 Christine Braegelmann
Christine Braegelmann Tanja Fetter
Tanja Fetter Dennis Niebel
Dennis Niebel Joerg Wenzel
Joerg Wenzel- Department of Dermatology and Allergy, University Hospital Bonn, Bonn, Germany
Interface dermatitis is a histopathological pattern mirroring a distinct cytotoxic immune response shared by a number of clinically diverse inflammatory skin diseases amongst which lichen planus and cutaneous lupus erythematosus are considered prototypic. Interface dermatitis is characterized by pronounced cytotoxic immune cell infiltration and necroptotic keratinocytes at the dermoepidermal junction. The initial inflammatory reaction is established by cytotoxic immune cells that express CXC chemokine receptor 3 and lesional keratinocytes that produce corresponding ligands, CXC motif ligands 9/10/11, recruiting the effector cells to the site of inflammation. During the resulting anti-epithelial attack, endogenous immune complexes and nucleic acids are released from perishing keratinocytes, which are then perceived by the innate immune system as danger signals. Keratinocytes express a distinct signature of pattern recognition receptors and binding of endogenous nucleic acid motifs to these receptors results in interferon-mediated immune responses and further enhancement of CXC chemokine receptor 3 ligand production. In this perspective article, we will discuss the role of innate nucleic acid sensing as a common mechanism in the perpetuation of clinically heterogeneous diseases featuring interface dermatitis based on own data and a review of the literature. Furthermore, we will introduce a keratinocyte-specific in vitro model of interface dermatitis as follows: Stimulation of human keratinocytes with endogenous nucleic acids alone and in combination with interferon gamma leads to pronounced production of distinct cytokines, which are essential in the pathogenesis of interface dermatitis. This experimental approach bears the capability to investigate potential therapeutics in this group of diseases with unmet medical need.
Introduction
Interface dermatitis (ID), also referred to as lichenoid tissue reaction, describes a histopathological pattern defined by morphological anomalies of the epidermal basal cell layer characterized by perishing keratinocytes labeled vacuolar or hydropic colloid bodies. Anti-epithelial activity of autoreactive cytotoxic lymphocytes is causative (1). The distinct pattern is observed in clinically heterogeneous skin diseases including autoimmune skin disorders [lichen planus (LP), lichen sclerosus (LS), cutaneous lupus erythematosus (CLE), dermatomyositis (DM)] and immunologic reactions against viruses, drugs and specific tumors (“lichenoid” keratosis) (2, 3). In 1995, for the first time, Fäh et al. detected MxA expression not only in virally infected tissue but also in dermatoses featuring ID (4). These findings are explained by MxA expression being directly induced by type-I and type-III IFNs (5). Today, it is accepted that activation of the interferon system resulting in a cellular immune response is a pathogenic key feature of the histologic “look-alikes” sharing ID (1).
Amongst the autoimmune skin disorders featuring ID, LP, and CLE are considered prototypic (6): LP may affect the skin including its appendages and both oral and genital mucosa (7). Classical LP presents with violaceous papules generally accompanied by extensive pruritus. When affecting the nails, thinning, scarring, and even complete loss of the nail is possible. Lichen planopilaris affecting the hair follicles may cause scarring and permanent baldness. Affected mucosa usually presents as erosive (8). The clinical spectrum of lupus erythematosus is broad ranging from systemic manifestations [systemic lupus erythematosus (SLE)] to manifestations solely affecting the skin. Cutaneous lupus erythematosus (CLE) may present itself as one symptom of SLE or may occur as an isolated skin disease (9, 10). CLE manifestation can be further subdivided into four main subsets (acute, subacute, intermittent, or chronic). Acute CLE may present either with a localized facial indurated erythematous lesion (malar rash) or with a widespread erythematous maculopapular rash. Subacute CLE is characterized by either annular/polycyclic or by papulosquamous skin lesions. Intermittent CLE shows non-scaling and non-scarring skin lesions. The last subset is chronic CLE which may be further subdivided into chronic discoid lupus erythematosus, chilblain lupus and lupus erythematosus profundus. Chronic discoid lupus erythematosus constitutes the largest group and features scarring erythrosquamous plaques in a disc-like shape. Chilblain lupus is a rare acral variant of chronic CLE whereas lupus erythematosus profundus affects the subcutaneous fat (11).
Despite clinical heterogeneity and although the initial stimulus may differ between diseases featuring ID, final common path is a cytotoxic anti-epithelial directed attack by autoreactive T lymphocytes (12–14) that are recruited to the site of inflammation by keratinocytes producing large amounts of C-X-C Motif Chemokine Ligands 9/10/11 (CXCL9/10/11) (15).
We herein summarize etiopathological mechanisms involved in ID and particularly outline the role of innate nucleic acid sensing in keratinocytes as a hallmark of perpetuation of the proceeding “pro-inflammatory vicious circle”. We, furthermore, present a human in vitro model that functions as a tool to evaluate potential therapeutic interventions and thus facilitate prediction of therapeutic response to novel treatment strategies in diseases featuring ID.
Interface Dermatitis—The Pathogenic Background
Interferon Signaling and Cellular Response in Interface Dermatitis
A multitude of genes is differentially expressed similarly in both LP and CLE skin when compared to healthy skin. In particular, distinct associations have been described for genes concerning interferon signaling as well as associated downstream cascades (16–18). The type-I [IFNalpha(a)/beta(b)/kappa(k)] (1, 19–21) and type-III interferon system [IFNlambda(λ)] (22) do not only participate in antiviral immune defense, but also play an important pathophysiological role in ID. Particularly, they are expressed by respective lesional keratinocytes. Via autocrine loops, IFNs bind to their corresponding receptor on keratinocytes and unleash pro-inflammatory downstream cascades via activation of the JAK-STAT pathway (23–25). Finally, inflamed keratinocytes express CXCL9 (22), CXCL10 (26, 27) and CXCL11 (28). The corresponding CXCR3 receptors are expressed on activated pDCs (29), T cells [Th1-type CD4+ T cells (30) and effector CD8+ T cells (31–33)] and macrophages (34, 35). Hereby attracted pDCs contribute to the inflammation via further type-I interferon production (36, 37) and Th1 lymphocytes create a specific inflammatory milieu via secretion of distinct cytokines. T cells isolated from lesional skin of LP and CLE patients revealed high frequency of IFNy (IFN gamma) and TNFa, two key cytokines of Th1 lymphocytes (16). The type-II interferon, IFNy, sparks downstream cascades which partly overlap with those of type-I interferons (38): It induces CXCR3 ligands and the differentiation of naïve T cells into Th1 cells and it activates macrophages (39). In response to stimulation with IFNy and TLR (Toll Like Receptor) ligands macrophages undergo classical “M1” activation (40): This pro-inflammatory M1 phenotype is prevalently seen in rheumatic diseases (41) and has specifically been described in lichen planus (42) and lupus skin (43). Cytotoxic lymphocytes represent the last group of CXCR3 receptor carrying immune cells and execute their anti-epidermal attack via cytotoxic granules and the perforine/granzyme pathway (44). Apart from upregulation of genes mediating direct cytotoxicity, enhanced expression of markers of apoptosis (FASL) and necroptosis (RIP3) have been detected in ID (16).
Nucleic Acid Sensing Induces Interferon Response and Mediates Inflammasome Activation as Well as Cell Death Cascades
Inflammatory cell death upon cytotoxic attack inevitably results in release of intracellular components, amongst them are endogenous nucleic acids (eNA). Nucleic acid sensing by the innate immune system functions via pattern recognition receptors (PRRs), that are activated by pathogen associated molecular patterns (PAMPs) or host molecules (damage associated molecular patterns, DAMPs) (45). Physiologically, PRRs enable sufficient immune response to either an invading pathogen or damage of host cells (46, 47). Sensing of self-RNA and self-DNA, however, also holds the potential to contribute to autoimmunity (45, 48, 49). In ID, the pro-inflammatory capacity of released nucleic acids is supposedly supported by the cathelicidin LL37 which has been shown to be overexpressed in CLE (50, 51), LP (52), and DM (53). Its complex formation with nucleic acids has been proven to enable transport of extracellular nucleic acid fragments into intracellular compartments (54). Specifically, our working group has previously shown that addition of LL37 enhances immunogenicity of nucleic acids in keratinocytes in vitro (50). Key features of downstream signaling of PRRs include induction of interferons (45, 46, 55) and inflammasome activation (56) as well as programmed cell death cascades (46).
The respective downstream mechanisms of important PRRs are as follows:
AIM2 (Absent In Melanoma 2) activates the inflammasome upon double stranded (ds) DNA sensing (57–59) which leads to Caspase 1 cleavage and finally maturation of the pro-inflammatory interleukins IL18 and IL1ß (60). Furthermore, activated Caspase 1 cleaves Gasdermin D which executes pyroptosis via pore formation in affected cell membranes (61). AIM2 is upregulated in skin samples of lichen planus patients (62). Inflammasome activity is enhanced in lupus erythematosus (63) and the inflammasome-activated cytokine IL18 is highly upregulated in the epidermis of CLE patients (64). The discovery of its dysregulation in autoimmunity suggests inhibition of inflammasome components as an interesting therapeutic approach, as postulated by Kahlenberg et al. (65).
Upon DNA binding to cGAS (Cyclic GMP-AMP synthase) an IFN response is unleashed (66): CGAS activates STING (Stimulator of IFN genes) (67, 68) which, in turn, interacts with TBK1 (TANK-binding kinase) resulting in phosphorylation of IRF3 (Interferon Regulatory Factor) and finally type-I interferon gene transcription (69). Furthermore, the cGAS-STING pathway has multiple functions in mediating cell death that are not fully elucidated, yet (46). Its activation by self-DNA is described as an important mechanism in autoimmunity which might constitute another promising target for therapeutic intervention (49).
IFI16 (Gamma-interferon-inducible protein 16) is a further key DNA sensor in human keratinocytes. It cooperates with cGAS in the activation of STING (70). Excessive IFI16-dependent production of IFN-I is considered an important mediator of autoimmune inflammation (71, 72) and has specifically been shown to contribute to SLE (73) and to cytokine induction in keratinocytes (74). Apart from STING-dependent type-I IFN production, IFI16 enables direct inflammasome activation (75, 76).
ZBP1 (Z-DNA Binding Protein) binds to ds nucleic acids when presenting in the unusual Z‐conformation (77). Activated ZBP1 recruits TBK1 and IRF3 (78) and triggers RIP3-dependent necroptosis (79) as well as NLRP3-dependent inflammasome activation (80). Specifically, aberrant sensing of endogenous nucleic acids by ZBP1 has been shown to induce inflammation in murine skin (81). Guo et al. have shown that ZBP1 activation also induces necroptosis in human cells (82) and sera from some SLE patients exhibit anti‐Z‐DNA autoantibodies (83). Thus, ZBP1 has been suggested as a potential therapeutic target that requires further research (84).
RIG-I-like receptors (RLRs) comprise three important sensors: RIG-I, MDA5 (Melanoma differentiation-associated protein), and LGP2 (Laboratory of Genetics and Physiology 2) (85) with the latter being considered a regulator of the others. RIG-I and MDA5 experience conformational changes upon cytosolic RNA sensing that result in exposure of their CARD domain and consecutive activation of IRF3 via phosphorylation of TBK1 and NFkB activation (85, 86). RLRs are also implicated in apoptosis and RIP3-mediated necroptosis (87, 88). Human keratinocytes constitutively express RIG-I and MDA5 (89). Challenge with IFNy or TNFa has induced RIG-I in a human keratinocyte cell line (90) and both RIG-I and MDA5 expression is increased in psoriatic skin (90). A specific single nucleotide polymorphism in the gene encoding MDA5 has been identified in autoimmune diseases, including SLE (91, 92).
Nucleic-acid-sensing TLRs are mainly expressed in the endosomes (93) and comprise TLR3 [recognizes dsRNA (94)], TLR7 and TLR8 [recognize ssRNA (95)] and TLR9 [recognizes unmethylated CpG-containing DNA motifs (96)]. Activation of these TLRs, with the exception of TLR3, incorporates MyD88 to the respective receptor complex which subsequently interacts with TRAF6 leading to nuclear translocation of NFkB (97) and type-I IFN induction (98). TLR3 alternatively signals via the adaptor TRIF which activates TBK1 and subsequently leads to type-I IFN induction via phosphorylation of IRF3 (97). Furthermore, TLR3 signaling can activate cell death cascades by engaging RIP1 and RIP3 (99). Keratinocytes, constitutively express TLR 3 and 9 and their stimulation with corresponding ligands results in induction of TNFa and type-I IFN as well as ICAM1 (100). Interestingly, in oral LP, induction of TLR9 has been described (101). Although keratinocytes do not constitutively express TLR7 or 8, several case reports describe individuals who have developed LP and LS upon use of Imiquimod, an agonist of TLR7/8 (102–104) which is possibly explained by keratinocytes expressing TLR7 under specific conditions (105).
An In Vitro Model to Study Interface Dermatitis
Background
In 2016, our working group has first established an in vitro model that mirrors our understanding of ID as being fueled by endogenous nucleic acids (106) and further characterized it within a study from 2017 (50). Stimulation with eNA results in a pronounced expression of typical ID-associated cytokines within different keratinocyte models. IFNy, mainly produced by lymphocytes, is known to play a pivotal role in the pathogenesis of diseases featuring ID (107–109), and has been shown to induce typical morphological changes in human epidermis equivalents, in vitro (110). Herein, we aim to deliver an in-depth analysis of differentially regulated genes in our ID model and furthermore present synergistic effects of endogenous nucleic acids in addition to IFNy on human keratinocytes.
Results
Cytosolic Localization of DNA Motifs in Interface Dermatitis Keratinocytes
DNA motifs in extranuclear compartments were significantly more present in ID than in healthy control samples (Figure 1A).
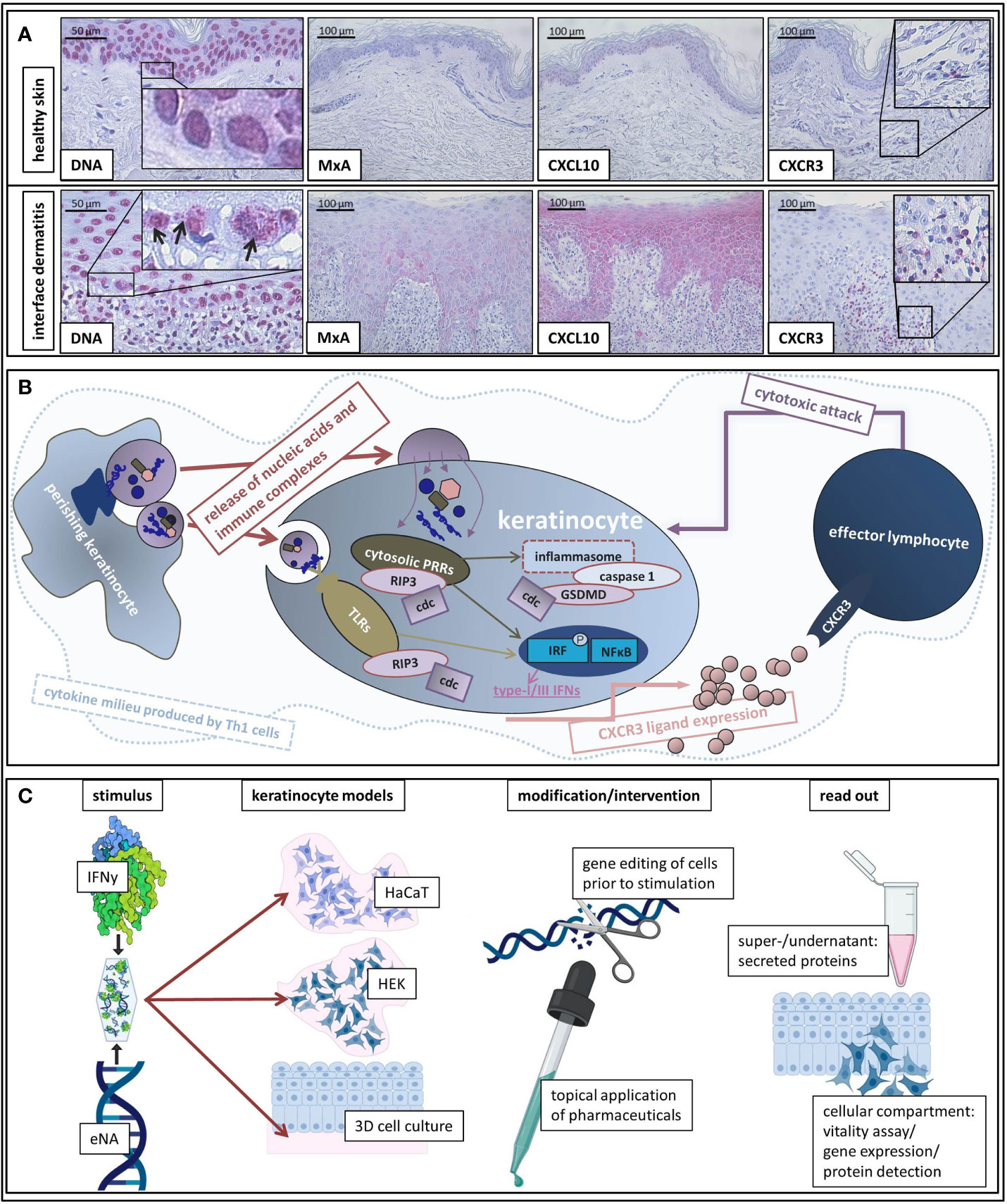
Figure 1 (A) Representative histological findings in interface dermatitis and healthy skin. Representative findings of DNA, MxA, CXCL10, and CXCR3 immunostaining in healthy skin and interface dermatitis (lichen planus). Original magnification x200 (x400 concerning DNA). Black arrows highlight extranuclear localization of DNA motifs. Boxes highlight digitally enlarged aspects. (B) Schematic presentation of assumed etiopathological mechanisms of interface dermatitis (as reviewed above). ID is characterized by a Th1 cytokine milieu in which endogenous nucleic acids activate PRRs. Downstream signaling unleashes cell death cascades (cdc) and leads to production of type-I and -III IFNs and pro-inflammatory cytokines as well as inflammasome activation. Interferon-inducible chemokines (produced by keratinocytes upon autocrine IFN-stimulation) recruit CXCR3 + effector cells into lesional skin, which induce keratinocyte perishing and thus release of pro-inflammatory cell components. (C) Schematic presentation of our in vitro model of interface dermatitis. Nucleic acids extracted from keratinocytes and IFNy are administered to different keratinocyte models (HaCaT, HEK, epiCS) as an ID-like stimulus. Genetic modification of the cells of interest can be made prior to stimulation in order to evaluate specific components of ID pathogenesis. Furthermore, the effect of innovative pharmaceuticals on ID-like stimulation can be analyzed. Super-/undernatants and the cellular compartment are available to read out methods.
CXCL10 and MxA are Expressed by Keratinocytes in Interface Dermatitis and the Majority of Infiltrating Immune Cells Express CXCR3 Receptors
Figure 1A depicts findings within a LP skin specimen that are representative for all examined samples: MxA (MX Dynamin Like GTPase A) and CXCL10 are expressed by keratinocytes and the majority of infiltrating immune cells carries CXCR3 receptors.
Stimulation With Endogenous Nucleic Acids Induces a Molecular Signature in Keratinocytes Resembling Interface Dermatitis
In normal human epidermal keratinocytes (HEK), stimulation with eNA significantly induces expression of genes encoding key drivers (111–117) of innate inflammatory pathways (IRFs, IFNs, STAT2, RELA, NFkB, CXCL9/10/11, Mx1, OASL), inflammasome activation (AIM2), cell death (RIP3) and factors mediating interaction between keratinocytes and T cells (ICAM1) as well as the adaptive immune system (BLyS) (Figure 2A).
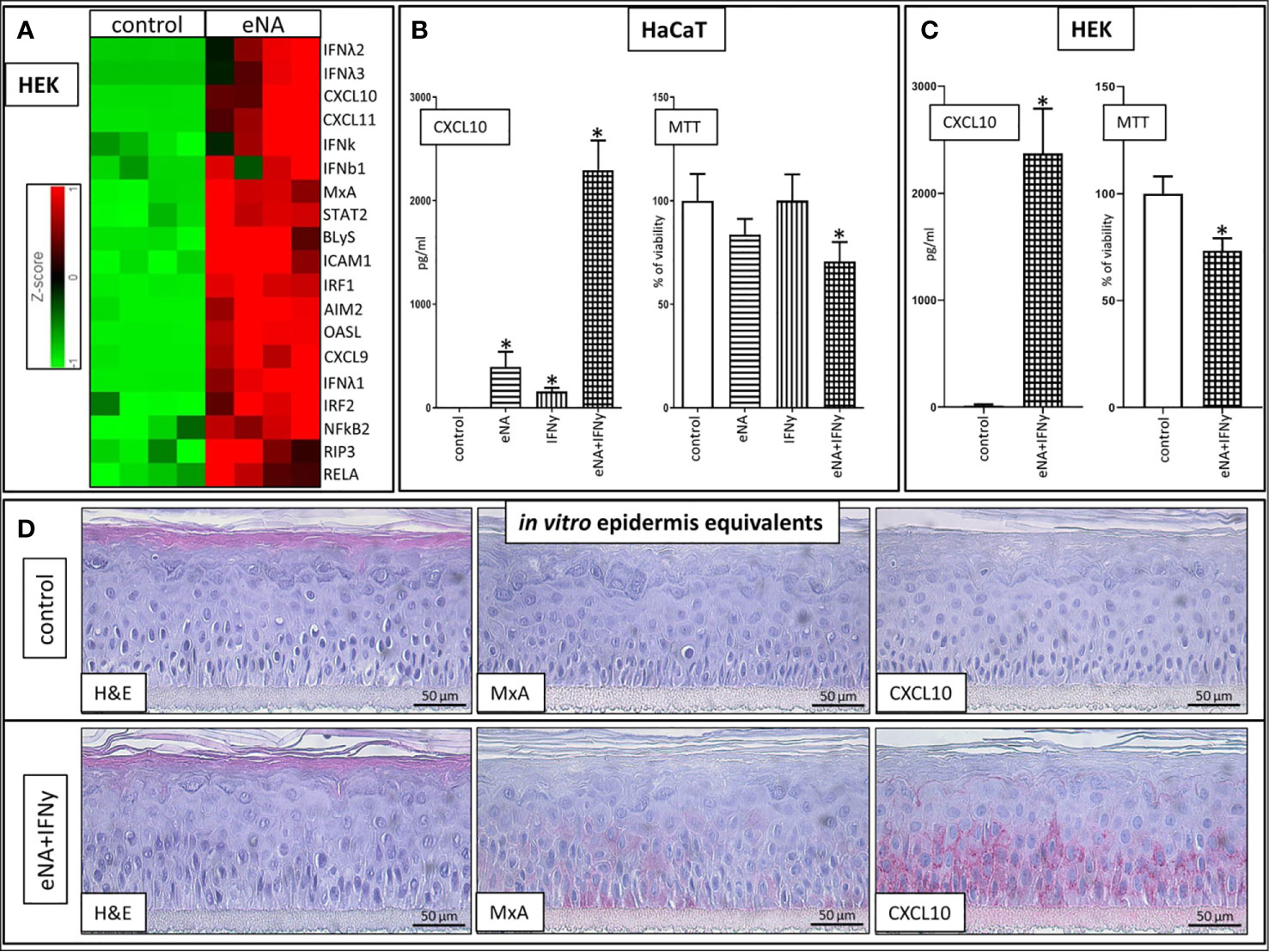
Figure 2 (A) Expression of upregulated genes involved in ID pathogenesis in HEK cells stimulated with eNA (12,5 µg/ml) for 24 h compared to HEK cells solely exposed to medium (control), (n = 4, fold change > 2, p < 0.01, Partek® Flow®). (B) CXCL10 levels within the supernatant of HaCaT cells stimulated with eNA (5 µg/ml) or IFNy (1 x 10^3 U/ml) or the combination of both for 20 h compared to HaCaT cells solely exposed to medium (control), MTT assay executed on the cells corresponding with the respective supernatant, mean of controls defined as 100%. Given are respective means with standard deviations indicated by error bars (n = 4, * indicates significance (p < 0.05), Mann Whitney test). (C) CXCL10 levels within the supernatant of HEK cells stimulated with the combination of eNA (5 µg/ml) and IFNy (1 x 10^3 U/ml) for 6.5 h compared to HEK cells solely exposed to medium (control), MTT assay executed on the cells corresponding with the respective supernatant, mean of controls defined as 100%. Given are respective means with standard deviations indicated by error bars (n = 4, * indicates significance (p < 0.05), Mann Whitney test). (D) Representative findings in 3D epidermis equivalents upon control (medium) settings and stimulation with eNA (5 µg/ml) and IFNy (1 x 10^3 U/ml) for 22 h. Hematoxylin and eosin stain, MxA, CXCL10. Original magnification ×400. (n = 3).
Stimulation With IFNy and Endogenous Nucleic Acids Induce CXCL10 and MxA Expression
Stimulation with IFNy or eNA respectively leads to significant expression of CXCL10 in HaCaT cells. Combined administration of IFNy and eNA results in an over-additive effect concerning CXCL10 release (Figure 2B). Strong CXCL10 expression upon concomitant stimulation could be confirmed in normal human epidermal keratinocytes (HEK, Figure 2C) and in reconstructed human epidermis equivalents (epiCS, Figure 2D). Furthermore, MxA expression is induced upon combined stimulation in epiCS whose staining resembles the pattern detected in ID (compare Figure 1A).
Concomitant Stimulation with IFNy and Endogenous Nucleic Acids Has a Direct Cytotoxic Effect on Keratinocytes
Cytotoxic effects in our approach were not significant upon stimulation with eNA or IFNy alone. Upon concomitant stimulation, however, the ability to reduce MTT reagent into its insoluble formazan was significantly impaired in both HaCaT (Figure 2B) and HEK (Figure 2C) serving as a marker for cell viability.
Methods
Please find a description of applied methods as a supplement to this main text.
Discussion
Our working group has previously described extranuclear DNA motifs being significantly more present in keratinocytes of CLE patients than in healthy skin (50). We herein present analogous findings in LP patients. In vitro, stimulation with endogenous nucleic acids induces a gene expression pattern in human keratinocytes which resembles key features of ID: We present IFNλ induction upon stimulation with eNA in keratinocytes, which is known to be significantly elevated in skin diseases featuring ID but neither in healthy controls nor other inflammatory skin diseases (22). The type-I IFNs, IFNb and IFNk, are both induced in keratinocytes upon stimulation with eNA. IFNb has been described to be expressed in basal epidermal layers of LP (118) skin. IFNk has been shown to be highly expressed in CLE (19) and LP skin but not in other inflammatory dermatoses (119) and is acknowledged to be a key regulator of IFN response in keratinocytes. Stimulation with eNA did not upregulate expression of IFNa. Although it has been detected in keratinocytes of the whole epidermis in LP skin (118), pDCs are considered to be the main producers of IFNa in vivo (17, 36, 120). Via autocrine loops, all type-I IFNs bind to IFNAR (23, 24, 121) and type-III IFNs signal via their receptor IFNLR (24). Activation of both receptors causes phosphorylation of JAK1 and TYK2 (25). Receptor bound STATs (STAT1 and STAT2) are subsequently phosphorylated leading to heterodimerization and formation of ISGF3 together with IRF9 (122). This complex translocates to the nucleus and induces expression of genes that exhibit specific ISREs (Interferon-sensitive response elements) to which the complex binds. Amongst such genes are OAS, MxA and multiple transcription factors, including IRFs (24, 38) which are induced upon eNA-stimulation in our experiments. While IFNAR is expressed on nearly all cell types and IFNLR is mainly restricted to epithelial cells, their downstream signaling is quite congruent (24). Type-I IFN dependency (24), however, is described for ISGF3-like complex formation, which consists of IRF9 and STAT2 homodimers and can reinstate inflammatory cascades in the absence of STAT1 (123).
In our approach, type-II IFN was not induced in keratinocytes upon eNA-stimulation, which is in accordance with the view that the pronounced presence of type-II IFN (IFNy) in ID skin derives from other sources than keratinocytes. Specifically, it is predominantly produced by natural killer cells, group 1 innate lymphoid cells, yδ T cells and CD8+ cytotoxic T cells as well as CD4+ Th1 cells [as reviewed in (124)]. Its receptor (IFNyR) signals via the JAK1/JAK2 and STAT1/STAT2 pathway (107, 125). Shao et al. describe that, in vitro, priming of keratinocytes with type-I IFNs, and to an even greater extent type-II IFNs, increases their susceptibility to MHC I-dependent, T-cell mediated cytotoxicity (107). Knock out of JAK2 or STAT1 inhibited this induction of MHC I in keratinocytes upon IFNy-stimulation whereas only minimal suppression was detected in JAK1 or STAT2 KO cells (107). The potential of human keratinocytes as nonprofessional antigen-presenting cells has recently been further underlined by Orlik et al. who have demonstrated the capacity of IFNy-pretreated keratinocytes to activate co-cultured naïve T-cells (126). ICAM1 is a further mediator supporting interaction between T lymphocytes and keratinocytes that is inducible by IFNy (127) and is overexpressed in diseases featuring ID (128). Our data, in turn, shows that ICAM1 is also induced upon stimulation with eNA. Furthermore, cell death cannot only be induced via activation of infiltrating immune cells but also via induction of keratinocytic apoptosis (cleaved caspase 3) and necroptosis (RIP3) as both factors have been shown to be overexpressed in keratinocytes of LP and CLE patients (16, 107). These markers can be induced by IFNy (107) and although stimulation with eNAs alone does not result in a significant reduction of cell viability as measured by vitality assay, cell death cascades are activated upon stimulation with eNA that mimic the ones described in ID as we detected induction of RIP3 and the inflammasome component AIM2. Furthermore, significant cytotoxicity is detectable upon concomitant stimulation of keratinocytes with eNA and IFNy. A cross-talk by keratinocytes to the adaptive immune system is mediated by BLyS, a B lymphocyte survival factor (129) which has been described to be overexpressed in CLE and LP (130). We herein show that it is induced upon stimulation with eNA. Another mediator implicated in immune and inflammatory responses is NFkB that has been shown to be among the top regulated genes shared by LP and CLE (16). This crucial transcriptional factor family comprises NFkB1, NFkB2, RELA, RELB, and C-REL (131): Stimulation with eNA induces this important mediator in keratinocytes. As reviewed above, expression of CXCR3 ligands by lesional keratinocytes is decisive for attraction of effector cytotoxic T cells. CXCL10 (26, 27) and CXCL11 (28) have been shown to be inducible by type-I interferons. CXCL9, on the other hand, has repetitively been described as truly dependent on IFNy (26, 132). Our group, however, has demonstrated CXCL9 induction in keratinocytes upon stimulation with the type-I interferon IFNk earlier (22). Furthermore, a recent study has detected CXCL9 expression in keratinocytes as a result of inflammasome activation (133). As outlined above, inflammasome activation is another major pathway upon sensing of nucleic acids that might explain expression of all three CXCR3 ligands by stimulation with eNA in the absence of externally administered IFNy.
Stimulation of keratinocytes with endogenous nucleic acids induces key mediators of ID. According to the mechanisms discussed above, we are convinced that addition of IFNy to the interface dermatitis model promotes an even more realistic imitation of in vivo scenarios. Concomitant stimulation of keratinocytes with endogenous nucleic acids and IFNy not only promoted direct cytotoxicity but also caused an overadditive effect on CXCL10 level elevation.
Outlook
Lichen planus as well as cutaneous lupus erythematosus go along with a high disease burden and are considered therapeutically challenging because current treatments often fail to achieve disease control (134–136). We are convinced that preclinical studies and clinical trials evaluating innovative future therapeutic approaches should not focus on one particular condition but rather on clusters of diseases featuring common immune response patterns. Our working group has recently successfully employed the here described model to elucidate the influence of JAK inhibition on keratinocytes in an interface-dermatitis-like context (121). In the herein described refined version of the model IFNy mimics the presence of a T-helper cell mediated cytokine milieu and together with eNA synergistically intensifies the resulting pro-inflammatory signature. Our model represents pathomechanistic key features of ID and thus enables evaluation of potential future pharmaceuticals. It might aid in predicting therapeutic response to novel treatment strategies in therapeutically challenging diseases featuring ID.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Medical Faculty of the University of Bonn, Venusberg-Campus 1, 53127 Bonn. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CB designed the study. CB, LD, and TF performed the experiments. CB and JW analyzed the data. DN and TB contributed essential resources during manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
CB receives a scholarship from the Medical Faculty of the University of Bonn (“Gerok-scholarship”, grant numbers 2019-1A-02 and 2020-1A-07).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all employees of the Next Generation Sequencing core facility from the University of Bonn and, particularly, Dr. rer. nat. André Heimbach as well as Morgan Conlon from the Bioinformatics core facility from the University of Bonn for their most valuable support. Figure 1C was created with the help of BioRender.com. Figure 1B was created with Microsoft PowerPoint 2010.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.622511/full#supplementary-material
Abbreviations
AIM2, Absent In Melanoma 2; cGAS, Cyclic GMP-AMP synthase; CLE, Cutaneous lupus erythematosus; DAMP, Damage associated molecular patterns; DM, Dermatomyositis; ds, double stranded; eNA, endogenous nucleic acids; ID, Interface dermatitis; IFI16, Gamma-interferon-inducible protein 16; IFNa, Interferon alpha; IFNb, Interferon beta; IFNy, Interferon gamma; IFNk, Interferon kappa; IFN λ, Interferon lambda; IRF1/2/3, Interferon Regulatory Factor 1/2/3; ISREs, Interferon-sensitive response elements; LGP2, (Laboratory of Genetics and Physiology 2); LP, Lichen planus; LS, Lichen sclerosus; MDA5, Melanoma differentiation-associated protein 5; MxA, MX Dynamin Like GTPase A; PAMP, Pathogen associated molecular pattern; pDC, plasmacytoid dendritic cell; PRR, Pattern recognition receptors; RLR, RIG-I-like receptors; SLE, Systemic lupus erythematosus; ss, single stranded; STING, Stimulator of IFN genes; TBK1, TANK-binding kinase 1; TLR, Toll Like Receptor; ZBP1, Z-DNA Binding Protein 1.
References
1. Wenzel J, Tüting T. An IFN-associated cytotoxic cellular immune response against viral, self-, or tumor antigens is a common pathogenetic feature in “interface dermatitis”. J Invest Dermatol (2008) 128(10):2392–402. doi: 10.1038/jid.2008.96
2. Patterson JW. The spectrum of lichenoid dermatitis. J Cutan Pathol (1991) 18(2):67–74. doi: 10.1111/j.1600-0560.1991.tb00130.x
3. LeBoit PE. Interface dermatitis. How specific are its histopathologic features? Arch Dermatol (1993) 129(10):1324–8. doi: 10.1001/archderm.129.10.1324
4. Fäh J, Pavlovic J, Burg G. Expression of MxA protein in inflammatory dermatoses. J Histochem Cytochem (1995) 43(1):47–52. doi: 10.1177/43.1.7822763
5. Alase AA, El-Sherbiny YM, Vital EM, Tobin DJ, Turner NA, Wittmann M. IFNλ Stimulates MxA Production in Human Dermal Fibroblasts via a MAPK-Dependent STAT1-Independent Mechanism. J Invest Dermatol (2015) 135(12):2935–43. doi: 10.1038/jid.2015.317
6. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives. J Invest Dermatol (2009) 129(5):1088–99. doi: 10.1038/jid.2009.42
7. Cassol-Spanemberg J, Blanco-Carrión A, Rodríguez-de Rivera-Campillo M-E, Estrugo-Devesa A, Jané-Salas E, López-López J. Cutaneous, genital and oral lichen planus: A descriptive study of 274 patients. Med Oral Patol Oral Cir Bucal (2019) 24(1):e1–7. doi: 10.4317/medoral.22656
9. Kuhn A, Wenzel J, Bijl M. Lupus erythematosus revisited. Semin Immunopathol (2016) 38(1):97–112. doi: 10.1007/s00281-015-0550-0
10. Stannard JN, Kahlenberg JM. Cutaneous Lupus Erythematosus: Updates on Pathogenesis and Associations with Systemic Lupus. Curr Opin Rheumatol (2016) 28(5):453–9. doi: 10.1097/BOR.0000000000000308
11. Wenzel J. Cutaneous lupus erythematosus: new insights into pathogenesis and therapeutic strategies. Nat Rev Rheumatol (2019) 15(9):519–32. doi: 10.1038/s41584-019-0272-0
12. Shiohara T, Mizukawa Y. The immunological basis of lichenoid tissue reaction. Autoimmun Rev (2005) 4(4):236–41. doi: 10.1016/j.autrev.2004.11.005
13. Iijima W, Ohtani H, Nakayama T, Sugawara Y, Sato E, Nagura H, et al. Infiltrating CD8+ T cells in oral lichen planus predominantly express CCR5 and CXCR3 and carry respective chemokine ligands RANTES/CCL5 and IP-10/CXCL10 in their cytolytic granules: a potential self-recruiting mechanism. Am J Pathol (2003) 163(1):261–8. doi: 10.1016/S0002-9440(10)63649-8
14. Wenzel J, Uerlich M, Wörrenkämper E, Freutel S, Bieber T, Tüting T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br J Dermatol (2005) 153(5):1011–5. doi: 10.1111/j.1365-2133.2005.06784.x
15. Meller S, Gilliet M, Homey B. Chemokines in the pathogenesis of lichenoid tissue reactions. J Invest Dermatol (2009) 129(2):315–9. doi: 10.1038/jid.2008.251
16. Lauffer F, Jargosch M, Krause L, Garzorz-Stark N, Franz R, Roenneberg S, et al. Type I Immune Response Induces Keratinocyte Necroptosis and Is Associated with Interface Dermatitis. J Invest Dermatol (2018) 138(8):1785–94. doi: 10.1016/j.jid.2018.02.034
17. Wenzel J, Scheler M, Proelss J, Bieber T, Tüting T. Type I interferon-associated cytotoxic inflammation in lichen planus. J Cutan Pathol (2006) 33(10):672–8. doi: 10.1111/j.1600-0560.2006.00527.x
18. Wenzel J, Schmidt R, Proelss J, Zahn S, Bieber T, Tüting T. Type I interferon-associated skin recruitment of CXCR3+ lymphocytes in dermatomyositis. Clin Exp Dermatol (2006) 31(4):576–82. doi: 10.1111/j.1365-2230.2006.02150.x
19. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis (2018) 77(11):1653–64. doi: 10.1136/annrheumdis-2018-213197
20. Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol (2005) 57(5):664–78. doi: 10.1002/ana.20464
21. Rönnblom L. The importance of the type I interferon system in autoimmunity. Clin Exp Rheumatol (2016) 34(4 Suppl 98):21–4.
22. Zahn S, Rehkämper C, Kümmerer BM, Ferring-Schmidt S, Bieber T, Tüting T, et al. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J Invest Dermatol (2011) 131(1):133–40. doi: 10.1038/jid.2010.244
23. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem (2017) 292(18):7285–94. doi: 10.1074/jbc.R116.774562
24. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front Immunol (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
25. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3(9):651–62. doi: 10.1038/nrm909
26. Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front Immunol (2018) 8:1970. doi: 10.3389/fimmu.2017.01970
27. Qian C, An H, Yu Y, Liu S, Cao X. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood (2007) 109(8):3308–15. doi: 10.1182/blood-2006-08-040337
28. Rani MR, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM. Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J Biol Chem (1996) 271(37):22878–84. doi: 10.1074/jbc.271.37.22878
29. Kohrgruber N, Gröger M, Meraner P, Kriehuber E, Petzelbauer P, Brandt S, et al. Plasmacytoid dendritic cell recruitment by immobilized CXCR3 ligands. J Immunol (2004) 173(11):6592–602. doi: 10.4049/jimmunol.173.11.6592
30. Jabbari A, Suárez-Fariñas M, Fuentes-Duculan J, Gonzalez J, Cueto I, Franks AG, et al. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J Invest Dermatol (2013) 134(1):87–95. doi: 10.1038/jid.2013.269
31. Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity (2012) 37(6):1091–103. doi: 10.1016/j.immuni.2012.08.016
32. Groom JR. Regulators of T-cell fate: Integration of cell migration, differentiation and function. Immunol Rev (2019) 289(1):101–14. doi: 10.1111/imr.12742
33. Wenzel J, Zahn S, Mikus S, Wiechert A, Bieber T, Tüting T. The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br J Dermatol (2007) 157(4):752–7. doi: 10.1111/j.1365-2133.2007.08137.x
34. Björkander S, Heidari-Hamedani G, Bremme K, Gunnarsson I, Holmlund U. Peripheral monocyte expression of the chemokine receptors CCR2, CCR5 and CXCR3 is altered at parturition in healthy women and in women with systemic lupus erythematosus. Scand J Immunol (2013) 77(3):200–12. doi: 10.1111/sji.12021
35. Torraca V, Cui C, Boland R, Bebelman J-P, van der Sar AM, Smit MJ, et al. CXCR3-CXCL11 signaling axis mediates macrophage recruitment and dissemination of mycobacterial infection. Dis Model Mech (2015) 8(3):253–69. doi: 10.1242/dmm.017756
36. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol (2001) 159(1):237–43. doi: 10.1016/S0002-9440(10)61689-6
37. Santoro A, Majorana A, Roversi L, Gentili F, Marrelli S, Vermi W, et al. Recruitment of dendritic cells in oral lichen planus. J Pathol (2005) 205(4):426–34. doi: 10.1002/path.1699
38. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol (2019) 20(12):1574–83. doi: 10.1038/s41590-019-0466-2
39. Achtman JC, Werth VP. Pathophysiology of cutaneous lupus erythematosus. Arthritis Res Ther (2015) 17(1):182. doi: 10.1186/s13075-015-0706-2
40. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
41. Laria A, Lurati A, Marrazza M, Mazzocchi D, Re KA, Scarpellini M. The macrophages in rheumatic diseases. J Inflammation Res (2016) 9:1–11. doi: 10.2147/JIR.S82320
42. Merry R, Belfield L, McArdle P, McLennan A, Crean S, Foey A. Oral health and pathology: a macrophage account. Br J Oral Maxillofac Surg (2012) 50(1):2–7. doi: 10.1016/j.bjoms.2010.10.020
43. Chong BF, Tseng L-C, Hosler GA, Teske NM, Zhang S, Karp DR, et al. A subset of CD163+ macrophages displays mixed polarizations in discoid lupus skin. Arthritis Res Ther (2015) 17:324. doi: 10.1186/s13075-015-0839-3
44. Kastelan M, Prpić Massari L, Gruber F, Zamolo G, Zauhar G, Coklo M, et al. The role of perforin-mediated apoptosis in lichen planus lesions. Arch Dermatol Res (2004) 296(5):226–30. doi: 10.1007/s00403-004-0512-1
45. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity (2013) 38(5):870–80. doi: 10.1016/j.immuni.2013.05.004
46. Maelfait J, Liverpool L, Rehwinkel J. Nucleic Acid Sensors and Programmed Cell Death. J Mol Biol (2020) 432(2):552–68. doi: 10.1016/j.jmb.2019.11.016
47. Handfield C, Kwock J, MacLeod AS. Innate Antiviral Immunity in the Skin. Trends Immunol (2018) 39(4):328–40. doi: 10.1016/j.it.2018.02.003
48. Günther C. Nucleic Acid Immunity in the Pathogenesis of Cutaneous Lupus Erythematosus. Front Immunol (2019) 10:1636. doi: 10.3389/fimmu.2019.01636
49. Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA (2015) 112(42):E5699–705. doi: 10.1073/pnas.1516465112
50. Scholtissek B, Zahn S, Maier J, Klaeschen S, Braegelmann C, Hoelzel M, et al. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J Invest Dermatol (2017) 137(7):1484–92. doi: 10.1016/j.jid.2017.03.018
51. Kreuter A, Jaouhar M, Skrygan M, Tigges C, Stücker M, Altmeyer P, et al. Expression of antimicrobial peptides in different subtypes of cutaneous lupus erythematosus. J Am Acad Dermatol (2011) 65(1):125–33. doi: 10.1016/j.jaad.2010.12.012
52. Davidopoulou S, Theodoridis H, Nazer K, Kessopoulou E, Menexes G, Kalfas S. Salivary concentration of the antimicrobial peptide LL-37 in patients with oral lichen planus. J Oral Microbiol (2014) 6:26156. doi: 10.3402/jom.v6.26156
53. Lu X, Tang Q, Lindh M, Dastmalchi M, Alexanderson H, Popovic Silwerfeldt K, et al. The host defense peptide LL-37 a possible inducer of the type I interferon system in patients with polymyositis and dermatomyositis. J Autoimmun (2017) 78:46–56. doi: 10.1016/j.jaut.2016.12.003
54. Chamilos G, Gregorio J, Meller S, Lande R, Kontoyiannis DP, Modlin RL, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood (2012) 120(18):3699–707. doi: 10.1182/blood-2012-01-401364
55. Ablasser A, Hur S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat Immunol (2020) 21(1):17–29. doi: 10.1038/s41590-019-0556-1
56. Man SM, Karki R, Kanneganti T-D. DNA-sensing inflammasomes: regulation of bacterial host defense and the gut microbiota. Pathog Dis (2016) 74(4):ftw028. doi: 10.1093/femspd/ftw028
57. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
58. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
59. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10(3):266–72. doi: 10.1038/ni.1702
60. Schroder K, Kanneganti T-D, Shao F, Broz P. Mechanisms and Consequences of Inflammasome Activation. J Mol Biol (2018) 430(2):131–2. doi: 10.1016/j.jmb.2017.12.005
61. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nat 2015; (7575) 526:660–5. doi: 10.1038/nature15514
62. Domingues R, Pietrobon AJ, Carvalho GC, Pereira NZ, Pereira NV, Sotto MN, et al. Lichen planus: altered AIM2 and NLRP1 expression in skin lesions and defective activation in peripheral blood mononuclear cells. Clin Exp Dermatol (2019) 44(4):e89–95. doi: 10.1111/ced.13859
63. Liu J, Berthier CC, Kahlenberg JM. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol (Hoboken NJ) (2017) 69(9):1840–9. doi: 10.1002/art.40166
64. Wittmann M, Purwar R, Hartmann C, Gutzmer R, Werfel T. Human keratinocytes respond to interleukin-18: implication for the course of chronic inflammatory skin diseases. J Invest Dermatol (2005) 124(6):1225–33. doi: 10.1111/j.0022-202X.2005.23715.x
65. Kahlenberg JM, Kaplan MJ. The Inflammasome and lupus- another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol (2014) 26(5):475–81. doi: 10.1097/BOR.0000000000000088
66. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet (2019) 20(11):657–74. doi: 10.1038/s41576-019-0151-1
67. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature (2013) 498(7454):380–4. doi: 10.1038/nature12306
68. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339(6121):826–30. doi: 10.1126/science.1229963
69. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science (2019) 363(6431):eaat8657. doi: 10.1126/science.aat8657
70. Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun (2017) 8:14392. doi: 10.1038/ncomms14392
71. Mondini M, Vidali M, Airò P, de Andrea M, Riboldi P, Meroni PL, et al. Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci (2007) 1110:47–56. doi: 10.1196/annals.1423.006
72. Bawadekar M, de Andrea M, Gariglio M, Landolfo S. Mislocalization of the interferon inducible protein IFI16 by environmental insults: implications in autoimmunity. Cytokine Growth Factor Rev (2015) 26(2):213–9. doi: 10.1016/j.cytogfr.2014.10.003
73. Caneparo V, Landolfo S, Gariglio M, de Andrea M. The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front Immunol (2018) 9:1180. doi: 10.3389/fimmu.2018.01180
74. Cao T, Shao S, Li B, Jin L, Lei J, Qiao H, et al. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci Rep (2016) 6:25381. doi: 10.1038/srep25381
75. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11(11):997–1004. doi: 10.1038/ni.1932
76. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma associated herpesvirus infection. Cell Host Microbe (2011) 9(5):363–75. doi: 10.1016/j.chom.2011.04.008
77. Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J (2017) 36(17):2529–43. doi: 10.15252/embj.201796476
78. Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA (2008) 105(14):5477–82. doi: 10.1073/pnas.0801295105
79. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature (2020) 580(7803):391–5. doi: 10.1038/s41586-020-2129-8
80. Kuriakose T, Zheng M, Neale G, Kanneganti T-D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J Immunol (2018) 200(4):1489–95. doi: 10.4049/jimmunol.1701538
81. Devos M, Tanghe G, Gilbert B, Dierick E, Verheirstraeten M, Nemegeer J, et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J Exp Med (2020) 217(7):e20191913. doi: 10.1084/jem.20191913
82. Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis (2018) 9(8):816. doi: 10.1038/s41419-018-0868-3
83. Lafer EM, Valle RP, Möller A, Nordheim A, Schur PH, Rich A, et al. Z-DNA-specific antibodies in human systemic lupus erythematosus. J Clin Invest (1983) 71(2):314–21. doi: 10.1172/JCI110771
84. Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, et al. ZBP1 mediates interferon-induced necroptosis. Cell Mol Immunol (2020) 17(4):356–68. doi: 10.1038/s41423-019-0237-x
85. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38(5):855–69. doi: 10.1016/j.immuni.2013.05.007
86. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5(7):730–7. doi: 10.1038/ni1087
87. Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest (2009) 119(8):2399–411. doi: 10.1172/JCI37155
88. Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J Immunol (2018) 200(8):2748–56. doi: 10.4049/jimmunol.1701492
89. Kalali BN, Köllisch G, Mages J, Müller T, Bauer S, Wagner H, et al. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J Immunol (2008) 181(4):2694–704. doi: 10.4049/jimmunol.181.4.2694
90. Kitamura H, Matsuzaki Y, Kimura K, Nakano H, Imaizumi T, Satoh K, et al. Cytokine modulation of retinoic acid-inducible gene-I (RIG-I) expression in human epidermal keratinocytes. J Dermatol Sci (2007) 45(2):127–34. doi: 10.1016/j.jdermsci.2006.11.003
91. Enevold C, Kjær L, Nielsen CH, Voss A, Jacobsen RS, Hermansen MLF, et al. Genetic polymorphisms of dsRNA ligating pattern recognition receptors TLR3, MDA5, and RIG-I. Association with systemic lupus erythematosus and clinical phenotypes. Rheumatol Int (2014) 34(10):1401–8. doi: 10.1007/s00296-014-3012-4
92. Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PloS Genet (2013) 9(2):e1003222. doi: 10.1371/journal.pgen.1003222
93. Kawai T, Akira S. TLR signaling. Semin Immunol (2007) 19(1):24–32. doi: 10.1016/j.smim.2006.12.004
94. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature (2001) 413(6857):732–8. doi: 10.1038/35099560
95. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Sci 2004; (5663) 303:1526–9. doi: 10.1126/science.1093620
96. Häcker G, Redecke V, Häcker H. Activation of the immune system by bacterial CpG-DNA. Immunology (2002) 105(3):245–51. doi: 10.1046/j.0019-2805.2001.01350.x
97. Balic JJ, Albargy H, Luu K, Kirby FJ, Jayasekara WSN, Mansell F, et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1β expression. Nat Commun (2020) 11(1):3816. doi: 10.1038/s41467-020-17669-5
98. Puig M, Tosh KW, Schramm LM, Grajkowska LT, Kirschman KD, Tami C, et al. TLR9 and TLR7 agonists mediate distinct type I IFN responses in humans and nonhuman primates in vitro and in vivo. J Leukoc Biol (2012) 91(1):147–58. doi: 10.1189/jlb.0711371
99. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
100. Lebre MC, van der Aar AMG, van Baarsen L, van Capel TMM, Schuitemaker JHN, Kapsenberg ML, et al. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J Invest Dermatol (2007) 127(2):331–41. doi: 10.1038/sj.jid.5700530
101. Siponen M, Kauppila JH, Soini Y, Salo T. TLR4 and TLR9 are induced in oral lichen planus. J Oral Pathol Med (2012) 41(10):741–7. doi: 10.1111/j.1600-0714.2012.01169.x
102. O’Mahony C, Yesudian PD, Stanley M. Imiquimod use in the genital area and development of lichen sclerosus and lichen planus. Int J STD AIDS (2010) 21(3):219–21. doi: 10.1258/ijsa.2009.009154
103. Domingues E, Chaney KC, Scharf MJ, Wiss K. Imiquimod reactivation of lichen planus. Cutis (2012) 89(6):276–7, 283.
104. Wang H-W, Miao F, Shi L, Lü T, Huang Z, Wang X-L. Imiquimod-induced localized vitiligo in wife and lichen planus in husband. Chin Med J (2013) 126(13):2593.
105. Li ZJ, Sohn K-C, Choi D-K, Shi G, Hong D, Lee H-E, et al. Roles of TLR7 in Activation of NF-κB Signaling of Keratinocytes by Imiquimod. PloS One (2013) 8(10):e77159. doi: 10.1371/journal.pone.0077159
106. Braegelmann C, Hölzel M, Ludbrook V, Dickson M, Turan N, Ferring-Schmitt S, et al. Spleen tyrosine kinase (SYK) is a potential target for the treatment of cutaneous lupus erythematosus patients. Exp Dermatol (2016) 25(5):375–9. doi: 10.1111/exd.12986
107. Shao S, Tsoi LC, Sarkar MK, Xing X, Xue K, Uppala R, et al. IFN-γ enhances cell-mediated cytotoxicity against keratinocytes via JAK2/STAT1 in lichen planus. Sci Transl Med (2019) 11(511):eaav7561. doi: 10.1126/scitranslmed.aav7561
108. Toro JR, Finlay D, Dou X, Zheng SC, LeBoit PE, Connolly MK. Detection of type 1 cytokines in discoid lupus erythematosus. Arch Dermatol (2000) 136(12):1497–501. doi: 10.1001/archderm.136.12.1497
109. Terlou A, Santegoets LAM, van der Meijden WI, Heijmans-Antonissen C, Swagemakers SMA, van der Spek PJ, et al. An autoimmune phenotype in vulvar lichen sclerosus and lichen planus: a Th1 response and high levels of microRNA-155. J Invest Dermatol (2012) 132(3 Pt 1):658–66. doi: 10.1038/jid.2011.369
110. Farley SM, Wood LJ, Iordanov MS. An Epidermotypic Model of Interface Dermatitis Reveals Individual Functions of Fas Ligand and Gamma Interferon in Hypergranulosis, Cytoid Body Formation, and Gene Expression. Am J Dermatopathol (2011) 33(3):244–50. doi: 10.1097/DAD.0b013e3181f1b200
111. Lee C-K, Bluyssen HAR. Editorial: STATs and IRFs in Innate Immunity: From Transcriptional Regulators to Therapeutic Targets. Front Immunol (2019) 10:1829. doi: 10.3389/fimmu.2019.01829
112. Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol (2019) 20(4):493–502. doi: 10.1038/s41590-019-0323-3
113. Basagoudanavar SH, Thapa RJ, Nogusa S, Wang J, Beg AA, Balachandran S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J Virol (2011) 85(6):2599–610. doi: 10.1128/JVI.02213-10
114. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol (2004) 25(6):280–8. doi: 10.1016/j.it.2004.03.008
115. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol (2015) 7(5):a016303. doi: 10.1101/cshperspect.a016303
116. Haller O, Staeheli P, Schwemmle M, Kochs G. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol (2015) 23(3):154–63. doi: 10.1016/j.tim.2014.12.003
117. Hornung V, Hartmann R, Ablasser A, Hopfner K-P. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol (2014) 14(8):521–8. doi: 10.1038/nri3719
118. Wenzel J, Peters B, Zahn S, Birth M, Hofmann K, Küsters D, et al. Gene expression profiling of lichen planus reflects CXCL9+-mediated inflammation and distinguishes this disease from atopic dermatitis and psoriasis. J Invest Dermatol (2008) 128(1):67–78. doi: 10.1038/sj.jid.5700945
119. Scarponi C, Nardelli B, Lafleur DW, Moore PA, Madonna S, de Pità O, et al. Analysis of IFN-kappa expression in pathologic skin conditions: downregulation in psoriasis and atopic dermatitis. J Interferon Cytokine Res (2006) 26(3):133–40. doi: 10.1089/jir.2006.26.133
120. Ito T, Kanzler H, Duramad O, Cao W, Liu Y-J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107(6):2423–31. doi: 10.1182/blood-2005-07-2709
121. Fetter T, Smith P, Guel T, Braegelmann C, Bieber T, Wenzel J. Selective Janus Kinase 1 Inhibition Is a Promising Therapeutic Approach for Lupus Erythematosus Skin Lesions. Front Immunol (2020) 11:344. doi: 10.3389/fimmu.2020.00344
122. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
123. Kraus TA, Lau JF, Parisien J-P, Horvath CM. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J Biol Chem (2003) 278(15):13033–8. doi: 10.1074/jbc.M212972200
124. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
125. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
126. Orlik C, Deibel D, Küblbeck J, Balta E, Ganskih S, Habicht J, et al. Keratinocytes costimulate naive human T cells via CD2: a potential target to prevent the development of proinflammatory Th1 cells in the skin. Cell Mol Immunol (2020) 17(4):380–94. doi: 10.1038/s41423-019-0261-x
127. Norris DA. Cytokine modulation of adhesion molecules in the regulation of immunologic cytotoxicity of epidermal targets. J Invest Dermatol (1990) 95:111S–20S. doi: 10.1111/1523-1747.ep12874977
128. Bennion SD, Middleton MH, David-Bajar KM, Brice S, Norris DA. In three types of interface dermatitis, different patterns of expression of intercellular adhesion molecule-1 (ICAM-1) indicate different triggers of disease. J Invest Dermatol (1995) 105:71S–9S. doi: 10.1111/1523-1747.ep12316107
129. Stohl W, Hilbert DM. The discovery and development of belimumab: the anti-BLyS-lupus connection. Nat Biotechnol (2012) 30(1):69–77. doi: 10.1038/nbt.2076
130. Wenzel J, Landmann A, Vorwerk G, Kuhn A. High expression of B lymphocyte stimulator in lesional keratinocytes of patients with cutaneous lupus erythematosus. Exp Dermatol (2018) 27(1):95–7. doi: 10.1111/exd.13419
131. Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflammation Res (2018) 11:407–19. doi: 10.2147/JIR.S140188
132. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
133. Ahn Y, Seo J, Lee EJ, Kim JY, Park M-Y, Hwang S, et al. ATP-P2X7-Induced Inflammasome Activation Contributes to Melanocyte Death and CD8+ T-Cell Trafficking to the Skin in Vitiligo. J Invest Dermatol (2020) 140(9):1794–804.e4. doi: 10.1016/j.jid.2019.12.035
134. Ni Riordain R, Christou J, Pinder D, Squires V, Hodgson T. Cost of illness of oral lichen planus in a U.K. population–a pilot study. J Oral Pathol Med (2016) 45(5):381–4. doi: 10.1111/jop.12415
135. Ogunsanya ME, Brown CM, Lin D, Imarhia F, Maxey C, Chong BF. Understanding the disease burden and unmet needs among patients with cutaneous lupus erythematosus: A qualitative study. Int J Womens Dermatol (2018) 4(3):152–8. doi: 10.1016/j.ijwd.2018.01.002
Keywords: interface dermatitis/lichenoid tissue reaction, nucleic acid sensing, damage associated molecular patterns (DAMPs), type I immunity, lupus erythematosus, lichen planus, dermatomyositis, in vitro model
Citation: Braegelmann C, Fetter T, Niebel D, Dietz L, Bieber T and Wenzel J (2021) Immunostimulatory Endogenous Nucleic Acids Perpetuate Interface Dermatitis—Translation of Pathogenic Fundamentals Into an In Vitro Model. Front. Immunol. 11:622511. doi: 10.3389/fimmu.2020.622511
Received: 28 October 2020; Accepted: 26 November 2020;
Published: 11 January 2021.
Edited by:
Vincenzo Torraca, University of London, United KingdomReviewed by:
Quang-Tien Phan, National University of Singapore, SingaporeMasanori Fujii, Kyoto Pharmaceutical University, Japan
Copyright © 2021 Braegelmann, Fetter, Niebel, Dietz, Bieber and Wenzel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine Braegelmann, Christine.Braegelmann@ukbonn.de