 Meihong Da
Meihong Da Luxia Chen†
Luxia Chen† Karsten Mahnke
Karsten Mahnke- Department of Dermatology, University Hospital Heidelberg, Heidelberg, Germany
Adenosine (Ado) has been shown to have immunosuppressive effects in a variety of diseases. It can either be released directly into the extracellular environment by cells, or it can be produced by degradation of ATP within the extracellular spaces. This extracellular pathway is facilitated by the concerted actions of the ectoenzymes CD39 and CD73. In a first step CD39 dephosphorylates ATP to ADP and AMP, respectively, and in a second step CD73 converts AMP to Ado. Thus, activity of CD73 on the cell surface of cells is the rate limiting step in the generation of extracellular Ado. Among T cells, CD73 is most abundantly expressed by regulatory T cells (Tregs) and is even upregulated after their activation. Functionally, the generation of Ado by CD73+ Tregs has been shown to play a role in immune suppression of dendritic cells, monocytes and T cells, and the defined expression of CD73 by Tregs in immunosuppressive environments, such as tumors, made CD73 a novel checkpoint inhibitor. Therefore, therapeutical intervention by anti-CD73 antibodies or by chemical inhibitors of the enzymatic function is currently under investigation in some preclinical animal models. In the following we summarize the expression pattern and the possible functions of CD73 in T cells and Tregs, and exemplify novel ways to manipulate CD73 functions in Tregs to stimulate anti-tumor immunity.
Introduction
For long CD73 has been identified as a major adenosine (Ado) producing ectoenzyme expressed by different cell types. As Ado has profound immunosuppressive effects in a variety of diseases, there is a clear link between CD73 expressing cells and their immunosuppressive functions. In some cancers this relation even led to a correlation of the amount of CD73 expression with the severity of the disease. The defined expression of CD73 by tumor invading immune suppressive leukocytes and/or the tumor itself made CD73 a novel immune checkpoint molecule that may be targeted by specific inhibitors.
Among T cells, CD73 is most abundantly expressed by CD4+ CD25+ regulatory T cells (Tregs) and the generation of Ado by CD73+ Tregs has been shown to play a role in their immune suppressive action against dendritic cells, monocytes and effector T cells (1, 2). Therefore, therapeutical intervention by anti-CD73 antibodies (Abs) or by chemical inhibitors of the enzymatic function is currently under investigation in some preclinical animal models. In the following we summarize the expression pattern and the possible functions of CD73 in vivo and exemplify novel ways to manipulate CD73 functions in Tregs to stimulate anti-tumor immunity.
The Structure of CD73
The ecto-5-nucleotidase, abbreviated as eN, e5NT or CD73 (gene NT5E, E.C. 3.1.3.5, UniProt P21589), is an enzyme that hydrolyses AMP. CD73 consists of a non-covalently linked homodimer and is attached to the outer cell membranes via glycosylphosphatidyl-inositol (GPI) anchors (3). The structure of human CD73 has been determined (4) and it revealed two distinct conformations (5). The binding of AMP occurs in an inactive (open) conformation, which enables the binding of the substrate to the C-terminal domain. The active (closed) conformation represents the active state where hydrolysis of the substrate occurs. This “closing” of the enzyme is accomplished by rotation of one domain (of nearly 100°), which brings the N- and C-terminal domains together forming the active site. By hydrolysis the phosphate group is cleaved from the ribose moiety and after switching back into the “open conformation” the product is released from CD73. Although this description is rather sketchy, it illustrates possible mechanisms of CD73 inhibition, as small molecules may be designed to interfere with the enzymatic activity by inhibiting the open–close process and/or binding to the defined region with higher affinity than the natural ligand AMP.
The Basic Functions of CD73 During Steady State and Inflammation
But why may it be important to develop CD73 inhibitors? The reason for developing strong and specific CD73 antagonists derives from its Ado-producing properties. Ado, which constitutes one product of the enzymatic degradation of AMP by CD73, is a potent immunosuppressor (6). In vivo, CD73+ Tregs frequently co-express CD39, yet another ectoenzyme, which has comparable functions as CD73. CD39, however, fuels the CD73-derived production of Ado as it degrades ATP and ADP to AMP, which acts as substrate for CD73. This functional interdependence of CD39 and CD73 makes Tregs special as these cells express both molecules in sizable numbers (7) making them self-sufficient to create an Ado-rich, immunosuppressive pericellular environment, independent from other CD39+ cells (8).
In “steady state” situations, i.e. in absence of tissue disruption and infection, the presence of CD73+ Tregs in the environment helps to maintain the state of tolerance by producing Ado from extracellular ATP (Figure 1A). This immune suppression is rather weak, as only trace amounts of ATP are present in the tissue environment (9–11). In the absence of tissue damage, ATP can be released by cells via several mechanisms ranging from exocytosis to ATP-specific membrane transporters (12, 13). For instance, the vesicular nucleotide transporters VNUT or SLC17A9 can accumulate ATP in intracellular vesicles first, which can then subsequently be transported by exocytosis to the outside of cells (14). Such vesicle-mediated ATP release has been shown to be of importance during nutrient deprivation of cancer cells (15). Of interest, the ATP receptor P2x7 can stimulate the release of vesicles (16), offering the possibility that extracellular ATP promotes its own replenishment by this self-sustaining cycle.
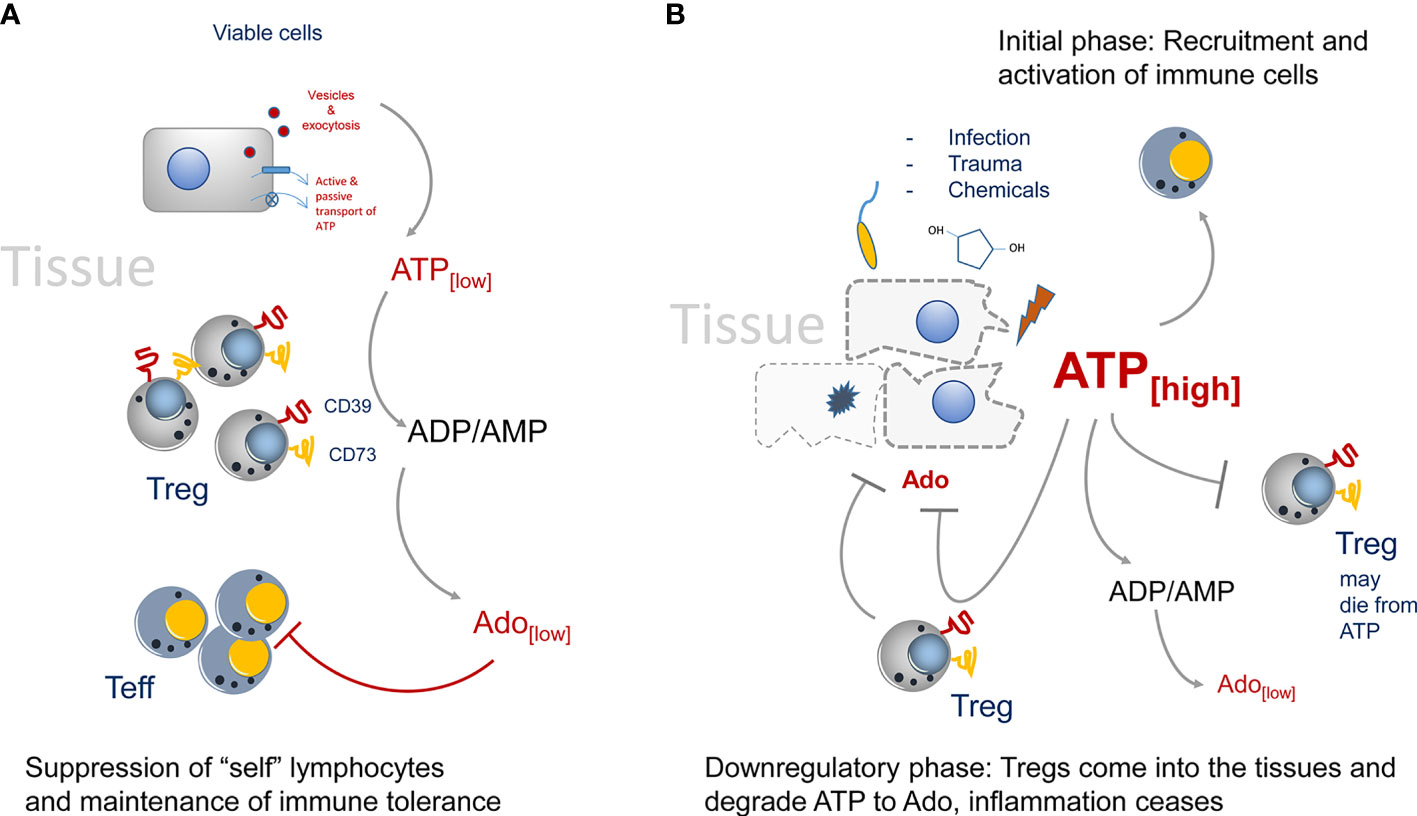
Figure 1 Regulation of CD73-mediated adenosine production in healthy and diseased tissues. (A) In the steady state only trace amounts of ATP are released by viable or by dying cells. It will be immediately degraded by CD73 to adenosine. Adenosine acts as immune suppressor and maintains immune tolerance by creating an immune suppressive environment. (B) The situation changes when infection, trauma and toxicity occur. Massive release of ATP cannot be degraded immediately by CD73. It activates lymphocytes and downregulates Treg populations, presumably by inducing cell death. Adenosine concentrations are kept low to maintain immunity. Later, more Tregs will enter the tissue side, degrading ATP and producing the immune suppressor adenosine.
Other non-exocytotic means of ATP release comprise ATP-binding cassette (ABC) transporters, eg. the multi drug resistance protein 1 (MDR-1), that hydrolyze ATP as energy source for the transmembrane movement of different molecules. These transporters do not only transport other molecules through membranes, they also can act as transporters for ATP (17). Finally, “classical” channels and pores in cells that allow ATP to cross the cell membrane are described. Among them are chloride ion channels (18), volume- and voltage-dependent anion channels (19), connexin hemichannels, pannexin 1, calcium homeostasis modulator 1, volume-regulated anion channels and maxi-anion channels (MACs) (20). Once outside of cells, ATP may act as a potent proinflammatory stimulus that engages P2x7 receptors, which can act as ATP channels themselves (21, 22), leading to activation of inflammasomes (23–25). Under homeostatic conditions extracellular ATP, although only present in trace amounts, needs to be readily degraded by CD39/CD73 to prevent the induction of an immune reaction. Additionally, the generated Ado may help even further to maintain a slightly immunosuppressive extracellular environment (26). Altogether this mechanism may be necessary to maintain a “healthy”, uninflamed tissue, as only few dying cells in the tissue must not induce an immune reaction (27).
However, the situation changes when trauma and/or infection in tissues occurs (Figure 1B). Now huge amounts of ATP are set free into the tissue environment and it will act as a potent immune activator (28). These relatively high amounts of ATP are necessary to trigger P2x7 ATP-receptors, which have an EC50 of approximately 1 mM. These concentrations can only be reached in diseased or necrotic tissues (29). Degradation of ATP to Ado lags behind and a potent immune reaction is generated. Only later in an inflammatory reaction Tregs will enter the tissue and due to their high expression of CD39 and CD73 they are strong producers of Ado, which will lead to downregulation of the inflammatory reaction. Of note, Tregs are highly sensitive to ATP concentrations. I.e., CD4+ CD25+ Tregs, as opposed to CD4+ effector T cells will die from high extracellular ATP levels (30). That way in the beginning of an inflammation immune reaction, the suppressive activity of Tregs may be switched off. That excess of extracellular ATP is indeed important to booster immunity can also be inferred from tumor bearing mice. It has been shown in tumor models that in P2x7 ATP-receptor deficient animals, more Tregs than effector T cells were recruited to the tumors, and that the Tregs were activated and highly suppressive as indicated by upregulation of the markers OX40, PD1 (31) and CD73. In summary, deletion of P2x7 signaling led to an immunosuppressive tissue environment (32, 33). However, at later stages of an inflammation, or after tumors have been largely eliminated, the extracellular ATP levels cease gradually by the activity of CD73+ and CD39+ cells. Among them are also double positive Tregs that will seed into the tissues and help to downregulate inflammation by ATP-to-Ado conversion, inducing the “healing” of the insult (34, 35).
How Do CD73+ Tregs Suppress?
The main suppressive function of CD73+ Tregs relies on their capacity to produce Ado. However, CD73 expression does not make other suppressive means obsolete, as CD73+ Tregs still express their prototypic set of inhibitory receptors and Ado production just adds another means to an already existing arsenal of suppressive mechanisms (36, 37). Ado seems rather unstable and is thought to be degraded in tissues by adenosine deaminase within minutes (38). However, that was only investigated in dog’s serum and the exact distribution and half-life in tissues, other than blood, is not clear yet. Nevertheless, due to several Ado degrading mechanisms in tissues, Ado seems to be a short-range immune suppressor and therefore aligns with several other suppressive mechanisms of Tregs that require close cell-to-cell contact (i.e. CTLA-4, GITR) (36). At the same time, the short half-life of Ado restricts its suppressive effects to the local environment. Systemic immune suppression is therefore avoided.
Ado suppresses cells by engaging Ado receptors (AdoR) that can be divided into four distinct receptors, A1, A2A, A2B, A3 (39). They all belong to the G-protein-coupled-receptor (GPCR) family, but their intracellular signaling differs. A2-type AdoR are Gαs-protein coupled receptors, with the A2B receptor additionally signaling via Gαq (40). After engaging AdoR, activated G protein complexes form at the inner cell membrane, leading to activation of the adenylate cyclase (AC) and to rising cAMP levels (in case of Gαs), finally activating protein kinase A (PKA) as secondary effector. On a molecular level this can directly be counteracted by engagement of A1 or A3 AdoR, which signal via Gαi/Gαq complexes, whereby the Gαi complex inhibits AC activity. However, the main pathway for signaling of A1 and A3 receptors is mediated by phospholipase C (PLC)-induced secondary messengers, eventually leading to a rise in intracellular Ca2+ and PKC activation. The common denominator of A2-type AdoR mediated suppression is cAMP as second messenger. Both A2-type AdoRs elevate cAMP levels by activating AC. Further downstream cAMP signals via PKA that regulates gene transcription via NF-κB, HIF-1α and CREB. In addition, A2B AdoR also acts on PLC, raising intracellular Ca2+ levels.
In a transcriptomic approach, elevated activity of AC was connected to inhibition of AKT signaling and to activation of PKA (41). PKA is important for host defense mechanisms, as inhibition of Salt induced kinases (SIK) by cAMP-activated PKA results in suppression of the pro-inflammatory cytokines IL-6, IL-12, and TNFα in innate immune cells (42–44). Another mediator of suppressive mechanisms along this PKA pathway is the CREB-regulated transcription co-activator 3 (CRTC3). Phosphorylation of CRTC3 is inhibited by the cAMP-activated PKA, leading to translocation of the non-phosphorylated CRTC3 into the nucleus, where interaction with activated CREB upregulates IL-10 gene transcription (43). Parallel to PKA activation, activity of AKT is downregulated by elevated cAMP levels, preventing translation of proteins by means of mTOR inhibition (45). The later pathway plays an important role as regulator of the antibacterial responses in monocytes, macrophages and primary dendritic cells (46, 47). Finally, AdoR-triggered production of cAMP, which is the major second messenger in Tregs after A2-type AdoR engagement does not only convey suppressive downstream signals, it also plays a major role as means of suppression (48). I.e. Tregs suppress other cells by forming gap junctions and transferring cAMP into target cells. Thus, elevated levels of cAMP triggered in Tregs by AdoR engagement may “fuel” the gap junctional suppression (49).
CD73 in Maintenance of Treg Functions
These suppressive actions of extracellular Ado are operative in almost all A2-type AdoR expressing cells, including Tregs. In particular the conjointly expression of CD39 and CD73 by Tregs enables them to create a self-inhibiting cellular environment, maintaining hyperresponsiveness and stabilizing their own population. For instance, engagement of A2-type AdoR induced expression of Foxp3 and LAG3 in conventional CD4+ CD25- T cells, suggesting induction of Tregs (50), and enhanced development of Tregs induced by TGF-β (51). But not only A2A AdoRs are important, as A2BR AdoR agonists have been demonstrated to play a role in generation of Tregs, whereas A2B AdoR-deficiency prevented Treg induction (52).
Also direct expansion of Tregs by A2A AdoR-dependent mechanisms has been observed to be important in inflammatory disorders, such as graft-versus-host disease and experimental autoimmune uveitis (51, 53). In in vitro cocultures of responder T cells and Tregs the addition of A2-type agonists strongly inhibited activation of cytotoxic effector T cells as expected, but of interest, at the same time significantly enhanced numbers of CD4+ Foxp3+ Tregs were induced. These cells were highly suppressive and expressed more CTLA-4 molecules (53). Along the same lines, treatment of Tregs with A2-type AdoR agonists before transferring them into an ischemia model enhanced their efficacy in vivo, and vice versa A2A AdoR-deficient Tregs were less able to prevent inflammatory damage (51, 54, 55). Even the expression of the Ado-producing enzymes CD39 and CD73 was upregulated by AdoR engagement via an E2F-1 and CREB induced pathway (56). Thus, CD73 derived Ado is not only a suppressive means acting on adjacent cells, it also promotes suppressive activity of existing Tregs and generation of “induced” Tregs, and it sustains its own production in a feedback loop by upregulation of CD73 in Tregs.
Other Than “Ado-Producing” Functions of CD73
T Cell Signaling
In addition to its enzymatic function, CD73 has been proposed to act as a co-stimulatory molecule for T cells. In conjunction with sub-mitogenic doses of anti-CD3 Abs or PMA, immobilized anti-CD73 antibodies induce proliferation of human peripheral blood T cell, CD25 expression and IL-2 production (57). This qualifies CD73 as a member of stimulatory molecules that transduce activation signals in T lymphocytes (58). Other researchers found that naive CD45RAhi CD45ROlo CD8+ T cells that express CD73 have low responsiveness to immobilized anti-CD3, which can be overcome by cross-linking CD3 and CD73 simultaneously (59). The CD73/TCR-signaling complex plays a key role in CD73-mediated activation. This is evident from investigations showing that CD73 transfected mutants of the human Jurkat cell line, which are defective in TCR signaling due to faulty p56JCK or CD45 expression, did not secrete IL-2 after engagement of CD73. In contrast, wild type Jurkat cells transfected with CD73 were activated in response to soluble CD73 mAb plus PMA (60). As for the signal transduction of surface bound CD73, the GPI anchor is not absolutely required, considering that the secreted levels of IL-2 by CD73+ Jurkat clones in response to stimulation with CD73 mAb plus PMA were well comparable to that obtained from clones expressing GPI-anchored CD73 (60, 61). As a natural ligand for CD73 has not yet been discovered in vivo, all experimental procedures rely on antibody mediated engagement of CD73, which makes it difficult to decipher the role of CD73-mediated activation of T cells in vivo. However, Tregs are naturally hyporesponsive, i.e. they do not proliferate, therefore it is not supposed that CD73 conveys mitogenic stimuli in Tregs at all (61).
Adhesion
CD73 expressed by endothelial cells has a function in mediating lymphocyte adhesion to the endothelium, since CD73-specific monoclonal Ab can inhibit the binding of lymphocytes to endothelial cells (62). Adherent lymphocytes in turn lead to CD73 inhibition on endothelial cells and thus impair the vascular barrier function and facilitate the subsequent steps of transmigration into the tissue (63). Moreover, engagement of CD73 on the surface of lymphocytes indirectly impacts adhesion, as anti-CD73 Ab treatment induces clustering of leukocyte integrin LFA-1 and thereby enhances the adhesion of lymphocytes to cultured endothelial cells (64). Additionally, the anti-CD73 Ab interfered with the binding of germinal center B cells to isolated follicular dendritic cells in vitro (65).
In particular in tumors CD73 seems to be important for migration and dissemination, as the CD73 inhibitor APCP significantly decreases cell adhesion and migration in different breast cancer and hepatocellular carcinoma models in vitro, while Ado reversed the effects of APCP (66, 67). However, it is not clear whether these effects are directly mediated by CD73 interactions with cellular ligands or whether CD73-derived Ado is mediating these effects. Nevertheless, although these experiments were performed with T cells or tumor cells in general (not Tregs in particular), it is still conceivable that high and constitutive expression of CD73 by Tregs may act as “homing molecule” for peripheral tissues and CD73+ tumors may utilize this mechanism to recruit Tregs.
Expression of CD73 by T Cell Subsets
Beyond Tregs, almost all T-cell subsets also express CD73, however, to varying degrees. In humans, CD73 is expressed on all CD4+ and CD8+ T cell subsets, but with higher expression in naïve T cells compared with memory T cells (59, 68). Anergic CD4+ T cells that persistently recognize peripheral self-peptide:MHCII complexes and are restrained in their activity through the suppressive actions of Treg cells, share the expression of CD73 with “bona fide” Tregs. This phenotypic trait of CD73 expression enables them to support Tregs to suppress dangerous immune responses to tissue-restricted self-antigens (69). Moreover, uncommitted T helper primed precursor (Thpp) cells were identified in C57BL/6 mice as being CD73+ Ly6A/E- CD4+ T cells. These cells are able to differentiate into Th1- or Th2- like populations (70), however, a suppressive function is still elusive. Gene expression and phenotypic analysis confirmed that CD73 was enriched in T follicular helper (Tfh) cells, but the utility of CD73 as markers for human Tfh cells has not been fully understood (71).
In vitro Th17 cells generated with IL-6 and TGF-β were found to express both CD39 and CD73, leading to Ado formation and subsequent suppression of CD4+ and CD8+ T cells by limiting IFN-γ and granzyme B production (72). Therefore, acquisition of CD73 expression by otherwise rather proinflammatory T cells, i.e. Th17 cells, can provide them regulatory means. But not only CD4+ T cells can acquire a suppressive phenotype by expression of CD73. In humans, naive CD8+ T cells express higher levels of CD73 than CD8+ memory T cells (59). In line with this, activated CD8+ T cells from human peripheral blood cells were shown to lose the membrane expression of CD73 by CD73-containing extracellular vesicles, which have AMPase activity and produced adenosine mediating immune suppression (73). That makes CD73 distribution in tissues independent from Tregs.
Also in mice CD73 is expressed on naïve and memory CD8+ T cells and downregulated during terminal effector differentiation (74). CD73+ memory T cells are prone to differentiate into cells expressing a tissue-resident memory phenotype since CD73 expression was more frequent by CD69+ CXCR6+ cells than by CD69− CXCR6− cells (68). Similar to Th17 cells, in vitro generated CD8+ IL-17-producing T cells (Tc17 cells) were reported to present high levels of CD73 and produce Ado. However, a direct suppressive activity on the proliferation of CD4+ T cells was not recorded, but it is involved in maintaining the stem-cell-like properties on Tc17 cells (75).
Finally, even natural killer T (NKT) cells, which are recognizing lipid antigens presented by non-classical class I MHC molecules, express CD39 and CD73 (76). This allows these cells to generate Ado, thus having immunosuppressive properties during tumor and infections (76–78). All of the studies point to a widespread expression of CD73 on T cells, establishing CD73 expression as a major hallmark of a suppressive phenotype of T cell.
Exosomes Derived From Tumors and Tregs Contain CD73
Exosomes are formed from late endosomes, by invagination of multivesicular body membranes, resulting in the formation of intraluminal vesicles (79). During this process, proteins and cytosolic components are taken up into the lumen of the vesicles and subsequently the vesicles are released by fusion with the cell membrane into the pericellular space, becoming “exosomes” (80, 81). Almost all cells produce exosomes for varying purposes. Typically, exosomes are highly enriched in a collection of proteins, lipids, and inhibitory nucleic acids (82), however, expression of tetraspanins, such as CD9, CD63, CD81, CD82 is prototypic for all kinds of exosome vesicles (EVs). Otherwise is the content dependent on the EV-releasing cell type. Different types of cancer cells have captured the mechanism of EV release for immune suppressive actions and CD73 has been identified quite frequently in these “suppressive” EVs (Figure 2).
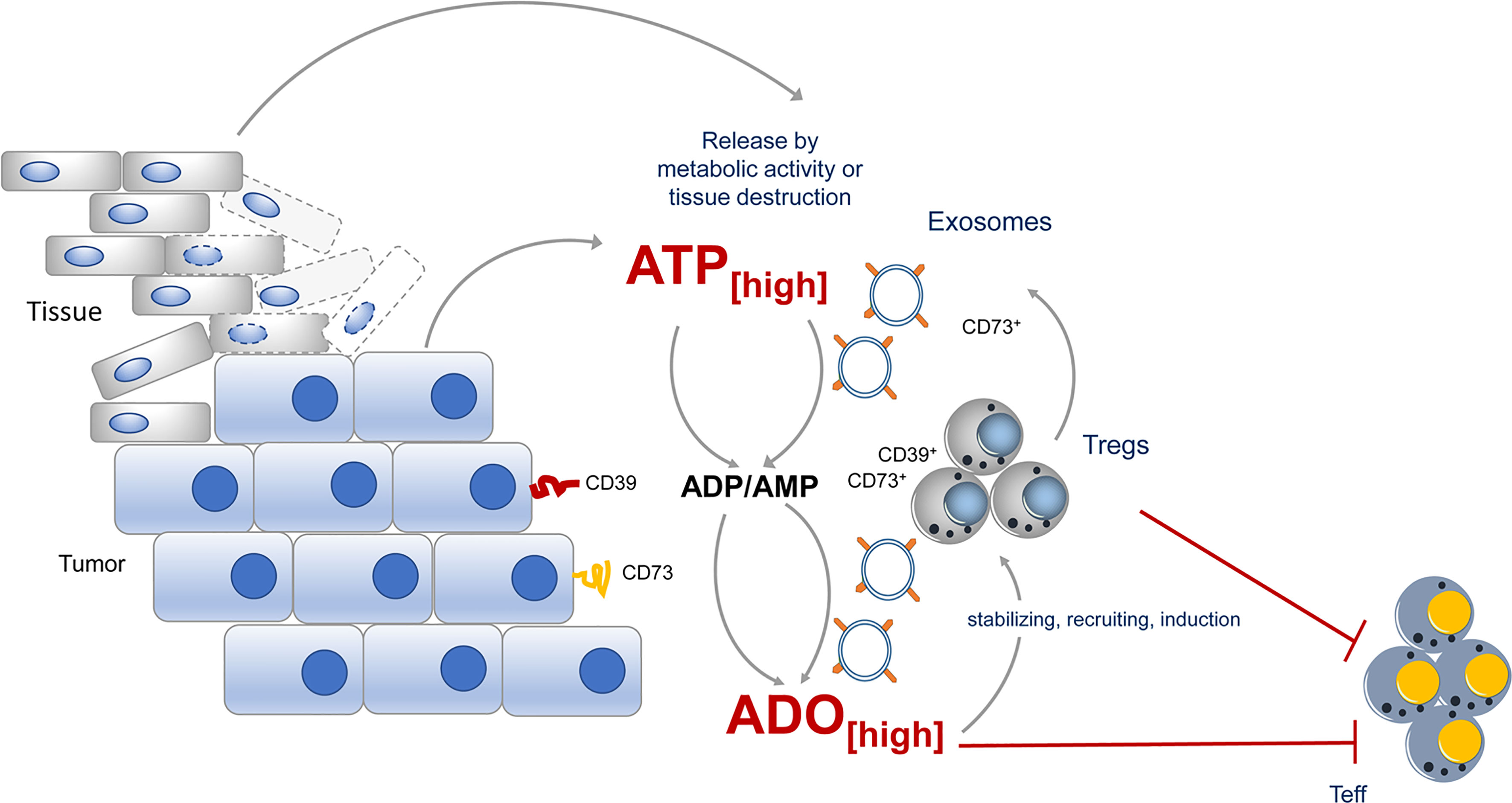
Figure 2 CD73 expression during tumor growth contributes to immune suppression. During tumor growth ATP is either actively release by metabolic active tumors or by dying tissue cells. ATP will quickly be degraded into immunosuppressive adenosine by CD73 expressed by the tumor itself, by incoming Tregs or by exosomes, derived from the tumor or tumor infiltrating lymphocytes. Adenosine maintains the suppressive activity of Tregs in the tumor vicinity and altogether an immunosuppressive environment is favored.
EVs derived from different cancer cell lines express CD73 and catalyze AMP to ado (83). Moreover, B cell-derived CD39+ CD73+ EVs are elevated in the serum of colon cancer patients and hydrolyze tumor-derived ATP to ado, impairing anti-tumor responses of CD8+ T cells (84). Comparably high levels of suppressive activity were also observed in exosomes from pleural fluid of pleural malignant mesothelioma patients. These exosomes trigger a cAMP response in A2A AdoR+, but not in A2A AdoR- cells. Significantly elevated cAMP was also induced in Jurkat cells by adding exosomes with ATP but not by adding exosomes or ATP alone (83), indicating that EVs contain the necessary Ado degradation machinery to exert suppression. But not only tumors, also Tregs were observed to release inhibitory EVs. These EVs contain several immunomodulatory molecules, including Treg-specific molecules, such as CD25 and CTLA-4, and of note CD73 (85, 86). As a consequence, proliferation of CD4+ T cells and production of IL-2 and IFN-γ was significantly inhibited when Treg-derived exosomes were added to T cell cultures. For these effects CD73 incorporated in EVs was instrumental (85), as exosomes derived from CD73-negative cells failed to convey suppression. Thus, by placing CD73 and other parts of the Ado-generating machinery from the cells surface into EVs, Tregs have developed a method to reach out for suppression and to “export” Ado beyond their immediate cell vicinity (85).
The Role of CD73 in Tumors and Tumor Infiltrating Tregs
As Tregs are highly positive for CD73, have immunosuppressive activity and are often recruited by tumors to impair anti-tumor immunity, CD73 and its contribution to tumor growth have been investigated thoroughly (Figure 2). The expression of CD73 has been found to be upregulated in several types of cancers, including ovarian carcinoma, melanoma, prostate cancer, breast cancer, colon cancer, head and neck cancer, leukemia and glioblastoma (87). Several studies reported that CD73 expression in cancer is associated with a poor prognosis, an increased risk of metastasis and resistance to chemotherapy (88 – 90). For the creation and immune suppressive environment of the tumor, it doesn’t matter whether CD73 is expressed directly by the tumor or by tumor infiltrating lymphocytes; and indeed, as most of the time the tumor is infiltrated by Tregs which express CD73, this Treg-derived “CD73” substantially contributes to elevated levels of immunosuppressive Ado in tumor tissues.
Tregs are CD73+ per se, but even further upregulation has been observed in tumors. This may be due to the hypoxic situation in tumors. For long it has been speculated that Tregs might be controlled by tissue oxygen tension. An initial correlation to eAdo is given by the presence of consensus sequences of hypoxia-responsive element (HIF-1α) and cAMP-responsive element in the promoter region of “anti-inflammatory” response genes (91). That could imply that hypoxia and Ado would be responsible for the regulation of CD73 and further immunosuppressive activity of Tregs. Although not definitely decided yet, it is clear that HIF-1α induce Foxp3 in T cells and increase Treg abundance. Notably, HIF-1α-dependent pathways induce upregulation of CD73 in epithelial barriers during hypoxia (92). Thus, the hypoxic tumor environment may provide “the soil” for incoming Tregs to upregulate CD73 and to create an eAdo-dependent immune-suppressive environment.
CD73 as Immune Checkpoint: Blockage of CD73 as Anti-Tumor Therapy
Due to the strong correlation of CD73 expression with a bad tumor prognosis, CD73 has become a novel immune checkpoint and a target for immunologic intervention. Immune checkpoint inhibitors comprise monoclonal antibodies or small molecule compounds that can target and block inhibitory receptors, and recently anti-CD73 Abs are tested in several (preclinical-) trials (Table 1). IPH5301 antibodies were designed to target human membrane bound or soluble forms of CD73, thereby efficiently blocking the degradation of AMP into Ado. Thus, immune cells that are normally targets for Ado-mediated suppression, such as T cells, dendritic cells and macrophages, remained activated and were able to dampen tumor growth in different clinical models (93).
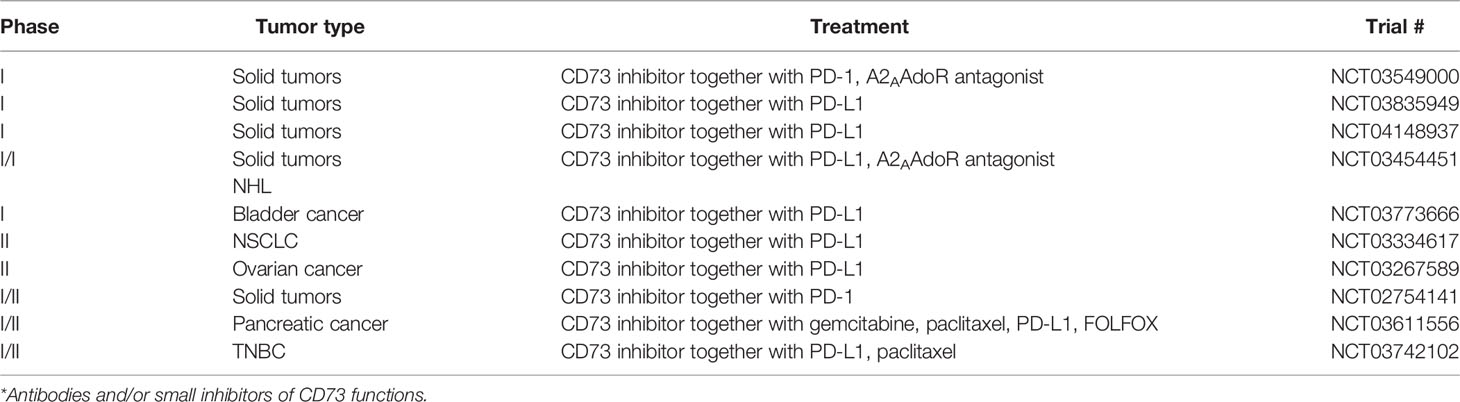
Table 1 Current clinical trials targeting CD73*.
Similar to other already used checkpoint inhibitors, which are applied in combinations, several trials started combinatorial therapies with anti-CD73 Abs (Table 1). Here, together with anti-CTLA-4 and anti-PD-1 Abs, anti-CD73 Abs showed significant efficacy in stimulating tumor immunity in several tumor models including colon, prostate, subcutaneous tumors, and breast cancer (94). Interestingly, anti-CD73 Abs preferentially synergized with anti-PD-1 Abs, as activation of A2A AdoR, presumably triggered by CD73-derived Ado, enhances PD-1 expression. Thus, blocking CD73 can interrupt this reciprocal upregulation of Ado and PD-1. However, CTLA-4 expression, on tumor-specific CD8+ T cells and CD4+ Foxp3+ Tregs was not affected by anti-CD73 treatment in this tumor (94). In contrast, anti-CD73 Abs significantly downregulated the expression of PD-1 and CTLA-4 by CD4+ and CD8+ T cells in a murine transgenic head and neck squamous cell carcinoma (HNSCC) model, resulting in substantial reduction of tumor growth (95). This indicates that different tumor entities may react differently to anti-CD73 treatment.
Yet another already established check point inhibitor, i.e. anti-4-1BB Abs, can profit from combination with anti-CD73 Abs. As anti-4-1BB (CD137) treatment preferentially drives CD73− effector T cell responses for tumor inhibition, combination with neutralizing anti-CD73 Abs induced more profound tumor regression and led to a great increase in survival as compared to anti-4-1BB therapy alone. Likewise, in B16-SIY tumors, combination therapy resulted in an increase in the percentage of tumor-infiltrating CD4+ T cells and CD8+ IFN-γ+ double positive T cells, whereas a significant decrease in the percentage of tumor infiltrating Foxp3+ Tregs was observed in anti-4-1BB/anti-CD73 Abs-treated mice (96).
However, anti-CD73 Abs in therapies have to be analyzed individually, as for instance the TGF-β-rich tumor milieu confers resistance to anti-4-1BB therapy by sustaining CD73 expression, primarily on infiltrating CD8+ T cells across several tumor models, and making them rather immunosuppressive. Therefore, even additional blockade of TGF-β may be beneficial for downregulation of CD73 expression on infiltrating T cells, and together with anti-CD73, Abs resistant tumors are rendered sensitive to agonistic anti-4-1BB therapy (96).
Beyond the mere blockade of Ado production by anti-CD73 therapy, murine tumor models also confirmed beneficial effects on the ability of tumors to metastasize and to induce tumor cell migration. For instance, in a mouse model of human breast cancer, anti-CD73 Abs changed cellular adhesion and migration patterns, by inducing clustering and internalization of surface-expressed CD73 (97). These effects were independent of hematopoetic cell involvement (97–101) and are reminiscent of “adhesion data” discussed in a previous section. Therefore, these studies point to a non-catalytic role of CD73 expressed on tumor cells and/or endothelial cells, and anti-CD73 Abs can positively influence tumor cell adhesion, extravasation and metastasis to support anti-tumor immunity.
Conclusion
CD73, the ecto-5-nucleotidase, is constitutively expressed on Tregs which can convert AMP to immunosuppressive Ado. A worse tumor prognosis has been found to be strongly associated with Ado production by CD73 in cancer. Thus, targeting CD73 becomes a novel method to improve anti-tumor immunity by combining to anti-CTLA-4, anti-PD-1 or anti-CD137 Abs.
Author Contributions
MD and LC wrote the first draft of the paper and prepared figures. SR and KM corrected the draft and the figures. AE drafted figures and tables and corrected the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft TR156 B03 and C04. MD and LC are supported by a scholarship CSC 201806090291 and 202006320072, respectively. Publication was supported by the Deutsche Forschungsgemeinschaft within the funding programme “Open Access Publikationskosten” as well as by Heidelberg University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
2. Ring S, Oliver SJ, Cronstein BN, Enk AH, Mahnke K. CD4+CD25+ Regulatory T Cells Suppress Contact Hypersensitivity Reactions Through a CD39, Adenosine-Dependent Mechanism. J Allergy Clin Immunol (2009) 123(6):1287–96.e2. doi: 10.1016/j.jaci.2009.03.022
3. Ogata S, Hayashi Y, Misumi Y, Ikehara Y. Membrane-Anchoring Domain of Rat Liver 5’-Nucleotidase: Identification of the COOH-Terminal Serine-523 Covalently Attached With a Glycolipid. Biochemistry (1990) 29(34):7923–7. doi: 10.1021/bi00486a021
4. Knapp K, Zebisch M, Pippel J, El-Tayeb A, Müller CE, Sträter N. Crystal Structure of the Human Ecto-5’-Nucleotidase (CD73): Insights Into the Regulation of Purinergic Signaling. Structure (2012) 20(12):2161–73. doi: 10.1016/j.str.2012.10.001
5. Scaletti E, Huschmann FU, Mueller U, Weiss MS, Sträter N. Substrate Binding Modes of Purine and Pyrimidine Nucleotides to Human Ecto-5’-Nucleotidase (CD73) and Inhibition by Their Bisphosphonic Acid Derivatives. Purinergic Signal (2021) 17(4):693–704. doi: 10.1007/s11302-021-09802-w
6. Whiteside TL, Mandapathil M, Schuler P. The Role of the Adenosinergic Pathway in Immunosuppression Mediated by Human Regulatory T Cells (Treg). Curr Med Chem (2011) 18(34):5217–23. doi: 10.2174/092986711798184334
7. Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular Purine Metabolism and Signaling of CD73-Derived Adenosine in Murine Treg and Teff Cells. Am J Physiol Cell Physiol (2011) 301(2):C530–9. doi: 10.1152/ajpcell.00385.2010
8. Linden J, Koch-Nolte F, Dahl G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol (2019) 37:325–47. doi: 10.1146/annurev-immunol-051116-052406
9. Onami K, Kimura Y, Ito Y, Yamauchi T, Yamasaki K, Aiba S. Nonmetal Haptens Induce ATP Release From Keratinocytes Through Opening of Pannexin Hemichannels by Reactive Oxygen Species. J Invest Dermatol (2014) 134(7):1951–60. doi: 10.1038/jid.2014.93
10. Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, et al. Rho Signaling Regulates Pannexin 1-Mediated ATP Release From Airway Epithelia. J Biol Chem (2011) 286(30):26277–86. doi: 10.1074/jbc.M111.260562
11. Woehrle T, Yip L, Elkhal A, Sumi Y, Chen Y, Yao Y, et al. Pannexin-1 Hemichannel-Mediated ATP Release Together With P2X1 and P2X4 Receptors Regulate T-Cell Activation at the Immune Synapse. Blood (2010) 116(18):3475–84. doi: 10.1182/blood-2010-04-277707
12. Vultaggio-Poma V, Sarti AC, Di Virgilio F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells (2020) 9(11):1–12. doi: 10.3390/cells9112496
13. Di Virgilio F, Adinolfi E. Extracellular Purines, Purinergic Receptors and Tumor Growth. Oncogene (2017) 36(3):293–303. doi: 10.1038/onc.2016.206
14. Bodin P, Burnstock G. Evidence That Release of Adenosine Triphosphate From Endothelial Cells During Increased Shear Stress is Vesicular. J Cardiovasc Pharmacol (2001) 38(6):900–8. doi: 10.1097/00005344-200112000-00012
15. Vultaggio-Poma V, Falzoni S, Chiozzi P, Sarti AC, Adinolfi E, Giuliani AL, et al. Extracellular ATP is Increased by Release of ATP-Loaded Microparticles Triggered by Nutrient Deprivation. Theranostics (2022) 12(2):859–74. doi: 10.7150/thno.66274
16. Pegoraro A, De Marchi E, Ferracin M, Orioli E, Zanoni M, Bassi C, et al. P2X7 Promotes Metastatic Spreading and Triggers Release of miRNA-Containing Exosomes and Microvesicles From Melanoma Cells. Cell Death Dis (2021) 12(12):1088. doi: 10.1038/s41419-021-04378-0
17. Schwiebert EM. ABC Transporter-Facilitated ATP Conductive Transport. Am J Physiol (1999) 276(1):C1–8. doi: 10.1152/ajpcell.1999.276.1.C1
18. Nejime N, Kagota S, Tada Y, Nakamura K, Hashimoto M, Kunitomo M, et al. Possible Participation of Chloride Ion Channels in ATP Release From Cancer Cells in Suspension. Clin Exp Pharmacol Physiol (2009) 36(3):278–82. doi: 10.1111/j.1440-1681.2008.05060.x
19. Okada SF, O’Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, et al. Voltage-Dependent Anion Channel-1 (VDAC-1) Contributes to ATP Release and Cell Volume Regulation in Murine Cells. J Gen Physiol (2004) 124(5):513–26. doi: 10.1085/jgp.200409154
21. Johnsen B, Kaschubowski KE, Nader S, Schneider E, Nicola JA, Fliegert R, et al. P2X7-Mediated ATP Secretion Is Accompanied by Depletion of Cytosolic ATP. Purinergic Signal (2019) 15(2):155–66. doi: 10.1007/s11302-019-09654-5
22. Brandao-Burch A, Key ML, Patel JJ, Arnett TR, Orriss IR. The P2X7 Receptor is an Important Regulator of Extracellular ATP Levels. Front Endocrinol (2012) 3:41. doi: 10.3389/fendo.2012.00041
23. Kepp O, Loos F, Liu P, Kroemer G. Extracellular Nucleosides and Nucleotides as Immunomodulators. Immunol Rev (2017) 280(1):83–92. doi: 10.1111/imr.12571
24. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP Release by Inflammatory Cells. Int J Mol Sci (2018) 19(4):1–16. doi: 10.3390/ijms19041222
25. Faigle M, Seessle J, Zug S, El Kasmi KC, Eltzschig HK. ATP Release From Vascular Endothelia Occurs Across Cx43 Hemichannels and is Attenuated During Hypoxia. PloS One (2008) 3(7):e2801. doi: 10.1371/journal.pone.0002801
26. Dwyer KM, Kishore BK, Robson SC. Conversion of Extracellular ATP Into Adenosine: A Master Switch in Renal Health and Disease. Nat Rev Nephrol (2020) 16(9):509–24. doi: 10.1038/s41581-020-0304-7
27. Greig AV, Linge C, Cambrey A, Burnstock G. Purinergic Receptors are Part of a Signaling System for Keratinocyte Proliferation, Differentiation, and Apoptosis in Human Fetal Epidermis. J Invest Dermatol (2003) 121(5):1145–9. doi: 10.1046/j.1523-1747.2003.12567.x
28. Lovászi M, Branco Haas C, Antonioli L, Pacher P, Haskó G. The Role of P2Y Receptors in Regulating Immunity and Metabolism. Biochem Pharmacol (2021) 187:114419. doi: 10.1016/j.bcp.2021.114419
29. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity (2017) 47(1):15–31. doi: 10.1016/j.immuni.2017.06.020
30. Ring S, Enk AH, Mahnke K. ATP Activates Regulatory T Cells In Vivo During Contact Hypersensitivity Reactions. J Immunol (2010) 184(7):3408–16. doi: 10.4049/jimmunol.0901751
31. Colombo MP. On OX40 and PD-1 Combination: Why Should OX40 Be First in Sequence? Clin Cancer Res (2017) 23(20):5999–6001. doi: 10.1158/1078-0432.CCR-17-2332
32. Adinolfi E, De Marchi E, Orioli E, Pegoraro A, Di Virgilio F. Role of the P2X7 Receptor in Tumor-Associated Inflammation. Curr Opin Pharmacol (2019) 47:59–64. doi: 10.1016/j.coph.2019.02.012
33. De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, et al. The P2X7 Receptor Modulates Immune Cells Infiltration, Ectonucleotidases Expression and Extracellular ATP Levels in the Tumor Microenvironment. Oncogene (2019) 38(19):3636–50. doi: 10.1038/s41388-019-0684-y
34. Borea PA, Gessi S, Merighi S, Varani K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol Sci (2016) 37(6):419–34. doi: 10.1016/j.tips.2016.02.006
35. Cekic C, Linden J. Purinergic Regulation of the Immune System. Nat Rev Immunol (2016) 16(3):177–92. doi: 10.1038/nri.2016.4
36. Lucca LE, Dominguez-Villar M. Modulation of Regulatory T Cell Function and Stability by Co-Inhibitory Receptors. Nat Rev Immunol (2020) 20(11):680–93. doi: 10.1038/s41577-020-0296-3
37. Ernst PB, Garrison JC, Thompson LF. Much Ado About Adenosine: Adenosine Synthesis and Function in Regulatory T Cell Biology. J Immunol (2010) 185(4):1993–8. doi: 10.4049/jimmunol.1000108
38. Moser GH, Schrader J, Deussen A. Turnover of Adenosine in Plasma of Human and Dog Blood. Am J Physiol (1989) 256(Pt 1):C799–806. doi: 10.1152/ajpcell.1989.256.4.C799
39. Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev (2018) 98(3):1591–625. doi: 10.1152/physrev.00049.2017
40. Huang SK, Pandey A, Tran DP, Villanueva NL, Kitao A, Sunahara RK, et al. Delineating the Conformational Landscape of the Adenosine A(2A) Receptor During G Protein Coupling. Cell (2021) 184(7):1884–94.e14. doi: 10.1016/j.cell.2021.02.041
41. Novak J, Fabrik I, Linhartova I, Link M, Cerny O, Stulik J, et al. Phosphoproteomics of cAMP Signaling of Bordetella Adenylate Cyclase Toxin in Mouse Dendritic Cells. Sci Rep (2017) 7(1):16298. doi: 10.1038/s41598-017-14501-x
42. Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, et al. Phosphorylation of CRTC3 by the Salt-Inducible Kinases Controls the Interconversion of Classically Activated and Regulatory Macrophages. Proc Natl Acad Sci USA (2012) 109(42):16986–91. doi: 10.1073/pnas.1215450109
43. MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, et al. PGE(2) Induces Macrophage IL-10 Production and a Regulatory-Like Phenotype via a Protein Kinase A-SIK-CRTC3 Pathway. J Immunol (2013) 190(2):565–77. doi: 10.4049/jimmunol.1202462
44. Sundberg TB, Choi HG, Song JH, Russell CN, Hussain MM, Graham DB, et al. Small-Molecule Screening Identifies Inhibition of Salt-Inducible Kinases as a Therapeutic Strategy to Enhance Immunoregulatory Functions of Dendritic Cells. Proc Natl Acad Sci USA (2014) 111(34):12468–73. doi: 10.1073/pnas.1412308111
45. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 Phosphorylation: A Novel Two-Step Mechanism. Genes Dev (1999) 13(11):1422–37. doi: 10.1101/gad.13.11.1422
46. Schmitz F, Heit A, Dreher S, Eisenacher K, Mages J, Haas T, et al. Mammalian Target of Rapamycin (mTOR) Orchestrates the Defense Program of Innate Immune Cells. Eur J Immunol (2008) 38(11):2981–92. doi: 10.1002/eji.200838761
47. Weichhart T, Hengstschlager M, Linke M. Regulation of Innate Immune Cell Function by mTOR. Nat Rev Immunol (2015) 15(10):599–614. doi: 10.1038/nri3901
48. Klein M, Bopp T. Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front Immunol (2016) 7:315. doi: 10.3389/fimmu.2016.00315
49. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic Adenosine Monophosphate Is a Key Component of Regulatory T Cell-Mediated Suppression. J Exp Med (2007) 204(6):1303–10. doi: 10.1084/jem.20062129
50. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A Receptor Signaling Promotes Peripheral Tolerance by Inducing T-Cell Anergy and the Generation of Adaptive Regulatory T Cells. Blood (2008) 111(1):251–9. doi: 10.1182/blood-2007-03-081646
51. Han KL, Thomas SV, Koontz SM, Changpriroa CM, Ha SK, Malech HL, et al. Adenosine a₂A Receptor Agonist-Mediated Increase in Donor-Derived Regulatory T Cells Suppresses Development of Graft-Versus-Host Disease. J Immunol (2013) 190(1):458–68. doi: 10.4049/jimmunol.1201325
52. Ehrentraut H, Westrich JA, Eltzschig HK, Clambey ET. Adora2b Adenosine Receptor Engagement Enhances Regulatory T Cell Abundance During Endotoxin-Induced Pulmonary Inflammation. PloS One (2012) 7(2):e32416. doi: 10.1371/journal.pone.0032416
53. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The Development and Immunosuppressive Functions of CD4(+) CD25(+) FoxP3(+) Regulatory T Cells Are Under Influence of the Adenosine-A2A Adenosine Receptor Pathway. Front Immunol (2012) 3:190. doi: 10.3389/fimmu.2012.00190
54. Lee DJ, Taylor AW. Both MC5r and A2Ar Are Required for Protective Regulatory Immunity in the Spleen of Post-Experimental Autoimmune Uveitis in Mice. J Immunol (2013) 191(8):4103–11. doi: 10.4049/jimmunol.1300182
55. Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, et al. Autocrine Adenosine Signaling Promotes Regulatory T Cell-Mediated Renal Protection. J Am Soc Nephrol: JASN (2012) 23(9):1528–37. doi: 10.1681/ASN.2012010070
56. Bao R, Shui X, Hou J, Li J, Deng X, Zhu X, et al. Adenosine and the Adenosine A2A Receptor Agonist, CGS21680, Upregulate CD39 and CD73 Expression Through E2F-1 and CREB in Regulatory T Cells Isolated From Septic Mice. Int J Mol Med (2016) 38(3):969–75. doi: 10.3892/ijmm.2016.2679
57. Klemens MR, Sherman WR, Holmberg NJ, Ruedi JM, Low MG, Thompson LF. Characterization of Soluble vs Membrane-Bound Human Placental 5′-Nucleotidase. Biochem Biophys Res Commun (1990) 172(3):1371–7. doi: 10.1016/0006-291X(90)91601-N
58. Resta R, Yamashita Y, Thompson LF. Ecto-Enzyme and Signaling Functions of Lymphocyte CD 7 3. Immunol Rev (1998) 161(1):95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x
59. Dianzani U, Redoglia V, Bragardo M, Attisano C, Bianchi A, Di Franco D, et al. Co-Stimulatory Signal Delivered by CD73 Molecule to Human CD45RAhiCD45ROlo (Naive) CD8+ T Lymphocytes. J Immunol (1993) 151(8):3961–70.
60. Resta R, Hooker SW, Laurent AB, Shuck JK, Misumi Y, Ikehara Y, et al. Glycosyl Phosphatidylinositol Membrane Anchor Is Not Required for T Cell Activation Through CD73. J Immunol (1994) 153(3):1046–53.
61. Resta R, Thompson LF. T Cell Signalling Through CD73. Cell Signal (1997) 9(2):131–9. doi: 10.1016/S0898-6568(96)00132-5
62. Airas L, Hellman J, Salmi M, Bono P, Puurunen T, Smith DJ, et al. CD73 is Involved in Lymphocyte Binding to the Endothelium: Characterization of Lymphocyte-Vascular Adhesion Protein 2 Identifies it as CD73. J Exp Med (1995) 182(5):1603–8. doi: 10.1084/jem.182.5.1603
63. Henttinen T, Jalkanen S, Yegutkin GG. Adherent Leukocytes Prevent Adenosine Formation and Impair Endothelial Barrier Function by Ecto-5′-Nucleotidase/CD73-Dependent Mechanism. J Biol Chem (2003) 278(27):24888–95. doi: 10.1074/jbc.M300779200
64. Airas L. CD73 and Adhesion of B-Cells to Follicular Dendritic Cells. Leuk Lymphoma (1998) 29(1-2):37–47. doi: 10.3109/10428199809058380
65. Airas L, Niemelä J, Jalkanen S. CD73 Engagement Promotes Lymphocyte Binding to Endothelial Cells via a Lymphocyte Function-Associated Antigen-1-Dependent Mechanism. J Immunol (2000) 165(10):5411–7. doi: 10.4049/jimmunol.165.10.5411
66. Cappellari AR, Vasques GJ, Bavaresco L, Braganhol E, Battastini AMO. Involvement of Ecto-5′-Nucleotidase/CD73 in U138MG Glioma Cell Adhesion. Mol Cell Biochem (2012) 359(1):315–22. doi: 10.1007/s11010-011-1025-9
67. Zhou P, Zhi X, Zhou T, Chen S, Li X, Wang L, et al. Overexpression of Ecto-5’-Nucleotidase (CD73) Promotes T-47D Human Breast Cancer Cells Invasion and Adhesion to Extracellular Matrix. Cancer Biol Ther (2007) 6(3):426–31. doi: 10.4161/cbt.6.3.3762
68. Fang F, Cao W, Zhu W, Lam N, Li L, Gaddam S, et al. The Cell-Surface 5′-Nucleotidase CD73 Defines a Functional T Memory Cell Subset That Declines With Age. Cell Rep (2021) 37(6):109981. doi: 10.1016/j.celrep.2021.109981
69. Kalekar LA, Mueller DL. Relationship Between CD4 Regulatory T Cells and Anergy In Vivo. J Immunol (2017) 198(7):2527–33. doi: 10.4049/jimmunol.1602031
70. Yang L, Kobie JJ, Mosmann TR. CD73 and Ly-6a/E Distinguish In Vivo Primed But Uncommitted Mouse CD4 T Cells From Type 1 or Type 2 Effector Cells. J Immunol (2005) 175(10):6458–64. doi: 10.4049/jimmunol.175.10.6458
71. Iyer SS, Latner DR, Zilliox MJ, McCausland M, Akondy RS, Penaloza-MacMaster P, et al. Identification of Novel Markers for Mouse CD 4+ T Follicular Helper Cells. Eur J Immunol (2013) 43(12):3219–32. doi: 10.1002/eji.201343469
72. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 Transcription Factors Control Th17 Cell Immunosuppressive Activity via the Regulation of Ectonucleotidase Expression. Immunity (2012) 36(3):362–73. doi: 10.1016/j.immuni.2011.12.019
73. Schneider E, Winzer R, Rissiek A, Ricklefs I, Meyer-Schwesinger C, Ricklefs FL, et al. CD73-Mediated Adenosine Production by CD8 T Cell-Derived Extracellular Vesicles Constitutes an Intrinsic Mechanism of Immune Suppression. Nat Commun (2021) 12(1):1–14. doi: 10.1038/s41467-021-26134-w
74. Briceño P, Rivas-Yañez E, Rosemblatt MV, Parra-Tello B, Farías P, Vargas L, et al. CD73 Ectonucleotidase Restrains CD8+ T Cell Metabolic Fitness and Anti-Tumoral Activity. Front Cell Dev Biol (2021) 9:185. doi: 10.3389/fcell.2021.638037
75. Flores-Santibáñez F, Fernández D, Meza D, Tejón G, Vargas L, Varela-Nallar L, et al. CD 73-Mediated Adenosine Production Promotes Stem Cell-Like Properties in Mouse Tc17 Cells. Immunology (2015) 146(4):582–94. doi: 10.1111/imm.12529
76. Beldi G, Wu Y, Banz Y, Nowak M, Miller L, Enjyoji K, et al. Natural Killer T Cell Dysfunction in CD39-Null Mice Protects Against Concanavalin A–Induced Hepatitis. Hepatology (2008) 48(3):841–52. doi: 10.1002/hep.22401
77. Berzofsky JA, Terabe M. The Contrasting Roles of NKT Cells in Tumor Immunity. Curr Mol Med (2009) 9(6):667–72. doi: 10.2174/156652409788970706
78. Mattarollo SR, Smyth MJ. Therapeutic Approaches Utilising NKT Cells. Natural Killer T Cells (2012) 111–28. doi: 10.1007/978-1-4614-0613-6_7
79. Minciacchi VR, Freeman MR, Di Vizio D. Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Semin Cell Dev Biol (2015) 40:41–51. doi: 10.1016/j.semcdb.2015.02.010
80. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev Cell (2011) 20(1):131–9. doi: 10.1016/j.devcel.2010.12.003
81. Record M. Intercellular Communication by Exosomes in Placenta: A Possible Role in Cell Fusion? Placenta (2014) 35(5):297–302. doi: 10.1016/j.placenta.2014.02.009
82. Wu Q, Duan W-Z, Chen J-B, Zhao X-P, Li X-J, Liu Y-Y, et al. Extracellular Vesicles: Emerging Roles in Developing Therapeutic Approach and Delivery Tool of Chinese Herbal Medicine for the Treatment of Depressive Disorder. Front Pharmacol (2022) 13. doi: 10.3389/fphar.2022.843412
83. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer Exosomes Express CD39 and CD73, Which Suppress T Cells Through Adenosine Production. J Immunol (2011) 187(2):676–83. doi: 10.4049/jimmunol.1003884
84. Schneider E, Rissiek A, Winzer R, Puig B, Rissiek B, Haag F, et al. Generation and Function of Non-Cell-Bound CD73 in Inflammation. Front Immunol (2019) 10:1729. doi: 10.3389/fimmu.2019.01729
85. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 Expression on Extracellular Vesicles Derived From CD4+ CD25+ Foxp3+ T Cells Contributes to Their Regulatory Function. Eur J Immunol (2013) 43(9):2430–40. doi: 10.1002/eji.201242909
86. Agarwal A, Fanelli G, Letizia M, Tung SL, Boardman D, Lechler R, et al. Regulatory T Cell-Derived Exosomes: Possible Therapeutic and Diagnostic Tools in Transplantation. Front Immunol (2014) 5:555. doi: 10.3389/fimmu.2014.00555
87. Allard B, Turcotte M, Stagg J. CD73-Generated Adenosine: Orchestrating the Tumor-Stroma Interplay to Promote Cancer Growth. J BioMed Biotechnol (2012) 2012:485156. doi: 10.1155/2012/485156
88. Turcotte M, Spring K, Pommey S, Chouinard G, Cousineau I, George J, et al. CD73 is Associated With Poor Prognosis in High-Grade Serous Ovarian Cancer. Cancer Res (2015) 75(21):4494–503. doi: 10.1158/0008-5472.CAN-14-3569
89. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc Natl Acad Sci USA (2013) 110(27):11091–6. doi: 10.1073/pnas.1222251110
90. Tahkola K, Ahtiainen M, Kellokumpu I, Mecklin J-P, Laukkarinen J, Laakkonen J, et al. Prognostic Impact of CD73 Expression and its Relationship to PD-L1 in Patients With Radically Treated Pancreatic Cancer. Virchows Archiv: Int J Pathol (2021) 478(2):209–17. doi: 10.1007/s00428-020-02888-4
91. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically Expanded Human Regulatory T-Cells Are Super-Suppressive Due to HIF1A Induced Expression of CD73. Commun Biol (2021) 4(1):1186. doi: 10.1038/s42003-021-02721-x
92. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-Nucleotidase (CD73) Regulation by Hypoxia-Inducible Factor-1 Mediates Permeability Changes in Intestinal Epithelia. J Clin Invest (2002) 110(7):993–1002. doi: 10.1172/JCI0215337
93. Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep (2019) 27(8):2411–25.e9. doi: 10.1016/j.celrep.2019.04.091
94. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 Enhances the Antitumor Activity of Anti-PD-1 and Anti-CTLA-4 Mabs. Clin Cancer Res (2013) 19(20):5626–35. doi: 10.1158/1078-0432.CCR-13-0545
95. Deng WW, Li YC, Ma SR, Mao L, Yu GT, Bu LL, et al. Specific Blockade CD73 Alters the “Exhausted” Phenotype of T Cells in Head and Neck Squamous Cell Carcinoma. Int J Cancer (2018) 143(6):1494–504. doi: 10.1002/ijc.31534
96. Chen S, Fan J, Zhang M, Qin L, Dominguez D, Long A, et al. CD73 Expression on Effector T Cells Sustained by TGF-β Facilitates Tumor Resistance to Anti-4-1BB/CD137 Therapy. Nat Commun (2019) 10(1):150. doi: 10.1038/s41467-018-08123-8
97. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-Human CD73 Monoclonal Antibody Inhibits Metastasis Formation in Human Breast Cancer by Inducing Clustering and Internalization of CD73 Expressed on the Surface of Cancer Cells. J Immunol (2013) 191(8):4165–73. doi: 10.4049/jimmunol.1301274
98. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. CD73 Has Distinct Roles in Nonhematopoietic and Hematopoietic Cells to Promote Tumor Growth in Mice. J Clin Invest (2011) 121(6):2371–82. doi: 10.1172/JCI45559
99. Xu Z, Gu C, Yao X, Guo W, Wang H, Lin T, et al. CD73 Promotes Tumor Metastasis by Modulating RICS/RhoA Signaling and EMT in Gastric Cancer. Cell Death Dis (2020) 11(3):1–15. doi: 10.1038/s41419-020-2403-6
100. Ma X-L, Shen M-N, Hu B, Wang B-L, Yang W-J, Lv L-H, et al. CD73 Promotes Hepatocellular Carcinoma Progression and Metastasis via Activating PI3K/AKT Signaling by Inducing Rap1-Mediated Membrane Localization of P110β and Predicts Poor Prognosis. J Hematol Oncol (2019) 12(1):1–17. doi: 10.1186/s13045-019-0724-7
Keywords: adenosine, regulatory T cells, immune suppression, CD73, immune checkpoint
Citation: Da M, Chen L, Enk A, Ring S and Mahnke K (2022) The Multifaceted Actions of CD73 During Development and Suppressive Actions of Regulatory T Cells. Front. Immunol. 13:914799. doi: 10.3389/fimmu.2022.914799
Received: 07 April 2022; Accepted: 03 May 2022;
Published: 31 May 2022.
Edited by:
Robert Zeiser, University of Freiburg, GermanyReviewed by:
Theresa L. Whiteside, University of Pittsburgh, United StatesElena Adinolfi, University of Ferrara, Italy
Copyright © 2022 Da, Chen, Enk, Ring and Mahnke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karsten Mahnke, karsten.mahnke@med.uni-heidelberg.de
†These authors have contributed equally to this work and share first authorship