 Csörsz Nagy
Csörsz Nagy Arvand Haschemi
Arvand Haschemi- Department of Laboratory Medicine (KILM), Medical University of Vienna, Vienna, Austria
A process is a function of time; in immunometabolism, this is reflected by the stepwise adaptation of metabolism to sustain the bio-energetic demand of an immune-response in its various states and shades. This perspective article starts by presenting an early attempt to investigate the physiology of inflammation, in order to illustrate one of the basic concepts of immunometabolism, wherein an adapted metabolism of infiltrating immune cells affects tissue function and inflammation. We then focus on the process of macrophage activation and aim to delineate the factor time within the current molecular context of metabolic-rewiring important for adapting primary carbohydrate metabolism. In the last section, we will provide information on how the pentose phosphate pathway may be of importance to provide both nucleotide precursors and redox-equivalents, and speculate how carbon-scrambling events in the non-oxidative pentose phosphate pathway might be regulated within cells by demand. We conclude that the adapted metabolism of inflammation is specific in respect to the effector-function and appears as a well-orchestrated event, dynamic by nature, and based on a functional interplay of signaling- and metabolic-pathways.
Concepts of Immunometabolism
The first concepts of immunometabolism date back to the pre-genomic age of biomedical research. As early as 1912, Levene and Meyer used dog blood-derived leukocytes to directly demonstrate that hexoses are converted into two molecules, each containing a chain of three carbons (1). They also provided further evidence that hexoses are the source of lactate and assumed that this process accounts for “synthethic purposes by the leukocytes.” This period is widely recognized as the onset of modern biochemistry and furthermore of immunometabolism.
Immunometabolism is also tightly linked to research on cancer metabolism, especially with regard to the pioneering work of Otto Warburg, wherein he further developed the concept of cellular physiology (2). It was revealed that exudate leukocytes have high aerobic glycolysis, while respiration was very low and it was concluded then that white blood cells must have a cancer metabolism (3, 4). However, they differentiated immune cell- and cancer metabolism in that cancer cells use aerobic glycolysis to live, while aerobic glycolysis in white blood cells is a sign of aging or dying off. With this background, Walter Kempner and Ernst Peschel, both from the Bergmann’sche Institut at the Charite in Berlin, published their work with the German title: “Stoffwechsel der Entzündung” (Metabolism of Inflammation) (5). In 1930, they formulated two fundamental questions: what are the specific reactions of inflammation? Which processes lead to cell migration and subsequently to tissue swelling or necrosis? They presumed that an adapted cellular metabolism of white blood cells may play a major role in these processes. They tested their hypothesis in a human in vivo model of sterile-inflammation and provided fundamental new insights, which are still of relevance for today’s concepts of immunometabolism. Kempner and Peschel used the beetle-juice (cantharidin)-induced skin blister model and metabolically defined the inflamed human tissue in order to examine the physiology of inflammation. They observed a disrupted equilibrium of oxygen, CO2, sugar, lactate, and bicarbonate as a result of inflammation and concluded that this was induced by the metabolism found in infiltrating immune cells. They expected this to happen as a function of time. They demonstrated a drop in glucose over a period of 6–90 h and pulsed oral glucose administrations indicating that glucose replenishment from healthy tissue was also gradually declining. Within the inflamed area (the blister) also oxygen concentration declined. This was again attributed to high cellular respiration of infiltrated cells and a reduced gas-exchange with the healthy tissue. In addition to that, they measured a time-dependent increase in lactate and a decrease in the bicarbonate levels, which together could explain the decrease in pH of inflamed tissue, previously observed by Schade (6). Kempner and Peschel identified metabolic changes in inflamed tissue as a function of time, which is actively established by infiltrating “injured” immune cells with an adapted cellular metabolism (5). Thereby, they delineated a complex interplay between cellular metabolism and the physiology of inflammation. In 2011, the cantharidin-induced skin blister was re-evaluated and recommended as an excellent human in vivo model to study inflammation (7). This report also reveals that the infiltrating cells in this model are mainly neutrophils and monocytes/macrophages; these cells were probably also the cause for the observation by Kempner and Peschel.
Since then, a new school of immunobiology has started to reveal the molecular mechanism behind the observed metabolic-adaptation in various immune cells and models of immunology. As an example, the action of the pentose phosphate pathway (PPP) and the power of redox-biology, including superoxide production, were identified as essential in forming the respiratory-burst of phagocytes (8, 9). Also amino acid and lipid metabolism, as well as their adaptations, were characterized as fundamental to properly fuel the function of an immune response (10, 11). In recent years, however, new concepts in immunometabolism have evolved and further mechanistic-details have surfaced that enable us to better understand how these metabolic-adaptions are reached and regulated.
Time Resolved Metabolic-Adaptations during Macrophage Activation
Macrophages are important immune cells, which regulate tissue homeostasis by sensing and interpreting cell injury and infection, the classic triggers of an inflammatory response (12). Today, macrophages are classified according to the activation stimuli into at least two polarization states, the classic M1 (representing a pro-inflammatory phenotype) and the alternative M2 macrophage (representing an anti-inflammatory or homeostasis inducing phenotype), in order to discriminate between the effector phenotypes resulting from the distinct activation signals (13). However, in vivo macrophages rather appear to blend into various “shades of activation,” while retaining some of their plasticity (14–18). Furthermore, macrophage populations and phenotypes can dramatically change over time, as exemplified by the finding that the inflammatory response is a spatially and temporally coordinated process. Recently, the polarization process of macrophages has been further associated with the reprograming of cellular metabolism (19–25). Information processing by signal-transduction pathways starts shortly after activation and is temporally coordinated, reflected by the phosphorylation and de-phosphorylation of signal transducers and effector molecules. The question arises how the reprograming of primary carbohydrate metabolism is timed in the process of macrophage activation. We would like to present more detailed and more importantly time-resolved information on key events, which appear to establish a pro-inflammatory M1-like metabolic-phenotype induced by lipopolysaccharide (LPS, Figure 1).
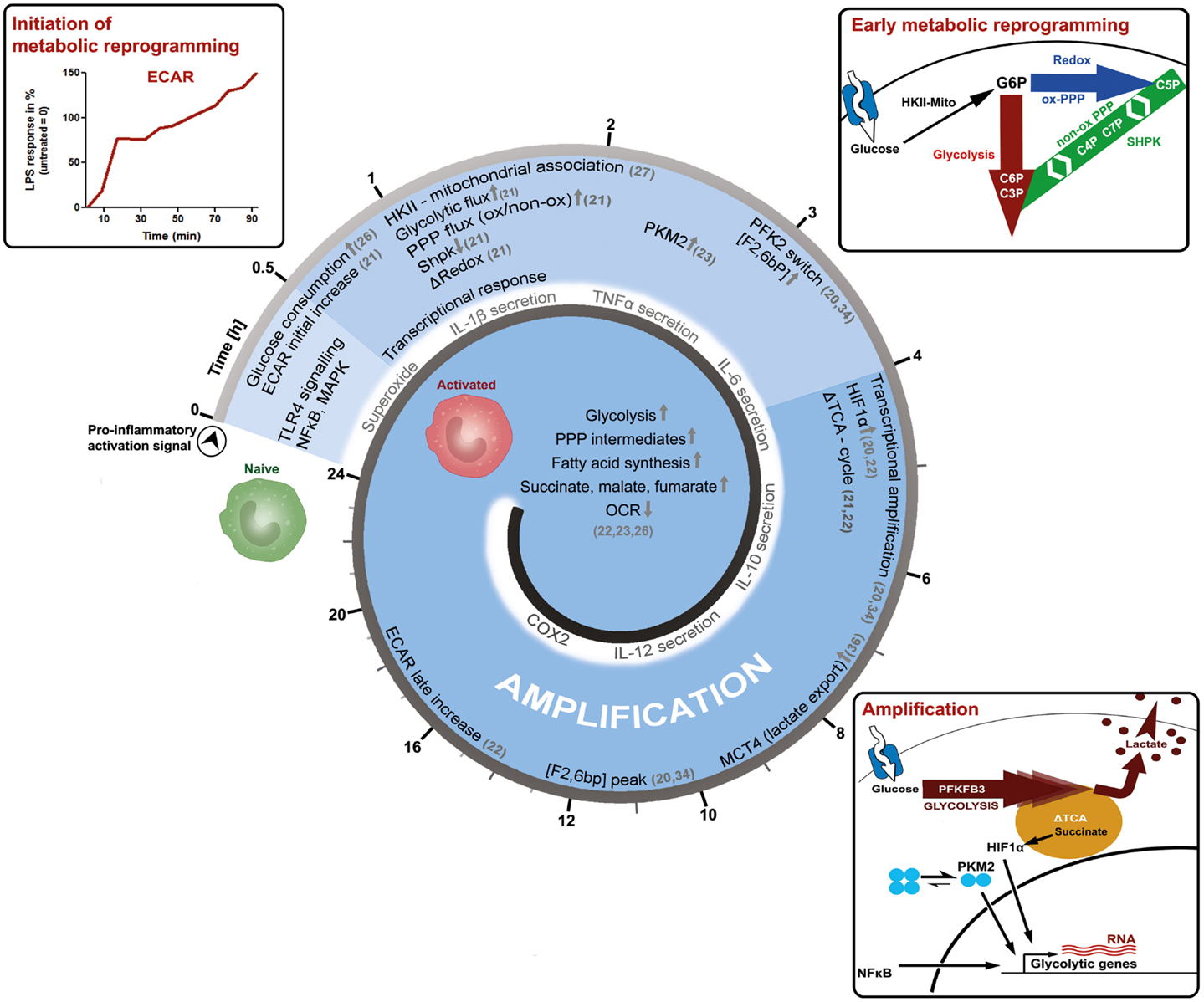
Figure 1. Time-resolved metabolic reprograming during pro-inflammatory macrophage polarization. This model illustrates the activation of a macrophage as a function of time and is based on the literature discussed in the main text. LPS-induced activation can be grouped into an initiation-, an early metabolic-reprograming,- and an amplification-phase. The initiation phase of the metabolic response is characterized by an increase in glucose consumption and in the extracellular acidification rate (ECAR). The early metabolic reprograming phase depicts the increase and rerouting of carbon flux through glycolysis and the PPP, events which also regulate the cellular redox-state. In this setting, the mitochondrial association of hexokinase-II (HKII) appears to provide sufficient levels of glucose 6-phosphate (G6P), while the downregulation of sedoheptulose kinase (Shpk, previously known as CARKL) appears to be necessary to maintain appropriate carbon flux at the interface of glycolysis and the PPP. During the amplification phase, this pro-glycolytic metabolic-phenotype is further strengthened. A switch toward the more active 6-phosphofructo-2-kinase (PFK2) enzyme PFKFB3 produces higher levels of fructose 2-bisphosphate [F2,6bP], thus allosterically activating PFK1 and enhancing glycolytic flux. Dimers of the pyruvate kinase M2 (PKM2), as well as accumulating succinate further augment metabolic reprograming by supporting HIF-1α dependent transcriptional induction of glycolytic genes. In the amplification phase, also the export of intracellular glycolysis-derived lactate through monocarboxylate transporter 4 (MCT4) becomes obligatory, which may otherwise inhibit PFK1. These initial events lead to more prominent metabolic changes observed 24 h after macrophages have encountered the pro-inflammatory stimuli. However, further time-resolved data is required to refine these processes and our current perspective, how cellular metabolism of macrophages adapts during activation.
After only 20 min of in vitro LPS-stimulation, simultaneously with prime signaling events, the glucose uptake of cells approximately doubles (26). At the same time, the extracellular acidification rate (ECAR), an indirect measure of aerobic glycolysis, also increases until reaching a certain plateau-state, to then adapt, and further increase (21). This response indicates that LPS leads to a rapid induction of glycolytic flux, which is modulated and amplified in multiple steps (Figure 1). The extension phase of ECAR is accompanied by a slow and marginal decrease in the oxygen consumption rate (OCR). The molecular mechanisms leading to these immediate early metabolic events, however, are not known and acidification may also result from sources other than the formation of lactic acid.
However, 1 h after LPS stimulation, the mRNA of the glucose transporter (GLUT1) is induced and the uptake of glucose further increases (26). After uptake of glucose, it becomes phosphorylated by hexokinases (HK) to glucose 6-phosphate (G6P), which can then be diverted into various catabolic and anabolic pathways. Non-stationary metabolic flux analysis, tracking the fate of intracellular glucose during macrophage activation, reveals that already 1 h after LPS-exposure a considerable amount of glucose is used by both, glycolysis and the PPP (21). In rat-Kupffer cells, which are specialized liver macrophages, as well as murine dendritic cells, HK-II was shown to associate with mitochondria within an hour after LPS stimulation (27, 28). A similar mechanism is observed in cancer cells, where mitochondrial matrix derived ATP is channeled to HK-II and thereby augmenting the glycolytic flux (29). Recently, the sedoheptulose kinase (Shpk, formerly known as CARKL) was characterized as a unique heptose kinase, phosphorylating sedoheptulose (a ketoheptose) to sedoheptulose 7-phosphate (S7P), which can then act as a reaction partner of glyceraldehyde 3-phosphate (G3P) in the non-oxidative PPP (21, 30–32). In macrophages, the mRNA of Shpk is rapidly down-regulated by LPS but not by interleukin (IL)-4 stimulation (21). Regulation of Shpk will be further discussed in the next section. Also, approximately after 1 h, LPS specifically induces pyruvate kinase M2 (PKM2) protein expression and phosphorylation, which becomes further augmented in the late phase of macrophage activation (23). Phosphorylation of PKM2 favors dimeric configuration and PKM2 translocation into the nucleus, where it acts together with hypoxia-inducible factor 1-alpha (HIF1α) as a transcriptional inducer of interleukin 1-beta (IL-1β) and more importantly of glycolytic genes like PFK, constituting an amplification loop in the intermediate and late phase of macrophage activation (23, 33). Within 2–4 h after activation by LPS, an isoform switchs from the liver-type 6-phosphofructo-2-kinase (PFKFB1 aka PFK2) to the ubiquitous and more active PFKFB3 occurs (34). This is also observed when LPS is used in combination with interferon gamma (IFNγ) to induce a pro-inflammatory macrophage activation (20). PFKFB3 produces augmented levels of fructose 2, 6-bisphosphate (F2,6bP), which then functions as an allosteric activator of 6-phosphofructo-1-kinase (PFK1) to further sustain the pro-glycolytic program (Figure 1). Interestingly in yeast, PFK1 derived F1,6bP allosterically activates PKM2, indicating the presence of metabolic feedback loops (35).
Approximately 4–6 h after macrophage activation, the export of glycolytic lactate appears to become mandatory for the activation process as indicated by the increased expression of monocarboxylate transporter 4 (MCT4) (36). Knockdown of MCT4 results in enhanced intracellular lactate accumulation, a decreased expression of LPS-induced glycolytic enzymes and an attenuated secretion of tumor necrosis factor-alpha (TNFα) and IL-6. Accumulating intracellular lactate might decrease glycolytic activity by inhibiting PFK1, an enzyme which may reach maximal activity in the later phase, as indicated by peaking F2,6bP concentrations and PFKFB3 mRNA levels at 6–12 h (34, 37). Also, approximately 4 h after initiation of macrophage polarization by LPS, the tricarboxylic acid (TCA) cycle changes its operational mode from a catabolic pathway to a partly anabolic system (21, 22). The TCA-cycle metabolite succinate accumulates in a macrophage cell line and bone marrow derived macrophages (BMDMs) (21, 22). Succinate, derived by glutamine-dependent anerplerosis and gamma-aminobutyric acid (GABA)-shunt, was shown to inhibit the prolyl hydroxylase-dependent degradation of HIF1α and to enhance IL-1β production (22). Increased succinate levels may also increase succinylation of metabolic enzymes such as glyceraldehyde 3-phosphate dehydrogenase (GAPDH), transaldolase (TALDO), and lactate dehydrogenase (LDH) A-chain, possibly further shaping late phase metabolic adaptations (22, 38). Succinate dependent HIF1α stabilization as well as increased succinylation are both suppressed by the inhibition of glycolysis, indicating that these processes are dependent on increased glycolytic flux (22, 23). Approximately 24 h after LPS-stimulation, metabolic reprograming is firmly established: glycolytic gene expression and metabolites are increased, as well as lactate and ECAR (22, 23). The TCA-cycle supports increased fatty acid synthesis as well as the formation of cycle intermediates (succinate, malate, fumarate), while OCR is reduced, indicating a significant decline in oxidative metabolism (22, 23).
To briefly summarize the overall consequences of the discussed adaptations: LPS stimulated macrophages increase aerobic glycolysis and PPP activity, reduce mitochondrial respiration, and reconfigure the TCA-cycle. In order to replenish NAD+ for glycolysis, lactate production and secretion are enhanced, leading to acidification of the environment. Such a pro-inflammatory metabolism is important for the generation of redox-equivalents as well as precursor molecules such as amino acids, lipids, and nucleotides, sustaining a burst in pro-inflammatory mediator production (39, 40). In reference to protein-signal-transduction leading to the observed metabolic adaptions, nuclear factor kappa-B (NFκB) and HIF1α are two well-characterized transcription factors, which increase the expression of glycolytic genes (41, 42). In contrast to an pro-inflammatory activation of macrophages, alternative activation (i.e., by IL-4) is associated with mitochondrial biogenesis as well as increased fatty acid oxidation and oxidative phosphorylation, primarily driven by lysosomal lipolysis of endocytosed lipoprotein particles (24, 43). In general, the M2 metabolic program mainly relies on STAT6, PPARγ, and its co-activator PGC1β to promote oxidative metabolism. The manifold metabolic changes during macrophage activation as well as their regulatory mechanisms have been recently discussed in detail in some excellent reviews (44–46).
The PPP Sustains the Metabolic Demand of Macrophages during Polarization
The PPP represents a prime example on how increased carbon-flux can contribute to mount the specific effector functions of LPS-activated macrophages by complementing their appropriate demands through supplying both redox-power and ribose moieties either at the same time or independently from each other. The PPP is divided into the oxidative (oxPPP) and non-oxidative branch (non-oxPPP). Briefly, the oxPPP, with glucose 6-phosphate dehydrogenase (G6PD) as its rate-limiting enzyme, is highly active in macrophages (21, 47), decarboxylates G6P, and forms ribose 5-phosphate (R5P) through three irreversible reactions, while simultaneously reducing two molecules of NADP+ to NADPH and liberating one molecule of CO2. The non-ox PPP consists of reversible reactions, which can either recycle R5P to glycolytic intermediates or use the latter to generate pentose phosphates (C5P) through reverse flux. The general aspects of PPP architecture and function have been reviewed elsewhere in great detail (48–50).
Oxidative PPP derived NADPH serves as a cofactor for NADPH-oxidase dependent reactive oxygen species (ROS) production, while also reducing oxidized redox-couples to simultaneously sustain an anti-oxidant response (i.e., glutathione and thioredoxin systems), thereby partly controlling the redox balance during macrophage activation (51, 52). Also, the function of many redox-sensitive signaling proteins, which are associated with the process of activation, are potentially dependent on increased flux through the oxPPP (53–59). Furthermore, NADPH is also critical for reductive biosynthesis serving activation associated membrane expansion and the production of lipid mediators such as prostaglandins. Remarkably, it was reported that NADPH levels undergo periodic oscillations in macrophages and neutrophils, which are tightly linked to superoxide oscillations and adapt, upon LPS stimulations to a higher frequency (60–63). These oscillations may depend on periodic glucose influx and PPP activity, and appear to encode information in their amplitude and frequency (64, 65). Severe G6PD deficient leukocytes have been associated with impairments in their oxidative burst, their bactericidal activity (66–68), their resistance to oxidative stress (69), as well as modified cytokine responses (70–72). Overexpression of G6PD in a macrophage cell line enhanced the activation of NFκB and p38-MAPK signaling pathways and potentiated the expression of pro-inflammatory cytokines as well as ROS production (73). In contrast to IL-1β, the production of TNFα and IL-6 does not appear to be directly dependent on aerobic glycolysis, as recently suggested by the activation of PKM2 and 2-deoxyglucose (2-DG) treatment, respectively (22, 23). Notably, 2-DG is a glycolytic inhibitor downstream from hexokinase and can therefore become phosphorylated to 2-deoxyglucose 6-phosphate, which is partly metabolized by the oxPPP in red blood cells (74). Whether this also occurs in macrophages or not remains to be investigated. Inhibition of G6PD or LDH, however, was shown to decrease TNFα and IL-6 levels, implicating that these cytokines are rather regulated by redox-state than simply by the increased glycolytic-flux (21). Apart from redox-power, the macrophage activation process also demands a large amount of pentose phosphates probably to sustain de novo nucleotide synthesis for their characteristic transcriptional response. In contrast to M2, M1 macrophages drastically change their transcriptional profile (40). Isotope distribution analysis of a non-stationary metabolic flux experiment with asymmetrically labeled glucose after 1 h of LPS-induced macrophage activation indicated that both ox- and non-oxPPP flux rates increase, while most of the pentose phosphates (C5) are derived from the non-ox branch (21).
The non-oxPPP relies on transketolase (TK) and TALDO catalyzed reversible transfer of keto-groups to various aldose acceptors. TK uses thiamine pyrophosphate as cofactor to transfer two carbon (C2)-units, while TALDO can transfer C3-units by forming Schiff base intermediates (75, 76). Thereby, this pathway interconverts carbohydrate-phosphates of different chain length (C3P to C7P), without the need of energy in form of ATP (carbon scrambling, Figure 2A). The regulation of non-oxPPP is complex due to its reversible nature and still not fully understood. The flux-rate and its direction are generally thought to depend on thermodynamics, which impose a major constraint on the structure of metabolic pathways (77). However, the recent identification of Shpk indicates additional regulatory mechanism, which was previously not considered (Figure 2B) (78). In contrast to TK or TALDO, Shpk is reported to be regulated differently during LPS- and IL-4 induced polarization (21). LPS stimulation leads to a rapid down-regulation of Shpk mRNA in the early phase of macrophage activation in mice and humans likewise and in vitro as well as in vivo. In contrast to LPS, IL-4 stimulation maintains or even slightly increases Shpk levels (21). Counterbalancing LPS-induced down-regulation of Shpk by overexpression in a macrophage cell line resulted in an accumulation of pentose phosphates and an imbalance of the cellular redox system, as indicated by the accumulation of oxidized redox couples as well as blunted LPS-induced intracellular superoxide production (21). In theory, Shpk, by the formation of rate-limiting S7P, should increase the shunting of glycolysis-derived G3P into the non-oxPPP (78) and regulate oxPPP activity through the formation or recycling of pentose phosphates (79). So far, we have no confirmed mode-of-action, how Shpk activity actually regulates carbon-flux through the non-oxPPP, and no information on its activity and local distribution during macrophage activation. Therefore, we can only speculate on the consequences of Shpk regulation for the process of metabolic-adaptation (Figure 2B). Shpk-derived S7P may act as a thermodynamic buffer to support a stable non-equilibrium, which drives (low S7P) or inhibits (high S7P) carbon-flux through the non-oxPPP. However, flux-direction seems to be determined by demand and by the presence of TK and TALDO (Figure 2B). In addition to that, high S7P levels can directly modulate glycolytic flux through the inhibition of hexose phosphate isomerase, as well as by competitively inhibiting fructose 6-phosphate (F6P) phosphorylation by PFK (80, 81). Therefore, the consequences of Shpk regulation appear as strictly context dependent, which is defined by the demand of metabolites (i.e., C5P) and the presence or absence of other enzymes. We know that Shpk only partially colocalizes with G6PD in the cytoplasm of cells, which points out that there are instances where the ox- and the non-oxPPP are coupled to and uncoupled from each other (21). Information on the function of TK and TALDO in the process of macrophage activation is rare; however, both enzymes were tightly linked to oxidative stress-defense in other cell types (82–84). Notably, yeast seems to lack a Shpk homolog but utilizes a specific sedoheptulose–bisphosphatase [dephosphorylates sedoheptulose 1,7-bisphosphate (S1,7bP) to S7P] for riboneogenesis when the demand for nucleotide precursors is high (85). S1,7bP was previously reported to also exists in rat liver tissue (86, 87); however, there appear to be some major differences in the architecture of heptose metabolism (heptolysis) between fungi and vertebrates (78, 85).
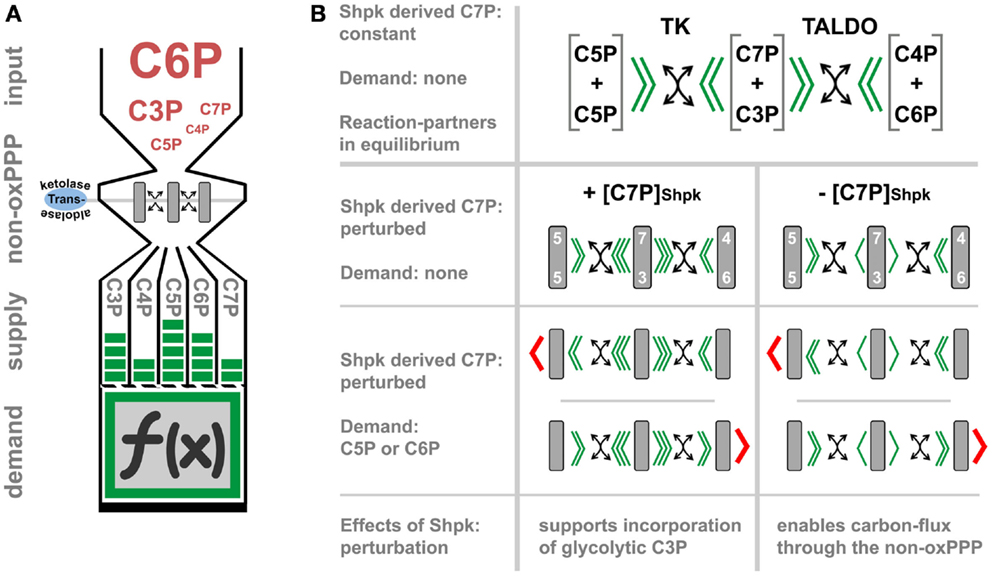
Figure 2. The function and regulation of the non-oxidative PPP. (A) represents a simplified model, which illustrates how transketolase (TK) and transaldolase (TALDO) may interconvert carbohydrate-phosphates of three to seven carbon-atoms in length (C3P to C7P) without the need of energy (carbon-scrambling) to account for the cellular demand, which in part defines cell function (indicated by the symbol f (x)). In (B), we theoretically evaluate the regulatory effect of Shpk-derived sedoheptulose 7-phosphate ([C7P]Shpk) on non-oxPPP flux in the presence of TK and TALDO. Flux through the non-oxPPP, by its reversible reactions, is dependent on the stoichiometry of the reactants (indicated by green arrowheads). In contrast to TK- and TALDO-derived S7P, the phosphorylation of free sedoheptulose to S7P by Shpk requires energy in form of ATP. Assuming a constant contribution by Shpk, this additional source of S7P may therefore act as a thermodynamic buffer, which can be actively regulated to induce a non-equilibrium. In theory, perturbation of Shpk can either increase the resistance (increased [C7P]Shpk) or lower it (decreased [C7P]Shpk) to support shunting through the non-oxPPP. However, in the presence of TK or TALDO, an increased [C7P]Shpk will promote the incorporation of glycolytic-G3P into the PPP. This model further illustrates that non-oxPPP flux-direction is also dependent on the demand of respective molecules (indicated by red arrowheads).
In summary, these findings indicate that during macrophage activation the cellular demands are covered by a precisely coordinated interplay of many pathways to sustain such profound polarization events. The PPP appears as a versatile hub to reroute carbon moieties within the network of primary carbohydrate metabolism while independently controlling cellular redox-states.
Conclusion
This collection of findings may support our perspective that time and demands are critical to understand the molecular events important to mount an immune response. Immunometabolism demonstrates its consequences for physiology at various levels including cells, tissues, organisms, and entire populations, as we currently experience with diseases like cancer, cardiovascular diseases, obesity, and diabetes to name but a few. Already, Kempner and Peschel considered diabetic patients in their investigations and noted a sustained glucose supply together with a prolonged inflammatory response compared to non-diabetics. Since then, many excellent studies further delineated the complex interplay of metabolism, the immune system and tissue function, and malfunction. At the molecular level, macrophages adapt their metabolism very early in the polarization process, which then become amplified over time. This highlights that we need to strongly consider the process leading to activation and not only the phenotypic “endpoints.” A macrophage located within a complex tissue microenvironment may go through multiple, subsequently occurring, activation events, which then may amplify or antagonize each other. It will be interesting to test in vivo if, and importantly how, subsequent or parallel cross-presentation of multiple activation-stimuli (i.e., pro- and anti-inflammatory signals such as LPS, IFNγ, TNFα, IL-6, IL-4, or IL-10) may skew and define the process of metabolic reprograming in macrophages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
3. Bakker A. Einige uebereinstimmungen im stoffwechsel der carcinomzellen und exsudatleukocyten. Klin Wochenschr (1927) 6:252–4. doi: 10.1007/BF01710710
6. Schade H, Claußen F, Häbler C, Hoff F, Mochizucki N, Birner M. Weitere untersuchungen der molekularpathologie der entzündung: die exsudate. Ztschr f d ges Exp Med (1926) 49(1):334–410. doi:10.1007/BF02622491
7. Dinh PH, Corraza F, Mestdagh K, Kassengera Z, Doyen V, Michel O. Validation of the cantharidin-induced skin blister as an in vivo model of inflammation. Br J Clin Pharmacol (2011) 72(6):912–20. doi:10.1111/j.1365-2125.2011.04020.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem (1959) 234(6):1355–62.
9. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J (1986) 239(1):121–5.
10. Newsholme P, Newsholme EA. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem J (1989) 261(1):211–8.
11. Murphy C, Newsholme P. Importance of glutamine metabolism in murine macrophages and human monocytes to l-arginine biosynthesis and rates of nitrite or urea production. Clin Sci (Lond) (1998) 95(4):397–407. doi:10.1042/CS19980194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi:10.1038/nature07201
13. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
14. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122(3):787–95. doi:10.1172/JCI59643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab (2013) 18(2):251–64. doi:10.1016/j.cmet.2013.06.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi:10.1038/nri2448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447(7147):972–8. doi:10.1038/nature05836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi:10.3389/fimmu.2014.00420
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14. doi:10.4049/jimmunol.0901698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15(6):813–26. doi:10.1016/j.cmet.2012.04.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi:10.1038/nature11986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab (2015) 21(1):65–80. doi:10.1016/j.cmet.2014.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4(1):13–24. doi:10.1016/j.cmet.2006.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112(5):645–57. doi:10.1016/S0092-8674(03)00154-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun (1996) 64(1):108–12.
27. Shulga N, Pastorino JG. Hexokinase II binding to mitochondria is necessary for Kupffer cell activation and is potentiated by ethanol exposure. J Biol Chem (2014) 289(38):26213–25. doi:10.1074/jbc.M114.580175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene (2006) 25(34):4777–86. doi:10.1038/sj.onc.1209603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Wamelink MM, Struys EA, Jansen EE, Levtchenko EN, Zijlstra FS, Engelke U, et al. Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: elucidation of the CARKL gene. Hum Mutat (2008) 29(4):532–6. doi:10.1002/humu.20685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kardon T, Stroobant V, Veiga-da-Cunha M, Schaftingen EV. Characterization of mammalian sedoheptulokinase and mechanism of formation of erythritol in sedoheptulokinase deficiency. FEBS Lett (2008) 582(23–24):3330–4. doi:10.1016/j.febslet.2008.08.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Touchman JW, Anikster Y, Dietrich NL, Maduro VV, McDowell G, Shotelersuk V, et al. The genomic region encompassing the nephropathic cystinosis gene (CTNS): complete sequencing of a 200-kb segment and discovery of a novel gene within the common cystinosis-causing deletion. Genome Res (2000) 10(2):165–73. doi:10.1101/gr.10.2.165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145(5):732–44. doi:10.1016/j.cell.2011.03.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ruiz-Garcia A, Monsalve E, Novellasdemunt L, Navarro-Sabate A, Manzano A, Rivero S, et al. Cooperation of adenosine with macrophage Toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene. J Biol Chem (2011) 286(22):19247–58. doi:10.1074/jbc.M110.190298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure (1998) 6(2):195–210. doi:10.1016/S0969-2126(98)00021-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Tan Z, Xie N, Banerjee S, Cui H, Fu M, Thannickal VJ, et al. The monocarboxylate transporter 4 is required for glycolytic reprogramming and inflammatory response in macrophages. J Biol Chem (2015) 290(1):46–55. doi:10.1074/jbc.M114.603589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Costa Leite T, Da Silva D, Guimaraes Coelho R, Zancan P, Sola-Penna M. Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J (2007) 408(1):123–30. doi:10.1042/BJ20070687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science (2011) 334(6057):806–9. doi:10.1126/science.1207861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab (2011) 13(5):540–9. doi:10.1016/j.cmet.2011.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol (2006) 177(10):7303–11. doi:10.4049/jimmunol.177.10.7303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol (2012) 22(11):557–66. doi:10.1016/j.tcb.2012.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol (2010) 345:105–20. doi:10.1007/82_2010_74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi:10.1038/ni.2956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493(7432):346–55. doi:10.1038/nature11862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38(4):633–43. doi:10.1016/j.immuni.2013.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol (2011) 6:275–97. doi:10.1146/annurev-pathol-011110-130138
47. Board M, Humm S, Newsholme EA. Maximum activities of key enzymes of glycolysis, glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J (1990) 265(2):503–9.
48. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc (2014). doi:10.1111/brv.12140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Kruger NJ, von Schaewen A. The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol (2003) 6(3):236–46. doi:10.1016/S1369-5266(03)00039-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis (2008) 31(6):703–17. doi:10.1007/s10545-008-1015-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal (2012) 24(5):981–90. doi:10.1016/j.cellsig.2012.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Pollak N, Dolle C, Ziegler M. The power to reduce: pyridine nucleotides – small molecules with a multitude of functions. Biochem J (2007) 402(2):205–18. doi:10.1042/BJ20061638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem (2002) 277(46):44548–56. doi:10.1074/jbc.M202970200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Chandel NS, Trzyna WC, McClintock DS, Schumacker PT. Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J Immunol (2000) 165(2):1013–21. doi:10.4049/jimmunol.165.2.1013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Perl A, Gergely P Jr, Puskas F, Banki K. Metabolic switches of T-cell activation and apoptosis. Antioxid Redox Signal (2002) 4(3):427–43. doi:10.1089/15230860260196227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity (2008) 29(1):21–32. doi:10.1016/j.immuni.2008.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Kumar S, Rabson AB, Gelinas C. The RxxRxRxxC motif conserved in all Rel/kappa B proteins is essential for the DNA-binding activity and redox regulation of the v-Rel oncoprotein. Mol Cell Biol (1992) 12(7):3094–106.
58. Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A (1991) 88(10):4328–32. doi:10.1073/pnas.88.10.4328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Wang F, Wang LY, Wright D, Parmely MJ. Redox imbalance differentially inhibits lipopolysaccharide-induced macrophage activation in the mouse liver. Infect Immun (1999) 67(10):5409–16.
60. Kindzelskii AL, Zhou MJ, Haugland RP, Boxer LA, Petty HR. Oscillatory pericellular proteolysis and oxidant deposition during neutrophil locomotion. Biophys J (1998) 74(1):90–7. doi:10.1016/S0006-3495(98)77770-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Olsen LF, Kummer U, Kindzelskii AL, Petty HR. A model of the oscillatory metabolism of activated neutrophils. Biophys J (2003) 84(1):69–81. doi:10.1016/S0006-3495(03)74833-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kindzelskii AL, Huang JB, Chaiworapongsa T, Fahmy RM, Kim YM, Romero R, et al. Pregnancy alters glucose-6-phosphate dehydrogenase trafficking, cell metabolism, and oxidant release of maternal neutrophils. J Clin Invest (2002) 110(12):1801–11. doi:10.1172/JCI200215973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Adachi Y, Kindzelskii AL, Ohno N, Yadomae T, Petty HR. Amplitude and frequency modulation of metabolic signals in leukocytes: synergistic role of IFN-gamma in IL-6- and IL-2-mediated cell activation. J Immunol (1999) 163(8):4367–74.
64. Petty HR, Kindzelskii AL, Chaiworapongsa T, Petty AR, Romero R. Oxidant release is dramatically increased by elevated glucose concentrations in neutrophils from pregnant women. J Matern Fetal Neonatal Med (2005) 18(6):397–404. doi:10.1080/14767050500361679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Slaby O, Lebiedz D. Oscillatory NAD(P)H waves and calcium oscillations in neutrophils? A modeling study of feasibility. Biophys J (2009) 96(2):417–28. doi:10.1016/j.bpj.2008.09.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Cooper MR, DeChatelet LR, McCall CE, LaVia MF, Spurr CL, Baehner RL. Complete deficiency of leukocyte glucose-6-phosphate dehydrogenase with defective bactericidal activity. J Clin Invest (1972) 51(4):769–78. doi:10.1172/JCI106871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Roos D, van Zwieten R, Wijnen JT, Gomez-Gallego F, de Boer M, Stevens D, et al. Molecular basis and enzymatic properties of glucose 6-phosphate dehydrogenase volendam, leading to chronic nonspherocytic anemia, granulocyte dysfunction, and increased susceptibility to infections. Blood (1999) 94(9):2955–62.
68. van Bruggen R, Bautista JM, Petropoulou T, de Boer M, van Zwieten R, Gomez-Gallego F, et al. Deletion of leucine 61 in glucose-6-phosphate dehydrogenase leads to chronic nonspherocytic anemia, granulocyte dysfunction, and increased susceptibility to infections. Blood (2002) 100(3):1026–30. doi:10.1182/blood.V100.3.1026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Efferth T, Fabry U, Osieka R. DNA damage and apoptosis in mononuclear cells from glucose-6-phosphate dehydrogenase-deficient patients (G6PD Aachen variant) after UV irradiation. J Leukoc Biol (2001) 69(3):340–2.
70. Wilmanski J, Siddiqi M, Deitch EA, Spolarics Z. Augmented IL-10 production and redox-dependent signaling pathways in glucose-6-phosphate dehydrogenase-deficient mouse peritoneal macrophages. J Leukoc Biol (2005) 78(1):85–94. doi:10.1189/jlb.0105010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Wilmanski J, Villanueva E, Deitch EA, Spolarics Z. Glucose-6-phosphate dehydrogenase deficiency and the inflammatory response to endotoxin and polymicrobial sepsis. Crit Care Med (2007) 35(2):510–8. doi:10.1097/01.CCM.0000254337.50361.2E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Sanna F, Bonatesta RR, Frongia B, Uda S, Banni S, Melis MP, et al. Production of inflammatory molecules in peripheral blood mononuclear cells from severely glucose-6-phosphate dehydrogenase-deficient subjects. J Vasc Res (2007) 44(4):253–63. doi:10.1159/000100903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Ham M, Lee JW, Choi AH, Jang H, Choi G, Park J, et al. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol (2013) 33(12):2425–35. doi:10.1128/MCB.01260-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Suzuki M, O’Dea JD, Suzuki T, Agar NS. 2-Deoxyglucose as a substrate for glutathione regeneration in human and ruminant red blood cells. Comp Biochem Physiol B (1983) 75(2):195–7. doi:10.1016/0305-0491(83)90312-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Zhao J, Zhong CJ. A review on research progress of transketolase. Neurosci Bull (2009) 25(2):94–9. doi:10.1007/s12264-009-1113-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Samland AK, Sprenger GA. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol (2009) 41(7):1482–94. doi:10.1016/j.biocel.2009.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Bar-Even A, Flamholz A, Noor E, Milo R. Thermodynamic constraints shape the structure of carbon fixation pathways. Biochim Biophys Acta (2012) 1817(9):1646–59. doi:10.1016/j.bbabio.2012.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Nagy C, Haschemi A. Sedoheptulose kinase regulates cellular carbohydrate metabolism by sedoheptulose 7-phosphate supply. Biochem Soc Trans (2013) 41(2):674–80. doi:10.1042/BST20120354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Price NE, Cook PF. Kinetic and chemical mechanisms of the sheep liver 6-phosphogluconate dehydrogenase. Arch Biochem Biophys (1996) 336(2):215–23. doi:10.1006/abbi.1996.0551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Karadsheh NS, Tejwani GA, Ramaiah A. Sedoheptulose-7-phosphate kinase activity of phosphofructokinase from the different tissues of rabbit. Biochim Biophys Acta (1973) 327(1):66–81. doi:10.1016/0005-2744(73)90104-6
81. Venkataraman R, Racker E. Mechanism of action of transaldolase. I. Crystalization and properties of yeast enzyme. J Biol Chem (1961) 236:1876–82.
82. Lassen N, Black WJ, Estey T, Vasiliou V. The role of corneal crystallins in the cellular defense mechanisms against oxidative stress. Semin Cell Dev Biol (2008) 19(2):100–12. doi:10.1016/j.semcdb.2007.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes (2008) 57(10):2809–17. doi:10.2337/db06-1003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Hanczko R, Fernandez DR, Doherty E, Qian Y, Vas G, Niland B, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest (2009) 119(6):1546–57. doi:10.1172/JCI35722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Clasquin MF, Melamud E, Singer A, Gooding JR, Xu X, Dong A, et al. Riboneogenesis in yeast. Cell (2011) 145(6):969–80. doi:10.1016/j.cell.2011.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Blackmore PF, Shuman EA. Regulation of hepatic altro heptulose 1,7-bisphosphate levels and control of flux through the pentose pathway by fructose 2,6-bisphosphate. FEBS Lett (1982) 142(2):255–9. doi:10.1016/0014-5793(82)80147-6
87. Belyaeva NF, Golubev MA, Grigorovich JA, Dubinsky ZV, Semenova NA, Pitkanen E, et al. The involvement of fructose 2,6-bisphosphate in substrate cycle control in the nonoxidative stage of the pentose phosphate pathway. A phosphorus magnetic resonance spectroscopy study. Experientia (1994) 50(8):780–4. doi:10.1007/BF01919382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: immunometabolism, inflammation, macrophage activation, metabolic reprograming, primary carbohydrate metabolism, pentose phosphate pathway, sedoheptulose kinase, time and demand
Citation: Nagy C and Haschemi A (2015) Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front. Immunol. 6:164. doi: 10.3389/fimmu.2015.00164
Received: 30 January 2015; Accepted: 26 March 2015;
Published online: 08 April 2015.
Edited by:
Christian Frezza, Hutchison/MRC Research Institute, UKReviewed by:
Mihai Netea, Radboud University Nijmegen Medical Center, NetherlandsGaurav K. Gupta, Massachusetts General Hospital and Harvard Medical School, USA
Copyright: © 2015 Nagy and Haschemi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arvand Haschemi, Department of Laboratory Medicine (KILM), Medical University of Vienna, Lazarettgasse 14, Vienna 1090, Austria e-mail: arvand.haschemi@meduniwien.ac.at