/s13277-013-1255-4
Wang, X., Zhang, F., Yang, X., Xue, M., Li, X., Gao, Y., et al. (2019). Secreted phosphoprotein 1 (SPP1) contributes to second-generation EGFR tyrosine kinase inhibitor resistance in non-small cell lung cancer. Oncol. Res. 27, 871–877. doi: 10.3727/096504018X15426271404407
Wang, X. F., Shi, J., Chen, Y., Wang, C. H., Shi, H. F., and Xie, X. F. (2019). Long noncoding RNA ANRIL knockdown increases sensitivity of non-small cell lung cancer to cisplatin by regulating the miR-656-3p/SOX4 axis. RSC Adv. 9, 38735–38744. doi: 10.1039/C9RA06993C
Wei, Z., Zhongqiu, T., Lu, S., Zhang, F., Xie, W., and Wang, Y. (2020). Gene coexpression analysis offers important modules and pathway of human lung adenocarcinomas. J. Cell. Physiol. 235, 454–464. doi: 10.1002/jcp.28985
Wu, K. L., Tsai, Y. M., Lien, C. T., Kuo, P. L., and Hung, A. J. (2019). The roles of MicroRNA in lung cancer. Int. J. Mol. Sci. 20:1611. doi: 10.3390/ijms20071611
Wu, S. G., Chang, T. H., Liu, Y. N., and Shih, J. Y. (2019). MicroRNA in lung cancer metastasis. Cancers 11:265. doi: 10.3390/cancers11020265
Xu, Z., Wang, Z., Sun, H., and Xin, H. (2020). Evaluation of exosomal miRNA in blood as a potential diagnostic biomarker for human non-small cell lung cancer. Med. Sci. Monit. 26:e924721. doi: 10.12659/MSM.924721
Yang, G., Zhang, Y., and Yang, J. (2019). Identification of potentially functional CircRNA-miRNA-mRNA regulatory network in gastric carcinoma using bioinformatics analysis. Med. Sci. Monit. 25, 8777–8796. doi: 10.12659/MSM.916902
Yao, S., Dong, S. S., Ding, J. M., Rong, Y., Zhang, Y. J., Chen, H., et al. (2019). Sex-specific SNP-SNP interaction analyses within topologically associated domains reveals ANGPT1 as a novel tumor suppressor gene for lung cancer. Genes Chromosomes Cancer 59, 13–22. doi: 10.1002/gcc.22793
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Yue, C., Ma, H., and Zhou, Y. (2019). Identification of prognostic gene signature associated with microenvironment of lung adenocarcinoma. PeerJ 7:e8128. doi: 10.7717/peerj.8128
Zhang, Y., Du, W., Chen, Z., and Xiang, C. (2017). Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. cell Res. 359, 449–457. doi: 10.1016/j.yexcr.2017.08.028
Zhang, Y. Q., Wang, W. Y., Xue, J. X., Xu, Y., Fan, P., Caughey, B. A., et al. (2016). MicroRNA expression profile on solid subtype of invasive lung adenocarcinoma reveals a panel of four miRNAs to be associated with poor prognosis in Chinese patients. J. Cancer 7, 1610–1620. doi: 10.7150/jca.14923
Zhao, Z., Partridge, V., Sousares, M., Shelton, S. D., Holland, C. L., Pertsemlidis, A., et al. (2018). microRNA-2110 functions as an onco-suppressor in neuroblastoma by directly targeting Tsukushi. PLoS One 13:e208777. doi: 10.1371/journal.pone.0208777
Zhong, Z., Hou, J., Zhang, Q., Zhong, W., Li, B., Li, C., et al. (2018). Circulating microRNA expression profiling and bioinformatics analysis of dysregulated microRNAs of patients with coronary artery disease. Medicine 97:e11428. doi: 10.1097/MD.0000000000011428
Zhou, X., Zhang, Z., and Liang, X. (2020). Regulatory network analysis to reveal important miRNAs and genes in non-small cell lung cancer. Cell J. 21, 459–466. doi: 10.22074/cellj.2020.6281
Zhu, J., Xu, Y., Liu, S., Qiao, L., Sun, J., and Zhao, Q. (2020). MicroRNAs associated with colon cancer: new potential prognostic markers and targets for therapy. Front. Bioeng. Biotechnol. 8:176. doi: 10.3389/fbioe.2020.00176
Keywords: lung adenocarcinoma, microRNAs, hub genes, bioinformatics, prognostic marker
Citation: Wang X-J, Gao J, Wang Z and Yu Q (2021) Identification of a Potentially Functional microRNA–mRNA Regulatory Network in Lung Adenocarcinoma Using a Bioinformatics Analysis. Front. Cell Dev. Biol. 9:641840. doi: 10.3389/fcell.2021.641840
Received: 15 December 2020; Accepted: 27 January 2021;
Published: 18 February 2021.
Edited by:
Liang Cheng, Harbin Medical University, ChinaReviewed by:
Tuantuan Zhao, Stanford University, United StatesGuiyou Liu, Chinese Academy of Sciences, China
Copyright © 2021 Wang, Gao, Wang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Yu, yuqin@lzu.edu.cn
†These authors have contributed equally to this work