Considering the Role of Extracellular Matrix Molecules, in Particular Reelin, in Granule Cell Dispersion Related to Temporal Lobe Epilepsy
 Jennifer Leifeld
Jennifer Leifeld Eckart Förster
Eckart Förster Gebhard Reiss3
Gebhard Reiss3  Mohammad I. K. Hamad
Mohammad I. K. Hamad- 1Department of Neuroanatomy and Molecular Brain Research, Medical Faculty, Ruhr University Bochum, Bochum, Germany
- 2Department of Biochemistry I—Receptor Biochemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Bochum, Germany
- 3Institute for Anatomy and Clinical Morphology, School of Medicine, Faculty of Health, Witten/ Herdecke University, Witten, Germany
The extracellular matrix (ECM) of the nervous system can be considered as a dynamically adaptable compartment between neuronal cells, in particular neurons and glial cells, that participates in physiological functions of the nervous system. It is mainly composed of carbohydrates and proteins that are secreted by the different kinds of cell types found in the nervous system, in particular neurons and glial cells, but also other cell types, such as pericytes of capillaries, ependymocytes and meningeal cells. ECM molecules participate in developmental processes, synaptic plasticity, neurodegeneration and regenerative processes. As an example, the ECM of the hippocampal formation is involved in degenerative and adaptive processes related to epilepsy. The role of various components of the ECM has been explored extensively. In particular, the ECM protein reelin, well known for orchestrating the formation of neuronal layer formation in the cerebral cortex, is also considered as a player involved in the occurrence of postnatal granule cell dispersion (GCD), a morphologically peculiar feature frequently observed in hippocampal tissue from epileptic patients. Possible causes and consequences of GCD have been studied in various in vivo and in vitro models. The present review discusses different interpretations of GCD and different views on the role of ECM protein reelin in the formation of this morphological peculiarity.
Introduction
The organization of neuronal cell bodies and fiber projections in ordered layers represents a fundamental principle of the mammalian cerebral cortex and is essential for proper brain function. Disruption of this ordered layering is often associated with functional deficits or a decreased threshold for the occurrence of epileptic seizures, which happens to be the case in the reeler mouse mutant, which lacks expression of the protein reelin. Reelin is best known as a protein that is secreted by Cajal-Retzius (CR) cells into the ECM, and is essential for the correct positioning of migrating neurons.
Epilepsy is a neurological disorder associated with cerebral seizures characterized by chronic overexcitation of neurons in the brain and represents one of the most common neurological diseases worldwide. The most common form of anatomically classifiable focal epilepsy is temporal lobe epilepsy (TLE), where epileptic discharges originate in the temporal lobe (TL), frequently being pharmaco-resistant. TLE is sometimes associated with a history of febrile seizures (FS) and the so-called Ammon’s horn or hippocampal sclerosis (HS), characterized by a loss of hippocampal neurons. HS, in turn, is often observed in association with astrogliosis or microgliosis, central nervous system (CNS) conditions characterized by proliferation of astrocytes and microglia, respectively, suggestive of CNS injury and inflammation. TLE can be associated with GCD, which is mostly considered as a morphological malformation of the dentate gyrus (DG), reflecting a pathological condition of hippocampal granule cells (GCs) leading to disintegration of their dense layering, reminiscent of the morphological malformations seen in the reeler mutant mouse. GCD has been reported to be associated with reelin deficiency. While the role of reelin for GCD and the clinical picture of TLE is under debate, a recent study suggests that GCD may represent a variation within the normal range rather than a pathological abnormality.
Temporal Lobe Epilepsy, Febrile Seizures, and Granule Cell Dispersion
It is estimated that epilepsy affects approximately 50 million people worldwide. Therefore, it can be considered as one of the most common neurological disorders (Dua et al., 2006; Guerreiro, 2016; Falco-Walter, 2020). A meta-analytic study of the prevalence (the number of existing cases) and incidence (new-onset cases) of epilepsy worldwide included a total of 222 studies (197 on prevalence and 48 on incidence) (Fiest et al., 2017). This study identified a prevalence of active epilepsy of 6.38 per 1,000 persons and a lifetime prevalence of 7.60 per 1,000 persons, which did not differ with respect to age or sex (Fiest et al., 2017). The annual cumulative incidence of epilepsy was found to be 67.77 per 100,000 persons (Fiest et al., 2017).
TLE is the most common form of epilepsy with focal seizures, characterized by recurrent, spontaneous seizures originating in the TL of the brain (Pascual, 2007; Nayak and Bandyopadhyay, 2021). The ILAE distinguishes two main types of TLE: mesial TLE (MTLE) and lateral TLE (LTLE) (Panayiotopoulos, 2005; Berg et al., 2010). The latter arises in the lateral part of the TL, meaning the lateral surface adjacent to the neocortex (Kutsy, 1999; Kim et al., 2003; O’muircheartaigh and Richardson, 2012). MTLE, on the other hand, originates in the MTL, i.e., in the hippocampus and nearby structures, such as the amygdala and the entorhinal cortex, and in structures surrounding it, such as the parahippocampal gyrus (Engel, 2001; Pascual, 2007; Nayak and Bandyopadhyay, 2021). At least 80% of all TLE cases originate in the hippocampus due to neuronal overexcitability (Tatum, 2012) making it one of the most susceptible areas for TLE and MTLE, respectively. Figure 1 depicts an overview of the hippocampus and the tri-synaptic circuit.
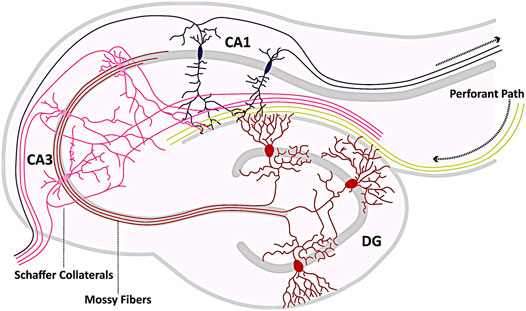
FIGURE 1. Hippocampal circuitry. Shown is a schematic representation of the hippocampal circuitry (tri-synaptic circuit). The hippocampus receives afferents from the neighboring entorhinal cortex, which enter the hippocampal formation primarily via the Perforant Path (yellow) and form synaptic contacts with both pyramidal cells (pink, purple) and partially with granule cells (GCs, red). The axons of the GCs (mossy fibers) innervate the pyramidal cells of the CA3 region (pink), from whose axons the Schaffer collaterals branch to form synapses with the dendrites of the CA1 pyramidal cells (purple). Axons of the pyramidal cells (pink, purple) eventually exit the hippocampus as efferent elements. The template for this figure was (Marín-Padilla, 1998; Twible et al., 2021).
HS and FS are associated primarily with TLE because retrospective studies or Magnetic Resonance Imaging (MRI) studies repeatedly found that many patients who underwent surgery for intractable TLE had FS in childhood and showed signs of HS. It has been speculated that FS lead to structural damages in the TL, as a result of which HS develops, eventually terminating in TLE (Mathern et al., 1997; Vanlandingham et al., 1998; Cendes, 2004). Another theory is that innate hippocampal abnormalities predispose the TL to be particularly susceptible to prolonged infantile FS, eventually leading to TLE (Annegers et al., 1987; Vanlandingham et al., 1998; Blümcke et al., 2002; Cendes, 2004). However, looking at the available literature, it is striking that there is no clarity regarding this relationship (Shinnar, 1998; Sloviter and Pedley, 1998; Kälviäinen and Salmenperä, 2002; Cendes, 2004; Blümcke et al., 2009). Thus, it is still controversial whether persistent FS lead to HS or MTLE. Retrospective studies performed by MRI or postoperative histopathology have reported many cases of adults with intractable MTLE had prolonged FS in childhood (Abou-Khalil et al., 1993; Cendes et al., 1993; French et al., 1993; Kuks et al., 1993; Trenerry et al., 1993; Maher and Mclachlan, 1995). Although HS is particularly associated with MTLE, it has also been observed post-mortem (PM) associated with other forms of epilepsy (Thom, 2014). The incidence for HS in PM series related to all forms of epilepsy is 30.5%–45% and is 56% related to MTLE (Novy et al., 2013; Thom, 2014). In a study with a total of 3,311 TLE patients, HS was detected in 48% (Blümcke, 2009; Blümcke et al., 2012). In another study, conducted on a total of 4,512 epilepsy patients in whom the reasons for surgical resection varied, HS was detected in 35.2% (Blümcke et al., 2002). Nevertheless, population-based and prospective studies have not necessarily been able to establish this relationship (Nelson and Ellenberg, 1976; Lee et al., 1981; Janszky et al., 2003; Tarkka et al., 2003). Limitations of retrospective studies include, for example, certainty about the exact diagnosis as well as the exact characteristics of the previous FS (Camfield et al., 1994). In addition, it is possible that patients had prior pathologies that led to FS or that these two conditions are simply not correlated (Tarkka et al., 2003; Thom, 2014). Moreover, retrospective studies usually examine only highly selected patients with drug-resistant MTLE (Tarkka et al., 2003; Cendes, 2004) with an epileptic seizure duration of an average of 23 years (Blümcke, 2009), and epilepsy-free controls are usually absent (or it is not stated) because control sections are often not available (Abou-Khalil et al., 1993; Blümcke et al., 2002). For example, in one study (Cendes et al., 1993), a total of 43 TLE patients (with LTLE and MTLE) who underwent temporal lobectomy were examined by MRI, and the sections were subsequently examined for HS. Of these, 25% had severe, 42% moderate, and 33% mild HS (Cendes et al., 1993). Prolonged FS was previously suffered by 35% of the patients (Cendes et al., 1993). Because no sections from the control group were available, they could not be examined for HS, but the authors found by means of MRI that the patients suffering from TLE had significant smaller volumes of amygdala and hippocampus compared to the controls (Cendes et al., 1993). In another study, a total of 32 adolescent patients with previously prolonged FS or an unprovoked FS after the first seizure was examined using MRI by a radiologist blinded to the patients’ history compared with 32 control patients selected for age, sex, and handedness who had previously had only a simple FS without a subsequent seizure, and none of the patients were found to show HS in the MTL, nor did hippocampal volume differ between the two groups (Tarkka et al., 2003). Drawbacks of this study are certainly the low number of (only adolescent) patients studied, with most temporal lobectomies being performed in older patients with more advanced MTLE (Blümcke et al., 2009). For a more detailed critical discussion of this study, see (Scott et al., 2003). In a population-based study examining the frequency of developing epilepsy after FS, 2% of 1,706 children studied who had suffered at least one FS and were followed until 7 years of age developed epilepsy, whereas children whose neurologic or developmental status was suspicious or abnormal before a seizure and whose first seizure was complex (i.e., >15 min, multiple, or focal seizures) were 18 times more likely than children without FS to develop epilepsy (Nelson and Ellenberg, 1976). Previously unremarkable children with a noncomplex first FS, developed epilepsy in 1.1% of cases, which was higher than in children without FS, so it was concluded that the children’s previous neurological and developmental status and characteristics of the first FS were important predictors of epilepsy after FS (Nelson and Ellenberg, 1976). In another retrospective study conducted on the resections of 243 TLE patients performed for intractable seizures, 32% of all patients had prehistoric FS, of whom, in turn, 52% also had HS, the latter observed in a total of 25% of all patients, suggesting a stronger association between FS and HS, but leading the authors to conclude that HS cannot be considered the exclusive cause of TLE, as HS was observed in most cases in association with other pathologies (Tassi et al., 2009).
In addition to FS, regarding the patient’s medical histories, there are a number of other putative risk factors that are commonly found in TLE patients including birth trauma, head injury, and meningitis (Blümcke et al., 2002; Wieser, 2004). Moreover, HS is not exclusively found in TLE patients, but can also be observed in other diseases, e.g., Alzheimer’s Disease (AD), Dementia with Lewy bodies (DLB), or can occur along with brain abnormalities such as white matter hyperintensities and is also frequently observed in the elderly associated with anoxic or ischemic injury (Zarow et al., 2008; Kotrotsou et al., 2015; Mak et al., 2016; Fiford et al., 2017; Yu et al., 2020). HS also appears to vary with age. For instance, in a study (Blumcke et al., 2017) of brain resections from 9,523 surgery-treated patients due to drug-resistant epilepsy, with the TL being involved in 71.9% of all cases, HS was diagnosed in 44.5% of adults and in 15% of children.
It should also be noted that MTLE may also be genetically predisposed, which is referred to as familial MTLE (Cendes, 2004). Thus, MRI abnormalities suggestive of developmental abnormalities of the medial temporal structures or hippocampus have been noted in unaffected and affected individuals with familial MTLE, some of whom have an appearance distinct from nonfamilial TLE (Depondt et al., 2002; Kobayashi et al., 2002; Kobayashi et al., 2003; Andrade-Valença et al., 2008; Crompton et al., 2010). Investigations of familial MTLE showed that MRI evidence of HS was not necessarily related to seizure severity and occurred even in individuals who never had a seizure (Kobayashi et al., 2002). Different possible gene loci for familial MTLE have been described previously, indicating complexity and genetic heterogeneity in familial focal epilepsy (Hedera et al., 2007; Chahine et al., 2013; Kasperaviciute et al., 2013; Sheilabi et al., 2020). For example, a meta-analysis revealed a genome-wide significant association for MTLE with HS in the α1-subunit of the voltage-gated sodium channel (VGSC) (Kasperaviciute et al., 2013) and further experiments have demonstrated a reduction in the expression profile of the β4-subunit of the VGSC at both the transcriptional and translational levels in sclerotic hippocampal tissue from patients with pharmaco-resistant MTLE (Sheilabi et al., 2020). Because the β4-subunit plays an important role in determining the gating of the VGSC, it has been suggested that mutation of the sodium channel subunit may not only alter channel gating possibly resulting in epileptic neuronal activity, but may also affect the binding sensitivities of pharmaceuticals, as they often act in a use-dependent manner (Sheilabi et al., 2020). In summary, the numbers with respect to TLE related phenomena like FS and HS vary in the existing studies conducted on this topic.
The different findings reported in the literature also result from the fact that the diagnosis of HS has not always been uniformly classified, which is why the ILAE agreed in 2017 to adopt new definitions for this pathology in TLE, with the main histopathological feature of TLE-associated HS being the segmental loss of pyramidal neurons, which can occur in every area of the Ammon’s horn (Cornu Ammonis, CA) (Blümcke et al., 2012; Blümcke et al., 2013; Nayak and Bandyopadhyay, 2021). According to the ILAE’s new definition, HS type 1 is defined as the severe segmental loss of pyramidal neurons in the CA1 and CA4 areas (Blümcke et al., 2013; Thom, 2014; McIntosh and Joe, 2021), type 2 features major cell loss in the CA1 region, and HS ILAE type 3 in the CA4 area (Blümcke et al., 2013; Thom, 2014; McIntosh and Joe, 2021). In addition, all three types display gliosis (Blümcke et al., 2013). HS is always associated with astrogliosis and is traditionally referred to as “Ammon’s horn sclerosis” (Sommer, 1880; Blümcke et al., 2012). Astrogliosis denotes the proliferation of reactive astrocytes in response to CNS injury and, as a result, the affected tissue hardens (Sofroniew and Vinters, 2010; Nayak and Bandyopadhyay, 2021). This CNS condition can exhibit a wide spectrum of alterations, from reversible cell hypertrophy with tissue preservation to persistent scarring accompanied by tissue remodeling (Blümcke et al., 2013; McIntosh and Joe, 2021).
Some neuroimaging studies of patients with TLE also showed that HS is not always localized (Bronen et al., 1995; Woermann et al., 1998), although this has not been confirmed in all studies (Vos et al., 2020). In a neuropathological study of the extent of HS in epilepsy along the longitudinal axis of the hippocampus, ten autopsies from patients with a long history of drug-refractory epilepsy and from three control subjects were systematically examined at seven coronal anatomical levels along the body to the tail (Thom et al., 2012). In this study, the pattern of sclerosis was the same at all levels in less than one-third of cases and showed marked longitudinal variability in most cases, leading the authors to suggest that studies of HS in sections derived exclusively from one coronal level cannot necessarily be considered fully representative of the pathology (Thom et al., 2012).
In addition to early-onset FS and HS, TLE is frequently associated with the putative pathogenic appearance of GCD, in which the GCs of the dentate gyrus (DG) abandon their “normally” dense layer and subsequently appear in the ML, resulting in an expansion of the GCL sometimes showing a bi-laminated pattern and in the loss of a clear boundary between the two cell layers (Houser, 1990; Lurton et al., 1997; Thom et al., 2005b; Gong et al., 2007; Koyama et al., 2012) (see Figure 2 for overview of possible GCD patterns). Yet, GCD has often been identified as a hallmark in the hippocampus not only in epilepsy patients, but also in sudden unexplained deaths in infants or children and in numerous animal models that are frequently used to study epilepsy (Koyama et al., 2012; Chai et al., 2014; Kinney et al., 2015; Hefti et al., 2016).
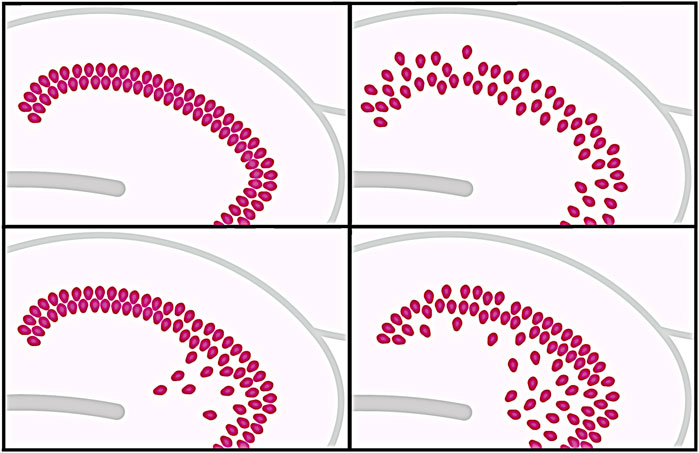
FIGURE 2. Different patterns of granule cell dispersion (GCD). The different possible patterns of GCD are illustrated. A granule cell layer (GCL) considered normal in the classical sense represents a compact band of cells with a relatively high cell density and is clearly delineated against the molecular layer (ML) (top left). In the disaggregated form of GCD, the GCL appears less densely packed and the boundary against the ML is blurred (top right). Ectopic granule cells, some of which can be found deep within the hilar region (bottom left), are shown in comparison to developmental GCD (bottom right), in which young differentiated GCs migrate from the hilar region to the GCL to ultimately build a compact GCL.
Two hypotheses exist for the occurrence of GCD. First, GCD could reflect a migratory defect during GC neurogenesis that persists into adulthood (Altman and Das, 1966; Eriksson et al., 1998; Kempermann et al., 1998), associated with early precipitating injuries before the age of 4 years, particularly FS, i.e., an extrinsic insult (Houser, 1990; Lurton et al., 1997). The second hypothesis assumes an intrinsic failure, such as a defect in the genetically determined developmental program (Houser, 1990; Harding and Thom, 2001). In contrast, other studies suggest that GCD is not a common consequence of recurrent seizures at a young age, but rather depends on the type of epileptic syndrome (Mathern et al., 1997). To date, this question has not been clearly confirmed or disproved, and now another hypothesis has been added, according to which GCD does not represent a pathological change at all, but is rather a natural non-specific variation (Roy et al., 2020). In this publication, GCD, previously commonly considered pathogenic, is described as an “erroneous dogma” (Roy et al., 2020). The authors (Roy et al., 2020) retrospectively examined 147 pediatric human hippocampi, including a total of 21 cases with epilepsy and 126 controls, and identical morphologic spectra of GCD were obtained in normal and seizure-affected brains. In addition, sections through the entire antero-posterior axis of a control hippocampus were examined, and the repeated occurrence of different morphologies of the GCL, that is, compact, focally disaggregated, and bilaminar morphology was observed (Roy et al., 2020) (Figure 2). Therefore, the authors conclude that the scatter of GCs represents a normal variation instead of a specific trait of epilepsy and suggest that sampling biases are responsible for a false dogma (Roy et al., 2020). In fact, many studies, especially those performed on human resected specimens, included few and sometimes no (appropriate) controls. For an overview of relevant studies, see (Roy et al., 2020). The number of controls examined from specimens of patients without epilepsy ranged from two to a maximum of eight in most studies (Houser, 1990; Lurton et al., 1997; El Bahh et al., 1999; Harding and Thom, 2001; Thom et al., 2002; Thom et al., 2005a; Blümcke et al., 2007; Abrahám et al., 2011); a total of four studies in which GCD was identified as a specific characteristic of epilepsy included no controls at all (Blümcke et al., 2009; Bae et al., 2010; Marucci et al., 2010; Freiman et al., 2011). In contrast, the number of examined specimens of epilepsy patients in the mentioned studies varies between ten (Freiman et al., 2011) and 206 (Thom et al., 2002) and is thus significantly higher than the number of included controls. A more recent study also found an association between GCD and TLE, particularly in the presence of HS, and the occurrence of GCD was additionally associated with an initial precipitating injury as well as with a higher number of epileptic focal seizures (Jardim et al., 2021). In the aforementioned study (Jardim et al., 2021), specimens from 108 TLE patients with unilateral HS were examined for histopathologic hippocampal changes compared with specimens from twelve individuals without epilepsy or other neurologic conditions, also significantly outnumbering the tested epilepsy specimens. In addition, there are two other studies not mentioned in the previously described publication (Roy et al., 2020) that also found a correlation between GCD and TLE (Duarte et al., 2018; Jeong et al., 2018). These found an increased probability of abnormal distribution of GCs in the DG in patients with longer duration of epilepsy, with 77 cases of epilepsy compared to twelve controls included in the study (Duarte et al., 2018). In the other study, quantification of the width of the GCL of patients diagnosed with TLE-HS (eight), normalized to the width in normal controls without HS (four), a significant increase in GCL width was observed (Jeong et al., 2018). In addition to the report from (Roy et al., 2020), only one further exists in which GCD was found in two neurologically normal pediatric patients, from which it was concluded that the appearance of GCD may not be exclusively related to epilepsy and may be a separate developmental disorder (Harding and Thom, 2001).
Considering the number of included controls compared to the amount of examined specimens of TLE patients in the performed studies, it might indeed be possible that sampling bias could be responsible for the association between GCD and TLE. But there are other possible causes that could have contributed to the different findings. One difference from many other studies is that the study (Roy et al., 2020) examined mainly pediatric specimens. During development, early generated GCs migrate from the hilus of the DG towards the marginal zone (MZ) to their final positions (Förster et al., 2006; Hayashi et al., 2015), so a transient distribution of newborn GCs in the hilus could reflect a natural stage of early postnatal development of the DG, which, conversely, could be rapidly mistaken for pathological GCD (Weninger et al., 2021). Figure 3 illustrates the histological organization of the DG. It has to be admitted that cell migration into the GCL occurs during the first eight postnatal months, when immature cells gradually disappear from the subgranular zone near the hilus (Seress, 1992; Seress et al., 2001). However, statistical analyses revealed no significant correlation between the occurrence of GCD and age at death, cell loss, PM interval, sex, clinical diagnosis, seizure, or other clinical history between control as well as seizure brains (Roy et al., 2020). Notably, gliosis was observed more frequently in some of the hippocampi affected by seizures but was independent of GCD, although the DG did not show increased GFAP-positive astrocytes in all epilepsy cases (Roy et al., 2020). Moreover, in contrast to the determination of HS, there is currently no consensus on the criteria for determining GCD, which depend in part on the type of measurement and range from complex morphometric analyses to subjective assessment of DG histology, what makes standardized identification and assessment of GCD difficult (Lurton et al., 1997; Mathern et al., 1997; Haas et al., 2002; Blümcke et al., 2009; Abrahám et al., 2011). Another discrepancy important to note is that there are significant differences between the aforementioned studies and the one performed by (Roy et al., 2020) in terms of the hippocampal specimens studied. For instance, all studies were limited to biopsies from (mostly adult) patients with intractable TLE and a usually long history of epileptic seizures (Houser, 1990; Lurton et al., 1997; El Bahh et al., 1999; Thom et al., 2002; Thom et al., 2005a; Blümcke et al., 2007; Blümcke et al., 2009; Bae et al., 2010; Marucci et al., 2010; Abrahám et al., 2011; Freiman et al., 2011; Duarte et al., 2018; Jeong et al., 2018; Jardim et al., 2021). Patients whose tissues were examined had an average duration of epilepsy of at least 17.5 years (Lurton et al., 1997) to a maximum of 24.8 years (Thom et al., 2002). In contrast, the average duration of epileptic seizures in the study conducted by (Roy et al., 2020) was only three years. Moreover, only the hippocampi of cadaveric epilepsy cases and controls, respectively, were examined (Roy et al., 2020). This also applies to the study performed by (Harding and Thom, 2001), who also found GCD in control hippocampi of pediatric cadaveric patients, and again the duration of epileptic seizures in the one epileptic patient examined lasted only about six months (Harding and Thom, 2001). These serious differences may well have affected the results of the different studies.
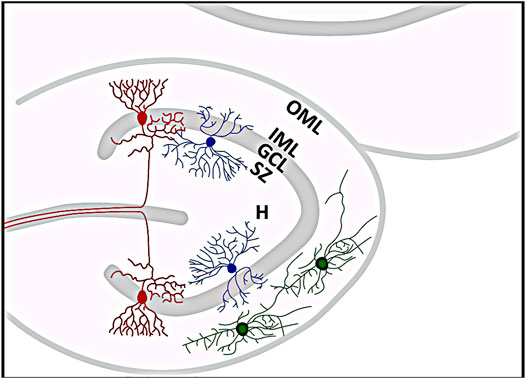
FIGURE 3. Histological organization of the dentate gyrus (DG). Red neurons represent granule cells whose somata form the granule cell layer (GCL). Just below is the subgranular zone (SZ), where reelin-expressing interneurons (blue) can be found, and above is the molecular layer, which can be further subdivided into an inner (IML) and an outer molecular layer (OML). The outer OML harbors reelin-synthesizing Cajal-Retzius cells (green). The hilar area (H) of the DG is located below the SZ. The template for this figure was (Marín-Padilla, 1998; Twible et al., 2021).
Investigating GCD as a cause or consequence of epilepsy is particularly difficult in surgical series on human specimens, as these typically involve resections of selected patients, most of whom have had preexisting hippocampal damage detected by MRI with an advanced disease stage often underlying a strong heterogeneity in terms of potentially epileptogenic injuries (Harding and Thom, 2001; Fisher et al., 2014; Jeong et al., 2018; Jardim et al., 2021). In parallel with the apparent lack of a suitable number of non-epileptic, age-matched human control brain samples, the study of epileptogenesis on human brain tissue is particularly limited by statistically valid numbers of homogeneous sample series in terms of clinical, pathological, and individual parameters, which is why research largely relies on animal models herein (Jimenez-Mateos et al., 2012; Dębski et al., 2016; Becker, 2018).
Animal Models Mirroring Human Temporal Lobe Epilepsy
Animal models but also in vitro models derived from animal tissue have been used in an attempt to at least partially compensate for the lack of human control hippocampi and to gain insight into early stages of epileptogenesis, for which human hippocampal tissue is usually unavailable (Becker, 2018; Nirwan et al., 2018). Moreover, animal models play an important role in drug discovery and development, and molecular changes observed in both focal epileptic lesions in humans and in corresponding animal models should be more likely to be of pathogenetic significance (Becker, 2018; Nirwan et al., 2018). In the most commonly used animal models, rodents, primarily mice and rats, are treated with chemoconvulsants, with kainate (KA) and pilocarpine (PL) being the most commonly used, inducing recurrent chronic seizures in treated animals that reflect important neuropathological changes of human TLE (Leite et al., 2002; Becker, 2018). In this regard, systemic or local administration of chemoconvulsants in rodents results in severe depolarization of neurons and subsequent epileptic seizures, with the focus of damage in the hippocampal formation (Sharma et al., 2007; Becker, 2018). Although many animal models can be used to reproduce the pathophysiological changes such as HS, GCD and spontaneous recurrent seizures, no single animal model shares all characteristics of TLE (Nirwan et al., 2018). For example, various parameters, such as sex, age, and weight, influence the sensitivity to KA (Nirwan et al., 2018). Intrahippocampal injection of KA or PL results in bilateral electrical spikes and after-discharges as well as behavioral seizures in rodents comparable to status epilepticus (SE) and subsequent chronic recurrent spontaneous seizures (Becker, 2018; Nirwan et al., 2018; Freiman et al., 2021; Moura et al., 2021). In chemoconvulsant-treated rodents, GCD also develops (Freiman et al., 2021; Moura et al., 2021), which may indicate that it is a concomitant of TLE rather than a natural variation. However, in research comparing KA- versus PL-injected animals, GCD was observed exclusively in KA-treated animals, but not in those treated with PL (Moura et al., 2021). This finding rather suggests that KA has an additional local effect with epileptic activity alone not being sufficient to cause GCD (Moura et al., 2021), especially since epileptic neuronal activity after KA treatment has also been observed on the side contralateral to the injection site (Moura et al., 2021). In contrast, previous studies have shown that the width of the GCL was significantly greater after PL treatment compared with untreated control animals, but exclusively in animals in which PL treatment induced SE (Mello et al., 1992; Mello et al., 1993) speaking against the conclusion that epileptic discharges and SE, respectively, are not sufficient to evoke GCD. In another model, animals are implanted unilaterally or bilaterally with two stimulating and two recording electrodes in the perforant pathway (Figure 1) and then electrically stimulated to induce seizures (Bumanglag and Sloviter, 2008; Nirwan et al., 2018). Although GCD is not subsequently manifested, degeneration of DG cells is observed after stimulation (Sloviter et al., 1996). In a study in which epileptic seizures were induced in rats using amygdala kindling, more ectopically located GCs were found in treated animals, but no change was observed between controls and electrically stimulated animals in terms of GCL volume or width (Fournier et al., 2010). Interestingly, another study found an increase in the width of the GCL one day after the last seizure triggered by amygdala kindling, and this change had regressed in animals examined one month later (Singh et al., 2013). In contrast, other morphological features often observed in other models of TLE, such as ectopic GCs, were not observed in kindled animals examined at either time point (Singh et al., 2013). In another animal model of experimental TLE, FS are triggered in rodents to resemble the state of human FS (Jiang et al., 1999; Bender et al., 2004; Koyama and Matsuki, 2010; Koyama et al., 2012). For this purpose, animals are kept hyperthermic at 39°C–41.5°C for 30 min, resulting in seizures lasting approximately 20 min (Bender et al., 2004). Similarities between experimentally induced hyperthermic seizures in rats and TLE in humans have been reported previously (Jiang et al., 1999). Using the described model, it was examined whether FS can induce GCD, finding that convulsive rats indeed displayed ectopic GCs in the hippocampus, but this was more likely triggered by the seizures themselves, and temperature elevation alone was not sufficient to evoke GCD (Koyama et al., 2012). The authors further demonstrated that abnormal migration of neonatal-generated GCs leads to GC ectopy, persisting into adulthood (Koyama et al., 2012), whereas another study concluded that fully differentiated GCs become motile after KA-induced epileptiform activity, resulting in the manifestation of GCD (Chai et al., 2014). In this regard, decreased expression of the ECM protein reelin, which is illuminated in more detail in the next section, was found after application of KA, both at the mRNA and protein levels, with interneurons in the hilus and the subgranular zone being particularly affected (Chai et al., 2014) (Figure 3). Results from studies using animal models have to be taken with caution as it is questionable to which extend, they reflect the respective human disease state and the findings may vary depending on the model used. Nevertheless, they are important to gain insight into pathological mechanisms underlying human TLE and drug design for which human tissue is not available. However, it is striking that GCD, unlike in human hippocampi, is not observed in the healthy rodent hippocampus. It is therefore conceivable that this phenomenon is a natural variation in humans but not in rodents.
Extracellular Matrix and the Extracellular Matrix Molecule Reelin
The ECM constitutes a three-dimensional, cell-free matrix surrounding neurons and glial cells in the CNS, which synthesize and secrete ECM molecules (Dityatev, 2010; Lau et al., 2013; Lillis, 2021). The ECM is rich in proteoglycans and hydrophilic glycosaminoglycans and also contains collagen, elastin, fibronectin, laminin, and other glycoproteins and accounts for around 20% of cerebral volume (Cragg, 1979; Lau et al., 2013; Theocharis et al., 2016). The structure of the ECM of the CNS is heterogeneous and depends on various factors, e.g., on the present cell types (Dityatev and Fellin, 2008). For example, the perineuronal network, which surrounds cell somata and proximal dendrites as well as at initial segments of axons of certain neurons and surrounds mainly parvalbumin-expressing GABAergic interneurons in the cerebral cortex and hippocampus, represents one form of ECM (Dityatev, 2010). Depending on its composition, this complex and diverse structure filling the extracellular space influences the structural plasticity of the tissue as well as cell-cell and cell-matrix interactions with a multitude of secreted growth factors (Dityatev, 2010). Thus, the ECM of the CNS forms a dynamic system and the constant adaptation of ECM structure to required conditions by regulating the expression, respectively the activity of extracellular proteases, is fundamental for the correct function of physiological processes (Ulbrich et al., 2021). Extracellular proteases include the plasmin system, the proteins ADAM (a disintegrin and metalloproteinase) and ADAMTS (ADAM with thrombospondin motifs), as well as matrix metalloproteases (MMPs) (Stevenson and Lawrence, 2018; Ulbrich et al., 2021). Besides degrading proteins, the latter is also capable of processing signaling molecules and in this way controls cellular signaling events (Rodríguez et al., 2010). The activity of MMPs is in turn regulated by tissue inhibitor of metalloproteinases (TIMPs) (Acar et al., 2015) and a disbalance between expression levels of MMPs and TIMPs has been associated with several pathologic conditions (Mizoguchi and Yamada, 2013). ECM molecules transmit signals by binding to cell surface receptors (Theocharis et al., 2016) and thereby influence higher-level processes, such as synaptic plasticity and brain development, the latter of which the ECM molecule reelin is particularly known to be responsible for (Dityatev et al., 2010).
Thus, the ECM is a fundamental component of neuronal processes, along with neurons and glial cells. Dysregulation of the ECM is associated with neurological diseases, including epilepsy (Dityatev, 2010). Injuries to the CNS, such as seizures, may activate the immune system, possibly leading to alteration of perivascular and perineuronal ECM, often accompanied by high expression and associated activity of matrix-remodeling extracellular proteases such as MMPs (Ulbrich et al., 2021). Activation of astrocytes enhances synthesis of ECM molecules and, together with increased expression of TIMP proteins, elicits aggregation of ECM molecules and activates signaling cascades involving various molecules such as integrins, Toll-like receptors, cell adhesion molecules, and ion channels (Ulbrich et al., 2021). Epileptic conditions in particular lead to aberrant expression of extracellular proteases, which in turn is associated with neurodegeneration, neuroinflammation, and concomitant altered synaptic plasticity (Konopacki et al., 2007; Lukasiuk et al., 2011; Mizoguchi and Yamada, 2013). The importance of the ECM is exemplified by hyaluronic acid (HA), a fundamental component of the ground substance of the ECM. Thus, enzymatic digestion of HA, by hyaluronidase is sufficient to trigger epileptic activity in vitro and in vivo (Vedunova et al., 2013; Balashova et al., 2019). Moreover, hyaluronan synthase ko mice exhibit spontaneous epileptic seizures (Arranz et al., 2014). Based on studies performed on animal models as well as on human tissue remnants, with respect to MMPs, MMP-9 is particularly associated with epilepsy, with epileptic activity leading to upregulation of MMP-9 (Szklarczyk et al., 2002; Konopacki et al., 2007; Wilczynski et al., 2008; Takács et al., 2010; Li et al., 2012; Acar et al., 2015; Pijet et al., 2018). In addition to HA and MMP-9, epileptic conditions lead to increased expression of other ECM molecules, such as tenascin R, tenascin C, and neuronal pentraxin 2, whereas other molecules, such as protein tyrosine phosphatase receptor type Z1 (phosphacan) are downregulated (Dityatev, 2010). Tenascin R molecules are, among others, present in perineuronal networks and, in di- or trimeric form, cross-link lecticans and thus have an ECM-stabilizing effect (Morawski et al., 2014). Tenascin R-deficient mice, although not epileptic, exhibit decreased perisomatic GABAergic inhibition and synaptic plasticity and show significantly increased neuronal activity in the CA1 region after PL treatment compared to wild-type mice (Saghatelyan et al., 2001; Brenneke et al., 2004; Pitkänen et al., 2014). These findings highlight the strong effect the ECM exerts on peripheral cells. Due to the importance of the ECM for proper CNS function, compounds modulating the effects of specific ECM molecules could serve as therapeutic interventions in various neurological conditions. For example, treatment with the MMP-9 inhibitor IPR-179 reduced the severity of seizures in the kindling model and reduced the number of spontaneous seizures in the KA model, with no side effects noted, and furthermore improved cognitive performance of treated rodents (Broekaart et al., 2021). Another MMP inhibitor that has been tested using the KA model is marimastat, also showing beneficial effects on seizure duration (Pijet et al., 2020). Another molecule that has already been studied intensively is the ECM molecule reelin.
Reelin is a large secreted ECM glycoprotein regulating many important processes in mammalian brain development, and dysregulation of reelin signaling has been linked to several brain diseases such as autism, schizophrenia, depression, AD, and, in particular, epilepsy (Ishii et al., 2016; Hirota and Nakajima, 2017; Santana and Marzolo, 2017; Wasser and Herz, 2017; Armstrong et al., 2019; Okugawa et al., 2020; Orcinha et al., 2021). The highly conserved human reelin gene is located on chromosome 7q22 and is circa 450 kbp long (Desilva et al., 1997; Manoharan et al., 2015). Composed of 3,461 amino acids, the reelin protein has a relative molecular mass of 388 kDa, and of 450 kDa in its glycosylated state (D’Arcangelo et al., 1995; Desilva et al., 1997; Jossin, 2020). With respect to its primary structure, reelin is subdivided into several major domains. An N-terminal signal peptide is followed by an F-spondin homology domain, which is joined by a unique region that displays no sequence similarity to previously known domains, featuring a series of reelin-specific repeats (Ichihara et al., 2001; Yasui et al., 2007; Manoharan et al., 2015). The C-terminal domain contains predominantly basic amino acids (D’Arcangelo et al., 1995; Jossin, 2020). Reelin is functional as a multimer and linkage is formed by disulfide bonds and by non-covalent interactions (Kubo et al., 2002; Yasui et al., 2011; Manoharan et al., 2015). After secretion into the ECM, reelin is proteolytically cleaved into smaller isoforms (Lambert de Rouvroit et al., 1999; Jossin et al., 2004), which may be important for the physiological function of the protein (Tinnes et al., 2011; Tinnes et al., 2013; Orcinha et al., 2016). In this process, reelin can be cleaved into different fragments, depending on which protease is active, although it is controversial which of the resulting reelin fragments is critical for the activation of the reelin signaling pathway (Lambert de Rouvroit et al., 1999; Lugli et al., 2003; Tinnes et al., 2013). Thus, some studies show that the N-terminal fragment is important for its biological function (Nakajima et al., 1997; Kubo et al., 2002; Orcinha et al., 2021), other studies focus on the C-terminal part (Nakano et al., 2007; Kohno et al., 2015) of the protein, while others suggest that a dimeric central fragment is signaling competent (Jossin et al., 2004; Yasui et al., 2007; Orcinha et al., 2021; Turk et al., 2021). Results of a recent study suggest an essential role for the dimeric central reelin fragment, but not the other reelin fragments or the monomeric central fragment, in the lipoprotein receptor-dependent activation of the canonical reelin pathway, whereas only full-length reelin is potent of stimulating both canonical and non-canonical reelin signaling cascade (Turk et al., 2021). Overall, current evidence implies full-length reelin to be more active than its processing-derived products (Nakano et al., 2007; Koie et al., 2014; Kohno et al., 2015; Sato et al., 2016; Dlugosz et al., 2019; Okugawa et al., 2020; Turk et al., 2021). However, it is also possible that the cleaved fragments diffuse to more distant regions to trigger downstream events (Jossin et al., 2007; Yasui et al., 2011). Reelin is processed by various enzymes at different sites of the protein and its availability therefore depends on the activity of the respective proteases (Jossin, 2020). These include ADAMTS-2, ADAMTS-3, ADAMTS-4, and ADAMTS-5, the serine protease tissue plasminogen activator, and meprin α as well as MMP-9, the latter of which triggers cleavage of reelin indirectly through the activation of ADAMTS-4 (Hisanaga et al., 2012; Krstic et al., 2012; Trotter et al., 2014; Sato et al., 2016; Ogino et al., 2017; Yamakage et al., 2019; Jossin, 2020). In contrast, the metalloproteinase inhibitors TIMP-1, TIMP-3, and α-2-macroglobulin inhibit reelin processing (Krstic et al., 2012; Jossin, 2020).
In the canonical reelin signaling pathway, reelin binds to receptors of the lipoprotein receptor family, apolipoprotein E receptor 2 (ApoER2) and very low-density lipoprotein receptor (VLDLR), and induces tyrosine phosphorylation of the intracellular adaptor protein Disabled-1 (Dab1) mediated by Src family kinases (D’Arcangelo et al., 1999; Howell et al., 1999; Arnaud et al., 2003; Förster, 2014; Jossin, 2020; Hattori and Kohno, 2021). Phosphorylated Dab1 in turn activates downstream signaling cascades, primarily influencing molecules affecting the actin and microtubule cytoskeleton but also influences adhesion molecules, ion channels, and neurotransmitter release (D’Arcangelo et al., 1999; Howell et al., 1999; Tissir and Goffinet, 2003; Jossin and Goffinet, 2007; Leemhuis and Bock, 2011; Bock and May, 2016; Ishii et al., 2016; Hirota and Nakajima, 2017; Wasser and Herz, 2017; Okugawa et al., 2020).
During cortical development, reelin is expressed by CR cells, a transient class of early-born neurons located in the MZ, the most superficial layer of the cerebral cortex (Nakajima et al., 1997; Marín-Padilla, 1998; Soriano and Del Río, 2005; Förster et al., 2006). CR cells are responsible for the proper lamination of the neocortex and hippocampus, respectively (Förster et al., 2006), by regulating the development of hippocampal connections (Del Río et al., 1997; Borrell et al., 2007), hippocampal dendrites and dendritic spines (Niu et al., 2004; Niu et al., 2008), as well as the proliferation and distribution of oligodendrocyte progenitor cells via the expression of reelin (Ogino et al., 2020). Furthermore, the presence and distribution of specific subtypes of CR cells during corticogenesis, as well as their timely death, appear to be important for the formation of cortical circuits (Griveau et al., 2010; Barber et al., 2015; Barber and Pierani, 2016; de Frutos et al., 2016; Causeret et al., 2021). After cortical development and demise of most CR cells, reelin then continues to be expressed primarily by interneurons in the adult brain (Alcántara et al., 1998; Drakew et al., 1998; Förster, 2014). Disruption of the reelin pathway leads to manifestation of the reeler phenotype (D’Arcangelo et al., 1995). In reeler mice, containing a genetic defect in the reelin gene, the characteristic inside-out sequence of neuronal layers in the neocortex, is reversed and neurons born later reveal a broader and irregular distribution (D’Arcangelo et al., 1995; Dekimoto et al., 2010; Boyle et al., 2011; Förster, 2014; Jossin, 2020). Reelin also regulates dendritic growth during embryonic development. For instance, dendritic growth of pyramidal cells in reeler mice (Pinto Lord and Caviness, 1979; Niu et al., 2004). The application of reelin in vitro has also been shown to increase dendritic growth of hippocampal neurons (Jossin and Goffinet, 2007; Matsuki et al., 2008). Conversely, a deficit of native reelin in vivo results in lower spine density on apical dendrites of hippocampal pyramidal neurons, which can be rescued by addition of recombinant reelin in vitro (Niu et al., 2008). In the developing neocortex, reelin promotes dendritic growth of pyramidal cells in early embryonic brain development (Nichols and Olson, 2010; Kupferman et al., 2014; Chai et al., 2015; Kohno et al., 2015; O'Dell et al., 2015). On the other hand, postnatally, reelin restricts dendritic growth of cortical pyramidal neurons (Chameau et al., 2009) and interneurons (Hamad et al., 2021b). As in the neocortex, reelin is also significantly involved in the proper lamination of the hippocampus (Stanfield and Cowan, 1979; Förster et al., 2006; Jossin, 2020). Thus, GCs in the DG of reeler mice do not display compact layering but are lesser in number and are diffusely distributed throughout the DG and the pyramidal cell layer is duplicated in the CA1 area (Stanfield and Cowan, 1979; Zhao et al., 2004; Förster et al., 2006; Boyle et al., 2011; Khalaf-Nazzal and Francis, 2013; Jossin, 2020). Mutants with defects in other molecules of the reelin signaling cascade display a phenotype comparable to that of the reeler mouse (Förster et al., 2006). Thus, mutants lacking the intracellular adaptor protein Dab1 or the reelin receptors VLDLR and ApoER2 show the same defects (Howell et al., 1997; Trommsdorff et al., 1999; Förster et al., 2006; Hirota et al., 2016). In contrast, mutants lacking only one of the two reelin receptors VLDLR or ApoER2 exhibit less severe migration defects and slightly different phenotypes, so that it is reasonable to assume that the two receptors exert different functions (Drakew et al., 2002; Förster et al., 2006). Findings of recent studies indicate that VLDLR suppresses neuronal invasion into the MZ, whereas ApoER2 promotes neuronal aggregation (Hirota et al., 2016; Hirota and Nakajima, 2020), speculating that the accumulation of neurons may be required for proper layer formation (Hirota and Nakajima, 2017). Mice lacking both, reelin and Dab1, have no additional defects compared to the reeler phenotype, indicating that the two proteins function in a linear pathway (Howell et al., 1999; Jossin, 2020). In the Orleans reeler strain, expression of a mutant reelin protein occurs that is not cleaved (Lambert de Rouvroit et al., 1999). Because the mutant protein is not secreted from the producing cells, it is assumed that cleavage of reelin occurs in the extracellular space (de Bergeyck et al., 1997; Hattori and Kohno, 2021). In humans, reelin deficiency leads to an autosomal recessive form of lissencephaly, characterized by abnormal neuronal lamination and lack or reduction of convolutions concomitant with cerebellar hypoplasia (Hong et al., 2000; Chang et al., 2007; Förster, 2014; Jossin, 2020). This malformation in humans is due to two different splicing mutations in the reelin gene, in both cases truncating the reelin gene, resulting in the absence of the highly basic C-terminus, which is required for normal secretion and function (D’Arcangelo et al., 1997; de Bergeyck et al., 1997; Hong et al., 2000; Manoharan et al., 2015).
Reelin in the Context of Granule Cell Dispersion and Temporal Lobe Epilepsy
Heterozygous mutations in the reelin gene in humans may cause autosomal dominant LTLE, likely due to decreased reelin secretion (Dazzo et al., 2015; Michelucci et al., 2017; Česká et al., 2019; Michelucci et al., 2020; Dazzo and Nobile, 2021). Moreover, GABABR-mediated mechanisms have been known for many years to be involved in the genesis and propagation of both typical (Crunelli and Leresche, 1991) and atypical absence seizures (Cortez et al., 2001). In this context it is interesting to note that reelin signaling has recently been shown to modulate GABAB receptor function in the neocortex (Hamad et al., 2021a).
In addition, TLE may be accompanied by GCD, reminiscent of GCD in reelin-deficient reeler mice (Houser, 1990; Frotscher et al., 2003; Haas and Frotscher, 2010; Orcinha et al., 2021). However, homozygous reeler mice do not have spontaneous seizures but exhibit increased seizure susceptibility (Patrylo et al., 2006). Dab1-deficient mice also lack spontaneous seizures, but these animals display interictal epileptiform abnormalities and a considerably shortened latency to chemoconvulsant-induced SE, in which these pro-epileptogenic changes occur with decreased neurogenesis and increased numbers of ectopic GCs (Korn et al., 2016). Other studies also found that postnatal loss of Dab1 or the expression of mutated Dab1 results in ectopically placed GCs (Teixeira et al., 2012; Arimitsu et al., 2021), accompanied by decreased Dab1 phosphorylation as well as impaired synapse formation and abnormal expression of transcription factors, suggesting that the reelin-Dab1 pathway is essential for neuronal migration as well as maturation and synaptogenesis in mice (Arimitsu et al., 2021). A common hypothesis is that ectopic GCs in the hilus may contribute to the manifestation of epilepsy (Scharfman and Pierce, 2012), however, relevant studies show that mice with ectopically located GCs are largely seizure-free and misplaced GCs alone are not sufficient to evoke seizures (Koyama et al., 2012; Myers et al., 2013; Korn et al., 2016). Nevertheless, the findings of the aforementioned studies do not rule out the possibility that ectopically placed GCs may act as “hub” cells within a seizure network, as it has already been shown that these cells are hyperexcitable and make aberrant connections (Scharfman et al., 2000; Morgan and Soltesz, 2008; Zhan and Nadler, 2009; Cameron et al., 2011; Althaus et al., 2015; Korn et al., 2016). It is evident that the scattered GCs in the reeler mouse represent a developmental defect, whereas GCD in epilepsy patients could also be a secondary effect related to seizure activity (Haas and Frotscher, 2010).
In a subtype of focal cortical dysplasia (FCD) in which abnormal cortical layering occurs in association with HS and GCD, loss of reelin has been identified as the pathogenetic basis, arguing for a link between reduced reelin expression and GCD in humans (Marucci et al., 2012). Moreover, in a study performed on human hippocampal sections from MTLE patients, the extent of GCD was found to be inversely correlated with the number of reelin-expressing CR cells, suggesting a link between reelin secretion and GCD in epilepsy patients (Haas et al., 2002). In this regard, epigenetic silencing by methylation of the reelin promoter may be an underlying pathogenetic mechanism of GCD, as it was found that methylation of the reelin promoter in TLE correlated with GCD in human TLE specimens (Kobow et al., 2009). In contrast, other studies found a correlation between a high number of persistent CR cells and early complex FS (Blümcke et al., 1999) or HS (Blumcke et al., 1996). Although these findings appear contrary, they suggest a link between the reelin pathway and TLE (Blümcke et al., 2002). In addition, different staining techniques were used in the different studies for histochemical visualization of CR cells, which may also have led to the conflicting results. Nevertheless, based on the similarities between the phenotype of the reeler mouse and the pathological findings in resections of TLE patients, it seems plausible that impaired reelin signaling results in increased susceptibility to develop epilepsy. For instance, there is evidence from rodent epilepsy models, that the formation of GCD can be triggered by the loss of reelin-producing cells (Heinrich et al., 2006; Gong et al., 2007; Antonucci et al., 2008; Duveau et al., 2011; Orcinha et al., 2016). In kainate-treated rodents, GCD formation was prevented in vivo by infusion of exogenous reelin into the hippocampus during epileptogenesis (Müller et al., 2009). Addition of recombinant reelin to KA-treated organotypic hippocampal slice cultures could prevent GCD (Orcinha et al., 2016; Orcinha et al., 2021). Conversely, antibody blockade of reelin function in the healthy mouse hippocampus resulted in local broadening of the GCL (Heinrich et al., 2006), which could also be shown in vitro (Orcinha et al., 2021). While these results suggest that reelin is not only important during development, but might play an important role in maintaining the lamination of GCs in the DG of adult mice (Haas and Frotscher, 2010), recent studies using adult conditional reeler ko mice did not confirm this hypothesis, since mice with conditionally induced reelin deficiency displayed a normal neocortical and hippocampal architecture with no GCD (Lane-Donovan et al., 2015). Selective inactivation of reelin in interneurons led to subtle changes in the DG but not in the neocortex, suggesting that interneuron derived reelin does not play a major role for the layering of GCs (Pahle et al., 2020).
GCD was found to occur in the absence of neurogenesis and thus was more likely due to the displacement of differentiated neurons (Haas and Frotscher, 2010). While Mathern et al. (2002) found that neurogenesis or proliferation of neurons was not increased in the hippocampus of TLE patients (Mathern et al., 2002), others found an increase correlating with GCD (Blümcke et al., 2001; Thom et al., 2005a; Crespel et al., 2005). Again, it cannot be excluded that the use of different markers led to the contradictory results, as bromodeoxyuridine cannot be used in resected human tissue (Haas and Frotscher, 2010). The question of whether reelin deficiency, in patients without mutations in the reelin gene, is a cause or consequence of epilepsy cannot be conclusively answered in studies on patient tissue samples. Therefore, the interpretation relies on animal models. For example, in a study performed on hippocampal slice cultures it was found that differentiated neurons became motile after induction of epileptic activity by KA application, which ultimately resulted in GCD and was also associated with decreased expression of reelin as evidenced by the loss of reelin-synthesizing cells (Chai et al., 2014). This suggests, on the one hand, that epileptic activity may be a trigger for GCD and is apparently related to decreased reelin expression. In line with this, after long-term amygdala kindling, rats showed a decrease of reelin-positive neurons accompanied by ectopic GCs in the hilus of the DG (Fournier et al., 2010). Consistently, further studies showed that proteolytic processing of reelin is essential for the maintenance of GC lamination in the DG, with reelin processing found to be disrupted by epileptic conditions (Duveau et al., 2011; Tinnes et al., 2011), although reelin synthesis and secretion was normal (Tinnes et al., 2011). In this regard, epileptic states induce the upregulation of endogenous TIMP-1, thereby inhibiting matrix metalloproteinase activity, ultimately resulting in the extracellular accumulation of un-cleaved reelin (Tinnes et al., 2013). In a more recent study, the association between the occurrence of GCD and FS was investigated, and a transient increase in temperature led to the induction of GCD in vitro, accompanied by partial degeneration of GCs, whereas reelin-expressing CR cells were preserved in the ML, although it could not be ruled out that reelin processing was abnormal (Weninger et al., 2021). In the aforementioned publication, the appearance of GCD after temperature elevation was accompanied by severe microgliosis, suggesting an immune response of the tissue, and it was proposed that microglia could serve as markers to distinguish pathological GCD from normal variation (Roy et al., 2020; Weninger et al., 2021). Concordant with this, the results of a transcriptome signature study indicate that macrophages and microglia in particular play a critical role in epileptogenesis (Chen et al., 2020).
A previous study found a correlation between GCD and the kainate-induced loss of reelin-expressing interneurons in the DG (Orcinha et al., 2016). Thus, it was hypothesized that interneuron-derived reelin keeps the GCL compact and a decrease in reelin concentration consequently evokes migration of mature GCs toward the hilus of the DG (Haas and Frotscher, 2010; Tinnes et al., 2013; Orcinha et al., 2016). Still, selective inactivation of reelin in interneurons in conditional reelin knockout mice does not lead to GCD (Pahle et al., 2020), arguing against the importance of interneuron-derived reelin in maintaining a compact GCL.
Currently, there is only one study examining changes in the hippocampal ECM in a TLE mouse model with respect to GCD. The authors found a sequential upregulation of the ECM molecules neurocan and tenascin C at the end of the latent period in the GCL of the DG (Heck et al., 2004). After onset of the first convulsions accompanying GCD, increased, but differentially localized, expression of phosphacan as well as DSD-1 chondroitin sulfate motif and HNK-1 oligosaccharide was detected, leading the authors to conclude GCD coincides with a general increase of the ECM (Heck et al., 2004). In contrast, the expression of laminin and fibronectin was unchanged (Heck et al., 2004). Further studies of this kind could help to unravel the emergence and thus the significance of GCD in the context of epilepsy.
Conclusion and Perspectives
All in all, the significance of GCD has not yet been clearly elucidated. Is it a pathological manifestation or the result of epileptic activity in rodents, whereas in humans it is merely a natural variation? Is it an “erroneous dogma”? Although the clinical appearance of GCD is often associated with TLE, particularly in patients with a history of FS, it has also been observed in pediatric patients without a history of FS or epilepsy. Therefore, it can be concluded that GCD is not always pathological but may also represent a non-specific naturally occurring variation. It would be important to know if this is also the case in adults, which is of course difficult to implement. Thus, for future studies, it could be helpful to differentiate disease-related GCD from normal variation. Ultimately, not only the significance of GCD, but also the relevance of FS remain controversial. The recently published large-scale study (Roy et al., 2020) has once again shown that the inclusion of adequate controls in sufficient quantities, which is difficult in the field of human epilepsy research, are of great importance for the interpretation of the results of the studies. Also, the association of FS and HS to TLE remains unclear to date, and the results of retrospective studies contrast with those of prospective studies. In addition, the role of the ECM molecule reelin in the clinical picture of TLE remains elusive. Since many studies point to a causal role of the reelin pathway in the development of GCD or TLE, molecules of this pathway could be a potential target for drug development to provide a cure for affected patients. However, the functions of reelin in the CNS are diverse, and experiments investigating the link of reelin to GCD and TLE obtained different and sometimes contradictory results. Hence, indirect mechanisms should also be considered, as reelin spins a complex network with a variety of different ECM molecules, influencing each other. An example is provided by MMP-9, known to be upregulated by epileptic activity and being able to process reelin indirectly via ADAMTS-4 as already described. Altogether, many questions remain unanswered on this topic and further studies, both on animal models and human tissue, will be needed to ultimately answer them.
Author Contributions
Conceptualization: JL, MH, EF, and GR; Writing—original draft: JL and MH; Writing—review and editing: MH, JL, EF, and GR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge support by the Open Access Publication Funds of the Ruhr-Universität Bochum.
References
Abou-Khalil, B., Andermann, E., Andermann, F., Olivier, A., and Quesney, L. F. (1993). Temporal Lobe Epilepsy after Prolonged Febrile Convulsions: Excellent Outcome after Surgical Treatment. Epilepsia 34, 878–883. doi:10.1111/j.1528-1157.1993.tb02105.x
Ábrahám, H., Richter, Z., Gyimesi, C., Horváth, Z., Janszky, J., Dóczi, T., et al. (2011). Degree and Pattern of Calbindin Immunoreactivity in Granule Cells of the Dentate Gyrus Differ in Mesial Temporal Sclerosis, Cortical Malformation- and Tumor-Related Epilepsies. Brain Res. 1399, 66–78. doi:10.1016/j.brainres.2011.05.010
Acar, G., Tanriover, G., Acar, F., and Demir, R. (2014). Increased Expression of Matrix Metalloproteinase-9 in Patients with Temporal Lobe Epilepsy. Turk. Neurosurg. 25, 749–756. doi:10.5137/1019-5149.Jtn.10738-14.0
Alcántara, S., Ruiz, M., D’Arcangelo, G., Ezan, F., De Lecea, L., Curran, T., et al. (1998). Regional and Cellular Patterns ofreelinmRNA Expression in the Forebrain of the Developing and Adult Mouse. J. Neurosci. 18, 7779–7799. doi:10.1523/jneurosci.18-19-07779.1998
Althaus, A. L., Sagher, O., Parent, J. M., and Murphy, G. G. (2015). Intrinsic Neurophysiological Properties of Hilar Ectopic and Normotopic Dentate Granule Cells in Human Temporal Lobe Epilepsy and a Rat Model. J. Neurophysiology 113, 1184–1194. doi:10.1152/jn.00835.2014
Altman, J., and Das, G. D. (1966). Autoradiographic and Histological Studies of Postnatal Neurogenesis. I. A Longitudinal Investigation of the Kinetics, Migration and Transformation of Cells Incoorporating Tritiated Thymidine in Neonate Rats, with Special Reference to Postnatal Neurogenesis in Some Brain Regions. J. Comp. Neurol. 126, 337–389. doi:10.1002/cne.901260302
Andrade-Valença, L. P. A., Valença, M. M., Velasco, T. R., Carlotti, C. G., Assirati, J. A., Galvis-Alonso, O. Y., et al. (2008). Mesial Temporal Lobe Epilepsy: Clinical and Neuropathologic Findings of Familial and Sporadic Forms. Epilepsia 49, 1046–1054. doi:10.1111/j.1528-1167.2008.01551.x
Annegers, J. F., Hauser, W. A., Shirts, S. B., and Kurland, L. T. (1987). Factors Prognostic of Unprovoked Seizures after Febrile Convulsions. N. Engl. J. Med. 316, 493–498. doi:10.1056/nejm198702263160901
Antonucci, F., Di Garbo, A., Novelli, E., Manno, I., Sartucci, F., Bozzi, Y., et al. (2008). Botulinum Neurotoxin E (BoNT/E) Reduces CA1 Neuron Loss and Granule Cell Dispersion, with No Effects on Chronic Seizures, in a Mouse Model of Temporal Lobe Epilepsy. Exp. Neurol. 210, 388–401. doi:10.1016/j.expneurol.2007.11.012
Arimitsu, N., Mizukami, Y., Shimizu, J., Takai, K., Suzuki, T., and Suzuki, N. (2021). Defective Reelin/Dab1 Signaling Pathways Associated with Disturbed hippocampus Development of Homozygous Yotari Mice. Mol. Cell. Neurosci. 112, 103614. doi:10.1016/j.mcn.2021.103614
Armstrong, N. C., Anderson, R. C., and Mcdermott, K. W. (2019). Reelin: Diverse Roles in Central Nervous System Development, Health and Disease. Int. J. Biochem. Cell. Biol. 112, 72–75. doi:10.1016/j.biocel.2019.04.009
Arnaud, L., Ballif, B. A., Förster, E., and Cooper, J. A. (2003). Fyn Tyrosine Kinase Is a Critical Regulator of Disabled-1 during Brain Development. Curr. Biol. 13, 9–17. doi:10.1016/s0960-9822(02)01397-0
Arranz, A. M., Perkins, K. L., Irie, F., Lewis, D. P., Hrabe, J., Xiao, F., et al. (2014). Hyaluronan Deficiency Due to Has3 Knock-Out Causes Altered Neuronal Activity and Seizures via Reduction in Brain Extracellular Space. J. Neurosci. 34, 6164–6176. doi:10.1523/jneurosci.3458-13.2014
Bae, E.-K., Jung, K.-H., Chu, K., Lee, S.-T., Kim, J.-H., Park, K.-I., et al. (2010). Neuropathologic and Clinical Features of Human Medial Temporal Lobe Epilepsy. J. Clin. Neurol. 6, 73–80. doi:10.3988/jcn.2010.6.2.73
El Bahh, B., Lespinet, V., Lurton, D., Coussemacq, M., Salle, G. L. G. L., and Rougier, A. (1999). Correlations between Granule Cell Dispersion, Mossy Fiber Sprouting, and Hippocampal Cell Loss in Temporal Lobe Epilepsy. Epilepsia 40, 1393–1401. doi:10.1111/j.1528-1157.1999.tb02011.x
Balashova, A., Pershin, V., Zaborskaya, O., Tkachenko, N., Mironov, A., Guryev, E., et al. (2019). Enzymatic Digestion of Hyaluronan-Based Brain Extracellular Matrix In Vivo Can Induce Seizures in Neonatal Mice. Front. Neurosci. 13, 1033. doi:10.3389/fnins.2019.01033
Barber, M., Arai, Y., Morishita, Y., Vigier, L., Causeret, F., Borello, U., et al. (2015). Migration Speed of Cajal-Retzius Cells Modulated by Vesicular Trafficking Controls the Size of Higher-Order Cortical Areas. Curr. Biol. 25, 2466–2478. doi:10.1016/j.cub.2015.08.028
Barber, M., and Pierani, A. (2016). Tangential Migration of Glutamatergic Neurons and Cortical Patterning during Development: Lessons from Cajal-Retzius Cells. Devel Neurobio 76, 847–881. doi:10.1002/dneu.22363
Becker, A. J. (2018). Review: Animal Models of Acquired Epilepsy: Insights into Mechanisms of Human Epileptogenesis. Neuropathol. Appl. Neurobiol. 44, 112–129. doi:10.1111/nan.12451
Bender, R. A., Dubé, C., and Baram, T. Z. (2004). “Febrile Seizures and Mechanisms of Epileptogenesis: Insights from an Animal Model,” in Recent Advances in Epilepsy Research. Editors D. K. Binder, and H. E. Scharfman (Boston, MA: Springer US), 213–225. doi:10.1007/978-1-4757-6376-8_15
Berg, A. T., Berkovic, S. F., Brodie, M. J., Buchhalter, J., Cross, J. H., Van Emde Boas, W., et al. (2010). Revised Terminology and Concepts for Organization of Seizures and Epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676–685. doi:10.1111/j.1528-1167.2010.02522.x
Blümcke, I., Beck, H., Suter, B., Hoffmann, D., Födisch, H. J., Wolf, H. K., et al. (1999). An Increase of Hippocampal Calretinin-Immunoreactive Neurons Correlates with Early Febrile Seizures in Temporal Lobe Epilepsy. Acta Neuropathol. 97, 31–39. doi:10.1007/s004010050952
Blümcke, I., Coras, R., Miyata, H., and Özkara, C. (2012). Defining Clinico-Neuropathological Subtypes of Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis. Brain Pathol. 22, 402–411. doi:10.1111/j.1750-3639.2012.00583.x
Blümcke, I., Kistner, I., Clusmann, H., Schramm, J., Becker, A. J., Elger, C. E., et al. (2009). Towards a Clinico-Pathological Classification of Granule Cell Dispersion in Human Mesial Temporal Lobe Epilepsies. Acta Neuropathol. 117, 535–544. doi:10.1007/s00401-009-0512-5
Blümcke, I. (2009). Neuropathology of Focal Epilepsies: a Critical Review. Epilepsy & Behav. 15, 34–39. doi:10.1016/j.yebeh.2009.02.033
Blümcke, I., Pauli, E., Clusmann, H., Schramm, J., Becker, A., Elger, C., et al. (2007). A New Clinico-Pathological Classification System for Mesial Temporal Sclerosis. Acta Neuropathol. 113, 235–244. doi:10.1007/s00401-006-0187-0
Blümcke, I., Schewe, J.-C., Normann, S., Brüstle, O., Schramm, J., Elger, C. E., et al. (2001). Increase of Nestin-Immunoreactive Neural Precursor Cells in the Dentate Gyrus of Pediatric Patients with Early-Onset Temporal Lobe Epilepsy. Hippocampus 11, 311–321. doi:10.1002/hipo.1045
Blumcke, I., Spreafico, R., Haaker, G., Coras, R., Kobow, K., Bien, C. G., et al. (2017). Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N. Engl. J. Med. 377, 1648–1656. doi:10.1056/NEJMoa1703784
Blümcke, I., Thom, M., Aronica, E., Armstrong, D. D., Bartolomei, F., Bernasconi, A., et al. (2013). International Consensus Classification of Hippocampal Sclerosis in Temporal Lobe Epilepsy: a Task Force Report from the ILAE Commission on Diagnostic Methods. Epilepsia 54, 1315–1329. doi:10.1111/epi.12220
Bock, H. H., and May, P. (2016). Canonical and Non-canonical Reelin Signaling. Front. Cell.. Neurosci. 10, 166. doi:10.3389/fncel.2016.00166
Borrell, V., Pujadas, L., Simó, S., Durà, D., Solé, M., Cooper, J. A., et al. (2007). Reelin and mDab1 Regulate the Development of Hippocampal Connections. Mol. Cell. Neurosci. 36, 158–173. doi:10.1016/j.mcn.2007.06.006
Boyle, M. P., Bernard, A., Thompson, C. L., Ng, L., Boe, A., Mortrud, M., et al. (2011). Cell-type-specific Consequences of Reelin Deficiency in the Mouse Neocortex, hippocampus, and Amygdala. J. Comp. Neurol. 519, 2061–2089. doi:10.1002/cne.22655
Brenneke, F., Bukalo, O., Dityatev, A., and Lie, A. A. (2004). Mice Deficient for the Extracellular Matrix Glycoprotein Tenascin-R Show Physiological and Structural Hallmarks of Increased Hippocampal Excitability, but No Increased Susceptibility to Seizures in the Pilocarpine Model of Epilepsy. Neuroscience 124, 841–855. doi:10.1016/j.neuroscience.2003.11.037
Broekaart, D. W. M., Bertran, A., Jia, S., Korotkov, A., Senkov, O., Bongaarts, A., et al. (2021). The Matrix Metalloproteinase Inhibitor IPR-179 Has Antiseizure and Antiepileptogenic Effects. J. Clin. Invest. 131, e138332. doi:10.1172/jci138332
Bronen, R. A., Fulbright, R. K., Kim, J. H., Spencer, S. S., Spencer, D. D., and Al-Rodhan, N. R. (1995). Regional Distribution of MR Findings in Hippocampal Sclerosis. AJNR Am. J. Neuroradiol. 16, 1193–1200. PMC8337833.
Bumanglag, A. V., and Sloviter, R. S. (2008). Minimal Latency to Hippocampal Epileptogenesis and Clinical Epilepsy after Perforant Pathway Stimulation-Induced Status Epilepticus in Awake Rats. J. Comp. Neurol. 510, 561–580. doi:10.1002/cne.21801
Cameron, M. C., Zhan, R.-Z., and Nadler, J. V. (2011). Morphologic Integration of Hilar Ectopic Granule Cells into Dentate Gyrus Circuitry in the Pilocarpine Model of Temporal Lobe Epilepsy. J. Comp. Neurol. 519, 2175–2192. doi:10.1002/cne.22623
Camfield, P., Camfield, C., Cordon, K., and Dooley, J. (1994). What Types of Epilepsy Are Preceded by Febrile Seizures? a Popilation-Based Study of Children. Dev. Med. Child. Neurol. 36, 887–892. doi:10.1111/j.1469-8749.1994.tb11779.x
Causeret, F., Moreau, M. X., Pierani, A., and Blanquie, O. (2021). The Multiple Facets of Cajal-Retzius Neurons. Development 148, dev199409. doi:10.1242/dev.199409
Cendes, F., Andermann, F., Dubeau, F., Gloor, P., Evans, A., Jones-Gotman, M., et al. (1993). Early Childhood Prolonged Febrile Convulsions, Atrophy and Sclerosis of Mesial Structures, and Temporal Lobe Epilepsy. Neurology 43, 1083. doi:10.1212/wnl.43.6.1083
Cendes, F. (2004). Febrile Seizures and Mesial Temporal Sclerosis. Curr. Opin. Neurology 17, 161–164. doi:10.1097/00019052-200404000-00013
Česká, K., Aulická, Š., Horák, O., Danhofer, P., Říha, P., Mareček, R., et al. (2019). Autosomal Dominant Temporal Lobe Epilepsy Associated with Heterozygous Reelin Mutation: 3 T Brain MRI Study with Advanced Neuroimaging Methods. Epilepsy & Behav. Case Rep. 11, 39–42. doi:10.1016/j.ebcr.2018.10.003
Chahine, L., Abou-Khalil, B., Siren, A., Andermann, F., Hedera, P., Ge, Q., et al. (2013). A New Locus for Familial Temporal Lobe Epilepsy on Chromosome 3q. Epilepsy Res. 106, 338–344. doi:10.1016/j.eplepsyres.2013.07.007
Chai, X., Fan, L., Shao, H., Lu, X., Zhang, W., Li, J., et al. (2015). Reelin Induces Branching of Neurons and Radial Glial Cells during Corticogenesis. Cereb. Cortex 25, 3640–3653. doi:10.1093/cercor/bhu216
Chai, X., Münzner, G., Zhao, S., Tinnes, S., Kowalski, J., Häussler, U., et al. (2014). Epilepsy-induced Motility of Differentiated Neurons. Cereb. Cortex 24, 2130–2140. doi:10.1093/cercor/bht067
Chameau, P., Inta, D., Vitalis, T., Monyer, H., Wadman, W. J., and Van Hooft, J. A. (2009). The N-Terminal Region of Reelin Regulates Postnatal Dendritic Maturation of Cortical Pyramidal Neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 7227–7232. doi:10.1073/pnas.0810764106
Chang, B. S., Duzcan, F., Kim, S., Cinbis, M., Aggarwal, A., Apse, K. A., et al. (2007). The Role ofRELN in Lissencephaly and Neuropsychiatric Disease. Am. J. Med. Genet. 144b, 58–63. doi:10.1002/ajmg.b.30392
Chen, Q. L., Xia, L., Zhong, S. P., Wang, Q., Ding, J., and Wang, X. (2020). Bioinformatic Analysis Identifies Key Transcriptome Signatures in Temporal Lobe Epilepsy. CNS Neurosci. Ther. 26, 1266–1277. doi:10.1111/cns.13470
Cortez, M. A., Mckerlie, C., and Snead, O. C. (2001). A Model of Atypical Absence Seizures: EEG, Pharmacology, and Developmental Characterization. Neurology 56, 341–349. doi:10.1212/wnl.56.3.341
Cragg, B. (1979). Brain Extracellular Space Fixed for Electron Microscopy. Neurosci. Lett. 15, 301–306. doi:10.1016/0304-3940(79)96130-5
Crespel, A., Rigau, V., Coubes, P., Rousset, M. C., De Bock, F., Okano, H., et al. (2005). Increased Number of Neural Progenitors in Human Temporal Lobe Epilepsy. Neurobiol. Dis. 19, 436–450. doi:10.1016/j.nbd.2005.01.020
Crompton, D. E., Scheffer, I. E., Taylor, I., Cook, M. J., Mckelvie, P. A., Vears, D. F., et al. (2010). Familial Mesial Temporal Lobe Epilepsy: a Benign Epilepsy Syndrome Showing Complex Inheritance. Brain 133, 3221–3231. doi:10.1093/brain/awq251
Crunelli, V., and Leresche, N. (1991). A Role for GABAB Receptors in Excitation and Inhibition of Thalamocortical Cells. Trends Neurosci. 14, 16–21. doi:10.1016/0166-2236(91)90178-w
D’Arcangelo, G., G. Miao, G., Chen, S.-C., Scares, H. D., Morgan, J. I., and Curran, T. (1995). A Protein Related to Extracellular Matrix Proteins Deleted in the Mouse Mutant Reeler. Nature 374, 719–723. doi:10.1038/374719a0
D’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T. (1999). Reelin Is a Ligand for Lipoprotein Receptors. Neuron 24, 471–479. doi:10.1016/s0896-6273(00)80860-0
D’Arcangelo, G., Nakajima, K., Miyata, T., Ogawa, M., Mikoshiba, K., and Curran, T. (1997). Reelin Is a Secreted Glycoprotein Recognized by the CR-50 Monoclonal Antibody. J. Neurosci. 17, 23–31. doi:10.1523/jneurosci.17-01-00023.1997
Dazzo, E., Fanciulli, M., Serioli, E., Minervini, G., Pulitano, P., Binelli, S., et al. (2015). Heterozygous Reelin Mutations Cause Autosomal-Dominant Lateral Temporal Epilepsy. Am. J. Hum. Genet. 96, 992–1000. doi:10.1016/j.ajhg.2015.04.020
Dazzo, E., and Nobile, C. (2021). Epilepsy-causing Reelin Mutations Result in Impaired Secretion and Intracellular Degradation of Mutant Proteins. Hum. Mol. Genet. 31, 665–673. doi:10.1093/hmg/ddab271
de Bergeyck, V., Nakajima, K., de Rouvroit, C. L., Naerhuyzen, B., Goffinet, A. M., Miyata, T., et al. (1997). A Truncated Reelin Protein Is Produced but Not Secreted in the 'Orleans' Reeler Mutation (Relnrl-Orl). Mol. Brain Res. 50, 85–90. doi:10.1016/s0169-328x(97)00166-6
de Frutos, C. A., Bouvier, G., Arai, Y., Thion, M. S., Lokmane, L., Keita, M., et al. (2016). Reallocation of Olfactory Cajal-Retzius Cells Shapes Neocortex Architecture. Neuron 92, 435–448. doi:10.1016/j.neuron.2016.09.020
Dębski, K. J., Pitkanen, A., Puhakka, N., Bot, A. M., Khurana, I., Harikrishnan, K., et al. (2016). Etiology Matters - Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy. Sci. Rep. 6, 25668. doi:10.1038/srep25668
Dekimoto, H., Terashima, T., and Katsuyama, Y. (2010). Dispersion of the Neurons Expressing Layer Specific Markers in the Reeler Brain. Dev. Growth Differ. 52, 181–193. doi:10.1111/j.1440-169X.2009.01153.x
Del Río, J. A., Heimrich, B., Borrell, V., Förster, E., Drakew, A., Alcántara, S., et al. (1997). A Role for Cajal-Retzius Cells and Reelin in the Development of Hippocampal Connections. Nature 385, 70–74. doi:10.1038/385070a0
Depondt, C., Van Paesschen, W., Matthijs, G., Legius, E., Martens, K., Demaerel, P., et al. (2002). Familial Temporal Lobe Epilepsy with Febrile Seizures. Neurology 58, 1429–1433. doi:10.1212/wnl.58.9.1429
Desilva, U., D’arcangelo, G., Braden, V. V., Chen, J., Miao, G. G., Curran, T., et al. (1997). The Human Reelin Gene: Isolation, Sequencing, and Mapping on Chromosome 7. Genome Res. 7, 157–164. doi:10.1101/gr.7.2.157
Dityatev, A., and Fellin, T. (2008). Extracellular Matrix in Plasticity and Epileptogenesis. Neuron Glia Biol. 4, 235–247. doi:10.1017/s1740925x09000118
Dityatev, A. (2010). Remodeling of Extracellular Matrix and Epileptogenesis. Epilepsia 51 (Suppl. 3), 61–65. doi:10.1111/j.1528-1167.2010.02612.x
Dityatev, A., Schachner, M., and Sonderegger, P. (2010). The Dual Role of the Extracellular Matrix in Synaptic Plasticity and Homeostasis. Nat. Rev. Neurosci. 11, 735–746. doi:10.1038/nrn2898
Dlugosz, P., Tresky, R., and Nimpf, J. (2019). Differential Action of Reelin on Oligomerization of ApoER2 and VLDL Receptor in HEK293 Cells Assessed by Time-Resolved Anisotropy and Fluorescence Lifetime Imaging Microscopy. Front. Mol. Neurosci. 12, 53. doi:10.3389/fnmol.2019.00053
Drakew, A., Frotscher, M., Deller, T., Ogawa, M., and Heimrich, B. (1998). Developmental Distribution of a Reeler Gene-Related Antigen in the Rat Hippocampal Formation Visualized by CR-50 Immunocytochemistry. Neuroscience 82, 1079–1086. doi:10.1016/s0306-4522(97)00326-6
Drakew, A., Deller, T., Heimrich, B., Gebhardt, C., Del Turco, D., Tielsch, A., et al. (2002). Dentate Granule Cells in Reeler Mutants and VLDLR and ApoER2 Knockout Mice. Exp. Neurol. 176, 12–24. doi:10.1006/exnr.2002.7918
Dua, T., De Boer, H. M., Prilipko, L. L., and Saxena, S. (2006). Epilepsy Care in the World: Results of an ILAE/IBE/WHO Global Campaign against Epilepsy Survey. Epilepsia 47, 1225–1231. doi:10.1111/j.1528-1167.2006.00595.x
Duarte, J. T. C., Jardim, A. P., Comper, S. M., De Marchi, L. R., Gaça, L. B., Garcia, M. T. F. C., et al. (2018). The Impact of Epilepsy Duration in a Series of Patients with Mesial Temporal Lobe Epilepsy Due to Unilateral Hippocampal Sclerosis. Epilepsy Res. 147, 51–57. doi:10.1016/j.eplepsyres.2018.08.009
Duveau, V., Madhusudan, A., Caleo, M., Knuesel, I., and Fritschy, J.-M. (2010). Impaired Reelin Processing and Secretion by Cajal-Retzius Cells Contributes to Granule Cell Dispersion in a Mouse Model of Temporal Lobe Epilepsy. Hippocampus 21, a-n. doi:10.1002/hipo.20793
Engel, J. (2001). A Proposed Diagnostic Scheme for People with Epileptic Seizures and with Epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 42, 796–803. doi:10.1046/j.1528-1157.2001.10401.x
Eriksson, P. S., Perfilieva, E., Björk-Eriksson, T., Alborn, A.-M., Nordborg, C., Peterson, D. A., et al. (1998). Neurogenesis in the Adult Human hippocampus. Nat. Med. 4, 1313–1317. doi:10.1038/3305
Falco-Walter, J. (2020). Epilepsy-Definition, Classification, Pathophysiology, and Epidemiology. Semin. Neurol. 40, 617–623. doi:10.1055/s-0040-1718719
Fiest, K. M., Sauro, K. M., Wiebe, S., Patten, S. B., Kwon, C.-S., Dykeman, J., et al. (2017). Prevalence and Incidence of Epilepsy. Neurology 88, 296–303. doi:10.1212/wnl.0000000000003509
Fiford, C. M., Manning, E. N., Bartlett, J. W., Cash, D. M., Malone, I. B., Ridgway, G. R., et al. (2017). White Matter Hyperintensities Are Associated with Disproportionate Progressive Hippocampal Atrophy. Hippocampus 27, 249–262. doi:10.1002/hipo.22690
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE Official Report: a Practical Clinical Definition of Epilepsy. Epilepsia 55, 475–482. doi:10.1111/epi.12550
Förster, E. (2014). Reelin, Neuronal Polarity and Process Orientation of Cortical Neurons. Neuroscience 269, 102–111. doi:10.1016/j.neuroscience.2014.03.004
Förster, E., Zhao, S., and Frotscher, M. (2006). Laminating the hippocampus. Nat. Rev. Neurosci. 7, 259–268. doi:10.1038/nrn1882
Fournier, N. M., Andersen, D. R., Botterill, J. J., Sterner, E. Y., Lussier, A. L., Caruncho, H. J., et al. (2009). The Effect of Amygdala Kindling on Hippocampal Neurogenesis Coincides with Decreased Reelin and DISC1 Expression in the Adult Dentate Gyrus. Hippocampus 20, NA. doi:10.1002/hipo.20653
Freiman, T. M., Eismann-Schweimler, J., and Frotscher, M. (2011). Granule Cell Dispersion in Temporal Lobe Epilepsy Is Associated with Changes in Dendritic Orientation and Spine Distribution. Exp. Neurol. 229, 332–338. doi:10.1016/j.expneurol.2011.02.017
Freiman, T. M., Häussler, U., Zentner, J., Doostkam, S., Beck, J., Scheiwe, C., et al. (2021). Mossy Fiber Sprouting into the Hippocampal Region CA2 in Patients with Temporal Lobe Epilepsy. Hippocampus 31, 580–592. doi:10.1002/hipo.23323
French, J. A., Williamson, P. D., Thadani, V. M., Darcey, T. M., Mattson, R. H., Spencer, S. S., et al. (1993). Characteristics of Medial Temporal Lobe Epilepsy: I. Results of History and Physical Examination. Ann. Neurol. 34, 774–780. doi:10.1002/ana.410340604
Frotscher, M., Haas, C. A., and Förster, E. (2003). Reelin Controls Granule Cell Migration in the Dentate Gyrus by Acting on the Radial Glial Scaffold. Cereb. Cortex 13, 634–640. doi:10.1093/cercor/13.6.634
Gong, C., Wang, T.-W., Huang, H. S., and Parent, J. M. (2007). Reelin Regulates Neuronal Progenitor Migration in Intact and Epileptic hippocampus. J. Neurosci. 27, 1803–1811. doi:10.1523/jneurosci.3111-06.2007
Griveau, A., Borello, U., Causeret, F., Tissir, F., Boggetto, N., Karaz, S., et al. (2010). A Novel Role for Dbx1-Derived Cajal-Retzius Cells in Early Regionalization of the Cerebral Cortical Neuroepithelium. PLoS Biol. 8, e1000440. doi:10.1371/journal.pbio.1000440
Guerreiro, C. M. (2016). Epilepsy: Is There Hope? Indian J. Med. Res. 144, 657–660. doi:10.4103/ijmr.IJMR_1051_16
Haas, C. A., Dudeck, O., Kirsch, M., Huszka, C., Kann, G., Pollak, S., et al. (2002). Role for Reelin in the Development of Granule Cell Dispersion in Temporal Lobe Epilepsy. J. Neurosci. 22, 5797–5802. doi:10.1523/jneurosci.22-14-05797.2002
Haas, C. A., and Frotscher, M. (2010). Reelin Deficiency Causes Granule Cell Dispersion in Epilepsy. Exp. Brain Res. 200, 141–149. doi:10.1007/s00221-009-1948-5
Hamad, M. I. K., Jbara, A., Rabaya, O., Petrova, P., Daoud, S., Melliti, N., et al. (2021a). Reelin Signaling Modulates GABA B Receptor Function in the Neocortex. J. Neurochem. 156, 589–603. doi:10.1111/jnc.14990
Hamad, M. I. K., Petrova, P., Daoud, S., Rabaya, O., Jbara, A., Melliti, N., et al. (2021b). Reelin Restricts Dendritic Growth of Interneurons in the Neocortex. Development 148, dev199718. doi:10.1242/dev.199718
Harding, B., and Thom, M. (2001). Bilateral Hippocampal Granule Cell Dispersion: Autopsy Study of 3 Infants. Neuropathol. Appl. Neurobiol. 27, 245–251. doi:10.1046/j.0305-1846.2001.00325.x
Hattori, M., and Kohno, T. (2021). Regulation of Reelin Functions by Specific Proteolytic Processing in the Brain. J. Biochem. 169, 511–516. doi:10.1093/jb/mvab015
Hayashi, K., Kubo, K.-i., Kitazawa, A., and Nakajima, K. (2015). Cellular Dynamics of Neuronal Migration in the hippocampus. Front. Neurosci. 9, 135. doi:10.3389/fnins.2015.00135
Heck, N., Garwood, J., Loeffler, J.-P., Larmet, Y., and Faissner, A. (2004). Differential Upregulation of Extracellular Matrix Molecules Associated with the Appearance of Granule Cell Dispersion and Mossy Fiber Sprouting during Epileptogenesis in a Murine Model of Temporal Lobe Epilepsy. Neuroscience 129, 309–324. doi:10.1016/j.neuroscience.2004.06.078
Hedera, P., Blair, M. A., Andermann, E., Andermann, F., D'agostino, D., Taylor, K. A., et al. (2007). Familial Mesial Temporal Lobe Epilepsy Maps to Chromosome 4q13.2-q21.3. Neurology 68, 2107–2112. doi:10.1212/01.wnl.0000261246.75977.89
Hefti, M. M., Cryan, J. B., Haas, E. A., Chadwick, A. E., Crandall, L. A., Trachtenberg, F. L., et al. (2016). Hippocampal Malformation Associated with Sudden Death in Early Childhood: a Neuropathologic Study. Forensic Sci. Med. Pathol. 12, 14–25. doi:10.1007/s12024-015-9731-3
Heinrich, C., Nitta, N., Flubacher, A., Müller, M., Fahrner, A., Kirsch, M., et al. (2006). Reelin Deficiency and Displacement of Mature Neurons, but Not Neurogenesis, Underlie the Formation of Granule Cell Dispersion in the Epileptic hippocampus. J. Neurosci. 26, 4701–4713. doi:10.1523/jneurosci.5516-05.2006
Hirota, Y., Kubo, K.-I., Fujino, T., Yamamoto, T. T., and Nakajima, K. (2016). ApoER2 Controls Not Only Neuronal Migration in the Intermediate Zone but Also Termination of Migration in the Developing Cerebral Cortex. Cereb. Cortex 28, 223–235. doi:10.1093/cercor/bhw369
Hirota, Y., and Nakajima, K. (2017). Control of Neuronal Migration and Aggregation by Reelin Signaling in the Developing Cerebral Cortex. Front. Cell. Dev. Biol. 5, 40. doi:10.3389/fcell.2017.00040
Hirota, Y., and Nakajima, K. (2020). VLDLR Is Not Essential for Reelin-Induced Neuronal Aggregation but Suppresses Neuronal Invasion into the Marginal Zone. Development 147, dev189936. doi:10.1242/dev.189936
Hisanaga, A., Morishita, S., Suzuki, K., Sasaki, K., Koie, M., Kohno, T., et al. (2012). A Disintegrin and Metalloproteinase with Thrombospondin Motifs 4 (ADAMTS-4) Cleaves Reelin in an Isoform-dependent Manner. FEBS Lett. 586, 3349–3353. doi:10.1016/j.febslet.2012.07.017
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O. B., et al. (2000). Autosomal Recessive Lissencephaly with Cerebellar Hypoplasia Is Associated with Human RELN Mutations. Nat. Genet. 26, 93–96. doi:10.1038/79246
Houser, C. R. (1990). Granule Cell Dispersion in the Dentate Gyrus of Humans with Temporal Lobe Epilepsy. Brain Res. 535, 195–204. doi:10.1016/0006-8993(90)91601-c
Howell, B. W., Hawkes, R., Soriano, P., and Cooper, J. A. (1997). Neuronal Position in the Developing Brain Is Regulated by Mouse Disabled-1. Nature 389, 733–737. doi:10.1038/39607
Howell, B. W., Herrick, T. M., and Cooper, J. A. (1999). Reelin-induced Tryosine Phosphorylation of Disabled 1 during Neuronal Positioning. Genes. & Dev. 13, 643–648. doi:10.1101/gad.13.6.643
Ichihara, H., Jingami, H., and Toh, H. (2001). Three Novel Repetitive Units of Reelin. Mol. Brain Res. 97, 190–193. doi:10.1016/s0169-328x(01)00307-2
Ingmar, I., Beck, H., Nitsch, R., Eickhoff, C., Scheffler, B., Celio, M. R., et al. (1996). Preservation of Calretinin-Immunoreactive Neurons in the Hippocampus of Epilepsy Patients with Ammonʼs Horn Sclerosis. J. Neuropathology Exp. Neurology 55, 329–341. doi:10.1097/00005072-199603000-00008
Ishii, K., Kubo, K.-i., and Nakajima, K. (2016). Reelin and Neuropsychiatric Disorders. Front. Cell. Neurosci. 10, 229. doi:10.3389/fncel.2016.00229
Janszky, J., Schulz, R., and Ebner, A. (2003). Clinical Features and Surgical Outcome of Medial Temporal Lobe Epilepsy with a History of Complex Febrile Convulsions. Epilepsy Res. 55, 1–8. doi:10.1016/s0920-1211(03)00087-1
Jardim, A. P., Duarte, J. T. C., Lancellotti, C. L. P., Carrete, H., Centeno, R. S., Scorza, C. A., et al. (2021). Granule Cell Dispersion Is Associated with Hippocampal Neuronal Cell Loss, Initial Precipitating Injury, and Other Clinical Features in Mesial Temporal Lobe Epilepsy and Hippocampal Sclerosis. Seizure 90, 60–66. doi:10.1016/j.seizure.2021.05.024
Jeong, K. H., Kim, S. H., Choi, Y. H., Cho, I., and Kim, W.-J. (2018). Increased Expression of WNK3 in Dispersed Granule Cells in Hippocampal Sclerosis of Mesial Temporal Lobe Epilepsy Patients. Epilepsy Res. 147, 58–61. doi:10.1016/j.eplepsyres.2018.09.006
Jiang, W., Duong, T. M., and Lanerolle, N. C. d. (1999). The Neuropathology of Hyperthermic Seizures in the Rat. Epilepsia 40, 5–19. doi:10.1111/j.1528-1157.1999.tb01982.x
Jimenez-Mateos, E. M., Engel, T., Merino-Serrais, P., Mckiernan, R. C., Tanaka, K., Mouri, G., et al. (2012). Silencing microRNA-134 Produces Neuroprotective and Prolonged Seizure-Suppressive Effects. Nat. Med. 18, 1087–1094. doi:10.1038/nm.2834
Jossin, Y., and Goffinet, A. M. (2007). Reelin Signals through Phosphatidylinositol 3-kinase and Akt to Control Cortical Development and through mTor to Regulate Dendritic Growth. Mol. Cell. Biol. 27, 7113–7124. doi:10.1128/mcb.00928-07
Jossin, Y., Gui, L., and Goffinet, A. M. (2007). Processing of Reelin by Embryonic Neurons Is Important for Function in Tissue but Not in Dissociated Cultured Neurons. J. Neurosci. 27, 4243–4252. doi:10.1523/jneurosci.0023-07.2007
Jossin, Y., Ignatova, N., Hiesberger, T., Herz, J., Lambert De Rouvroit, C., and Goffinet, A. M. (2004). The Central Fragment of Reelin, Generated by Proteolytic Processing In Vivo, Is Critical to its Function during Cortical Plate Development. J. Neurosci. 24, 514–521. doi:10.1523/jneurosci.3408-03.2004
Jossin, Y. (2020). Reelin Functions, Mechanisms of Action and Signaling Pathways during Brain Development and Maturation. Biomolecules 10, 964. doi:10.3390/biom10060964
Kälviäinen, R., and Salmenperä, T. (2002). Do recurrent Seizures Cause Neuronal Damage? A Series of Studies with MRI Volumetry in Adults with Partial Epilepsy. Prog. Brain Res. 135, 279–295. doi:10.1016/s0079-6123(02)35026-x
Kasperavičiūtė, D., Catarino, C. B., Matarin, M., Leu, C., Novy, J., Tostevin, A., et al. (2013). Epilepsy, Hippocampal Sclerosis and Febrile Seizures Linked by Common Genetic Variation Around SCN1A. Brain 136, 3140–3150. doi:10.1093/brain/awt233
Kempermann, G., Kuhn, H. G., and Gage, F. H. (1998). Experience-induced Neurogenesis in the Senescent Dentate Gyrus. J. Neurosci. 18, 3206–3212. doi:10.1523/jneurosci.18-09-03206.1998
Khalaf-Nazzal, R., and Francis, F. (2013). Hippocampal Development - Old and New Findings. Neuroscience 248, 225–242. doi:10.1016/j.neuroscience.2013.05.061
Kim, Y. K., Lee, D. S., Lee, S. K., Kim, S. K., Chung, C. K., Chang, K. H., et al. (2003). Differential Features of Metabolic Abnormalities between Medial and Lateral Temporal Lobe Epilepsy: Quantitative Analysis of (18)F-FDG PET Using SPM. J. Nucl. Med. 44, 1006–1012.
Kinney, H. C., Cryan, J. B., Haynes, R. L., Paterson, D. S., Haas, E. A., Mena, O. J., et al. (2015). Dentate Gyrus Abnormalities in Sudden Unexplained Death in Infants: Morphological Marker of Underlying Brain Vulnerability. Acta Neuropathol. 129, 65–80. doi:10.1007/s00401-014-1357-0
Kobayashi, E., D’Agostino, M. D., Lopes-Cendes, I., Berkovic, S. F., Li, M. L., Andermann, E., et al. (2003). Hippocampal Atrophy and T2-Weighted Signal Changes in Familial Mesial Temporal Lobe Epilepsy. Neurology 60, 405–409. doi:10.1212/wnl.60.3.405
Kobayashi, E., Li, L. M., Lopes-Cendes, I., and Cendes, F. (2002). Magnetic Resonance Imaging Evidence of Hippocampal Sclerosis in Asymptomatic, First-Degree Relatives of Patients with Familial Mesial Temporal Lobe Epilepsy. Arch. Neurol. 59, 1891–1894. doi:10.1001/archneur.59.12.1891
Kobow, K., Jeske, I., Hildebrandt, M., Hauke, J., Hahnen, E., Buslei, R., et al. (2009). Increased Reelin Promoter Methylation Is Associated with Granule Cell Dispersion in Human Temporal Lobe Epilepsy. J. Neuropathol. Exp. Neurol. 68, 356–364. doi:10.1097/NEN.0b013e31819ba737
Kohno, T., Honda, T., Kubo, K.-i., Nakano, Y., Tsuchiya, A., Murakami, T., et al. (2015). Importance of Reelin C-Terminal Region in the Development and Maintenance of the Postnatal Cerebral Cortex and its Regulation by Specific Proteolysis. J. Neurosci. 35, 4776–4787. doi:10.1523/jneurosci.4119-14.2015
Koie, M., Okumura, K., Hisanaga, A., Kamei, T., Sasaki, K., Deng, M., et al. (2014). Cleavage within Reelin Repeat 3 Regulates the Duration and Range of the Signaling Activity of Reelin Protein. J. Biol. Chem. 289, 12922–12930. doi:10.1074/jbc.M113.536326
Konopacki, F. A., Rylski, M., Wilczek, E., Amborska, R., Detka, D., Kaczmarek, L., et al. (2007). Synaptic Localization of Seizure-Induced Matrix Metalloproteinase-9 mRNA. Neuroscience 150, 31–39. doi:10.1016/j.neuroscience.2007.08.026
Korn, M. J., Mandle, Q. J., and Parent, J. M. (2016). Conditional Disabled-1 Deletion in Mice Alters Hippocampal Neurogenesis and Reduces Seizure Threshold. Front. Neurosci. 10, 63. doi:10.3389/fnins.2016.00063
Kotrotsou, A., Schneider, J. A., Bennett, D. A., Leurgans, S. E., Dawe, R. J., Boyle, P. A., et al. (2015). Neuropathologic Correlates of Regional Brain Volumes in a Community Cohort of Older Adults. Neurobiol. Aging 36, 2798–2805. doi:10.1016/j.neurobiolaging.2015.06.025
Koyama, R., and Matsuki, N. (2010). Novel Etiological and Therapeutic Strategies for Neurodiseases: Mechanisms and Consequences of Febrile Seizures: Lessons from Animal Models. J. Pharmacol. Sci. 113, 14–22. doi:10.1254/jphs.09r19fm
Koyama, R., Tao, K., Sasaki, T., Ichikawa, J., Miyamoto, D., Muramatsu, R., et al. (2012). GABAergic Excitation after Febrile Seizures Induces Ectopic Granule Cells and Adult Epilepsy. Nat. Med. 18, 1271–1278. doi:10.1038/nm.2850
Krstic, D., Rodriguez, M., and Knuesel, I. (2012). Regulated Proteolytic Processing of Reelin through Interplay of Tissue Plasminogen Activator (tPA), ADAMTS-4, ADAMTS-5, and Their Modulators. PLoS One 7, e47793. doi:10.1371/journal.pone.0047793
Kubo, K.-i., Mikoshiba, K., and Nakajima, K. (2002). Secreted Reelin Molecules Form Homodimers. Neurosci. Res. 43, 381–388. doi:10.1016/s0168-0102(02)00068-8
Kuks, J. B., Cook, M. J., Fish, D. R., Stevens, J. M., and Shorvon, S. D. (1993). Hippocampal Sclerosis in Epilepsy and Childhood Febrile Seizures. Lancet 342, 1391–1394. doi:10.1016/0140-6736(93)92754-h
Kupferman, J. V., Basu, J., Russo, M. J., Guevarra, J., Cheung, S. K., and Siegelbaum, S. A. (2014). Reelin Signaling Specifies the Molecular Identity of the Pyramidal Neuron Distal Dendritic Compartment. Cell. 158, 1335–1347. doi:10.1016/j.cell.2014.07.035
Kutsy, R. L. (1999). Focal Extratemporal Epilepsy: Clinical Features, EEG Patterns, and Surgical Approach. J. Neurological Sci. 166, 1–15. doi:10.1016/s0022-510x(99)00107-0
Lambert de Rouvroit, C., De Bergeyck, V., Cortvrindt, C., Bar, I., Eeckhout, Y., and Goffinet, A. M. (1999). Reelin, the Extracellular Matrix Protein Deficient in Reeler Mutant Mice, Is Processed by a Metalloproteinase. Exp. Neurol. 156, 214–217. doi:10.1006/exnr.1998.7007
Lane-Donovan, C., Philips, G. T., Wasser, C. R., Durakoglugil, M. S., Masiulis, I., Upadhaya, A., et al. (2015). Reelin Protects against Amyloid β Toxicity In Vivo. Sci. Signal. 8, ra67. doi:10.1126/scisignal.aaa6674
Lau, L. W., Cua, R., Keough, M. B., Haylock-Jacobs, S., and Yong, V. W. (2013). Pathophysiology of the Brain Extracellular Matrix: a New Target for Remyelination. Nat. Rev. Neurosci. 14, 722–729. doi:10.1038/nrn3550
Lee, K., Diaz, M., and Melchior, J. C. (1981). Temporal Lobe Epilepsy - Not a Consequence of Childhood Febrile Convulsions in Denmark. Acta Neurol. Scand. 63, 231–236. doi:10.1111/j.1600-0404.1981.tb00776.x
Leemhuis, J., and Bock, H. H. (2011). Reelin Modulates Cytoskeletal Organization by Regulating Rho GTPases. Commun. Integr. Biol. 4, 254–257. doi:10.4161/cib.4.3.14890
Leite, J. P., Garcia-Cairasco, N., and Cavalheiro, E. A. (2002). New Insights from the Use of Pilocarpine and Kainate Models. Epilepsy Res. 50, 93–103. doi:10.1016/s0920-1211(02)00072-4
Li, S., Yu, S., Zhang, C., Shu, H., Liu, S., An, N., et al. (2012). Increased Expression of Matrix Metalloproteinase 9 in Cortical Lesions from Patients with Focal Cortical Dysplasia Type IIb and Tuberous Sclerosis Complex. Brain Res. 1453, 46–55. doi:10.1016/j.brainres.2012.03.009
Lillis, K. P. (2021). The (Extracellular) Matrix Reloaded: Imaging Matrix Metalloproteinase Activity. Epilepsy Curr. 21, 120–121. doi:10.1177/1535759720988547
Lugli, G., Krueger, J. M., Davis, J. M., Persico, A. M., Keller, F., and Smalheiser, N. R. (2003). Methodological Factors Influencing Measurement and Processing of Plasma Reelin in Humans. BMC Biochem. 4, 9. doi:10.1186/1471-2091-4-9
Lukasiuk, K., Wilczynski, G. M., and Kaczmarek, L. (2011). Extracellular Proteases in Epilepsy. Epilepsy Res. 96, 191–206. doi:10.1016/j.eplepsyres.2011.08.002
Lurton, D., Sundstrom, L., Brana, C., Bloch, B., and Rougier, A. (1997). Possible Mechanisms Inducing Granule Cell Dispersion in Humans with Temporal Lobe Epilepsy. Epilepsy Res. 26, 351–361. doi:10.1016/s0920-1211(96)01002-9
Maher, J., and Mclachlan, R. S. (1995). Febrile Convulsions. Brain 118 (Pt 6), 1521–1528. doi:10.1093/brain/118.6.1521
Mak, E., Su, L., Williams, G. B., Watson, R., Firbank, M., Blamire, A., et al. (2016). Differential Atrophy of Hippocampal Subfields: A Comparative Study of Dementia with Lewy Bodies and Alzheimer Disease. Am. J. Geriatric Psychiatry 24, 136–143. doi:10.1016/j.jagp.2015.06.006
Manoharan, M., Muhammad, S. A., and Sowdhamini, R. (2015). Sequence Analysis and Evolutionary Studies of Reelin Proteins. Bioinform Biol. Insights 9, BBI.S26530–193. doi:10.4137/bbi.S26530
Marín-Padilla, M. (1998). Cajal-Retzius Cells and the Development of the Neocortex. Trends Neurosci. 21, 64–71. doi:10.1016/s0166-2236(97)01164-8
Marucci, G., Farnedi, A., and Giulioni, M. (2012). Reelin: a Possible Link between Hippocampal Sclerosis and Cortical Dyslamination in the Setting of FCD Type IIIa. Neurol. Sci. 33, 1479–1481. doi:10.1007/s10072-011-0895-7
Marucci, G., Rubboli, G., and Giulioni, M. (2010). Role of Dentate Gyrus Alterations in Mesial Temporal Sclerosis. Np 29, 32–35. doi:10.5414/npp29032
Mathern, G. W., Kuhlman, P. A., Mendoza, D., and Pretorius, J. K. (1997). Human Fascia Dentata Anatomy and Hippocampal Neuron Densities Differ Depending on the Epileptic Syndrome and Age at First Seizure. J. Neuropathology Exp. Neurology 56, 199–212. doi:10.1097/00005072-199702000-00011
Mathern, G. W., Leiphart, J. L., De Vera, A., Adelson, P. D., Seki, T., Neder, L., et al. (2002). Seizures Decrease Postnatal Neurogenesis and Granule Cell Development in the Human Fascia tDentata. Epilepsia 43 (Suppl. 5), 68–73. doi:10.1046/j.1528-1157.43.s.5.28.x
Matsuki, T., Pramatarova, A., and Howell, B. W. (2008). Reduction of Crk and CrkL Expression Blocks Reelin-Induced Dendritogenesis. J. Cell. Sci. 121, 1869–1875. doi:10.1242/jcs.027334
Mcintosh, W. C., and Joe, M. D. (2021). “Temporal Seizure,” in StatPearls (Treasure Island (FL): StatPearls Publishing). Copyright © 2021, StatPearls Publishing LLC.
Md, I. B., MRCPath, M. T., and Wiestler, O. D. (2002). Ammon's Horn Sclerosis: a Maldevelopmental Disorder Associated with Temporal Lobe Epilepsy. Brain Pathol. 12, 199–211. doi:10.1111/j.1750-3639.2002.tb00436.x
Mello, L. E., Cavalheiro, E. A., Tan, A. M., Pretorius, J. K., Babb, T. L., and Finch, D. M. (1992). Granule Cell Dispersion in Relation to Mossy Fiber Sprouting, Hippocampal Cell Loss, Silent Period and Seizure Frequency in the Pilocarpine Model of Epilepsy. Epilepsy Res. Suppl. 9, 51–60. discussion 59-60.
Mello, L. E. A. M., Cavalheiro, E. A., Tan, A. M., Kupfer, W. R., Pretorius, J. K., Babb, T. L., et al. (1993). Circuit Mechanisms of Seizures in the Pilocarpine Model of Chronic Epilepsy: Cell Loss and Mossy Fiber Sprouting. Epilepsia 34, 985–995. doi:10.1111/j.1528-1157.1993.tb02123.x
Michelucci, R., Dazzo, E., Volpi, L., Pasini, E., Riguzzi, P., Minardi, R., et al. (2020). Autosomal Dominant Lateral Temporal Lobe Epilepsy Associated with a Novel Reelin Mutation. Epileptic Disord. 22, 443–448. doi:10.1684/epd.2020.1176
Michelucci, R., Pulitano, P., Di Bonaventura, C., Binelli, S., Luisi, C., Pasini, E., et al. (2017). The Clinical Phenotype of Autosomal Dominant Lateral Temporal Lobe Epilepsy Related to Reelin Mutations. Epilepsy & Behav. 68, 103–107. doi:10.1016/j.yebeh.2016.12.003
Mizoguchi, H., and Yamada, K. (2013). Roles of Matrix Metalloproteinases and Their Targets in Epileptogenesis and Seizures. Clin. Psychopharmacol. Neurosci. 11, 45–52. doi:10.9758/cpn.2013.11.2.45
Morawski, M., Dityatev, A., Hartlage-Rübsamen, M., Blosa, M., Holzer, M., Flach, K., et al. (2014). Tenascin-R Promotes Assembly of the Extracellular Matrix of Perineuronal Nets via Clustering of Aggrecan. Phil. Trans. R. Soc. B 369, 20140046. doi:10.1098/rstb.2014.0046
Morgan, R. J., and Soltesz, I. (2008). Nonrandom Connectivity of the Epileptic Dentate Gyrus Predicts a Major Role for Neuronal Hubs in Seizures. Proc. Natl. Acad. Sci. U.S.A. 105, 6179–6184. doi:10.1073/pnas.0801372105
Moura, D. M. S., De Sales, I. R. P., Brandão, J. A., Costa, M. R., and Queiroz, C. M. (2021). Disentangling Chemical and Electrical Effects of Status Epilepticus-Induced Dentate Gyrus Abnormalities. Epilepsy & Behav. 121, 106575. doi:10.1016/j.yebeh.2019.106575
Müller, M. C., Osswald, M., Tinnes, S., Häussler, U., Jacobi, A., Förster, E., et al. (2009). Exogenous Reelin Prevents Granule Cell Dispersion in Experimental Epilepsy. Exp. Neurol. 216, 390–397. doi:10.1016/j.expneurol.2008.12.029
Myers, C. E., Bermudez-Hernandez, K., and Scharfman, H. E. (2013). The Influence of Ectopic Migration of Granule Cells into the Hilus on Dentate gyrus-CA3 Function. PLoS One 8, e68208. doi:10.1371/journal.pone.0068208
Nakajima, K., Mikoshiba, K., Miyata, T., Kudo, C., and Ogawa, M. (1997). Disruption of Hippocampal Development In Vivo by CR-50 mAb against Reelin. Proc. Natl. Acad. Sci. U.S.A. 94, 8196–8201. doi:10.1073/pnas.94.15.8196
Nakano, Y., Kohno, T., Hibi, T., Kohno, S., Baba, A., Mikoshiba, K., et al. (2007). The Extremely Conserved C-Terminal Region of Reelin Is Not Necessary for Secretion but Is Required for Efficient Activation of Downstream Signaling. J. Biol. Chem. 282, 20544–20552. doi:10.1074/jbc.M702300200
Nayak, C. S., and Bandyopadhyay, S. (2021). “Mesial Temporal Lobe Epilepsy,” in StatPearls (Treasure Island (FL): StatPearls Publishing). Copyright © 2021, StatPearls Publishing LLC.
Nelson, K. B., and Ellenberg, J. H. (1976). Predictors of Epilepsy in Children Who Have Experienced Febrile Seizures. N. Engl. J. Med. 295, 1029–1033. doi:10.1056/nejm197611042951901
Nichols, A. J., and Olson, E. C. (2010). Reelin Promotes Neuronal Orientation and Dendritogenesis during Preplate Splitting. Cereb. Cortex 20, 2213–2223. doi:10.1093/cercor/bhp303
Nirwan, N., Vyas, P., and Vohora, D. (2018). Animal Models of Status Epilepticus and Temporal Lobe Epilepsy: a Narrative Review. Rev. Neurosci. 29, 757–770. doi:10.1515/revneuro-2017-0086
Niu, S., Renfro, A., Quattrocchi, C. C., Sheldon, M., and D’arcangelo, G. (2004). Reelin Promotes Hippocampal Dendrite Development through the VLDLR/ApoER2-Dab1 Pathway. Neuron 41, 71–84. doi:10.1016/s0896-6273(03)00819-5
Niu, S., Yabut, O., and D’arcangelo, G. (2008). The Reelin Signaling Pathway Promotes Dendritic Spine Development in Hippocampal Neurons. J. Neurosci. 28, 10339–10348. doi:10.1523/jneurosci.1917-08.2008
Novy, J., Belluzzo, M., Caboclo, L. O., Catarino, C. B., Yogarajah, M., Martinian, L., et al. (2013). The Lifelong Course of Chronic Epilepsy: the Chalfont Experience. Brain 136, 3187–3199. doi:10.1093/brain/awt117
O'Dell, R. S., Cameron, D. A., Zipfel, W. R., and Olson, E. C. (2015). Reelin Prevents Apical Neurite Retraction during Terminal Translocation and Dendrite Initiation. J. Neurosci. 35, 10659–10674. doi:10.1523/jneurosci.1629-15.2015
O’muircheartaigh, J., and Richardson, M. P. (2012). Epilepsy and the Frontal Lobes. Cortex 48, 144–155. doi:10.1016/j.cortex.2011.11.012
Ogino, H., Hisanaga, A., Kohno, T., Kondo, Y., Okumura, K., Kamei, T., et al. (2017). Secreted Metalloproteinase ADAMTS-3 Inactivates Reelin. J. Neurosci. 37, 3181–3191. doi:10.1523/jneurosci.3632-16.2017
Ogino, H., Nakajima, T., Hirota, Y., Toriuchi, K., Aoyama, M., Nakajima, K., et al. (2020). The Secreted Glycoprotein Reelin Suppresses the Proliferation and Regulates the Distribution of Oligodendrocyte Progenitor Cells in the Embryonic Neocortex. J. Neurosci. 40, 7625–7636. doi:10.1523/jneurosci.0125-20.2020
Okugawa, E., Ogino, H., Shigenobu, T., Yamakage, Y., Tsuiji, H., Oishi, H., et al. (2020). Physiological Significance of Proteolytic Processing of Reelin Revealed by Cleavage-Resistant Reelin Knock-In Mice. Sci. Rep. 10, 4471. doi:10.1038/s41598-020-61380-w
Orcinha, C., Kilias, A., Paschen, E., Follo, M., and Haas, C. A. (2021). Reelin Is Required for Maintenance of Granule Cell Lamination in the Healthy and Epileptic Hippocampus. Front. Mol. Neurosci. 14, 730811. doi:10.3389/fnmol.2021.730811
Orcinha, C., Münzner, G., Gerlach, J., Kilias, A., Follo, M., Egert, U., et al. (2016). Seizure-Induced Motility of Differentiated Dentate Granule Cells Is Prevented by the Central Reelin Fragment. Front. Cell. Neurosci. 10, 183. doi:10.3389/fncel.2016.00183
Pahle, J., Muhia, M., Wagener, R. J., Tippmann, A., Bock, H. H., Graw, J., et al. (2020). Selective Inactivation of Reelin in Inhibitory Interneurons Leads to Subtle Changes in the Dentate Gyrus but Leaves Cortical Layering and Behavior Unaffected. Cereb. Cortex 30, 1688–1707. doi:10.1093/cercor/bhz196
Panayiotopoulos, C. P. (2005). in The Epilepsies: Seizures, Syndromes and Management (Oxfordshire (UK): Bladon Medical Publishing). Copyright © 2005, Bladon Medical Publishing, an imprint of Springer Science+Business Media.).
Patrylo, P. R., Browning, R. A., and Cranick, S. (2006). Reeler Homozygous Mice Exhibit Enhanced Susceptibility to Epileptiform Activity. Epilepsia 47, 257–266. doi:10.1111/j.1528-1167.2006.00417.x
Pijet, B., Konopka, A., Rejmak, E., Stefaniuk, M., Khomiak, D., Bulska, E., et al. (2020). The Matrix Metalloproteinase Inhibitor Marimastat Inhibits Seizures in a Model of Kainic Acid-Induced Status Epilepticus. Sci. Rep. 10, 21314. doi:10.1038/s41598-020-78341-y
Pijet, B., Stefaniuk, M., Kostrzewska-Ksiezyk, A., Tsilibary, P.-E., Tzinia, A., and Kaczmarek, L. (2018). Elevation of MMP-9 Levels Promotes Epileptogenesis after Traumatic Brain Injury. Mol. Neurobiol. 55, 9294–9306. doi:10.1007/s12035-018-1061-5
Pinto Lord, M. C., and Caviness, V. S. (1979). Determinants of Cell Shape and Orientation: a Comparative Golgi Analysis of Cell-Axon Interrelationships in the Developing Neocortex of Normal and Reeler Mice. J. Comp. Neurol. 187, 49–69. doi:10.1002/cne.901870104
Pitkänen, A., Ndode-Ekane, X. E., Łukasiuk, K., Wilczynski, G. M., Dityatev, A., Walker, M. C., et al. (2014). Neural ECM and Epilepsy. Prog. Brain Res. 214, 229–262. doi:10.1016/b978-0-444-63486-3.00011-6
Pascual, M. R. Q. (2007). Temporal Lobe Epilepsy: Clinical Semiology and Neurophysiological Studies. Seminars Ultrasound, CT MRI 28, 416–423. doi:10.1053/j.sult.2007.09.004
Rodríguez, D., Morrison, C. J., and Overall, C. M. (2010). Matrix Metalloproteinases: what Do They Not Do? New Substrates and Biological Roles Identified by Murine Models and Proteomics. Biochimica Biophysica Acta (BBA) - Mol. Cell. Res. 1803, 39–54. doi:10.1016/j.bbamcr.2009.09.015
Roy, A., Millen, K. J., and Kapur, R. P. (2020). Hippocampal Granule Cell Dispersion: a Non-specific Finding in Pediatric Patients with No History of Seizures. Acta Neuropathol. Commun. 8, 54. doi:10.1186/s40478-020-00928-3
Saghatelyan, A. K., Dityatev, A., Schmidt, S., Schuster, T., Bartsch, U., and Schachner, M. (2001). Reduced Perisomatic Inhibition, Increased Excitatory Transmission, and Impaired Long-Term Potentiation in Mice Deficient for the Extracellular Matrix Glycoprotein Tenascin-R. Mol. Cell. Neurosci. 17, 226–240. doi:10.1006/mcne.2000.0922
Santana, J., and Marzolo, M.-P. (2017). The Functions of Reelin in Membrane Trafficking and Cytoskeletal Dynamics: Implications for Neuronal Migration, Polarization and Differentiation. Biochem. J. 474, 3137–3165. doi:10.1042/bcj20160628
Sato, Y., Kobayashi, D., Kohno, T., Kidani, Y., Prox, J., Becker-Pauly, C., et al. (2016). Determination of Cleavage Site of Reelin between its Sixth and Seventh Repeat and Contribution of Meprin Metalloproteases to the Cleavage. J. Biochem. 159, mvv102–312. doi:10.1093/jb/mvv102
Scharfman, H. E., Goodman, J. H., and Sollas, A. L. (2000). Granule-like Neurons at the hilar/CA3 Border after Status Epilepticus and Their Synchrony with Area CA3 Pyramidal Cells: Functional Implications of Seizure-Induced Neurogenesis. J. Neurosci. 20, 6144–6158. doi:10.1523/jneurosci.20-16-06144.2000
Scharfman, H. E., and Pierce, J. P. (2012). New Insights into the Role of Hilar Ectopic Granule Cells in the Dentate Gyrus Based on Quantitative Anatomic Analysis and Three-Dimensional Reconstruction. Epilepsia 53 (Suppl. 1), 109–115. doi:10.1111/j.1528-1167.2012.03480.x
Scott, R. C., King, M. D., Gadian, D. G., Neville, B. G., and Connelly, A. (2003). Hippocampal Abnormalities after Prolonged Febrile Convulsion: a Longitudinal MRI Study. Brain 126, 2551–2557. doi:10.1093/brain/awg262
Seress, L. (1992). Morphological Variability and Developmental Aspects of Monkey and Human Granule Cells: Differences between the Rodent and Primate Dentate Gyrus. Epilepsy Res. Suppl. 7, 3–28.
Seress, L., Ábrahám, H., Tornóczky, T., and Kosztolányi, G. (2001). Cell Formation in the Human Hippocampal Formation from Mid-gestation to the Late Postnatal Period. Neuroscience 105, 831–843. doi:10.1016/s0306-4522(01)00156-7
Sharma, A. K., Reams, R. Y., Jordan, W. H., Miller, M. A., Thacker, H. L., and Snyder, P. W. (2007). Mesial Temporal Lobe Epilepsy: Pathogenesis, Induced Rodent Models and Lesions. Toxicol. Pathol. 35, 984–999. doi:10.1080/01926230701748305
Sheilabi, M. A., Takeshita, L. Y., Sims, E. J., Falciani, F., and Princivalle, A. P. (2020). The Sodium Channel B4-Subunits Are Dysregulated in Temporal Lobe Epilepsy Drug-Resistant Patients. Ijms 21, 2955. doi:10.3390/ijms21082955
Shinnar, S. (1998). Prolonged Febrile Seizures and Mesial Temporal Sclerosis. Ann. Neurol. 43, 411–412. doi:10.1002/ana.410430402
Singh, S. P., He, X., Mcnamara, J. O., and Danzer, S. C. (2013). Morphological Changes Among Hippocampal Dentate Granule Cells Exposed to Early Kindling-Epileptogenesis. Hippocampus 23, 1309–1320. doi:10.1002/hipo.22169
Sloviter, R. S., Dean, E., Sollas, A. L., and Goodman, J. H. (1996). Apoptosis and Necrosis Induced in Different Hippocampal Neuron Populations by Repetitive Perforant Path Stimulation in the Rat. J. Comp. Neurol. 366, 516–533. doi:10.1002/(sici)1096-9861(19960311)366:3<516::Aid-cne10>3.0.Co;2-n
Sloviter, R. S., and Pedley, T. A. (1998). Subtle Hippocampal Malformation: Importance in Febrile Seizures and Development of Epilepsy. Neurology 50, 846–849. doi:10.1212/wnl.50.4.846
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: Biology and Pathology. Acta Neuropathol. 119, 7–35. doi:10.1007/s00401-009-0619-8
Sommer, W. (1880). Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch. F. Psychiatr. 10, 631–675. doi:10.1007/BF02224538
Soriano, E., and del Río, J. A. (2005). The Cells of Cajal-Retzius: Still a Mystery One Century after. Neuron 46, 389–394. doi:10.1016/j.neuron.2005.04.019
Stanfield, B. B., and Cowan, W. M. (1979). The Morphology of the hippocampus and Dentate Gyrus in Normal and Reeler Mice. J. Comp. Neurol. 185, 393–422. doi:10.1002/cne.901850302
Stevenson, T. K., and Lawrence, D. A. (2018). Characterization of Tissue Plasminogen Activator Expression and Trafficking in the Adult Murine Brain. eNeuro 5. doi:10.1523/eneuro.0119-18.2018
Szklarczyk, A., Lapinska, J., Rylski, M., Mckay, R. D. G., and Kaczmarek, L. (2002). Matrix Metalloproteinase-9 Undergoes Expression and Activation during Dendritic Remodeling in Adult hippocampus. J. Neurosci. 22, 920–930. doi:10.1523/jneurosci.22-03-00920.2002
Takács, E., Nyilas, R., Szepesi, Z., Baracskay, P., Karlsen, B., Røsvold, T., et al. (2010). Matrix Metalloproteinase-9 Activity Increased by Two Different Types of Epileptic Seizures that Do Not Induce Neuronal Death: a Possible Role in Homeostatic Synaptic Plasticity. Neurochem. Int. 56, 799–809. doi:10.1016/j.neuint.2010.03.003
Tarkka, R., Paakko, E., Pyhtinen, J., Uhari, M., and Rantala, H. (2003). Febrile Seizures and Mesial Temporal Sclerosis: No Association in a Long-Term Follow-Up Study. Neurology 60, 215–218. doi:10.1212/01.wnl.0000037482.55894.b1
Tassi, L., Meroni, A., Deleo, F., Villani, F., Mai, R., Lo Russo, G., et al. (2009). Temporal Lobe Epilepsy: Neuropathological and Clinical Correlations in 243 Surgically Treated Patients. Epileptic Disord. 11, 281–292. doi:10.1684/epd.2009.0279
Tatum, W. O. (2012). Mesial Temporal Lobe Epilepsy. J. Clin. Neurophysiol. 29, 356–365. doi:10.1097/WNP.0b013e31826b3ab7
Teixeira, C. M., Kron, M. M., Masachs, N., Zhang, H., Lagace, D. C., Martinez, A., et al. (2012). Cell-autonomous Inactivation of the Reelin Pathway Impairs Adult Neurogenesis in the hippocampus. J. Neurosci. 32, 12051–12065. doi:10.1523/jneurosci.1857-12.2012
Theocharis, A. D., Skandalis, S. S., Gialeli, C., and Karamanos, N. K. (2016). Extracellular Matrix Structure. Adv. Drug Deliv. Rev. 97, 4–27. doi:10.1016/j.addr.2015.11.001
Thom, M. (2014). Review: Hippocampal Sclerosis in Epilepsy: a Neuropathology Review. Neuropathol. Appl. Neurobiol. 40, 520–543. doi:10.1111/nan.12150
Thom, M., Liagkouras, I., Martinian, L., Liu, J., Catarino, C. B., and Sisodiya, S. M. (2012). Variability of Sclerosis along the Longitudinal Hippocampal axis in Epilepsy: a Post Mortem Study. Epilepsy Res. 102, 45–59. doi:10.1016/j.eplepsyres.2012.04.015
Thom, M., Martinian, L., Williams, G., Stoeber, K., and Sisodiya, S. M. (2005a). Cell Proliferation and Granule Cell Dispersion in Human Hippocampal Sclerosis. J. Neuropathol. Exp. Neurol. 64, 194–201. doi:10.1093/jnen/64.3.194
Thom, M., Sisodiya, S. M., Beckett, A., Martinian, L., Lin, W.-R., Harkness, W., et al. (2002). Cytoarchitectural Abnormalities in Hippocampal Sclerosis. J. Neuropathol. Exp. Neurol. 61, 510–519. doi:10.1093/jnen/61.6.510
Thom, M., Zhou, J., Martinian, L., and Sisodiya, S. (2005b). Quantitative Post-mortem Study of the hippocampus in Chronic Epilepsy: Seizures Do Not Inevitably Cause Neuronal Loss. Brain 128, 1344–1357. doi:10.1093/brain/awh475
Tinnes, S., Ringwald, J., and Haas, C. A. (2013). TIMP‐1 Inhibits the Proteolytic Processing of Reelin in Experimental Epilepsy. FASEB J. 27, 2542–2552. doi:10.1096/fj.12-224899
Tinnes, S., Schäfer, M. K. E., Flubacher, A., Münzner, G., Frotscher, M., and Haas, C. A. (2011). Epileptiform Activity Interferes with Proteolytic Processing of Reelin Required for Dentate Granule Cell Positioning. FASEB J. 25, 1002–1013. doi:10.1096/fj.10-168294
Tissir, F., and Goffinet, A. M. (2003). Reelin and Brain Development. Nat. Rev. Neurosci. 4, 496–505. doi:10.1038/nrn1113
Trenerry, M. R., Jack, C. R., Sharbrough, F. W., Cascino, G. D., Hirschorn, K. A., Marsh, W. R., et al. (1993). Quantitative MRI Hippocampal Volumes: Association with Onset and Duration of Epilepsy, and Febrile Convulsions in Temporal Lobectomy Patients. Epilepsy Res. 15, 247–252. doi:10.1016/0920-1211(93)90062-c
Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., et al. (1999). Reeler/Disabled-like Disruption of Neuronal Migration in Knockout Mice Lacking the VLDL Receptor and ApoE Receptor 2. Cell. 97, 689–701. doi:10.1016/s0092-8674(00)80782-5
Trotter, J. H., Lussier, A. L., Psilos, K. E., Mahoney, H. L., Sponaugle, A. E., Hoe, H.-S., et al. (2014). Extracellular Proteolysis of Reelin by Tissue Plasminogen Activator Following Synaptic Potentiation. Neuroscience 274, 299–307. doi:10.1016/j.neuroscience.2014.05.046
Turk, L. S., Kuang, X., Dal Pozzo, V., Patel, K., Chen, M., Huynh, K., et al. (2021). The Structure-Function Relationship of a Signaling-Competent, Dimeric Reelin Fragment. Structure 29, 1156–1170. e1156. doi:10.1016/j.str.2021.05.012
Twible, C., Abdo, R., and Zhang, Q. (2021). Astrocyte Role in Temporal Lobe Epilepsy and Development of Mossy Fiber Sprouting. Front. Cell. Neurosci. 15, 725693. doi:10.3389/fncel.2021.725693
Ulbrich, P., Khoshneviszadeh, M., Jandke, S., Schreiber, S., and Dityatev, A. (2021). Interplay between Perivascular and Perineuronal Extracellular Matrix Remodelling in Neurological and Psychiatric Diseases. Eur. J. Neurosci. 53, 3811–3830. doi:10.1111/ejn.14887
Vanlandingham, K. E., Heinz, E. R., Cavazos, J. E., and Lewis, D. V. (1998). Magnetic Resonance Imaging Evidence of Hippocampal Injury after Prolonged Focal Febrile Convulsions. Ann. Neurol. 43, 413–426. doi:10.1002/ana.410430403
Vedunova, M., Sakharnova, T., Mitroshina, E., Perminova, M., Pimashkin, A., Zakharov, Y., et al. (2013). Seizure-like Activity in Hyaluronidase-Treated Dissociated Hippocampal Cultures. Front. Cell. Neurosci. 7, 149. doi:10.3389/fncel.2013.00149
Vos, S. B., Winston, G. P., Goodkin, O., Pemberton, H. G., Barkhof, F., Prados, F., et al. (2020). Hippocampal Profiling: Localized Magnetic Resonance Imaging Volumetry and T2 Relaxometry for Hippocampal Sclerosis. Epilepsia 61, 297–309. doi:10.1111/epi.16416
Wasser, C. R., and Herz, J. (2017). Reelin: Neurodevelopmental Architect and Homeostatic Regulator of Excitatory Synapses. J. Biol. Chem. 292, 1330–1338. doi:10.1074/jbc.R116.766782
Weninger, J., Meseke, M., Rana, S., and Förster, E. (2021). Heat-Shock Induces Granule Cell Dispersion and Microgliosis in Hippocampal Slice Cultures. Front. Cell. Dev. Biol. 9, 626704. doi:10.3389/fcell.2021.626704
Wieser, H. G. (2004). Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis. Epilepsia 45, 695–714. doi:10.1111/j.0013-9580.2004.09004.x
Wilczynski, G. M., Konopacki, F. A., Wilczek, E., Lasiecka, Z., Gorlewicz, A., Michaluk, P., et al. (2008). Important Role of Matrix Metalloproteinase 9 in Epileptogenesis. J. Cell. Biol. 180, 1021–1035. doi:10.1083/jcb.200708213
Woermann, F. G., Barker, G. J., Birnie, K. D., Meencke, H. J., and Duncan, J. S. (1998). Regional Changes in Hippocampal T2 Relaxation and Volume: a Quantitative Magnetic Resonance Imaging Study of Hippocampal Sclerosis. J. Neurology, Neurosurg. Psychiatry 65, 656–664. doi:10.1136/jnnp.65.5.656
Yamakage, Y., Kato, M., Hongo, A., Ogino, H., Ishii, K., Ishizuka, T., et al. (2019). A Disintegrin and Metalloproteinase with Thrombospondin Motifs 2 Cleaves and Inactivates Reelin in the Postnatal Cerebral Cortex and hippocampus, but Not in the Cerebellum. Mol. Cell. Neurosci. 100, 103401. doi:10.1016/j.mcn.2019.103401
Yasui, N., Kitago, Y., Beppu, A., Kohno, T., Morishita, S., Gomi, H., et al. (2011). Functional Importance of Covalent Homodimer of Reelin Protein Linked via its Central Region. J. Biol. Chem. 286, 35247–35256. doi:10.1074/jbc.M111.242719
Yasui, N., Nogi, T., Kitao, T., Nakano, Y., Hattori, M., and Takagi, J. (2007). Structure of a Receptor-Binding Fragment of Reelin and Mutational Analysis Reveal a Recognition Mechanism Similar to Endocytic Receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 9988–9993. doi:10.1073/pnas.0700438104
Yu, L., Boyle, P. A., Dawe, R. J., Bennett, D. A., Arfanakis, K., and Schneider, J. A. (2020). Contribution of TDP and Hippocampal Sclerosis to Hippocampal Volume Loss in Older-Old Persons. Neurology 94, e142–e152. doi:10.1212/wnl.0000000000008679
Zarow, C., Sitzer, T. E., and Chui, H. C. (2008). Understanding Hippocampal Sclerosis in the Elderly: Epidemiology, Characterization, and Diagnostic Issues. Curr. Neurol. Neurosci. Rep. 8, 363–370. doi:10.1007/s11910-008-0057-3
Zhan, R.-Z., and Nadler, J. V. (2009). Enhanced Tonic GABA Current in Normotopic and Hilar Ectopic Dentate Granule Cells after Pilocarpine-Induced Status Epilepticus. J. Neurophysiology 102, 670–681. doi:10.1152/jn.00147.2009
Keywords: temporal lobe epilepsy, granule cell dispersion, reelin, febrile seizures, hippocampal sclerosis, extracellular matrix
Citation: Leifeld J, Förster E, Reiss G and Hamad MIK (2022) Considering the Role of Extracellular Matrix Molecules, in Particular Reelin, in Granule Cell Dispersion Related to Temporal Lobe Epilepsy. Front. Cell Dev. Biol. 10:917575. doi: 10.3389/fcell.2022.917575
Received: 11 April 2022; Accepted: 23 May 2022;
Published: 06 June 2022.
Edited by:
Shinobu Matsuura, Boston University, United StatesReviewed by:
Mitsuharu Hattori, Nagoya City University, JapanNathalie Berube, Western University, Canada
Copyright © 2022 Leifeld, Förster, Reiss and Hamad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer Leifeld, Jennifer.Leifeld@ruhr-uni-bochum.de; Eckart Förster, eckart.foerster@rub.de