 Clément Kerneur
Clément Kerneur Carla E. Cano
Carla E. Cano Daniel Olive
Daniel Olive- 1ImCheck Therapeutics, Marseille, France
- 2Team Immunity and Cancer, Centre de Recherche en Cancérologie de Marseille (CRCM), Inserm U1068, CNRS UMR7258, Institut Paoli Calmettes, Marseille, France
Macrophages play an important role in tissue homeostasis, tissue remodeling, immune response, and progression of cancer. Consequently, macrophages exhibit significant plasticity and change their transcriptional profile and function in response to environmental, tissue, and inflammatory stimuli resulting in pro- and anti-tumor effects. Furthermore, the categorization of tissue macrophages in inflammatory situations remains difficult; however, there is an agreement that macrophages are predominantly polarized into two different subtypes with pro- and anti-inflammatory properties, the so-called M1-like and M2-like macrophages, respectively. These two macrophage classes can be considered as the extreme borders of a continuum of many intermediate subsets. On one end, M1 are pro-inflammatory macrophages that initiate an immunological response, damage tissue integrity, and dampen tumor progression by fostering robust T and natural killer (NK) cell anti-tumoral responses. On the other end, M2 are anti-inflammatory macrophages involved in tissue remodeling and tumor growth, that promote cancer cell proliferation, invasion, tumor metastasis, angiogenesis and that participate to immune suppression. These decisive roles in tumor progression occur through the secretion of cytokines, chemokines, growth factors, and matrix metalloproteases, as well as by the expression of immune checkpoint receptors in the case of M2 macrophages. Moreover, macrophage plasticity is supported by stimuli from the Tumor Microenvironment (TME) that are relayed to the nucleus through membrane receptors and signaling pathways that result in gene expression reprogramming in macrophages, thus giving rise to different macrophage polarization outcomes. In this review, we will focus on the main signaling pathways involved in macrophage polarization that are activated upon ligand-receptor recognition and in the presence of other immunomodulatory molecules in cancer.
Introduction
Tumor Associated Macrophages can be polarized into pro-inflammatory (M1-like) and anti-inflammatory (M2-like) phenotypes
Tumor-Associated Macrophages
TME is the result of complex interactions between different cell types, including tumor cells, immune cells, endothelial cells, and fibroblasts. TAMs, which represent 50–80% of non-tumor stromal cells, are critical components of the TME. After recruitment of circulating monocytes via the chemokine ligand 2 (CCL2)- chemokine-receptor 2 (CCR2) axis and many others chemokines and cytokines such as macrophage colony-stimulating factor (M-CSF), chemokine (C-X3-C motif) ligand 1 (CX3CL1), CCL3, CCL5, and vascular endothelial growth factor A (VEGF-A) (1), TME will favor their polarization either towards anti-inflammatory or pro-inflammatory macrophages, that will be referred to as M1-like or M2-like TAMs, respectively. The two main macrophage polarization states described above and detailed below constitute most of the TAMs compartment, with a large predominance of the M2-like phenotype. However, this TAMs dichotomy is a simplistic way to represent their complex functions in the microenvironment. Recent data obtained using techniques such as single cell RNA sequencing may contribute to better identify macrophage subpopulations and have shown previously unknown complexity in macrophage polarization, going much beyond the traditional dogma of the binary “M1-M2” system (2–4). Nevertheless, regarding the fact that the categorization of tissue macrophages in inflammatory situations remains difficult and variable among different indications and patients, there is an agreement that TAMs are predominantly polarized into the two main subtypes, M1-like and M2-like TAMs. Indeed, TAMs will change from one type to another depending on the environment in which they reside further described in next sections (1.b. and 1.c.).
In addition, it has been well established that TAMs prevalence in many solid tumors, such as breast, ovarian, cervical, and prostate cancers, is associated with poor prognosis, tumor progression, and metastasis (5–7). Indeed, TAMs promote tumor development by several ways:
i. Anti-inflammatory TAMs control immune responses by secreting immunosuppressive cytokines and enzymes resulting in promotion of tumor growth and survival (8). In addition, TAMs also impair tumor-infiltrating lymphocytes’ (TILs’) capacity to migrate and interact with malignant cells (9).
ii. TAMs also enhance cancer stem cell-like properties of tumor cells (10) that promote invasive capability and metastasis of later stages cancers including formation of the pre-metastatic niche in secondary site, extravasation, and early colonization (11, 12) and favors angiogenesis by secreting proangiogenic factors including, placental growth factor (PIGF), TGF-β, CCL2, and CXCL12 (13–15).
iii. Furthermore, TAMs contribute to cancer resistance and relapse after treatment in radiotherapy and with several chemotherapeutic agents via releasing survival factors leading to malignant cells activation of anti-apoptotic signaling pathways (16–18). Additionally, direct contact of myeloma cells with macrophages can also induce chemoresistance (19). Indeed, relapse from anticancer treatment can be due to TAMs polymorphism during tumor development (20), for example in breast cancer treated with Taxol (21) or hepatocellular carcinoma treated with Sorafenib (22) both in vitro and in vivo (mice model).
Targeting TAMs could thus be an effective and promising therapeutic strategy for enhancing anti-cancer immunity. Current development of potentially but not yet effective medicines that target TAMs are based on TAM depletion, enhancement of TAM phagocytosis, inhibition of monocyte recruitment, and reprogramming of anti-inflammatory M2-like TAMs into pro-inflammatory M1-like TAMs. Therefore, it is essential to understand better the molecular and biological mechanisms leading to macrophage recruitment and polarization in cancer, and which functions are impacted.
Pro-inflammatory Macrophages (M1-like)
IFN-γ was the first macrophage-activating factor discovered in 1970 (23). IFN-γ is a soluble cytokine produced by activated CD4+ T helper (Th) 1 cells, CD8+ T cytotoxic cells, and NK cells that drives resting macrophages into potent cells with enhanced antigen–presenting capacity, increased synthesis of proinflammatory cytokines and toxic mediators, and enhanced complement–mediated phagocytosis among other functions. This early description of macrophage activation came to be known as classical activation. Since classically activated macrophages result from Th1 (cytotoxic) T cell responses, these macrophages were therefore renamed M1 macrophages. As well as IFN-γ, M1 macrophages can be differentiated by other Th1 cytokines, like TNF-α, and bacterial components such as lipopolysaccharide (LPS). Granulocyte-macrophage colony stimulating factor (GM-CSF) also activates M1 responses and antitumoral activity (17). Conversely, M1 macrophages stimulate the response of Th1 and cytotoxic cells, which in turn release IFN-γ, thereby reinforcing M1 polarization in a feedback loop (23–26). M1 macrophages release significant amounts of pro-inflammatory cytokines including IL-1β, IL-6, IL-12, IL-23, IFN-β and TNF-α that concomitantly drive Th1 responses (27, 28). M1 macrophages are also a main source of IL-12, while producing little or no IL-10, further amplifying M1 polarization.
The chemokines produced by M1 macrophages are mostly of the CC type (CCL2, CCL3, CCL4, CCL5, CCL8, CCL19, CCL20), moreover IFN-γ also increases the production of CXCL10 and CXCL9 leading to IFN-β expression. Overall, these chemokines contribute to the establishment of a type 1 immune response (29), resulting in protection against intracellular pathogens and tumor progression.
It is crucial to highlight that pro-inflammatory M1 macrophages can act as tumor suppressors in many ways:
i. Destroying malignant cells. Pro-inflammatory Macrophages produce reactive oxygen species (ROS) and enhance their production by tumor cells through secretion of stimuli like TNF-α. ROS production by macrophages as a mean of killing tumor cells is well documented. Macrophages rapid generation of superoxide is driven predominantly by NAPDH oxidase resulting in hydrogen peroxide synthesis (30). In addition, during inflammation, nitric oxide (NO) synthetized by the inducible nitric oxide synthase (iNOS) from L-arginine reacts with superoxide to produce peroxinitrite radicals that are similar in their activity to hydroxyl radicals, and contribute to direct tumor cell apoptosis (31, 32). M1 also play an important role in tumor suppression through the generation of cytotoxic molecules such as TNF-α-related apoptosis inducing ligand (TRAIL) or FAS ligand (FASL), or via the Antibody-Dependent Cellular Phagocytosis (ADCP) or Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) (24, 25).
ii. Stimulating anti-tumor immunity. The crosstalk between NK cells and macrophages constitutes a line of defense against tumors. Indeed, M1 macrophages express CD48, which binds to the 2B4 receptor at the plasma membrane of NK cells inducing IFN-γ production, which in turn supports M1 phenotype. In addition, release of IL-1β, IFN-β, and/or IL-23 by M1 will promote the expression of natural killer cell p44-related protein (NKp44) and natural killer group 2 member D (NKG2D). Consequently, NK cells will carry cytotoxic responses against target cells that express their ligands, such as stress ligands (33, 34). These cells are also able to activate CD8+ cytotoxic T cells (35), which are the most potent anticancer immune effectors and constitute the foundation of current effective cancer immunotherapies (36) through the secretion of the chemokines previously listed (CCL2, CCL3, CCL4, CCL5 and CXCL10) (37). M1 macrophages also express major histocompatibility complex (MHC)-II molecules and CD80/CD86 costimulatory molecules that are essential for T-cell activation, cytokine production, proliferation, and differentiation (38–41). M1-secreted CCL2, CCL5 or IFN-β may increase neutrophil infiltration to the tumor location and contribute to pro-inflammatory neutrophil-targeted tumor regression (42, 43).
Several reports indicate that the highest the M1/M2 ratio, the longest the survival in cancer patients. Indeed, lower proportion of M2-like TAMs in the TME enhances the prognosis of patients in multiple different cancer indications such as ovarian cancer (44, 45), multiple myeloma (46), pediatric classical Hodgkin lymphoma (47), colorectal cancer (CRC) (48) and gastric cancer (49). M1-like TAMs are also correlated with better efficacy of currently used chemotherapy in ovarian and lung cancer (50, 51). M1-like abundancy also leads to a tumor immunological status which, when combined with chemotherapy, aids in tumor growth control (50).
Anti-inflammatory Macrophages (M2-like)
In contrast to M1, M2 macrophages have anti-inflammatory and tumor promoting properties. M2-like TAMs predominate in advanced tumors, functioning as critical regulators in response to TME. Several studies have shown that M2-like macrophages represent most of TAMs in many cancer indications, which is associated with poor prognosis in several cancer patients, consistent with their abilities to favor tumor growth, to promote cancer invasion and metastasis, to participate in neovascularization, and to contribute to the formation of the immunosuppressive TME (25, 52–63).
M2 macrophages were first found in the setting of helminth infection, distinguishing from monocytic progenitors under the influence of IL-4 and IL-13 in a severely Th2-polarized response. Although Th2 cytokines are required for M2 recruitment, M2 macrophages also secrete cytokines that mediate Th2 responses and regulate inflammatory responses (64–67).
The primary agents that cause M2 type activation include cytokines (IL-4, IL-10, IL-34 and IL-13), vitamin D3, TGF-β, prostaglandin E2 (PGE2), VEGF, EGF, glucocorticoids (68) and M-CSF (69). M2-like TAMs are further sub-categorized into IL-4/13–activated M2a, immune complex–activated M2b, IL-10–deactivated M2c and IL-6/M-CSF loop induced M2d (70) macrophages based on the stimulation of different cytokines. Those subtypes will not be discussed here but are well described in a review by Mantovani et al. (29) and Duluc et al. (70).
M2 macrophages act as tumor promoters in several ways:
i. Promoting cancer cell proliferation and invasion. EGF secreted by M2-like TAMs directly stimulates cancer cell proliferation and invasion. M2 macrophages can also contribute directly to cell proliferation, invasiveness, and migration in breast cancer by suppressing IFN regulatory factor (IRF)7 expression through MicroRNAs mIR-1587 (71). Moreover, several studies have shown that microRNAs play critical roles in macrophage activation, polarization, tissue infiltration, and inflammation resolution (miR-155, miR-181, and miR-451 are implicated in M1 macrophage polarization whereas miR-146a, miR-125a, and miR-145-5p are involved in M2 macrophage polarization) (72).
ii. In contrast to M1-like macrophages, NO and iNOS production appears diminished after TAMs switch to an M2-like phenotype leading to abolishment of M1 direct killing of tumor cells (73).
iii. Promoting angiogenesis. VEGF secretion by M2-like TAMs promotes angiogenesis in tumor sites. In turn, VEGF receptors on the surface of TAMs activate an autocrine loop, reinforcing their pro-angiogenic and immunosuppressive properties (25) but also their M2 phenotype. TAMs also secrete pro-angiogenic chemokines (e.g., IL-8) and proteolytic enzymes (e.g., MMPs and cathepsins induced by IL-4) (74), which breakdown the extracellular matrix (ECM), causing the release of angiogenic components such as TGF-β, VEGF, and fibroblast growth factor (FGF) that had previously been stored in the ECM in an inactive state (75–77). MMP-2 and MMP-9, were also shown to increase VEGF expression by cancer cells (76), and their expression has been linked to greater tumor invasiveness and a poor prognosis.
iv. Dampening the activity of tumor killer cells. M2 macrophages also play a role on inhibition of NK cell function through secretion of inhibitory factors and through cell-cell contact (78). By secreting PGE2, M2-like macrophages suppress NK cell cytotoxicity through downregulation of NKp30, NKp44, NKp46, and NKG2D by binding to E-prostanoid 2 (EP2) and EP4 receptors, leading to immunosuppressive cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling in NK cells. PGE2 has also a direct impact on macrophages that we will discuss later. MMPs secreted by M2-like macrophages cleave FASL expressed at the NK cell surface thus inhibiting tumor cell apoptosis mediated by FAS : FASL ligation, which plays a critical role in immunosuppression. In addition, M2-like TAMs produce large amounts of IL-10, which inhibits both the production of IL-12 by intratumoral dendritic cells (DCs) and CD8+ T cell-mediated anti-tumor responses (79). IL-10, like TGF-β, suppresses CD8+ T cell and NK cell cytotoxicity by direct transcriptional repression of genes encoding functional mediators, such as perforins, granzymes, and cytotoxins. TGF-β contributes to metastasis allowing epithelial-to-mesenchymal transition of cancer cells, which empowers them with more invasive potential related to metastasis formation (80). TGF-β is associated with poor prognosis in multiple tumor types and has been demonstrated to be a major contributor of CD8+ T cell exclusion from tumors (81). M2 macrophages can also suppress T cell activation through depletion of tryptophan du to elevated expression of indoleamine 2,3-dioxygenase (IDO) and local depletion of L-arginine through Arginase1 (ARG1) expression (82).
v. Promoting resistance to therapy. The ability of TAMs to limit the efficacy of immune checkpoint blockade therapy has been reported in several studies (83). Indeed, M2-like TAMs express high levels of both PD-1 and PD-L1, thus directly participating to immune suppression, as well as high levels of FcγRs that can quench therapeutic antibodies used for immune checkpoint blocking (84). In addition, as reviewed by C. Anfray (2019) (84) and A.Mantovani (2015) (85), TAMs seem to inhibit most anti-tumor treatments frequently used in clinical practice, including conventional chemotherapy, anti-angiogenic therapy, and radiation.
vi. Hence, high proportion of M2-like macrophages in TAMs might be a causative factor for poor prognosis and shorter survival of patients in several cancers such as chronic lymphocytic leukemia (86), T cell leukemia/lymphoma (60), oral cancer, thyroid cancer, bladder cancer (87) non-small-cell lung cancer (88) gastric cancer (50, 88, 89), clear cell renal cell carcinoma (89), ovarian cancer (46, 50, 88), breast cancer (7, 88) CRC (90–92) and laryngeal cancer patients (93). Moreover, in CRC patients, an increase in the proportion of M2 type TAMs was associated with an increase in liver metastases (94).
In summary, M1-like TAMs that promote inflammatory responses against tumor cells mostly have an anticancer impact, whereas M2-like TAMs have anti-inflammatory activity that provides them with a pro-tumoral effect (25).
Macrophage receptors and signaling pathways that regulate macrophage polarization in cancer
Receptor signaling pathways leading to pro-inflammatory M1-like macrophages
Knowing that the TME plays a major role in macrophage polarization and that the interaction of chemokine/cytokine receptors with their ligands leads to the polarization of macrophages, we will describe in the next section the main signaling pathways involved in M1-macrophage polarization in humans through the different ligand-receptor interaction.
IFNγ receptors
IFNγR1 and IFNγR2 chains are members of the class II cytokine receptor family. Two chains of the IFNγR1 will first bind to an IFN-γ homodimer inducing subsequent recruitment of the two IFNγR2 chains. Both IFNγR chains must connect with a signaling machinery to transduce their signals. The intracellular domain of IFNγR1 has binding sites for Janus Tyrosine Kinase (JAK)1 and the latent cytosolic factor, signal transducer and activator of transcription (STAT)1. The JAK1- and STAT1-binding motifs are required for receptor phosphorylation, signal transduction, and induction of biological response (Figure 1). In the same way, the intracellular region of IFNγR2 contains a binding motif for recruitment of JAK2 kinase or Tyrosine Kinase 2 (TYK2) depending on ligand type for participation in signal transduction.
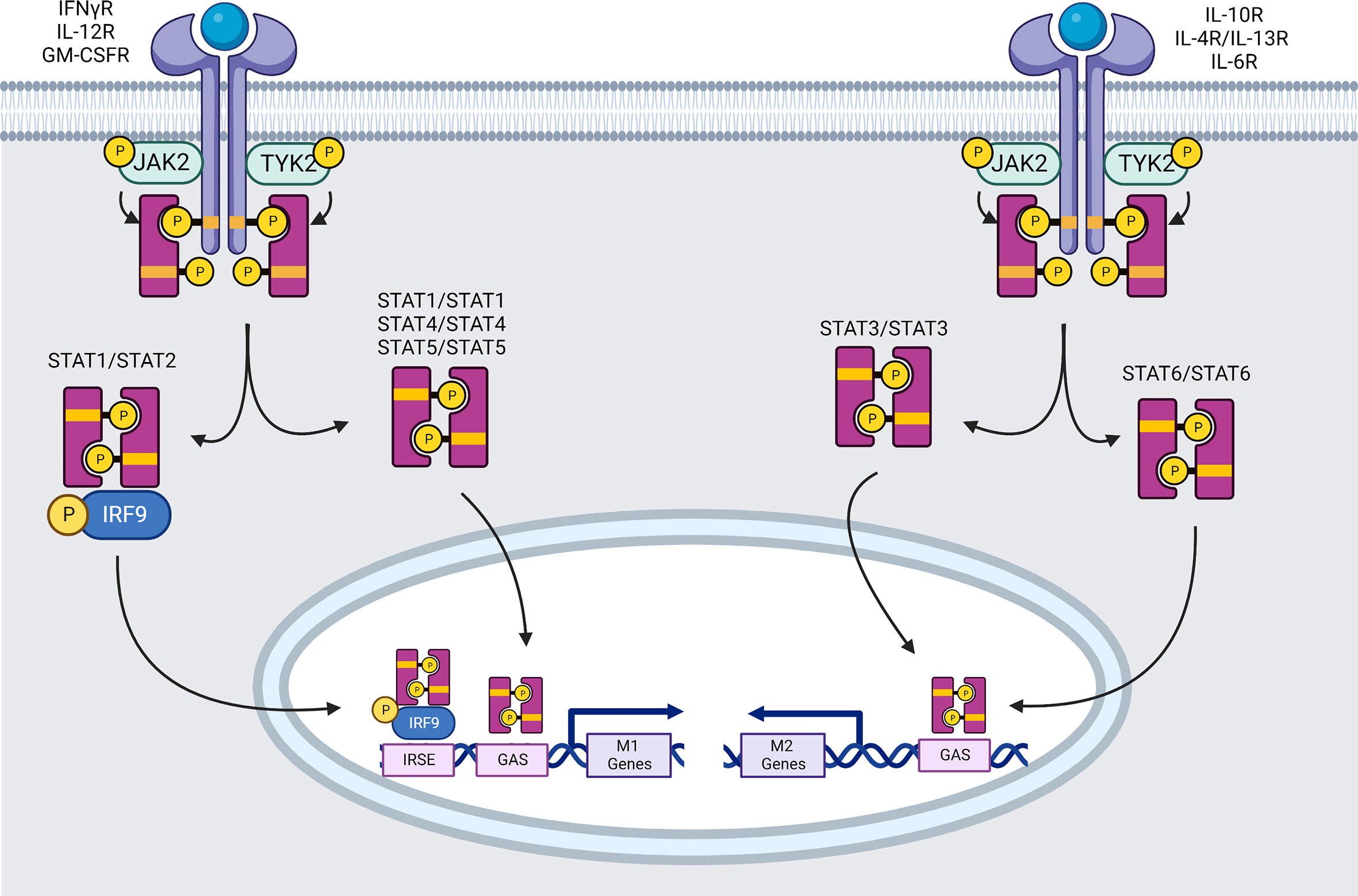
Figure 1 Signaling pathways mediated by the JAK family of tyrosine kinases (created using BioRender®).
Within 1 minute of pro-inflammatory IFN-γ-therapy, the four essential downstream tyrosine kinases (JAK1, JAK2, TYK2, and STAT1) are phosphorylated (95). Ligand interaction leads to auto-phosphorylation and activation of JAK2, allowing JAK2 to transphosphorylate JAK1 (96). Activated JAK1 phosphorylates functionally important Y440 residue of each IFNγR1 chain, forming two contiguous docking sites for latent STAT1’s SRC homology 2 (SH2) domains (97). STAT1 pair is phosphorylated near the C-terminus at Y701, most likely by JAK2. Phosphorylation causes a STAT1 homodimer to dissociate from the receptor (98). The dissociated STAT1 homodimer reaches the nucleus and attaches to promoter regions, allowing or inhibiting transcription of IFN-regulated genes. The mechanism of STAT1 translocation into the nucleus remains unknown, however the participation of importin-1 (NPI-1) is suggested (99). STAT1 homodimers bind DNA at GAS sites in the TTCN (24, 25, 52) GAA consensus sequence (100) leading to expression of several genes. Two to five hundred genes are activated in response to the IFNs, including iNOS, MIG (=CXCL9), intercellular adhesion molecule 1 (ICAM-1), IRF1 and STAT1 himself (101). For example, ICAM-1 is known to inhibit M2 macrophage polarization (102), and its expression appears to interfere with M2-related phosphoinositide 3-kinase (PI3K)/Ak strain transforming (AKT) pathway reducing phosphorylation and expression of phosphatase and tensin homolog (PTEN) and AKT (103). iNOS and IRF1 are highly associated with inflammation and preferentially expressed in M1 phenotype (104).
STAT1 is also able to form heterodimers with STAT2 when type I and III IFN (α and λ) bind to IFNαR1:IFNαR2 or IFNλR1:IL-10R2 respectively. STAT2 is recruited following its activation on the tyrosine 466 (Y466) of IFNαR1, which has already been phosphorylated by TYK2. STAT2 then recruits STAT1, which can only be activated by phosphorylation on Y701 (105). STAT1-STAT2 heterodimers will join forces with IRF9 to create the Interferon-stimulated gene factor 3 (ISGF3) transcription complex. ISGF3 recognizes the IFN stimulated response elements (ISRE) (consensus region: YAGTTTC(A/T)YTTTYCC) promoter elements of pro-inflammatory macrophage specific genes induced by IFNs such as hypoxia-inducible factors (HIF)-1α (101, 106).
STAT1 phosphorylation at Serine (S)727 is also required for maximum transcriptional activation of target genes. STAT1 serine phosphorylation is induced by a variety of stimuli, including types I and II IFN, LPS, IL-2, IL-12, TNF-α, and platelet-derived growth factor. In macrophages, for example, LPS signaling enhances STAT1 S727 phosphorylation independently of Y701 phosphorylation, enhancing cellular responses to IFN-γ (107) thus contributing to polarization into M1 macrophages.
Il-12R
IL-12 has been mainly described for activating naïve Lymphocytes and inducing their differentiation. However, IL-12 also promotes M1 macrophage polarization by inducing IFN-γ production by Th1 cells, which in turn boosts anti-tumor cytotoxic CD8+ T and NK cells (108). IL-12p40 and IL-12p35 bind to IL-12R-β1 and -β2 respectively, resulting in transphosphorylation of associated JAKs (JAK2 and TYK2) and then lead to the activation and translocation of STAT4 homodimer into the nucleus where they bind to STAT binding sites in the IFN-γ promoter of targeted M1 genes (109, 110).
Furthermore, IL-12 acts as a M1 classical activating factor through an indirect mechanism. Indeed, IL-12 promotes antigen presentation by increasing the expression of MHC-I on tumor cells (111), promoting polarization to M1 macrophages, and recruiting effector immune cells by increasing the expression of the chemokines CXCL9, CXCL10, and CXCL11 (112).
IL-12 has been also studied in pathological context showing again its ability to favor M1 polarization. In fact, it has been shown that IL-12R/STAT4 drives obesity-associated insulin resistance through pro-inflammatory M1 macrophage polarization (113).
Toll like receptors and IL-1R
The evolutionarily conserved Toll-like receptors (TLRs) family is one of the most studied families of pattern recognition receptor (PRRs) and plays a crucial role in early host defense against invading pathogens (40).
Among the 13 members of the TLR family including transmembrane TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, and endosomal TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13, the most relevant TLRs related to macrophage polarization in cancer context are TLR2/TLR6 (114), TLR3 (114, 115), TLR4 (115, 116) and TLR7/8 (117). TLR4, for example, activates signaling cascades (NF-κB and MAPK) that result in production of proinflammatory cytokines (TNFα, IL-1, IL-6, IL-12) and type-I interferons, which are essential for the propagation of the inflammatory response.
TLRs are type I integral membrane glycoproteins that belong to a wider superfamily that includes the IL-1 receptors, due to significant similarities in their cytoplasmic region known as the Toll/IL-1R (TIR) domain. TLRs/IL-1Rs dimerize following ligand binding and undergo the conformational shift necessary for the recruitment of pro-inflammatory downstream signaling molecules. These include the adaptor molecule myeloid differentiation primary-response protein 88 (MyD88), IL-1R-associated kinases (IRAKs), TGF-activated kinase (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TNFR-receptor-associated factor 6 (TRAF6) (118) (Figure 2).
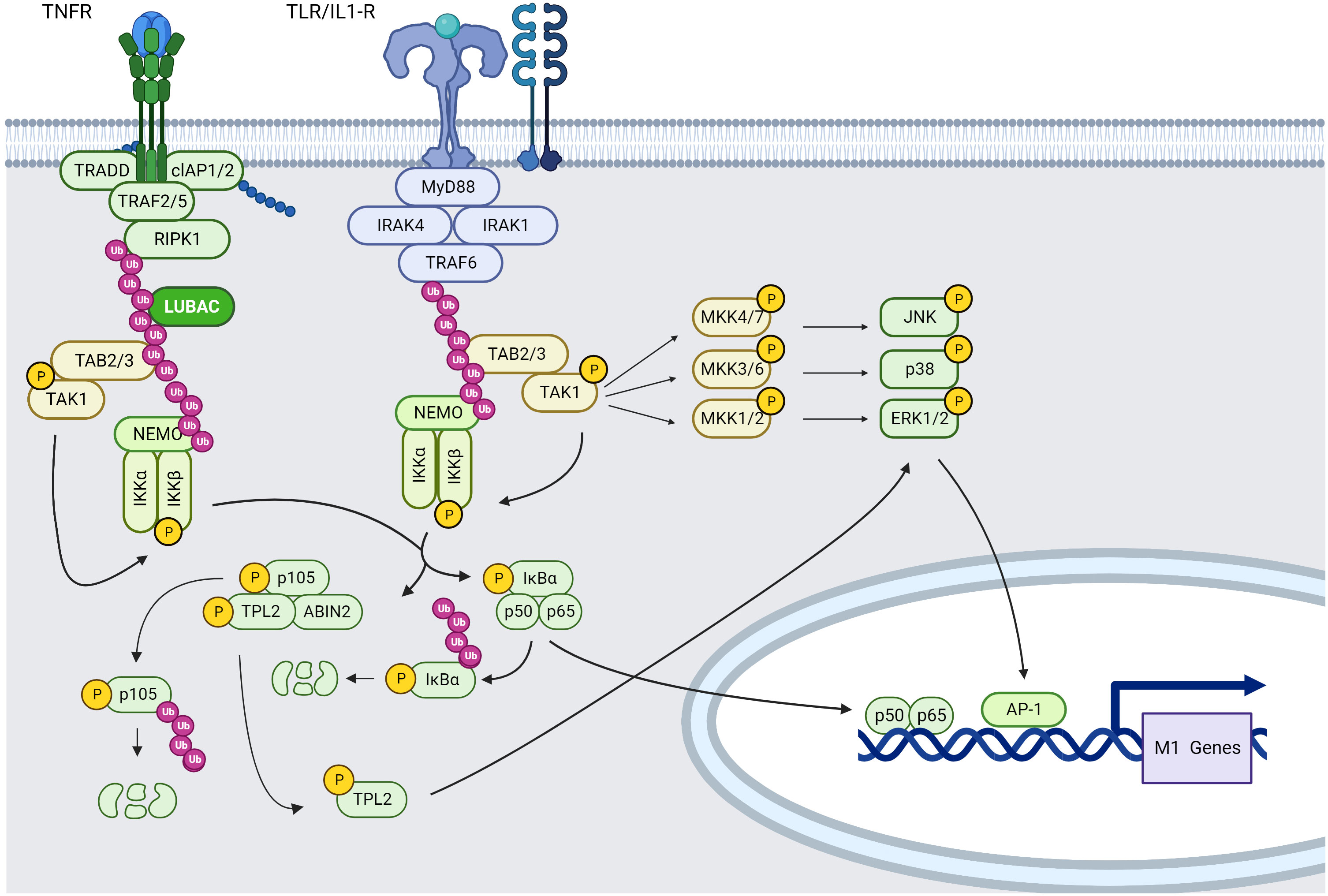
Figure 2 TNFR and TLR/IL-1R, NF-κB and MAPK signaling pathways in macrophages (created using BioRender®).
TLR4, TLR2, and TLR2/TLR6 recruit MyD88 via the bridging adaptor MAL, whereas TLR7/TLR8 recruit MyD88 directly. TIR-domain-containing adapter-inducing interferon-β (TRIF) is also recruited indirectly to TLR4 via the bridging adaptor TRIF-related adaptor molecule (TRAM), whereas TLR3 recruits TRIF directly. TLR4 is the only receptor that uses TRIF, TRAM, MyD88, and MyD88 adaptor-like protein (MAL/TIRAP). As a result, it serves as a model for both the TRIF- and MyD88-dependent pathways (119).
i. MYD88-dependent TLR signaling pathway
MyD88 is recruited to the cytoplasmic TIR domain, where it enhances IRAK4-receptor complex attachment via a homophilic Death Domain (DD) interaction. MyD88 binding to IRAK4 enhances IRAK4-mediated phosphorylation of S376 and T387 residues of IRAK1 and subsequent autophosphorylation, resulting in IRAK1 kinase activity (120, 121).
IRAK1 interacts then with TRAF6. TRAF6, in collaboration with the ubiquitin-conjugating enzymes UBC13 and UEV1A, promotes K63-linked polyubiquitination of several target proteins including TRAF6 itself, Inhibitor of kB kinase gamma (IKKγ/NEMO) and the TAK1 protein kinase complex. Regarding TAK1, TRAF2/5 (through TNF-α stimulation) or TRAF6-mediated K63 ubiquitination leads to recruitment of TAK1-binding protein (TAB)2 or TAB3 in order to form the TAK1 protein kinase complex composed of TAK1, TAB2 or TAB3, which phosphorylates and activates the inhibitory kappa B kinase alpha/beta (IKKα:IKKβ)–NF-κB complex and MAPK kinases (122–124), resulting in the expression of M1 type pro-inflammatory cytokines. Moreover, IKK, MAPK signaling and cytokines in TAK1-deficient murine peritoneal macrophages stimulated with LPS are slightly reduced compared with wild-type controls (122). More precisely, the TAB2:TAB3:TAK1 complex will phosphorylate mitogen-activated protein kinase Kik 4 (MKK4) and MMK7 leading to c-Jun N-terminal kinases (JNKs) phosphorylation, thus contributing to the acquisition of a M1 macrophage phenotype. Moreover, TAK1 can phosphorylate MKK3 and MKK6 leading to P38α phosphorylation and then MK2 phosphorylation resulting into pro-inflammatory cytokine production such as IL-1β and TNF-α. TAK1 can be also activated by many stimuli, including IL-1β, TNF-α, TLR ligands, and B and T cell receptor ligation (125).
MAP/ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway activation occurs preferentially via the activation of the MAP3K tumor progression locus 2 (TPL2; also known as MAP3K8) (126). Indeed, TPL2 is activated via IκB kinase (IKK)-induced proteolysis of the NF-κB subunit precursor protein p105, IKKβ phosphorylation of p105 causes ubiquitination and partial degradation of p105, resulting in the release of TPL2. Free TPL2 will phosphorylate MKK1 and MKK2 (also called respectively MEK1 and MEK2) and subsequently activates ERK signaling pathway. IKKβ also directly phosphorylates TPL2 on S400, which is essential for LPS-mediated pro-inflammatory activation of macrophages via ERK (127).
These observations show a direct link between MAPK activation and NF-κB activation in IL-1R and TLR-stimulated macrophages. Moreover, MAPK and IKK signaling in macrophages plays a pro-inflammatory role, suggesting that modulation of NF-κB, JNK, ERK and P38 pathways could constitute a potential therapeutic target regulating tumor homeostasis and favoring immunostimulatory context.
ii. TRIF-dependent TLR signaling pathways
TRIF-dependent signaling leads to Type-I IFNs production. TRIF can recruit both TRAF3 or TRAF6, TRAF3 recruits then the IKK-related kinases TBK1 and IKKε, as well as NEMO, to phosphorylate IRF3. IRF3 then forms a dimer and translocate from the cytoplasm into the nucleus, where it stimulates the transcription of type I IFN genes (128).The recruitment of TRAF6 activates the TAK1 complex leading to downstream signaling pathways previously described.
It has been recently shown that STAT3 acts directly on TLR4 signaling pathways via interaction with TRAF6 and TBK-1, resulting in non-canonical STAT3 S727 phosphorylation but not Y705, thus inducing metabolic M1 reprogramming and inflammation (129).
In the TME, cellular components released during necrosis act as DAMPs, that can also be ligands for TLRs and assist the establishment of a pre-programmed pro-inflammatory M1 or “classically activated” phenotype. This is accomplished via enhanced activation of signaling pathways including NF-κB, P38 MAPK, and others, which govern the production of pro-inflammatory cytokines (e.g., IL-1, IL-6, and IL-12) (130–132).
Meanwhile, stimulation of TLR3 and TLR4 may also activate a MyD88-independent pathway that induces IFN-β secretion. This pathway is mediated by Toll/IL-1 receptor (TIR) domain-containing adaptor and knockdown of TIR led to blockade of TLR4 mediated inflammation (119, 133, 134).
All ligands for each TLR and related functions are detailed in S. Akira and K. Takeda review (135).
Tnfr
TNF receptor superfamily TNFRSF is made up of 27 physically similar receptors that bind one or more molecules from the TNF superfamily (TNFSF), composed of more than 20 structurally related proteins (ligands). TNFSF ligands are membrane-anchored or soluble trimers that cluster their cognate cell surface receptors to start signal transduction. TNFSF ligands and receptors have distinct structural properties that relate them to cell growth, survival, or death, while some molecules can activate both inflammatory and cell death pathways, depending on target cell types and other external stimuli. Many of the TNFRSF molecules, such as CD40, TNFR1 and TNFR2 for example, are expressed in immune cells, indicating that they may have a role in autoimmune and inflammatory illnesses, as well as in cancer.
Moreover, TNFα is a positive regulator of M1 polarization via the NF-κB pathway. Therefore, among the superfamily of TNFR, we will describe more precisely TNFR1 and TNFR2 that are TNFα receptor subunits and are highly expressed in TAMs (136).
TNFR1 and 2 are single-spanning type I transmembrane proteins characterized by several cysteine-rich domains (CRDs) in their extracellular domain that are not directly involved in ligand binding but mediate inactive self-association in the absence of ligand. Regarding the cytoplasmic moiety, TNFR1 harbors a DD allowing protein-protein interaction with cytoplasmic proteins also harboring a DD domain (137).
After binding TNF-α, TNFR1-associated death domain (TRADD) and receptor-interacting serine/threonine-protein kinase 1 (RIPK1) are recruited by the TNFR1 via DD-DD interactions inducing the recruitment of TRAF2 homotrimers and E3 ligase, leading to NF-κB classical signaling. Indeed, TRAF2:TRAF2 homodimers (or TRAF3:TRAF3) already form complexes in the cytoplasm with the E3 ligases cIAP1 and cIAP2 and are recruited on TRADD. RIPK1 will be modified by addition of K63-linked ubiquitin chains that will serve as docking site for the LUBAC complex. This complex will ubiquitinate NEMO, a member of the IKK complex that interacts with TRAF2 via its IKK2 subunit, the TAK1-TAB2 complex interacts with K63-ubiquitin modified RIP1 via the TAB2 K63-ubiquitin binding subunit. Activated TAK1 then phosphorylates IKK2 leading to phosphorylation of iκBα and its subsequential K48-ubiquitination and degradation. This degradation will allow p50/p65 NF-κB dimer translocation to the nucleus inducing pro-inflammatory gene expression (138) (Figure 2).
TNFR1 signaling complex starts internalizing and this comes along with the release of the TNFR1-bound signaling molecules that trigger apoptosis through caspase-8 or necroptosis through RIPK3:RIPK1 complex formation. Apoptotic cells produce membrane-enclosed apoptotic vesicles carrying the dying cell’s material, which are removed by macrophages during the resolution of inflammation. Necroptosis, on the other hand, is a lytic form of cell death that causes inflammation by releasing intracellular DAMPs and proinflammatory cytokines (139).
TNFR2 engagement also results in activation of the classical NF-κB pathway and to the recruitment of TRAF2-cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) and TRAF1-TRAF2-cIAP1/2 complexes. This phenomenon can lead to a significant depletion of these complexes in the cytoplasm and may affect other activities of these molecules inducing activation of the alternative NF-κB pathway through TRAF3: MAP3K NF-κB-inducing kinase (NIK) complex formation (140).
cIAP1/2 are also implicated into alternative NF-κB pathways through degradation of NIK which activates IKKα, then phosphorylates p100 on NF-κB2 (p52/p100), leading to p100 proteasomal degradation and release of p52. After binding to RelB, p52 is translocated to the nucleus to control gene transcription (139).
TNFR1 also triggers the MAPK cascades leading to the activation of ERK, JNK (141) and P38 (142). TNFR1 activates a MAP3K called apoptosis-signaling kinase-1 (ASK-1) that associates with TRAF2 in the TRADD-RIPK1-TRAF2 complex, activating MAP2Ks, JNK/ERK kinase-1 (SEK1, also known as MAPK-kinase (MAP2K)-4), and MAP2K-7, which in turn activates JNKs, MAP2K-3 and MAP2K-6 which activate P38 MAPK (143). TNFRs also activate the ERK1/2 signaling pathway via activation of TPL2-MAP2K1/2 (also called TPL2-MEK1/2-ERK) pathway through activation of NF-κB pathway as described before (144). It has also been documented that TRAF2 initiates P38 activation by binding two proximal protein kinases: GCK and RIP. GCK and RIP, in turn, signals by binding MAP3Ks upstream of the JNKs and P38s. The signaling cascade downstream of TRAF2 is well known to regulate and mediate proinflammatory responses leading to macrophage production of cytokines and type I IFN (145).
Another interesting receptor from TNFR family in terms of macrophage polarization is the trans-membrane costimulatory receptor CD40. CD40 is expressed on macrophages and was shown to play crucial roles in autoimmune and infectious diseases, transplant rejection and tumor regression. CD40 can transduce signals that regulate a wide range of cellular responses, from proliferation and differentiation to growth repression and cell death. The ligand of CD40, CD154 (=CD40L or GP39), has a bidirectional effect on antigen-presenting cells. Indeed, CD40-CD154 interaction activates ERK1/2 leading to synthesis of anti-inflammatory IL-10 and phosphorylation of P38 that results in pro-inflammatory IL-12 production depending on signaling strength. It has also been shown that CD40-CD154 interactions can activate macrophages and is required for production of NO (146).
CD40 triggers TRAF6 pathway leading to SRC/MEK/ERK signaling (147) previously described and can activate others MAPKs. As the other members of TNFR family, CD40 can trigger NF-κB pathway through TRAF6 (148). Transduction downstream of CD40 also involves the JAK3/STAT3 and PI3K/AKT pathway activation (149) known as pro M2-type signaling pathways.
All these observations show that TNFR family might be a major player in M1 to M2 macrophage balance in the TME.
Gm-csfr
GM-CSF interaction with its receptor (GM-CSFR) triggers M1 polarization of macrophages, with the generation of proinflammatory mediators such as TNF-α, IL-6, IL-12, and IL-23 (150).
GM-CSF can interact with the two receptor subunits of GM-CSFR. The first component is the ligand-specific α-chain (GM-CSFRα), which forms a complex with the b-chain (GM-CSFRβ) to form GMC-SFR. The β-chain is also shared by IL-3 and IL-5. These complexes of GM-CSF–GM-CSFRα–GM-CSFRβ create first hexameric and then dodecameric ligand–receptor complexes, which will trigger JAK/STAT pathway and especially the JAK2–STAT5 (151) pathway that favor pro-inflammatory polarization of macrophages (152), but also NF-κB, PI3K/AKT, and growth factor receptor-bound protein 2 (GRB2)/MEK/ERK signaling pathways (151, 153) (Figure 3), which will be described later in this review.
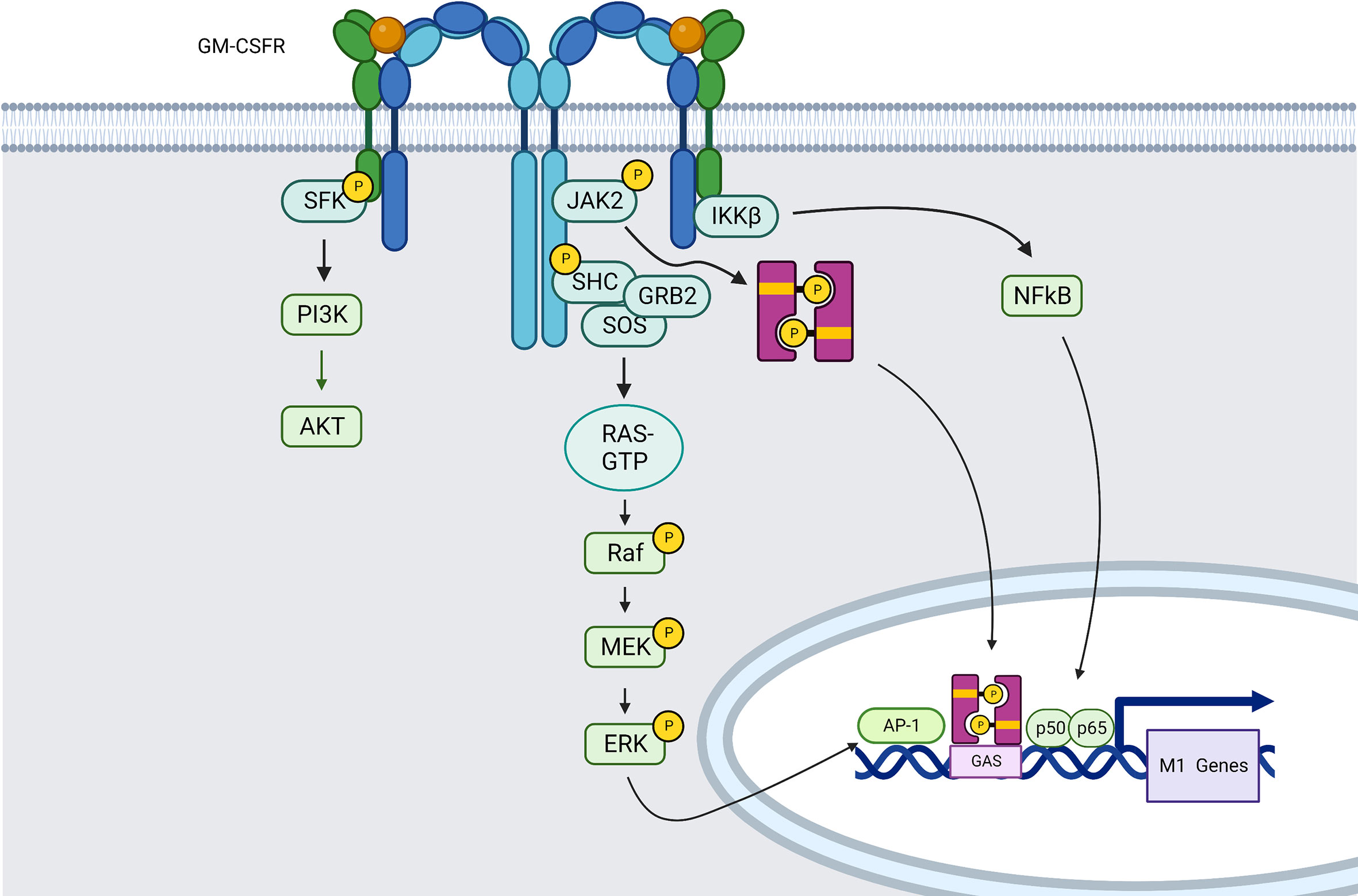
Figure 3 GM-CSFR induced signaling pathways in macrophages (created using BioRender®).
Indeed, MAPK pathways can also be directly activated by receptor tyrosine kinases (RTKs) such as GM-CSFR, among others. Ligand-induced receptor dimerization increases receptor activation and Tyrosine autophosphorylation in the intracellular region. In the case of MEK/ERK pathway for example, the phosphorylated residues serve as binding sites for proteins like GRB2 that have SH2 or phosphotyrosine-binding (PTB) domains. Son of sevenless (SOS), a guanine nucleotide exchange factor (GEF) is recruited from the cytosol to the plasma membrane by GRB2, where it drives the exchange of guanosine diphosphate (GDP) bound to RAS by guanosine triphosphate (GTP), which is necessary for positive control of RAS activity. RAS may interact directly with its target effectors, one of which is RAF, thanks to this nucleotide exchange. Activated RAF binds to and phosphorylates the dual-specificity kinases MEK1/2, which then phosphorylate ERK1/2 in their activation loop via a conserved Thr-Glu-Tyr (TEY) motif (121) (Figure 3).
NOTCH receptor
The Notch signaling pathway is widely acknowledged to have a critical role in controlling development and assisting in the regulation of the response to various stimuli. Hitherto, four NOTCH receptors and five NOTCH ligands have been identified. NOTCH receptors attach to members of their ligand families, Delta-like proteins (DLLs) and Jagged (JAG) proteins, inducing the release of NOTCH intracellular domain (NICD) into the cell nucleus, then binding to recombination signal binding protein for immunoglobulin kappa J (RBP-J) to form a transcriptional IRF8 complex, thus promoting target M1 gene expression (154). In a non-canonical way, Notch can also activate mechanistic target of rapamycin (mTORC2)/AKT and NF-κB through its NICD (155).
NOTCH1 expression is increased in M1 macrophages, and its inhibition was shown to enhance M2 polarization (156). Furthermore, in a mouse model of breast cancer (mouse mammary tumor virus-polyoma middle tumor-antigen (MMTV-PyMT)), hyperactivation of Notch signaling, particularly in TAMs, was found to inhibit tumor development (157).
Moreover, in the literature both M1 and M2 phenotypes could be generated in TAMs through Notch pathway depending on the upstream ligand-Receptor involved. For example, in vitro coincubation of macrophages with cells expressing DLL4 can lead to activation of NOTCH1 signaling and result in pro-inflammatory genes expression, such as IL-12 and iNOS (158, 159). Moreover, the blockade of DLL4 using an antibody reduced pro-inflammatory macrophage accumulation in inflammatory lesions and attenuated atherosclerosis and metabolic disease, while NOTCH1/JAG1 signaling was shown to regulate anti-inflammatory macrophage activation and IL-10-producing TAMs (160).
Research about how Notch signaling is controlled in TAMs and translated into pro- or anti-tumor actions is still in its early stages (158).
Trem-1
Triggering receptor expressed on myeloid cells 1 (TREM-1) is found mostly on myeloid cells such as monocytes/macrophages and granulocytes. TREM-1 exists in two forms: as a membrane-bound receptor and/or as a soluble protein. In its membrane bound form, TREM1 is composed by three different domains: an immunoglobulin-like domain that is responsible for ligand binding, a transmembrane portion, and a cytoplasmic tail that interacts with the immunoreceptor tyrosine-based activation motif (ITAM) of adaptor molecule DAP12. This interaction leads to DAP12 tyrosine phosphorylation and results in downstream signal transduction through ζ-associated protein of 70 kD (ZAP70) and spleen tyrosine kinase (SYK) recruitment. SYK then recruits and tyrosine phosphorylates adaptor complexes including Casitas B-lineage Lymphoma (CBL), SOS and GRB2, resulting in downstream signal transduction through the PI3K, phospholipase-C-gamma (PLC-γ), and ERK pathways. These pathways result in transcription factor activation, including ETS domain-containing protein (Elk1), nuclear factor of activated T-cells (NFAT), AP-1 (c-Fos and c-Jun), and NF-κB, which trigger transcription of genes encoding pro-inflammatory cytokines such IL-1β, chemokines such as CCL2, and cell-surface molecules such as CD86 and MHC class II (161, 162).
Receptor signaling pathways leading to anti-inflammatory M2-like macrophages
As for M1 macrophages, the interaction of chemokine/cytokine receptors with their ligands present in the TME is the main actor of M2 macrophage polarization through receptor signal transduction and downstream signaling pathways. Therefore, we will describe in the next part, these main mechanisms implicated in M2 polarization in a tumor context.
Mcs-r
Also known as CSF1R, this receptor binds M-CSF also named CSF-1. MCSF-R is also activated upon IL-34 engagement, thus representing the only RTK known to be activated by two ligands of unrelated sequence. MCSF and IL-34 support the expression of CD11b and of chemokines, cytokines, and other plasma membrane markers characteristic of M2 polarization (163).
M-CSF binding to MCSF-R causes fast dimerization of the receptor, a first wave of tyrosine phosphorylation of MCSF-R, and the creation of complexes between the CSF-1R and SFK, CBL, the regulatory subunit of PI3K (p85) and p110δ, GRB2, and other signaling molecules, many of which get tyrosine-phosphorylated (Figure 4).
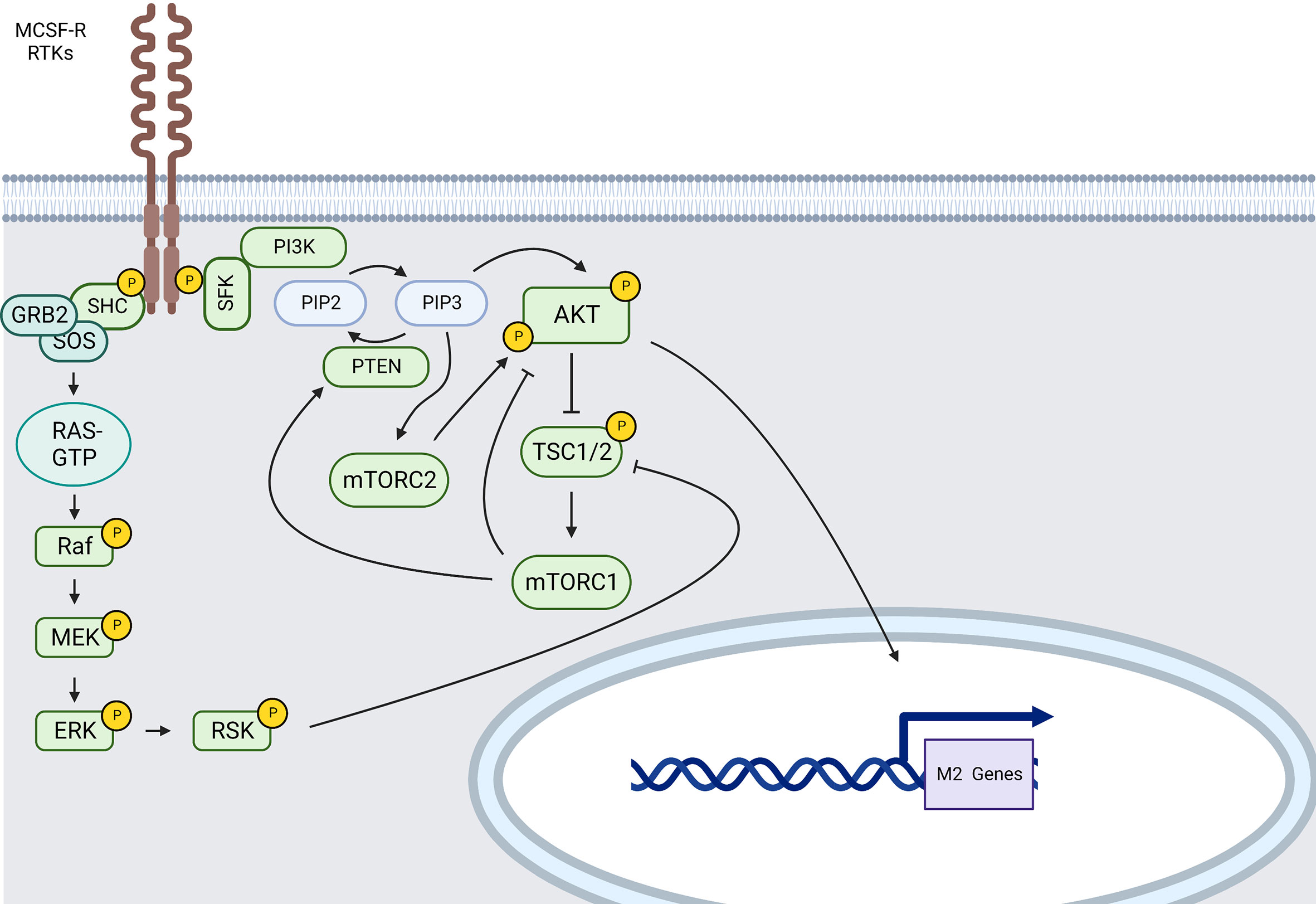
Figure 4 MCSF-R and RTKs main signaling pathways involved in M2-like polarization (created using BioRender® ).
Indeed, after engagement of the MCSF-R, a cascade of downstream signaling molecules, including those involved in the PI3K/AKT and MAPKs signaling pathways (164), is activated, boosting survival and differentiation of M2-like macrophages.
Recruitment of PI3K will then induce phosphorylation of phosphatidylinositol-4, 5-bisphosphate (PIP2) to catalyze phosphatidylinositol-3, 4, 5-triphosphate (PIP3) at the plasma membrane; PIP3 further recruits AKT through is pleckstrin homology domain (PH-domain) causing a conformational change and the phosphorylation of AKT at T308 by 3-Phosphoinositide-dependent protein kinase 1 (PDK1) (165). PIP3 also recruits the mTORC2 complex and enhances AKT activation by mTORC2 via S473 phosphorylation. When AKT is activated through its phosphorylation, it subsequently phosphorylates and inactivates the tuberous sclerosis complex (TSC) 1/2. TSC1/2 inhibition by AKT activates mTORC1 via Ras homolog enriched in brain (Rheb) suppression (166). mTORC1, such as JNK1 (167), can then induce PTEN and subsequent AKT pathway inhibition. AKT inhibition abrogates the upregulation of several M2 genes (168). Nevertheless, individual AKT2 isoforms also contribute to M1 polarization (169). Indeed, AKT is a serine-threonine protein kinase family composed of three isoforms expressed from independent genes (AKT1/PKBα, AKT2/PKBβ and AKT3/PKBγ) (170) and there is data suggesting that the outcome of macrophage polarization upon AKT activation depends on the AKT isoform involved. Indeed, AKT1 ablation results in M1 polarization whereas AKT2 ablation results in M2 polarization (169). Reciprocally, AKT2-deficient macrophages present with enhanced IL-10 secretion upon LPS stimulation (171).
AKT isoform-specific effects on macrophage function have been also reported for PI3K and isoform-specific effects on macrophage function have been reported both for AKT and PI3K (166, 170, 172, 173). Indeed, PI3K p110δ, p110β, p110γ isoforms seem to be associated with M2 macrophage polarization whereas p110α seems to lead to M1 macrophage polarization (170). The most characterized and expressed isoform in myeloid cells is p110γ. In fact, it has been shown that P110γ/AKT/mTOR-mediated immune suppression promotes tumor growth and that P110γ inhibition suppressed myeloid cell adhesion and recruitment into tumors. p110γ inhibition also reverses tumor growth and mediated immune suppression by inducing proinflammatory gene expression in TAMs through NF-κB inhibition (174, 175). However, more data would be needed to accurately assess the roles of the other different PI3K isoforms in macrophage polarization.
Other important actors implicated into PI3K/AKT/mTOR pathway have been identified. For example, the lipid phosphatase PTEN is a negative regulator preventing overactivation of the AKT/mTOR pathway. Mice treated with a myeloid-specific PTEN deletion present a larger number of M2 macrophages releasing less TNF-α and more IL-10 in response to TLR ligands (176). AMPK is an inhibitor of mTOR activity resulting in a stronger reduction of the anti-inflammatory cytokine production (IL-10) as well (177).
During macrophage response to M-CSF, ROS promotes AKT1, P38 and JNK activation (178, 179). PI3K regulates ERK phophorylation in macrophages treated with M-CSF resulting in VEGF production (180, 181). Internalized MCSF-R induces by CBL ubiquitination mediates sustained ERK1/2 and AKT signaling (182). GRB2 direct recruitment is also involved in MCSF-R-mediated transient ERK activation when it is associated with the GEF, SOS (183). ERK can activate ribosomal S6 kinase (RSK) that inhibits TSC1/2 leading to mTORC1 activation. These observations show that MAPKs and more particularly ERK1/2 activation can be linked to both M1 and M2 polarization of macrophages.
Among the several signaling cascades described as downstream of MCSF-R, PI3K/AKT is the most relevant and its downstream targets are critical in M2 macrophage polarization. This pathway is also important in limiting pro-inflammatory responses and increasing anti-inflammatory responses in TLR-stimulated macrophages, and was identified as a negative regulator of TLR and NF-κB signaling in macrophages (184).
All MCSF-R intracellular phosphorylation sites and related functions are detailed in E. Richard Stanley and Se Hwan Mun review (185).
Interleukin Receptors
Il-10R
IL-10 receptor is composed of at least two subunits, IL-10Rα and IL-10Rβ, which are members of the interferon receptor (IFNR) family. IL-10R signaling is mainly relayed by the JAK/STAT system (Figure 1). Indeed, IL-10Rα is constitutively associated to JAK1, whereas IL-10Rβ is constitutively associated to TYK2 (186). Upon IL-10 engagement, these kinases phosphorylate transcription factor of the STAT family. STAT3 is a transcriptional regulator acting downstream of IL-10 signal, a crucial anti-inflammatory cytokine. IL-10 has also been proven to be a key mediator in the resolution of inflammation. Conditional genetic inactivation of STAT3 in mouse macrophages revealed the role of STAT3 in the regulation of inflammation since these animals presented with decreased bactericidal activity and increased production of pro-inflammatory cytokines IL-12, IL-6, TNF-α, IL-1β, IFN-γ in response to LPS, and were refractory to IL-10 treatment (187, 188). Myeloid cells exhibit robust STAT3 activation in response to recombinant IL-10 variants across a wide range of IL-10Rβ–binding affinities (188). Moreover, IL-10-mediated inhibition of IFN-induced gene transcription (CXCL10, ISG54, ICAM-1) in human monocytes correlates with inhibition of IFN-induced STAT1 activation and tyrosine phosphorylation (189). Levels of IL-6, IL-8, and TNF-α produced by primary human monocyte-derived macrophages stimulated with LPS were strongly decreased upon IL-10 treatment (188).
Furthermore, induction of IL-10 in macrophages by proinflammatory signals requires activation of AKT (190) and IL-10 also promotes M2 polarization through the induction of p50 NF-κB homodimer, c-Maf, and STAT3 activities (191). Finally, IL-10 inhibit LPS-Induced MAPK activation (192).
Il-4R/Il-13R
There are two types of IL-4 receptors, namely Type I receptors, which are composed by the IL-4 receptor α-chain (IL-4Rα) and the common gamma chain (γc), and type II receptors, which are composed by IL-4Rα and the IL-13 receptor α-chain 1 (IL-13Rα1). IL-4 initially binds to IL-4Rα with high affinity, which then recruits γc or IL-13Rα1 to create type I or type II ternary ligand-receptor complexes, respectively.
The activation of the receptor-associated JAKs results in the phosphorylation of tyrosine residues in the cytoplasmic domain of the IL-4Rα, which then serve as docking sites for the recruitment of other signaling molecules, including STAT6, the primary STAT protein activated in response to IL-4 stimulation. JAKs can tyrosine-phosphorylate STAT6, causing it to disengage from the receptor and dimerize via reciprocal contacts between its SH2 domain and the phosphotyrosine 641 (Y641) on another STAT6 molecule (193). STAT6 homodimers translocate to the nucleus, where they bind to specific DNA patterns inside the promoters of responsive target genes and begin M2 gene transcription (57) (Figure 1).
Alternatively, IL-4 can signal by recruiting insulin receptor substrate (IRS) proteins to specific phosphotyrosine residues on the IL-4Rα, where IRS (mainly IRS2) can be phosphorylated and recruit other signaling molecules such as the p85 subunit of PI3K. PI3K activation has been identified as a critical step in macrophage M2 activation in response to IL-4 and it has been shown that a crosstalk between the STAT6 and PI3K pathway is required for IL-4–induced M2 macrophage activation in SHIP-deficient macrophages (193). IRS recruitment of PI3K leads to the activation of the downstream protein serine/threonine kinase AKT/mTOR pathways. IRS can also interact with GRB2, which is complexed to SOS and causes Ras and the downstream MAPK pathway activation.
With the development of genomic technology during the last decade, data on gene expression patterns in IL-4-stimulated macrophages has gathered. Hao-Wei Wang and Johanna A Joyce have summarized IL-4-induced gene sets in TAMs in mouse and human in their review named Alternative activation of tumor-associated macrophages by IL-4 Priming for protumoral functions (193).
The characterization of these cellular interactions may lead to novel strategies for disarming TAMs’ tumor-promoting functions by targeting either upstream regulators (e.g., IL-4) or downstream effectors (e.g., cathepsins, EGF signaling), and could have potential as monotherapies or complements to conventional anticancer therapies.
Il-6R
IL-6 is a prominent proinflammatory cytokine released during infection or tissue injury that contributes to both innate and adaptive immune responses (194). It is worth noting that only a few cell types, namely macrophages, neutrophils, CD4+ T cells, podocytes and hepatocytes, express IL-6R on their cell surface and may thus respond directly to IL-6. In particular, it is known that IL-6 modulates monocyte differentiation towards macrophages and DCs (195).
IL-6 requires two distinct receptors to trigger signaling, IL-6R and gp130. IL-6 can attach to membrane IL-6R in a classic manner or to soluble IL-6R in a trans-signaling manner or be presented by T cells via DC expressing IL-6R. A hexamer complex with gp130 is produced in all three modalities of IL-6R signaling. The NF-κB and JAK/STAT3 pathways are activated by IL-6 binding and influence the polarization of macrophages to M2-Arg1 expressing macrophages (196) (Figure 1).
In cancer, TAMs were reported to secrete IL-6 via STAT3 pathway leading to expansion of cancer stem cells and breast cancer progression. Furthermore, IL-6 promotes M2 macrophage polarization, while concurrently stimulating TNBC stemness and tumor progression (197–199).
TGFβR
The transforming growth factor (TGF-β) superfamily includes 32 secreted proteins as well as three receptors. These proteins are implicated in the development of a variety of fibrotic diseases. Moreover, TGF-β is important for immune suppression in the TME, being involved in tumor immune evasion and poor response to cancer immunotherapy. In addition of being secreted by M2-like TAMs, TGF-β promotes M2-like phenotype and IL-10 secretion and decreases TNF-α and IL-12 cytokine secretion via SNAIL signal transduction (200).
TGF-β binds to three isoforms of the TGF-β receptor (TβR). TβRI and RII are both serine/threonine and tyrosine kinases, while TβRIII does not have any kinase activity. Phosphorylation of TβRI/RII is necessary for activating canonical or noncanonical signaling pathways, as well as for regulating the activation of other signaling pathways (201) (Figure 5).
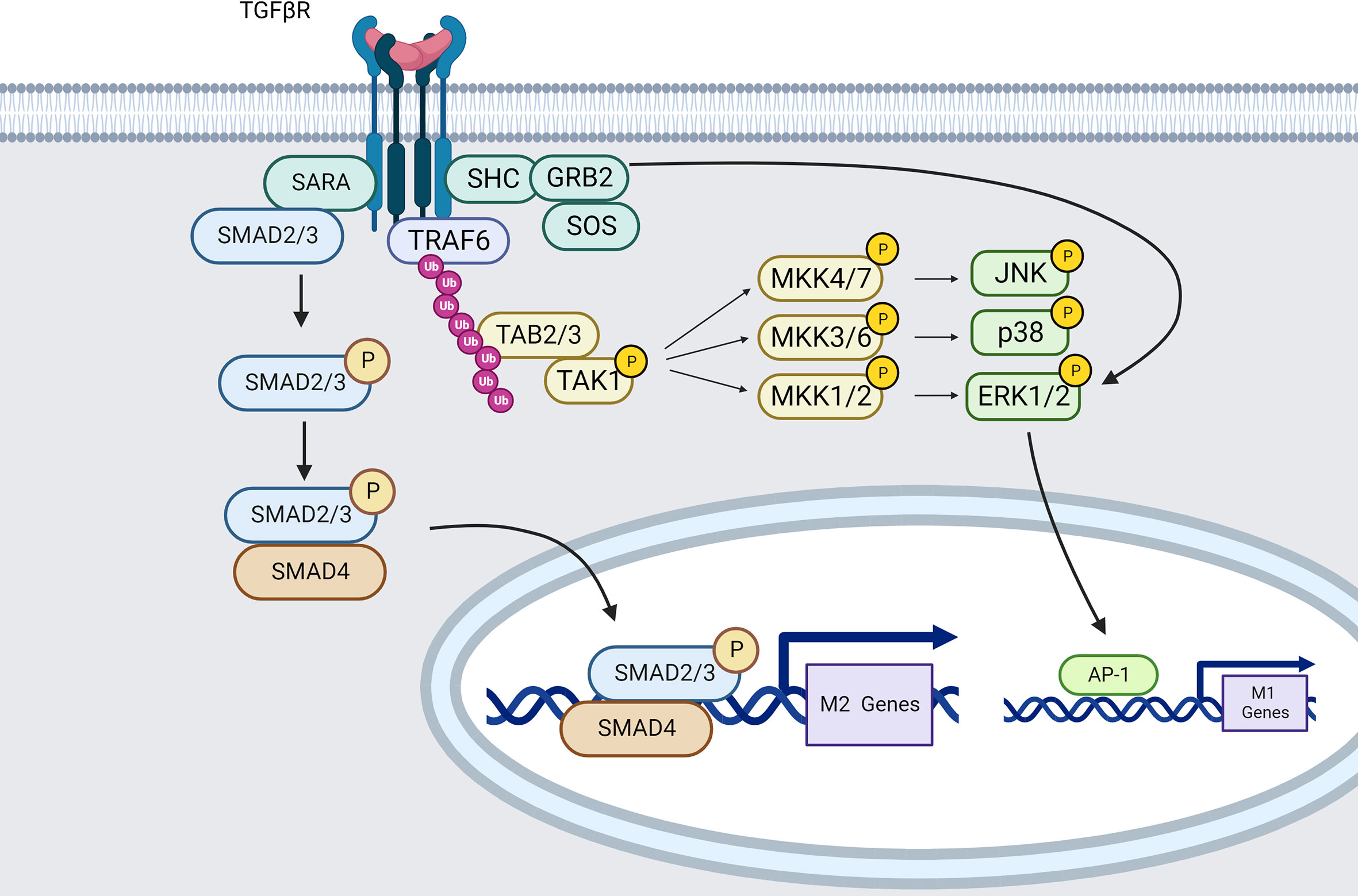
Figure 5 TGFβR signaling pathways in macrophages (created using BioRender® ).
The most extensively studied downstream mediators of TGF-β signaling are SMAD-dependent pathways. The TβRII subunit autophosphorylates and phosphorylates TβRI upon TGF-β engagement. As a result, the TβRI kinase domain interacts with the transcription factors SMAD2 and SMAD3 via the SMAD anchor for receptor activation (SARA) (202). TRβI then phosphorylates and activates SMAD2/3. When SMAD2/3 becomes active, it attaches to SMAD4, and the SMAD2/3/4 complex is translocated into the nucleus leading to ARG1 expression (203).
Data from Zhang et al. also demonstrate that blockade of the SMAD2/3 pathway reverses the immunophenotype of TGF-β induced macrophages from an anti-inflammatory M2-like phenotype to a pro-inflammatory M1 phenotype. TGF-β can also induce M2-macrophage polarization by up-regulating SNAIL expression through SMAD2/3 and PI3K/AKT signaling pathways (200).
In opposition, TGF-β signaling has also been characterized as using SMAD-independent pathways. Indeed, TRAF6 can bind TGF-β receptors to TAK1 leading to P38 MAPK, JNK and ERK-mediated M1 phenotype (204, 205).
Tie2
Angiopoietins (ANG)-1 and -2, as well as their receptor, the Tek tyrosine kinase receptor TIE2, are essential for controlling angiogenic processes throughout development, homeostasis, cancer, inflammation, and tissue repair. TIE2 is expressed by macrophages and has been proposed to contribute to solid tumor development by supporting immunosuppressive activities associated with M2 macrophage polarization.
TAMs expressing TIE2 have been named TIE2 expressing macrophages (TEM). TIE2 is a receptor for ANG1–4. In breast cancer and glioma, intratumoral TEMs are found near to nascent tumor vasculature and were found to have proangiogenic activities (206).
But interestingly, TIE2 signaling outcome can result in either a pro-inflammatory or an anti-inflammatory program according to the ligand engaged. ANG1 binding to TIE2 leads to receptor autophosphorylation on several tyrosine residues leading to signaling. Ang1 induces a pro-inflammatory phenotype in macrophages during differentiation, probably linked with P38 and ERK1/2 early activation but independent of AKT (207). However, TIE2-dependent phosphorylation of AKT prevented macrophages from apoptosis (208).
ANG2 was shown to convert macrophages to an anti-inflammatory or M2-polarized state (207, 209). Moreover, in cancer for example, when ANG2 is abundant, it binds to TIE2 and maintains TIE2 phosphorylation in an autocrine way, upregulates the expression of M2-type associated genes such as IL-10, Mannose Receptor C-Type 1 (MRC1) and CCL17 and pro-angiogenic genes such as thymidine phosphorylase (TP) and cathepsin B (CTSB) (209). However, ANG2/TIE2 interaction does not result in phosphorylation of the receptor, and instead, it acts as a competitive inhibitor preventing ANG1 binding.
G-protein-coupled receptors
Many extracellular signals are detected by GPCRs and transduced to heterotrimeric G proteins, which then transduce these signals intracellularly to suitable downstream effectors, playing a key part in numerous signaling cascades.
When activated by a ligand, GPCR proteins change conformation and subsequently activates the G proteins by increasing the exchange of GDP/GTP associated with the G subunit. This results in the dissociation of the G/G dimer from G. Both moieties are then free to engage on their downstream effectors and create distinct intracellular signaling responses.
The activation of membrane receptors (mostly GPCRs) that activate cellular adenylyl cyclases (AC) converting ATP to cAMP, causes the generation of cAMP (210). As shown by using cAMP-inducing drugs, cAMP is able to decrease the secretion of TNF-α, IL-12, leukotriene B4 (LTB4), IL-1, and chemokines such as CCL3, CXCL1, CCL2, CCL4, and CCL11. cAMP also induces activation of STAT3 and STAT6 that lead to M2 polarization. In addition, activation of Epac1/2 by cAMP inhibits the production of pro-inflammatory cytokines through NF-κB pathway (211, 212) (Figure 6).
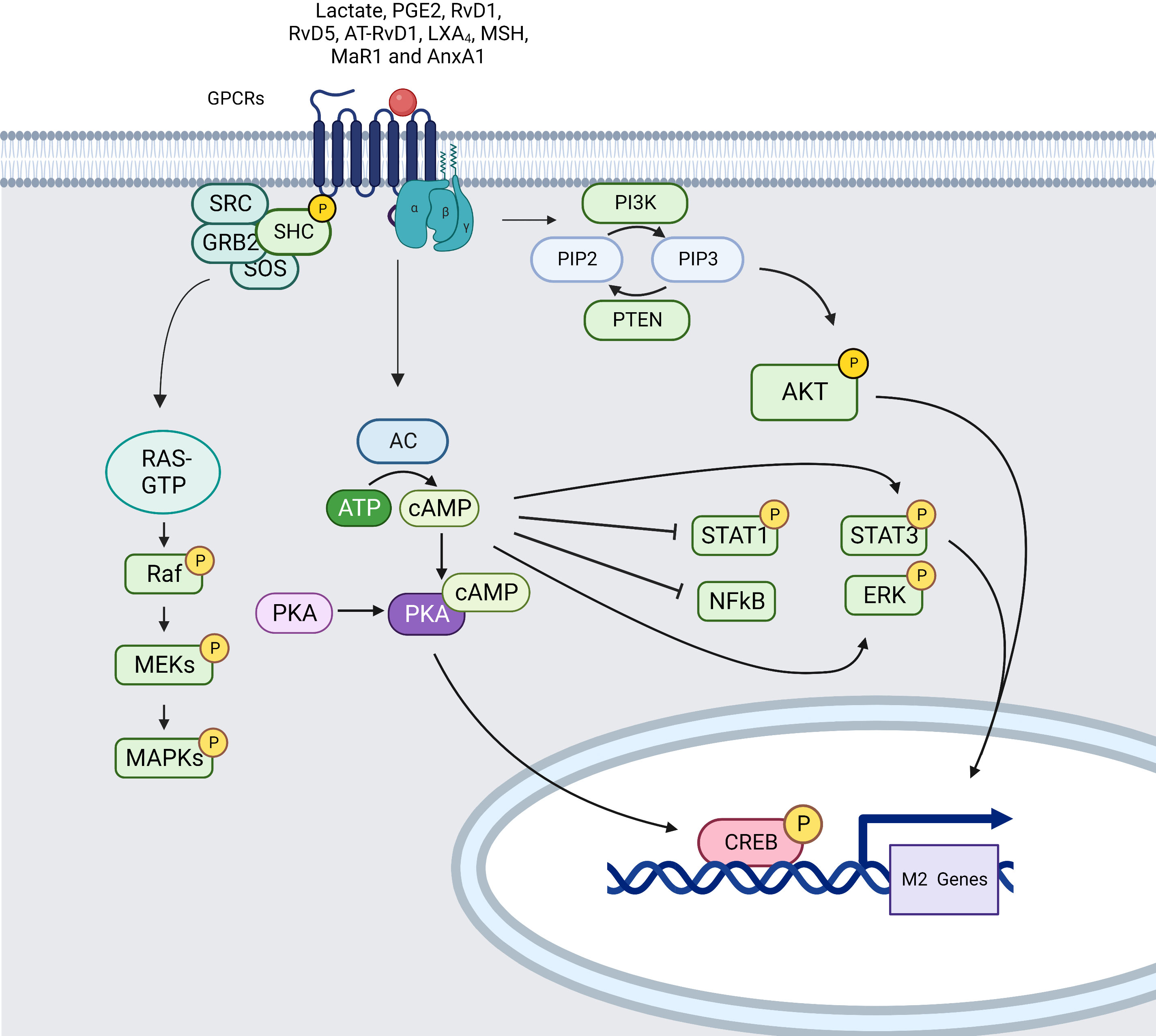
Figure 6 Simplified signaling pathway induced by GPCRs in macrophages (created using BioRender® ).
Several different mediators can lead to cAMP activation through GPCRs binding such as tumor-derived lactate (213), Resolvins (RvD1, RvD5 and AT-RvD1), lipoxins (LXA4), melanocortins (MSH), maresin 1 (MaR1), adenosine and potentially Annexin A1 (AnxA1) increasing AC activity through binding to GPCRs with subunits Gαs. Through activation of EP2–NF-κB signaling pathway, mediators such as PGE2 lead to cAMP induction triggering subsequent protein kinase A (PKA) activation inducing phosphorylation and nucleus translocation of cAMP-responsive element binding protein (CREB). CREB translocation promotes the production of anti-inflammatory cytokines and stimulate macrophage polarization. Moreover, PKA can inhibit NF-κB activity leading to diminution of inflammatory gene expression and activate ERK1/2 mediating the secretion of CCL2. cAMP drives M2 polarization by phosphorylating STAT3 and it also re-educates M1 macrophages towards an M2-like phenotype by lowering STAT1 phosphorylation via PKA (214, 215).
MAPK are also activated by ligands for heterotrimeric GPCRs mainly through the recruitment of GRB2/SOS complex that leads to RAS/RAF GTPase pathway activation and MEK/ERK signal transduction (216).
Chemokine receptors belong to the class A family of GPCRs and cluster of five chemokine receptors (CCR2, CCR5, CCR7, CX3CR1, and FPR1) is strongly expressed on myeloid cells. The chemokines CCL2, CXCL12 and the chemokine-like protein migration inhibitory factor (MIF) play a particular role in TAM polarization. Numerous models have dissected the pro-inflammatory axis between CCL2 and its corresponding receptor CCR2. In fact, blocking CCL2 during macrophage polarization upregulated the M1-associated HIF1α gene and enhanced the production of CXCL8 (217).
Sierra-Filardi et al. clearly demonstrate the anti-inflammatory intracellular signaling initiated by CCR2 ligation and indicate that CCL2 directs macrophage polarization toward the development of a M2 profile. This mechanism seems to be mediated through different pathways such as P38, HSP27 (an P38 downstream effector), ERK1/2, MSK1/2 (an ERK downstream kinase), JNK, STAT5a/b (218).
Another well studied chemokine in cancer is CCL5, which greatly promotes carcinogenesis, stroma formation, cancer progression and metastasis (219). Recent studies showed that CCL5 directly promotes M2 type polarization and that blocking CCL5-CCR5 binding led to M2 to M1 repolarization. Moreover, this modulation seems to appear through the JAK/STAT pathway (220). Inhibition of CCR5 (CCL5 receptor) using an antagonist antibody or drugs leads to repolarization of M2 to M1 tumor-associated macrophages (221, 222).
Others GPCRs expressed in macrophages and leading to macrophage activation are referenced into Wang et al. review (223).
SIRPα
The signal regulatory protein α (SIRPα)/CD47 axis has emerged as an important innate immune checkpoint that allows cancer cells to escape phagocytosis by macrophages. SIRPα is an immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing receptor of the SIRP family expressed on all myeloid cell types including monocytes, macrophages, DCs, and neutrophils, and found to be strongly expressed in the TME. Extracellular ligation of SIRPα of its ligand CD47 alone does not significantly increase phosphorylation of SIRPα’s ITIMs but comes to counteract the activation induced by phosphorylation of ITAM-containing activatory receptors such as FcγRs.
A recent paper demonstrates that SIRPα signaling partially represses NF-κB, MAPK and STAT1 activation and potently inhibits PI3K-induced AKT2 activation in IFN-γ/LPS–treated macrophages leading to inhibition of pro-inflammatory M1 macrophage polarization. In parallel, in the same model, depletion of SIRPα induces overactivation of NF-κB, MAPK, and STAT1 pathways and moreover, SIRPα exposition to CD47 drastically decreases but also shortens the AKT2 phosphorylation through SHP1 recruitment (224).
In addition, another recent article showed that SIRPα and Notch Signal-Mediated Macrophage Polarization are probably linked. Indeed, Notch activation has been documented to repress SIRPα transcription directly through HES1-binding sites in its promoter region. Notch signal altered macrophage polarization in part by controlling the expression of SIRPα (225).
Indeed, SHP1 (PTPN6) and SHP2 (PTPN11) are paralog cytoplasmic PTPases that are crucial for a wide range of cellular functions. Through phosphotyrosine-based motifs such as ITIM and immunoreceptor tyrosine-based switch motif (ITSM), a significant number of inhibitory receptors like SIRPα recruit SHP1 and/or SHP2, tandem-SH2-containing phosphatases. SHP2 and SHP1 appear to be involved in a variety of signal transduction processes, such as the GRB2/Ras/Raf/MAPK, JAK/STAT, and PI3K pathways through direct interaction with signaling intermediates such as GRB2, FRS-2, JAK2, p85 subunit of PI3K, IRS1, GAB1 and GAB2 (226, 227).
Hence, by counteracting signaling cascades involved in pro-inflammatory responses, SIRPα may play a key role in modifying macrophage polarization in cancer in addition to its role as phagocytosis inhibitor.
Lilrb2
The leukocyte immunoglobulin-like receptor (LILR) family is a group of paired immunomodulatory receptors found in human myeloid and lymphoid cells. LILR subfamily A (LILRA) members connect with membrane adaptors to signal via ITAMs, whereas LILR subfamily B (LILRB) members signal via numerous cytoplasmic ITIMs (228). More interestingly, LILRBs are documented as being negative regulators of myeloid cell activation. Such as SIRPα, the ITIM domain of LILRB has the ability to bind SHP1/SHP2. Blocking antibodies targeting LILRB2 show reduction in receptor-mediated activation of SHP1/2 resulting in upregulation to pro-inflammatory pathways such as MAPK P38 and ERK, NF-κB or STAT1, and downregulation of AKT and STAT6 pathways leading to reprogramming towards a M1-like phenotype (229).
Egfr
The EGF receptor (EGFR), also known as ErbB1/HER1, is the prototype of the EGFR family, which also includes ErbB2/HER2/Neu, ErbB3/HER3, and ErbB4/HER4. With the ability to form homo- and heterodimers, these family members may construct a total of 28 distinct combinations with one another (230). The interaction of an EGF ligand-EGFR causes receptor dimerization, receptor trans-autophosphorylation on the C-terminal domain, and the recruitment of signaling proteins or adaptors. The phosphorylated C-terminal domain contacts SHC and GRB2. As described before, GRB2 SH3 domain recruits SOS or GAB1 proline-rich domains to initiate ERK MAPK or PI3K/AKT signaling, respectively. EGFR can also bind SCR and PLCy leading to activation of downstream well-known signals (231).
In addition to EGF-secreted action on tumor cell proliferation and survival, studies shows that secreted EGF also plays a crucial role in M2 polarization in cancer (232, 233).
Conclusion and perspectives
TAMs have emerged as an interesting candidate population for innovative anti-tumor therapies and several emergent treatment approaches have been tested to reduce TAMs in tumors with, so far, limited efficacy. More recently, reprogramming M2-like TAMs into immunostimulatory and anti-tumor M1-like cells has appeared as an appealing approach in cancer therapy with encouraging preclinical and preliminary clinical data using antibodies targeting M2-like transmembrane proteins such as MARCO, CLEVER1 and ILT4 (234–236). Therefore, it is pivotal to better understand the mechanisms at the origin of the plasticity of this population.
In most of the cases, a stimulus from TME will trigger one or several of the JAK/STATs, MAPK, PI3K/AKT, NOTCH and NF-κB signaling pathways thus resulting in TAMs polarization. There is enough evidence establishing the association between M2 polarization and the activation of several signaling pathways including PI3K/AKT, JAK/STAT6 or STAT3, TGF-β/SMAD-dependent pathways. In addition, it is also possible to link directly JNK, P38, NF-κB p65, JAK/STAT1 signaling pathways and M1 polarization.
Indeed, the more currently advanced drugs targeting TAMs such as CSF1/CSF1R axis (237), MEK/STAT3 inhibitors (238), antibodies against IL-4, IL-4Rα, and IL-13 (239), IFN-γ (240, 241), CD40 agonists (242), inhibitors of PI3Kγ/mTOR (237) and agonists of TLR4/7/8/9 (237) all have an impact on macrophages signaling. For example, PI3Kγ inhibitor (ipi-549, phase II in combination with nivolumab), in addition to impact PI3K/AKT pathway will enhance drastically NF-κB phosphorylation after treatment (237). Moreover, patients with advanced malignancies are currently being tested with the STAT3 inhibitor (TTI-101) in a phase I clinical trial (NCT03195699). Indeed, targeting JAK2, the main activator of STAT3 in myeloid cells, is a crucial strategy for inhibiting STAT3. Another example is the use of anti-ILT4 mAbs, which was also found to activate P38, ERK, NF-κB and STAT1 while inhibiting the activation of STAT6 and AKT in the presence of M-CSF and IL-4 within 30 minutes of anti-ILT4 treatment (229). It has also been demonstrated that ILT4 blocking can both activate monocytes from PBMCs as well as acting in M2 macrophages to trigger M2-to-M1 reversion (229).
However, the M1 vs. M2 dichotomy is much less evident regarding other signaling pathways, such as NOTCH, ERK and even NF-κB, for which different studies demonstrated an implication in both polarization outcomes. Similarly, some receptors, which will be discussed further in the discussion section, can be associated with one phenotype, while others, depending on the stimulus, can lead to two different phenotypes. Moreover, some of them may operate a balance between the two phenotypes depending on time of exposure, timing of activation, specific serine/tyrosine phosphorylation, ligand type, multivalency and/or strength of stimuli in TME. Indeed, we cannot depict a black and white model that unequivocally defines whether the ligand-receptor engagement will be favorable to either M2 or M1 phenotype, and most likely, the integration of the different stimuli and the balance between their signals will determine the fate of macrophage polarization.
Besides, the analysis of TAMs using traditional, scRNA-seq, or time-of-flight (CyTOF) mass cytometry methods has shown the presence of several macrophage cell clusters with unique transcriptome and proteomic profiles. These methods have been essential in identifying the variety of TAMs outside the traditional M1-like or M2-like dichotomy in lung cancer, non-small cell lung cancer and brain tumors (243–245). Nevertheless, it remains clear that some sub-populations of TAMs either promote or limit cancer development and, as such, the M1-like and M2-like categories continue to have communicative value when the limitations of the macrophage classification are taken into account (246). A good illustration that could lead to better understanding of this paradoxes is the implication of signaling pathways that probably leads to more complex outcome than just M1/M2 dichotomy. However, the difficulty of characterizing different subpopulations of TAMs and of generating them in vitro under the extremely variableconditions of the TME has not yet made possible to study the involvement of signaling pathways in a more complex setting.
Additionally, this great heterogenicity in TAM could potentially explain limitations of targeting TAMs for tumor treatment. Indeed, in anti-CLEVER1 monotherapy, 7 of 30 patients with different pathology were classified as benefitting from the therapy by RECIST 1.1 (PR or SD response in target or non-target lesions) (247). Concerning anti-ILT4, clinical trial showed 1 partial response (PR) over 50 treated patients and 22% SD while anti-ILT4 in combination with pembrolizumab show encouraging results (21% PR, 26% SD and 3% complete response (CR)) (234, 235). This illustrates that targeting TAMs in combination with conventional immunotherapy treatments could significantly improve their effectiveness in the near future in the fight against cancer. Identification of TAM diversity at the single-cell level may open new perspectives on depletion strategy. This could justify the development of specific treatment against TAMs multiple subsets to improve clinic benefits of this kind of approaches.
Discussion
One possible explanation for the ability of cell surface receptors to mediate opposite biological responses is that cell surface receptors may regulate signaling pathways quantitatively differently to mediate specific biological responses: low levels of receptor occupancy may result in low levels of receptor signaling, whereas high levels of receptor occupancy may result in high levels of receptor signaling. For example, in the case of the GM-CSF/GM-CSFR axis, the intrinsically activated signaling pathways vary depending on GM-CSF concentration (248). Indeed, a very low dose of GM-CSF seems rather to be at the origin of the activation of pathways such as PI3K/AKT/mTOR, whereas a higher dose could rather favor the JAK2/STAT5 and RAS/MAPK pathway. This phenomenon is directly linked with another relevant point regarding specific serine/tyrosine residues phosphorylation dynamics. Indeed, the activation of JAK2/STAT5 and ERK pathway by higher dose of GM-CSF requires Y577 phosphorylation thus providing a SHC-binding domain, whereas low doses of GM-CSF induce S585 phosphorylation thus providing a binding domain for p85 subunit of PI3K, leading to its recruitment and inducing PI3K/AKT/mTOR pathway activation.
Additionally, this duality of signaling pathways outcome depending on phosphorylation site is also illustrated by STAT3. Indeed, STAT3 is phosphorylated on Y705 after IL-10 stimulation of macrophages leading to M2 phenotype. In the opposite way, TLR4 stimulation by LPS can lead to STAT3 S727 phosphorylation inducing STAT3 mitochondrial translocation altering ROS production (129), thus favoring the polarization of pro-inflammatory macrophages.
There are also disparities due to the differential impact of kinase isoforms on phenotype outcome. Indeed, it is well known that PI3K activates AKT. However, it is unknown how the expression and activation of AKT isoforms are regulated in macrophages. PI3K/AKT pathway has convincingly been associated with M2 polarization, however, there are some evidences showing that PI3K/AKT pathway activation can lead both to M1 (PI3K/AKT2 activation) or M2 (PI3K/AKT1 activation) according to AKT isoform (169). There are also evidences that the activation of different PI3K isoforms may have an inverse impact on macrophage polarization, but further research on this topic would allow us to better understand these mechanisms (170).
Upregulation of HIFs in response to oxygen stress can also lead to opposite effects on macrophage polarization when involving HIF-1α or HIF-2α. Indeed, HIF-1α expression in macrophages is induced by Th1 cytokines such as IFNγ leading to M1 macrophage polarization, whereas HIF-2α is induced by Th2 cytokines leading to M2 polarization (66, 162).
The duration and timing of pathway activation has also been found to regulate pleiotropically the subsequent biological responses. For example, the ‘transient versus sustained’ MAPK ERK1/2 signaling can lead to diverse phenotypes. Indeed, while many studies show that ERK pathway is involved in the inflammatory response of macrophages leading to the secretion of IL-1β, TNF-α, IL-6 or advanced glycation end products (AGEs) (249), it has also been described to be activated during M2 polarization. Indeed, the duration of ERK activation can be either transient and short-lived, or sustained and lasting several hours (250). For example, it has been shown in human macrophages that IL-4 causes an increase in ERK1/2 phosphorylation between 2 and 8 hours but not between 10 and 30 minutes, leading to M2 polarization (249). Furthermore, stimulation of macrophages with M-CSF induces sustained activation of ERK1/2 (182), leading to the same outcome than IL-4 stimulation. In fact, early and rapidly diminishing activation of ERK1/2 was mainly associated with M1 promotion (249, 251–254), as opposed to late and more persistent activation that seems to lead to M2 phenotype (182, 249).
Notch and TGF-β are also a perfect example of signaling pathway bipolarity given NOTCH receptor binding to different ligands from DLL and JAG families, and the capacity of TGFβR to induce different pathways from the same ligand. On one hand, NOTCH/JAG1 and TGF-β canonical pathways favor M2 type polarization whereas NOTCH/DLL and TGF-β non canonical pathways seem to favor M1 polarization.
Another balanced mechanism concerns NF-κB signaling pathway. Many studies showed that NF-κB is activated by proinflammatory cytokines such as TNF-α, IL-1 and pathogen-associated molecular pattern molecules leading to expression of genes involved in immunological and inflammatory responses (e.g IL-1β, TNF-α, iNOS, ICAM-1, IL-12 and CCL2), due to the nuclear translocation of NF-κB p50/c-Rel or p65/p50 dimers (255). However, formation and nuclear accumulation of other dimers such as p50/p50 is essential for M2 polarization (256, 257).
Another interesting point is the existence of redundant or parallel signaling pathways. Indeed, these notions have been highlighted in the context of resistance to cancer therapies, especially when using drugs against proliferative pathways. “Parallel” signaling pathways are defined as functionally and evolutionarily distinct, such as Notch, JAK, PI3K and MAPK, but are all capable of promoting cell proliferation. These signaling pathways are commonly considered redundant as they can fulfil the role of cell proliferation by substituting for the original pathway that has been repressed. Indeed, when drugs block one pathway, resistance may arise through the activation of other signaling pathways that are redundant or parallel. Furthermore, redundant signaling pathways are defined by the use of the same downstream signaling targets, such as K-RAS, H-RAS and N-RAS that can all active the MEK/ERK pathway (258). However, given the enormous diversity of stimuli present in the TME, which drive macrophage plasticity through the activation/repression of many different signaling pathways, this redundancy may play a key role in macrophage polarization and should attract our attention for a better understanding of these mechanisms and better thoughts on therapeutic approaches.
All these observations show that the plasticity of macrophages is tightly linked to the balance between the activation and inhibition of many different signaling pathways and remind us that a deep understanding of these mechanisms must be coupled with phenotype and functional characterization for macrophage polarization understanding and subsequent development of potent cancer therapies.
Author contributions
CK conceived of and wrote the review. CEC and DO reviewed the review. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work was sponsored by ImCheck Therapeutics and CRCM. We thank our colleagues from ImCheck Therapeutics for providing insight and knowledge that substantially helped for writing the review. We gratefully acknowledge, Etienne Foucher, Sophie Agaugué and Loui Madakamutil for suggestions that significantly improved the text as well as Aude De Gassart for assistance with figure conception.
Conflict of interest
CK and CEC are ImCheck Therapeutics employees. DO is a co-founder of ImCheck Therapeutics, Alderaan Biotechnology, and Emergence Therapeutics and has research funds from ImCheck Therapeutics, Alderaan Biotechnology, Cellectis, and Emergence Therapeutics.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lee H-W, Choi H-J, Ha S-J, Lee K-T, Kwon Y-G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim Biophys Acta (2013) 1835:170–9. doi: 10.1016/j.bbcan.2012.12.007
2. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell (2020) 181:442–59.e29. doi: 10.1016/j.cell.2020.03.048
3. Yang Q, Zhang H, Wei T, Lin A, Sun Y, Luo P, et al. Single-cell RNA sequencing reveals the heterogeneity of tumor-associated macrophage in non-small cell lung cancer and differences between sexes. Front Immunol (2021) 12:756722. doi: 10.3389/fimmu.2021.756722
4. Lantz C, Radmanesh B, Liu E, Thorp EB, Lin J. Single-cell RNA sequencing uncovers heterogenous transcriptional signatures in macrophages during efferocytosis. Sci Rep (2020) 10:14333. doi: 10.1038/s41598-020-70353-y
5. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer (2004) 4:71–8. doi: 10.1038/nrc1256
6. Tiainen S, Tumelius R, Rilla K, Hämäläinen K, Tammi M, Tammi R, et al. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology (2015) 66:873–83. doi: 10.1111/his.12607
7. Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F, et al. Prognostic significance of tumor-associated macrophages in breast cancer: a meta-analysis of the literature. Oncotarget (2017) 8:30576–86. doi: 10.18632/oncotarget.15736
8. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
9. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
10. Fan Q-M, Jing Y-Y, Yu G-F, Kou X-R, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett (2014) 352:160–8. doi: 10.1016/j.canlet.2014.05.008
11. Doak GR, Schwertfeger KL, Wood DK. Distant relations: Macrophage functions in the metastatic niche. Trends Cancer (2018) 4:445–59. doi: 10.1016/j.trecan.2018.03.011
12. Wu H, Zhang X, Han D, Cao J, Tian J. Tumour-associated macrophages mediate the invasion and metastasis of bladder cancer cells through CXCL8. PeerJ (2020) 8:e8721. doi: 10.7717/peerj.8721
13. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res (2007) 67:5064–6. doi: 10.1158/0008-5472.CAN-07-0912
14. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol (2014) 5:75. doi: 10.3389/fphys.2014.00075
15. Fu L-Q, Du W-L, Cai M-H, Yao J-Y, Zhao Y-Y, Mou X-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol (2020) 353:104119. doi: 10.1016/j.cellimm.2020.104119
16. Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology (2019) 8:1596004. doi: 10.1080/2162402X.2019.1596004
17. Mehraj U, Dar AH, Wani NA, Mir MA. Tumor microenvironment promotes breast cancer chemoresistance. Cancer Chemother Pharmacol (2021) 87:147–58. doi: 10.1007/s00280-020-04222-w
18. Mir MA, Mehraj U. Double-crosser of the immune system: Macrophages in tumor progression and metastasis. Curr Immunol Rev Discontin (2019) 15:172–84. doi: 10.2174/1573395515666190611122818
19. Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood (2009) 114:3625–8. doi: 10.1182/blood-2009-05-220285
20. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther (2021) 6:1–12. doi: 10.1038/s41392-021-00484-9
21. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev (2011) 25:2465–79. doi: 10.1101/gad.180331.111
22. Zhou S-L, Zhou Z-J, Hu Z-Q, Huang X-W, Wang Z, Chen E-B, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology (2016) 150:1646–1658.e17. doi: 10.1053/j.gastro.2016.02.040
23. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med (1983) 158:670–89. doi: 10.1084/jem.158.3.670
24. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
25. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
26. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
27. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol (2015) 36:229–39. doi: 10.1016/j.it.2015.02.004
28. Yang H-T, Wang Y, Zhao X, Demissie E, Papoutsopoulou S, Mambole A, et al. NF-κB1 inhibits TLR-induced IFN-β production in macrophages through TPL-2-dependent ERK activation. J Immunol Baltim Md 1950 (2011) 186:1989–96. doi: 10.4049/jimmunol.1001003
29. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
30. Liou G-Y, Storz P. Reactive oxygen species in cancer. Free Radic Res (2010) 44:479–96. doi: 10.3109/10715761003667554
31. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi: 10.1146/annurev.immunol.15.1.323
32. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
33. Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E, et al. Priming of human resting NK cells by autologous M1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol Baltim Md 1950 (2015) 195:2818–28. doi: 10.4049/jimmunol.1500325
34. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
35. Pozzi L-AM, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol Baltim Md 1950 (2005) 175:2071–81. doi: 10.4049/jimmunol.175.4.2071
36. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
37. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
38. Alegre M-L, Frauwirth KA, Thompson CB. T-Cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. doi: 10.1038/35105024
39. Medzhitov R, Janeway C. Innate immune recognition: mechanisms and pathways. Immunol Rev (2000) 173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x
40. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
41. Taylor PR, Martinez-Pomares L, Stacey M, Lin H-H, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol (2005) 23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816
42. Capucetti A, Albano F, Bonecchi R. Multiple roles for chemokines in neutrophil biology. Front Immunol (2020) 11:1259. doi: 10.3389/fimmu.2020.01259
43. Eriksson F, Tsagozis P, Lundberg K, Parsa R, Mangsbo SM, Persson MAA, et al. Tumor-specific bacteriophages induce tumor destruction through activation of tumor-associated macrophages. J Immunol (2009) 182:3105–11. doi: 10.4049/jimmunol.0800224
44. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res (2014) 7:19. doi: 10.1186/1757-2215-7-19
45. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol (2017) 147:181–7. doi: 10.1016/j.ygyno.2017.07.007
46. Chen X, Chen J, Zhang W, Sun R, Liu T, Zheng Y, et al. Prognostic value of diametrically polarized tumor-associated macrophages in multiple myeloma. Oncotarget (2017) 8:112685–96. doi: 10.18632/oncotarget.22340
47. Barros MHM, Segges P, Vera-Lozada G, Hassan R, Niedobitek G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PloS One (2015) 10:e0124531. doi: 10.1371/journal.pone.0124531
48. Edin S, Wikberg ML, Dahlin AM, Rutegård J, Öberg 𝛽Å, Oldenborg P-A, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PloS One (2012) 7:e47045. doi: 10.1371/journal.pone.0047045
49. Pantano F, Berti P, Guida FM, Perrone G, Vincenzi B, Amato MMC, et al. The role of macrophages polarization in predicting prognosis of radically resected gastric cancer patients. J Cell Mol Med (2013) 17:1415–21. doi: 10.1111/jcmm.12109
50. Macciò A, Gramignano G, Cherchi MC, Tanca L, Melis L, Madeddu C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci Rep (2020) 10:6096. doi: 10.1038/s41598-020-63276-1
51. Li Y, Cao F, Li M, Li P, Yu Y, Xiang L, et al. Hydroxychloroquine induced lung cancer suppression by enhancing chemo-sensitization and promoting the transition of M2-TAMs to M1-like macrophages. J Exp Clin Cancer Res CR (2018) 37:259. doi: 10.1186/s13046-018-0938-5
52. Cassetta L, Cassol E, Poli G. Macrophage polarization in health and disease. ScientificWorldJournal (2011) 11:2391–402. doi: 10.1100/2011/213962
53. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22:6995. doi: 10.3390/ijms22136995
54. Domagala M, Laplagne C, Leveque E, Laurent C, Fournié J-J, Espinosa E, et al. Cancer cells resistance shaping by tumor infiltrating myeloid cells. Cancers (2021) 13:165. doi: 10.3390/cancers13020165
55. Hamada I, Kato M, Yamasaki T, Iwabuchi K, Watanabe T, Yamada T, et al. Clinical effects of tumor-associated macrophages and dendritic cells on renal cell carcinoma. Anticancer Res (2002) 22:4281–4.
56. Hollmén M, Roudnicky F, Karaman S, Detmar M. Characterization of macrophage - cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci Rep (2015) 5:9188. doi: 10.1038/srep09188
57. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
58. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol (2002) 196:254–65. doi: 10.1002/path.1027
59. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/s1471-4906(02)02302-5
60. Komohara Y, Niino D, Saito Y, Ohnishi K, Horlad H, Ohshima K, et al. Clinical significance of CD163+ tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci (2013) 104:945–51. doi: 10.1111/cas.12167
61. Komohara Y, Horlad H, Ohnishi K, Fujiwara Y, Bai B, Nakagawa T, et al. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci (2012) 103:2165–72. doi: 10.1111/cas.12015
62. Atanasov G, Pötner C, Aust G, Schierle K, Dietel C, Benzing C, et al. TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget (2018) 9:29715–26. doi: 10.18632/oncotarget.25690
63. Vasievich EA, Huang L. The suppressive tumor microenvironment: A challenge in cancer immunotherapy. Mol Pharm (2011) 8:635–41. doi: 10.1021/mp1004228
64. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity (2010) 32:593–604. doi: 10.1016/j.immuni.2010.05.007
65. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
66. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflammation (2015) 2015:816460. doi: 10.1155/2015/816460
67. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi: 10.3389/fimmu.2014.00614
68. Josephs DH, Bax HJ, Karagiannis SN. Tumour-associated macrophage polarisation and re-education with immunotherapy. Front Biosci Elite Ed (2015) 7:293–308. doi: 10.2741/E735
69. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19:1264–72. doi: 10.1038/nm.3337
70. Duluc D, Delneste Y, Tan F, Moles M-P, Grimaud L, Lenoir J, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood (2007) 110:4319–30. doi: 10.1182/blood-2007-02-072587
71. Tu D, Dou J, Wang M, Zhuang H, Zhang X. M2 macrophages contribute to cell proliferation and migration of breast cancer. Cell Biol Int (2021) 45:831–8. doi: 10.1002/cbin.11528
72. Curtale G, Rubino M, Locati M. MicroRNAs as molecular switches in macrophage activation. Front Immunol (2019) 10:799. doi: 10.3389/fimmu.2019.00799
73. Weigert A, Brüne B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide Biol Chem (2008) 19:95–102. doi: 10.1016/j.niox.2008.04.021
74. Gocheva V, Wang H-W, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev (2010) 24:241–55. doi: 10.1101/gad.1874010
75. Krstic J, Santibanez JF. Transforming growth factor-beta and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci World J (2014) 2014:1–14. doi: 10.1155/2014/521754
76. Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol (2015) 44–46:94–112. doi: 10.1016/j.matbio.2015.04.004
77. Yang S-F, Lin C-Y, Yang P-Y, Chao S-C, Ye Y-Z, Hu D-N. Increased expression of gelatinase (MMP-2 and MMP-9) in pterygia and pterygium fibroblasts with disease progression and activation of protein kinase <g>C.</g>. Invest Ophthalmol Vis Sci (2009) 50:4588–96. doi: 10.1167/iovs.08-3147
78. Krneta T, Gillgrass A, Poznanski S, Chew M, Lee AJ, Kolb M, et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J Leukoc Biol (2017) 101:285–95. doi: 10.1189/jlb.3A1215-552R
79. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
80. Wendt MK, Allington TM, Schiemann WP. Mechanisms of epithelial-mesenchymal transition by TGF-β. Future Oncol Lond Engl (2009) 5:1145–68. doi: 10.2217/fon.09.90
81. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
82. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol (2020) 11:938. doi: 10.3389/fimmu.2020.00938
83. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med (2017) 9:eaal3604. doi: 10.1126/scitranslmed.aal3604
84. Anfray C, Ummarino A, Andón FT, Allavena P. Current strategies to target tumor-Associated-Macrophages to improve anti-tumor immune responses. Cells (2019) 9:E46. doi: 10.3390/cells9010046
85. Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med (2015) 212:435–45. doi: 10.1084/jem.20150295
86. Domagala M, Ysebaert L, Ligat L, Lopez F, Fournié J-J, Laurent C, et al. IL-10 rescues CLL survival through repolarization of inflammatory nurse-like cells. Cancers (2021) 14:16. doi: 10.3390/cancers14010016
87. Zhang Q, Liu L, Gong C, Shi H, Zeng Y, Wang X, et al. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PloS One (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
88. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang C-L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp Ther Med (2019) 18:4490–8. doi: 10.3892/etm.2019.8068
89. Shen H, Liu J, Chen S, Ma X, Ying Y, Li J, et al. Prognostic value of tumor-associated macrophages in clear cell renal cell carcinoma: A systematic review and meta-analysis. Front Oncol (2021) 11:657318. doi: 10.3389/fonc.2021.657318
90. Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci (2013) 104:437–44. doi: 10.1111/cas.12096
91. Yang C, Wei C, Wang S, Shi D, Zhang C, Lin X, et al. Elevated CD163+/CD68+ ratio at tumor invasive front is closely associated with aggressive phenotype and poor prognosis in colorectal cancer. Int J Biol Sci (2019) 15:984–98. doi: 10.7150/ijbs.29836
92. Kim H-D, Kim SY, Kim J, Kim JE, Hong YS, Han B, et al. Dynamic increase of M2 macrophages is associated with disease progression of colorectal cancers following cetuximab-based treatment. Sci Rep (2022) 12:1678. doi: 10.1038/s41598-022-05694-x
93. Goswami KK, Barik S, Banerjee S, Bhowmick AK, Biswas J, Bose A, et al. Supraglottic laryngeal tumor microenvironmental factors facilitate STAT3 dependent pro-tumorigenic switch in tumor associated macrophages to render utmost immune evasion. Immunol Lett (2013) 156:7–17. doi: 10.1016/j.imlet.2013.09.003
94. Cui Y-L, Li H-K, Zhou H-Y, Zhang T, Li Q. Correlations of tumor-associated macrophage subtypes with liver metastases of colorectal cancer. Asian Pac J Cancer Prev APJCP (2013) 14:1003–7. doi: 10.7314/apjcp.2013.14.2.1003
95. Igarashi K, Garotta G, Ozmen L, Ziemiecki A, Wilks AF, Harpur AG, et al. Interferon-gamma induces tyrosine phosphorylation of interferon-gamma receptor and regulated association of protein tyrosine kinases, Jak1 and Jak2, with its receptor. J Biol Chem (1994) 269:14333–6. doi: 10.1016/S0021-9258(17)36621-8
96. Briscoe J, Rogers NC, Witthuhn BA, Watling D, Harpur AG, Wilks AF, et al. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J (1996) 15:799–809. doi: 10.1002/j.1460-2075.1996.tb00415.x
97. Heim MH, Kerr IM, Stark GR, Darnell JE. Contribution of STAT SH2 groups to specific interferon signaling by the jak-STAT pathway. Science (1995) 267:1347–9. doi: 10.1126/science.7871432
98. Greenlund AC, Morales MO, Viviano BL, Yan H, Krolewski J, Schreiber RD. Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity (1995) 2:677–87. doi: 10.1016/1074-7613(95)90012-8
99. Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J (1997) 16:7067–77. doi: 10.1093/emboj/16.23.7067
100. Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (1997) 17:121–34. doi: 10.1089/jir.1997.17.121
101. Der SD, Zhou A, Williams BRG, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci (1998) 95:15623–8. doi: 10.1073/pnas.95.26.15623
102. Yang M, Liu J, Piao C, Shao J, Du J. ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis (2015) 6:e1780. doi: 10.1038/cddis.2015.144
103. Hamaï A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F, et al. ICAM-1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res (2008) 68:9854–64. doi: 10.1158/0008-5472.CAN-08-0719
104. Langlais D, Barreiro LB, Gros P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med (2016) 213:585–603. doi: 10.1084/jem.20151764
105. Mogensen KE, Lewerenz M, Reboul J, Lutfalla G, Uzé G. The type I interferon receptor: structure, function, and evolution of a family business. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (1999) 19:1069–98. doi: 10.1089/107999099313019
106. Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J (2013) 32:2751–63. doi: 10.1038/emboj.2013.203
107. Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci U.S.A. (1999) 96:13956–61. doi: 10.1073/pnas.96.24.13956
108. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages In vitro and In vivo. J Immunol (2007) 178:1357–62. doi: 10.4049/jimmunol.178.3.1357
109. Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev (2003) 14:361–8. doi: 10.1016/s1359-6101(03)00043-1
110. Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: Room for discovery. Immunity (2019) 50:851–70. doi: 10.1016/j.immuni.2019.03.011
111. Suzuki S, Umezu Y, Saijo Y, Satoh G, Abe Y, Satoh K, et al. Exogenous recombinant human IL-12 augments MHC class I antigen expression on human cancer cells. vitro. Tohoku J Exp Med (1998) 185:223–6. doi: 10.1620/tjem.185.223
112. Yu X, Wu B, Lin Y, Xiong H-Y, Xie C, Liu C, et al. Overexpression of IL-12 reverses the phenotype and function of M2 macrophages to M1 macrophages (2016). Available at: https://www.semanticscholar.org/paper/Overexpression-of-IL-12-reverses-the-phenotype-and-Yu-Wu/1e825aa8cf139d8f51bad72831241d95d9615703 (Accessed May 24, 2022).
113. O’Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
114. Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature (2009) 457:102–6. doi: 10.1038/nature07623
115. Vidyarthi A, Khan N, Agnihotri T, Negi S, Das DK, Aqdas M, et al. TLR-3 stimulation skews M2 macrophages to M1 through IFN-αβ signaling and restricts tumor progression. Front Immunol (2018) 9:1650. doi: 10.3389/fimmu.2018.01650
116. Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-c is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med (2009) 15:774–80. doi: 10.1038/nm.1987
117. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat BioMed Eng (2018) 2:578–88. doi: 10.1038/s41551-018-0236-8
118. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
119. Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol (2016) 100:27–45. doi: 10.1189/jlb.2RI1115-531R
120. Kollewe C, Mackensen A-C, Neumann D, Knop J, Cao P, Li S, et al. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem (2004) 279:5227–36. doi: 10.1074/jbc.M309251200
121. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000prime Rep (2014) 6:97. doi: 10.12703/P6-97
122. Ajibade AA, Wang Q, Cui J, Zou J, Xia X, Wang M, et al. TAK1 negatively regulates NF-κB and p38 MAP kinase activation in gr-1+CD11b+ neutrophils. Immunity (2012) 36:43–54. doi: 10.1016/j.immuni.2011.12.010
123. Ajibade AA, Wang HY, Wang R-F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol (2013) 34:307–16. doi: 10.1016/j.it.2013.03.007
124. Ain QU, Batool M, Choi S. TLR4-targeting therapeutics: Structural basis and computer-aided drug discovery approaches. Molecules (2020) 25:627. doi: 10.3390/molecules25030627
125. Dai L, Aye Thu C, Liu X-Y, Xi J, Cheung PCF. TAK1, more than just innate immunity. IUBMB Life (2012) 64:825–34. doi: 10.1002/iub.1078
126. Gantke T, Sriskantharajah S, Sadowski M, Ley SC. IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol Rev (2012) 246:168–82. doi: 10.1111/j.1600-065X.2012.01104.x
127. Roget K, Ben-Addi A, Mambole-Dema A, Gantke T, Yang H-T, Janzen J, et al. IκB kinase 2 regulates TPL-2 activation of extracellular signal-regulated kinases 1 and 2 by direct phosphorylation of TPL-2 serine 400. Mol Cell Biol (2012) 32:4684–90. doi: 10.1128/MCB.01065-12
128. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
129. Balic JJ, Albargy H, Luu K, Kirby FJ, Jayasekara WSN, Mansell F, et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1β expression. Nat Commun (2020) 11:3816. doi: 10.1038/s41467-020-17669-5
130. Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of macrophage plasticity in the tumor environment: Manipulating activation state to improve outcomes. Front Immunol (2021) 12:642285. doi: 10.3389/fimmu.2021.642285
131. Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis (2019) 10:273. doi: 10.1038/s41419-019-1435-2
132. Kuriakose S, Onyilagha C, Singh R, Olayinka-Adefemi F, Jia P, Uzonna JE. TLR-2 and MyD88-dependent activation of MAPK and STAT proteins regulates proinflammatory cytokine response and immunity to experimental trypanosoma congolense infection. Front Immunol (2019) 10:2673. doi: 10.3389/fimmu.2019.02673
133. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301:640–3. doi: 10.1126/science.1087262
134. Lu Y-C, Yeh W-C, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine (2008) 42:145–51. doi: 10.1016/j.cyto.2008.01.006
135. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511. doi: 10.1038/nri1391
136. Kratochvill F, Neale G, Haverkamp JM, de Velde L-AV, Smith AM, Kawauchi D, et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep (2015) 12:1902–14. doi: 10.1016/j.celrep.2015.08.033
137. Park HH, Lo Y-C, Lin S-C, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol (2007) 25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656
138. Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J (2011) 278:862–76. doi: 10.1111/j.1742-4658.2011.08015.x
139. Kearney CJ, Martin SJ. An inflammatory perspective on necroptosis. Mol Cell (2017) 65:965–73. doi: 10.1016/j.molcel.2017.02.024
140. Sun S-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17:545–58. doi: 10.1038/nri.2017.52
141. Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, et al. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science (1997) 275:200–3. doi: 10.1126/science.275.5297.200
142. Yuasa T, Ohno S, Kehrl JH, Kyriakis JM. Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38.s. J Biol Chem (1998) 273:22681–92. doi: 10.1074/jbc.273.35.22681
143. Dérijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, et al. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science (1995) 267:682–5. doi: 10.1126/science.7839144
144. Das S, Cho J, Lambertz I, Kelliher MA, Eliopoulos AG, Du K, et al. Tpl2/cot signals activate ERK, JNK, and NF-kappaB in a cell-type and stimulus-specific manner. J Biol Chem (2005) 280:23748–57. doi: 10.1074/jbc.M412837200
145. Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun (2015) 6:5930. doi: 10.1038/ncomms6930
146. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
147. Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol Baltim Md 1950 (2005) 174:1081–90. doi: 10.4049/jimmunol.174.2.1081
148. Pype S, Declercq W, Ibrahimi A, Michiels C, Van Rietschoten JG, Dewulf N, et al. TTRAP, a novel protein that associates with CD40, tumor necrosis factor (TNF) receptor-75 and TNF receptor-associated factors (TRAFs), and that inhibits nuclear factor-kappa b activation. J Biol Chem (2000) 275:18586–93. doi: 10.1074/jbc.M000531200
149. Eliopoulos AG, Davies C, Knox PG, Gallagher NJ, Afford SC, Adams DH, et al. CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol Cell Biol (2000) 20:5503–15. doi: 10.1128/MCB.20.15.5503-5515.2000
150. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and m-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol (2009) 86:411–21. doi: 10.1189/jlb.1108702
151. Fujita Y, Matsuoka N, Temmoku J, Furuya-Yashiro M, Asano T, Sato S, et al. JAK inhibitors impair GM-CSF-mediated signaling in innate immune cells. BMC Immunol (2020) 21:35. doi: 10.1186/s12865-020-00365-w
152. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol (2008) 8:533–44. doi: 10.1038/nri2356
153. Martinez-Moczygemba M, Huston DP. Biology of common beta receptor-signaling cytokines: IL-3, IL-5, and GM-CSF. J Allergy Clin Immunol (2003) 112:653–65. doi: 10.1016/j.jaci.2003.08.015
154. Kopan R, Ilagan M. The canonical notch signaling pathway: Unfolding the activation mechanism. Cell (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
155. Zeng C, Xing R, Liu J, Xing F. Role of CSL-dependent and independent notch signaling pathways in cell apoptosis. Apoptosis Int J Program Cell Death (2016) 21:1–12. doi: 10.1007/s10495-015-1188-z
156. Singla RD, Wang J, Singla DK. Regulation of notch 1 signaling in THP-1 cells enhances M2 macrophage differentiation. Am J Physiol-Heart Circ Physiol (2014) 307:H1634–42. doi: 10.1152/ajpheart.00896.2013
157. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344:921–5. doi: 10.1126/science.1252510
158. Palaga T, Wongchana W, Kueanjinda P. Notch signaling in macrophages in the context of cancer immunity. Front Immunol (2018) 9:652. doi: 10.3389/fimmu.2018.00652
159. Fung E, Tang S-MT, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation (2007) 115:2948–56. doi: 10.1161/CIRCULATIONAHA.106.675462
160. Liu H, Wang J, Zhang M, Xuan Q, Wang Z, Lian X, et al. Jagged1 promotes aromatase inhibitor resistance by modulating tumor-associated macrophage differentiation in breast cancer patients. Breast Cancer Res Treat (2017) 166:95–107. doi: 10.1007/s10549-017-4394-2
161. Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature (2001) 410:1103–7. doi: 10.1038/35074114
162. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther (2017) 177:81–95. doi: 10.1016/j.pharmthera.2017.02.043
163. Foucher ED, Blanchard S, Preisser L, Garo E, Ifrah N, Guardiola P, et al. IL-34 induces the differentiation of human monocytes into immunosuppressive macrophages. antagonistic effects of GM-CSF and IFNγ. PloS One (2013) 8:e56045. doi: 10.1371/journal.pone.0056045
164. Novak U, Harpur AG, Paradiso L, Kanagasundaram V, Jaworowski A, Wilks AF, et al. Colony-stimulating factor 1-induced STAT1 and STAT3 activation is accompanied by phosphorylation of Tyk2 in macrophages and Tyk2 and JAK1 in fibroblasts. Blood (1995) 86:2948–56. doi: 10.1182/blood.V86.8.2948.2948
165. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase balpha. Curr Biol CB (1997) 7:261–9. doi: 10.1016/s0960-9822(06)00122-9
166. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and akt signaling. Semin Immunol (2015) 27:286–96. doi: 10.1016/j.smim.2015.08.001
167. Linton MF, Moslehi JJ, Babaev VR. Akt signaling in macrophage polarization, survival, and atherosclerosis. Int J Mol Sci (2019) 20:2703. doi: 10.3390/ijms20112703
168. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun (2013) 4:2834. doi: 10.1038/ncomms3834
169. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA (2012) 109:9517–22. doi: 10.1073/pnas.1119038109
170. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
171. Nandan D, Camargo de Oliveira C, Moeenrezakhanlou A, Lopez M, Silverman JM, Subek J, et al. Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3β downstream of phosphatidylinositol-3 kinase. J Immunol Baltim Md 1950 (2012) 188:367–78. doi: 10.4049/jimmunol.1100076
172. Lv J, Liu C, Chen F-K, Feng Z-P, Jia L, Liu P-J, et al. M2−like tumour−associated macrophage−secreted IGF promotes thyroid cancer stemness and metastasis by activating the PI3K/AKT/mTOR pathway. Mol Med Rep (2021) 24:604. doi: 10.3892/mmr.2021.12249
173. Günzl P, Bauer K, Hainzl E, Matt U, Dillinger B, Mahr B, et al. Anti-inflammatory properties of the PI3K pathway are mediated by IL-10/DUSP regulation. J Leukoc Biol (2010) 88:1259–69. doi: 10.1189/jlb.0110001
174. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
175. Schmid MC, Kang SW, Chen H, Paradise M, Ghebremedhin A, Kaneda MM, et al. PI3Kγ stimulates a high molecular weight form of myosin light chain kinase to promote myeloid cell adhesion and tumor inflammation. Nat Commun (2022) 13:1768. doi: 10.1038/s41467-022-29471-6
176. Yue S, Rao J, Zhu J, Busuttil RW, Kupiec-Weglinski JW, Lu L, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol Baltim Md 1950 (2014) 192:5343–53. doi: 10.4049/jimmunol.1400280
177. Sloot YJE, Rabold K, Netea MG, Smit JWA, Hoogerbrugge N, Netea-Maier RT. Effect of PTEN inactivating germline mutations on innate immune cell function and thyroid cancer-induced macrophages in patients with PTEN hamartoma tumor syndrome. Oncogene (2019) 38:3743–55. doi: 10.1038/s41388-019-0685-x
178. Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, et al. The role of the NADPH oxidase complex, p38 MAPK, and akt in regulating human Monocyte/Macrophage survival. Am J Respir Cell Mol Biol (2007) 36:68–77. doi: 10.1165/rcmb.2006-0165OC
179. Himes SR, Sester DP, Ravasi T, Cronau SL, Sasmono T, Hume DA. The JNK are important for development and survival of macrophages. J Immunol Baltim Md 1950 (2006) 176:2219–28. doi: 10.4049/jimmunol.176.4.2219
180. Bhatt NY, Kelley TW, Khramtsov VV, Wang Y, Lam GK, Clanton TL, et al. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J Immunol Baltim Md 1950 (2002) 169:6427–34. doi: 10.4049/jimmunol.169.11.6427
181. Uemura Y, Kobayashi M, Nakata H, Kubota T, Bandobashi K, Saito T, et al. Effects of GM-CSF and m-CSF on tumor progression of lung cancer: roles of MEK1/ERK and AKT/PKB pathways. Int J Mol Med (2006) 18:365–73. doi: 10.3892/ijmm.18.2.365
182. Huynh J, Kwa MQ, Cook AD, Hamilton JA, Scholz GM. CSF-1 receptor signalling from endosomes mediates the sustained activation of Erk1/2 and akt in macrophages. Cell Signal (2012) 24:1753–61. doi: 10.1016/j.cellsig.2012.04.022
183. Mun SH, Park PSU, Park-Min K-H. The m-CSF receptor in osteoclasts and beyond. Exp Mol Med (2020) 52:1239–54. doi: 10.1038/s12276-020-0484-z
184. Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol (2003) 24:358–63. doi: 10.1016/s1471-4906(03)00139-x
185. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
186. Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J (1997) 16:5894–903. doi: 10.1093/emboj/16.19.5894
187. Bode JG, Ehlting C, Häussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal (2012) 24:1185–94. doi: 10.1016/j.cellsig.2012.01.018
188. Saxton RA, Tsutsumi N, Su LL, Abhiraman GC, Mohan K, Henneberg LT, et al. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science (2021) 371:eabc8433. doi: 10.1126/science.abc8433
189. Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, et al. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood (1999) 93:1456–63. doi: 10.1182/blood.V93.5.1456
190. Pengal RA, Ganesan LP, Wei G, Fang H, Ostrowski MC, Tridandapani S. Lipopolysaccharide-induced production of interleukin-10 is promoted by the serine/threonine kinase akt. Mol Immunol (2006) 43:1557–64. doi: 10.1016/j.molimm.2005.09.022
191. Ambade A, Satishchandran A, Saha B, Gyongyosi B, Lowe P, Kodys K, et al. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology (2016) 5:e1221557. doi: 10.1080/2162402X.2016.1221557
192. Geng Y, Gulbins E, Altman A, Lotz M. Monocyte deactivation by interleukin 10 via inhibition of tyrosine kinase activity and the ras signaling pathway. Proc Natl Acad Sci U.S.A. (1994) 91:8602–6. doi: 10.1073/pnas.91.18.8602
193. Wang H-W, Joyce JA. Alternative activation of tumor-associated macrophages by IL-4: priming for protumoral functions. Cell Cycle Georget Tex (2010) 9:4824–35. doi: 10.4161/cc.9.24.14322
194. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
195. Mitani H, Katayama N, Araki H, Ohishi K, Kobayashi K, Suzuki H, et al. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br J Haematol (2000) 109:288–95. doi: 10.1046/j.1365-2141.2000.02020.x
196. Chen L, Wang S, Wang Y, Zhang W, Ma K, Hu C, et al. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget (2018) 9:17443–54. doi: 10.18632/oncotarget.24734
197. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147:1393–404. doi: 10.1053/j.gastro.2014.08.039
198. Radharani NNV, Yadav AS, Nimma R, Kumar TVS, Bulbule A, Chanukuppa V, et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via stat-3 pathway. Cancer Cell Int (2022) 22:122. doi: 10.1186/s12935-022-02527-9
199. Manore SG, Doheny DL, Wong GL, Lo H-W. IL-6/JAK/STAT3 signaling in breast cancer metastasis: Biology and treatment. Front Oncol (2022) 12:866014. doi: 10.3389/fonc.2022.866014
200. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
201. Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature (2003) 425:577–84. doi: 10.1038/nature02006
202. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev (2005) 19:2783–810. doi: 10.1101/gad.1350705
203. Raes G, Brys L, Dahal B, Brandt J, Grooten J, Brombacher F, et al. Macrophage galactose-type c-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol (2005) 77:321–7. doi: 10.1189/jlb.0304212
204. Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol (2008) 10:1199–207. doi: 10.1038/ncb1780
205. Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, et al. TGF-beta activates erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J (2007) 26:3957–67. doi: 10.1038/sj.emboj.7601818
206. De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol (2007) 28:519–24. doi: 10.1016/j.it.2007.09.004
207. Seok SH, Heo J-I, Hwang J-H, Na Y-R, Yun J-H, Lee EH, et al. Angiopoietin-1 elicits pro-inflammatory responses in monocytes and differentiating macrophages. Mol Cells (2013) 35:550–6. doi: 10.1007/s10059-013-0088-8
208. Chen L, Li J, Wang F, Dai C, Wu F, Liu X, et al. Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res (2016) 76:6828–38. doi: 10.1158/0008-5472.CAN-16-1114
209. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
210. Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol (2006) 362:623–39. doi: 10.1016/j.jmb.2006.07.045
211. Kamthong PJ, Wu M. Inhibitor of nuclear factor-κB induction by cAMP antagonizes interleukin-1-induced human macrophage-colony-stimulating-factor expression. Biochem J (2001) 356:525–30. doi: 10.1042/bj3560525
212. Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, et al. Suppression of LPS-induced TNF-α production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal (2009) 2:ra28. doi: 10.1126/scisignal.2000202
213. Vadevoo SMP, Gunassekaran GR, Lee C, Lee N, Lee J, Chae S, et al. The macrophage odorant receptor Olfr78 mediates the lactate-induced M2 phenotype of tumor-associated macrophages. Proc Natl Acad Sci (2021) 118:e2102434118. doi: 10.1073/pnas.2102434118
214. Negreiros-Lima GL, Lima KM, Moreira IZ, Jardim BLO, Vago JP, Galvão I, et al. Cyclic AMP regulates key features of macrophages via PKA: Recruitment, reprogramming and efferocytosis. Cells (2020) 9:E128. doi: 10.3390/cells9010128
215. Tavares LP, Negreiros-Lima GL, Lima KM, E Silva PMR, Pinho V, Teixeira MM, et al. Blame the signaling: Role of cAMP for the resolution of inflammation. Pharmacol Res (2020) 159:105030. doi: 10.1016/j.phrs.2020.105030
216. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev MMBR (2011) 75:50–83. doi: 10.1128/MMBR.00031-10
217. Takeda N, O’Dea EL, Doedens A, Kim J, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev (2010) 24:491–501. doi: 10.1101/gad.1881410
218. Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and m-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol Baltim Md 1950 (2014) 192:3858–67. doi: 10.4049/jimmunol.1302821
219. Singh SK, Mishra MK, Eltoum I-EA, Bae S, Lillard JW, Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep (2018) 8:1323. doi: 10.1038/s41598-018-19643-0
220. Zhuang Y, Zhao X, Yuan B, Zeng Z, Chen Y. Blocking the CCL5-CCR5 axis using maraviroc promotes M1 polarization of macrophages cocultured with irradiated hepatoma cells. J Hepatocell Carcinoma (2021) 8:599–611. doi: 10.2147/JHC.S300165
221. Zhuang Y, Zhao X, Yuan B, Zeng Z, Chen Y. Blocking the CCL5–CCR5 axis using maraviroc promotes M1 polarization of macrophages cocultured with irradiated hepatoma cells. J Hepatocell Carcinoma (2021) 8:599–611. doi: 10.2147/JHC.S300165
222. Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell (2016) 29:587–601. doi: 10.1016/j.ccell.2016.03.005
223. Wang X, Iyer A, Lyons AB, Körner H, Wei W. Emerging roles for G-protein coupled receptors in development and activation of macrophages. Front Immunol (2019) 10:2031. doi: 10.3389/fimmu.2019.02031
224. Shi L, Bian Z, Kidder K, Liang H, Liu Y. Non-Lyn src family kinases activate SIRPα–SHP-1 to inhibit PI3K–Akt2 and dampen proinflammatory macrophage polarization. J Immunol (2021). 207:1–9. doi: 10.4049/jimmunol.2100266
225. Lin Y, Zhao J-L, Zheng Q-J, Jiang X, Tian J, Liang S-Q, et al. Notch signaling modulates macrophage polarization and phagocytosis through direct suppression of signal regulatory protein α expression. Front Immunol (2018) 9:1744. doi: 10.3389/fimmu.2018.01744
226. Varone A, Spano D, Corda D. Shp1 in solid cancers and their therapy. Front Oncol (2020) 10:935. doi: 10.3389/fonc.2020.00935
227. Qu CK. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res (2000) 10:279–88. doi: 10.1038/sj.cr.7290055
228. van der Touw W, Chen H-M, Pan P-Y, Chen S-H. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother CII (2017) 66:1079–87. doi: 10.1007/s00262-017-2023-x
229. Chen H-M, van der Touw W, Wang YS, Kang K, Mai S, Zhang J, et al. Blocking immunoinhibitory receptor LILRB2 reprograms tumor-associated myeloid cells and promotes antitumor immunity. J Clin Invest (2018) 128:5647–62. doi: 10.1172/JCI97570
230. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
231. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (2017) 9:E52. doi: 10.3390/cancers9050052
232. Zhang W, Chen L, Ma K, Zhao Y, Liu X, Wang Y, et al. Polarization of macrophages in the tumor microenvironment is influenced by EGFR signaling within colon cancer cells. Oncotarget (2016) 7:75366–78. doi: 10.18632/oncotarget.12207
233. Lian G, Chen S, Ouyang M, Li F, Chen L, Yang J. Colon cancer cell secretes EGF to promote M2 polarization of TAM through EGFR/PI3K/AKT/mTOR pathway. Technol Cancer Res Treat (2019) 18:1533033819849068. doi: 10.1177/1533033819849068
234. Virtakoivu R, Rannikko JH, Viitala M, Vaura F, Takeda A, Lönnberg T, et al. Systemic blockade of clever-1 elicits lymphocyte activation alongside checkpoint molecule downregulation in patients with solid tumors: Results from a phase I/II clinical trial. Clin Cancer Res (2021) 27:4205–20. doi: 10.1158/1078-0432.CCR-20-4862
235. Siu LL, Wang D, Hilton J, Geva R, Rasco D, Perets R, et al. First-in-Class anti-immunoglobulin–like transcript 4 myeloid-specific antibody MK-4830 abrogates a PD-1 resistance mechanism in patients with advanced solid tumors. Clin Cancer Res (2021) 28:57–70. doi: 10.1158/1078-0432.CCR-21-2160
236. Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
237. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
238. Datta J, Lamichhane P, Dai X, Mehra S, Dosch AR, Messaggio F, et al. Combined MEK and STAT3 inhibition reprograms the tumor microenvironment to overcome immunotherapy resistance in pancreatic cancer. Gastroenterology (2021) 2021:03. doi: 10.1101/2021.03.07.434236
239. Weng S-Y, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. eBioMedicine (2018) 29:92–103. doi: 10.1016/j.ebiom.2018.01.028
240. Colombo N, Peccatori F, Paganin C, Bini S, Brandely M, Mangioni C, et al. Anti-tumor and immunomodulatory activity of intraperitoneal IFN-gamma in ovarian carcinoma patients with minimal residual tumor after chemotherapy. Int J Cancer (1992) 51:42–6. doi: 10.1002/ijc.2910510109
241. Pujade-Lauraine E, Guastalla JP, Colombo N, Devillier P, François E, Fumoleau P, et al. Intraperitoneal recombinant interferon gamma in ovarian cancer patients with residual disease at second-look laparotomy. J Clin Oncol Off J Am Soc Clin Oncol (1996) 14:343–50. doi: 10.1200/JCO.1996.14.2.343
242. Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS, et al. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol Res (2017) 5:1109–21. doi: 10.1158/2326-6066.CIR-17-0258
243. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019) 50:1317–1334.e10. doi: 10.1016/j.immuni.2019.03.009
244. Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell (2021) 39:1594–1609.e12. doi: 10.1016/j.ccell.2021.10.009
245. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell (2020) 181:1643–1660.e17. doi: 10.1016/j.cell.2020.05.007
246. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov (2022) 1–22. doi: 10.1038/s41573-022-00520-5
247. Bono P, Ekström J, Karvonen MK, Mandelin J, Koivunen J. Modelling treatment benefit for bexmarilimab (an anti-Clever-1 antibody and a novel macrophage checkpoint inhibitor) using phase I first-in-man trial data. J Clin Oncol (2021) 39:e14530–0. doi: 10.1200/JCO.2021.39.15_suppl.e14530
248. Zhan Y, Lew AM, Chopin M. The pleiotropic effects of the GM-CSF rheostat on myeloid cell differentiation and function: More than a numbers game. Front Immunol (2019) 10:2679. doi: 10.3389/fimmu.2019.02679
249. He S, Hu Q, Xu X, Niu Y, Chen Y, Lu Y, et al. Advanced glycation end products enhance M1 macrophage polarization by activating the MAPK pathway. Biochem Biophys Res Commun (2020) 525:334–40. doi: 10.1016/j.bbrc.2020.02.053
250. Muta Y, Matsuda M, Imajo M. Divergent dynamics and functions of ERK MAP kinase signaling in development, homeostasis and cancer: Lessons from fluorescent bioimaging. Cancers (2019) 11:513. doi: 10.3390/cancers11040513
251. Shi L, Kishore R, McMullen MR, Nagy LE. Lipopolysaccharide stimulation of ERK1/2 increases TNF-α production via egr-1. Am J Physiol-Cell Physiol (2002) 282:C1205–11. doi: 10.1152/ajpcell.00511.2001
252. Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-α transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol (2000) 67:885–93. doi: 10.1002/jlb.67.6.885
253. Xagorari A, Roussos C, Papapetropoulos A. Inhibition of LPS-stimulated pathways in macrophages by the flavonoid luteolin. Br J Pharmacol (2002) 136:1058. doi: 10.1038/sj.bjp.0704803
254. Hashimoto M, Nasser H, Chihara T, Suzu S. Macropinocytosis and TAK1 mediate anti-inflammatory to pro-inflammatory macrophage differentiation by HIV-1 nef. Cell Death Dis (2014) 5:e1267. doi: 10.1038/cddis.2014.233
255. Dorrington MG, Fraser IDC. NF-κB signaling in macrophages: Dynamics, crosstalk, and signal integration. Front Immunol (2019) 10:705. doi: 10.3389/fimmu.2019.00705
256. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Liberto DD, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc Natl Acad Sci USA (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
257. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: Intracellular mechanisms of reprogramming and M3 macrophage “Switch” phenotype. BioMed Res Int (2015) 2015:341308. doi: 10.1155/2015/341308
Keywords: signaling/signaling pathways, cancer biology, TAMs (tumor associated myeloid cells), TME (tumor microenvironment), macrophages polarization
Citation: Kerneur C, Cano CE and Olive D (2022) Major pathways involved in macrophage polarization in cancer. Front. Immunol. 13:1026954. doi: 10.3389/fimmu.2022.1026954
Received: 24 August 2022; Accepted: 23 September 2022;
Published: 17 October 2022.
Edited by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyReviewed by:
Ramon Mohanlal, BeyondSpring Pharmaceuticals, Inc., United StatesByungheon Lee, Kyungpook National University, South Korea
Manzoor A. Mir, University of Kashmir, India
Copyright © 2022 Kerneur, Cano and Olive. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clément Kerneur, clement.kerneur@imcheck.fr; Carla E. Cano, carla.cano@imcheck.fr; Daniel Olive, daniel.olive@inserm.fr