 Yi-kai Lin
Yi-kai Lin Ping Zhu3†
Ping Zhu3† Wang-sheng Wang
Wang-sheng Wang Kang Sun
Kang Sun- 1Center for Reproductive Medicine, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 2Shanghai Key Laboratory for Assisted Reproduction and Reproductive Genetics, Shanghai, China
- 3Department of Obstetrics and Gynecology, No.971 Hospital of the PLA Navy, Qingdao, China
Serum amyloid A (SAA) is one of the acute phase proteins released primarily from the liver in response to infection, inflammation and trauma. Emerging evidence indicates that SAA may function as a host-derived damage-associated molecular pattern (DAMP) protein to sense danger signals in pregnancy. The plasma SAA levels in maternal circulation are significantly increased in normal parturition, particularly in postpartum, as well as in gestational disorders such as premature preterm rupture of membranes, pre-eclampsia, gestational diabetes, and recurrent spontaneous abortion. It is likely that SAA acts as a non-specific DAMP molecule in response to inflammation and trauma experienced under these conditions. Notably, SAA can also be synthesized locally in virtually all gestational tissues. Within these gestational tissues, under the induction by bacterial products, pro-inflammatory cytokines and stress hormone glucocorticoids, SAA may exert tissue-specific effects as a toll-like receptor 4 (TLR4)-sensed DAMP molecule. SAA may promote parturition through stimulation of inflammatory reactions via induction of pro-inflammatory cytokines, chemokines, adhesion molecules and prostaglandins in the uterus, fetal membranes and placenta. In the fetal membranes, SAA may also facilitate membrane rupture through induction of matrix metalloproteases (MMPs)- and autophagy-mediated collagen breakdown and attenuation of lysyl oxidase-mediated collagen cross-linking. SAA synthesized in extravillous trophoblasts may promote their invasiveness into the endometrium in placentation. Here, we summarized the current understanding of SAA in pregnancy with an aim to stimulate in-depth investigation of SAA in pregnancy, which may help better understand how inflammation is initiated in gestational tissues in both normal and abnormal pregnancies.
1 Introduction
Inflammation plays a key role in maintaining tissue homeostasis. Both exaggerated or inadequate inflammation can lead to diseases. In pregnancy, intricately-controlled inflammation is required in gestational tissues for implantation, gestational maintenance, and parturition (1–3). Dysregulation of inflammatory reactions in gestational tissues can lead to reproductive disorders, including infertility, spontaneous abortion, preterm birth, pre-eclampsia and gestational diabetes (1–4). Therefore, understanding the regulatory mechanism of inflammatory reactions in gestational tissues may help understand the normal process of pregnancy as well as the pathogenesis of diseases associated with pregnancy. The role of classical inflammatory mediators such as cytokines, chemokines, and prostaglandins in pregnancy has been extensively covered in a bunch of excellent review papers (1–7). However, how inflammation is launched in gestational tissues is still not very well understood.
Damage-associated molecular patterns (DAMPs) are molecules released from damaged or dying cells due to trauma or infection as a component of the innate immune response. DAMPs serve as a warning sign for the organism by launching inflammatory responses (8). Emerging evidence indicates that the acute phase response (APR) protein serum amyloid A (SAA) may function as a DAMP molecule in pregnancy. SAA released into the circulation or synthesized locally in the gestational tissues may help the mother and fetus sense danger signals in pregnancy by launching both specific and non-specific effects in gestational tissues. SAA may be associated with placentation, membrane rupture, parturition, postpartum recovery as well as gestational disorders. Herein, we reviewed the current understanding of SAA in pregnancy by summarizing the available publications in the literature as well as our own studies. To aid in the better understanding of SAA in pregnancy, we will first introduce the general aspects of SAA in the acute phase response briefly.
2 SAA in the acute phase response
Our body responds to infection, inflammation, and trauma by implementing APR, an innate body defense response which comprises a range of well-orchestrated physiological and biochemical reactions, to prevent ongoing tissue damage, to destroy the invading organism, and to return the body to normal function (9–11). Central to these reactions is the hepatic production of acute-phase proteins, particularly C-reactive protein (CRP), mannan-binding lectin (MBL) and serum amyloid A (SAA) (11–15). It is known that one of the major functions of CRP and MBL in APR is to act as soluble opsonin-like pattern-recognition receptors so that microorganisms can be tagged for phagocytosis. By contrast, the function of SAA in APR is not entirely understood. However, SAA must be crucial for individual survival because SAA has been conserved across all mammals for several million years (16, 17), and moreover, the plasma levels of SAA is increased by 1000-fold from basal 20-50 μg/mL to 1 mg/mL (80 mM) in APR within 24 hours (18). Over decades of investigations, we are getting to know more about SAA in APR. Several isoforms of SAA with diversified functions and multiple mediating receptors have been identified.
2.1 SAA isoforms
SAA family consists of members coded by different genes which are remarkably conserved across species (13, 16). In humans, there are three SAA-encoding genes (SAA1, SAA2, and SAA4) and a pseudo gene SAA3P which contains an early stop codon (19–21). These four genes are clustered in a 150-kb region on the short arm of chromosome 11 (13, 19) (Figure 1A). Of the SAA-encoding genes, SAA1 and SAA2 are the acute phase responsive genes which are highly inducible, while SAA4 is constitutively expressed (13, 20, 22). It is widely accepted that the liver is the primary source of plasma SAA1 and SAA2 in the acute phase response (13, 16, 20), which can be induced by pro-inflammatory cytokines like interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interferon-γ (IFN-γ) (13, 23–27). Notably, the stress hormone glucocorticoids are also capable of inducing SAA expression in the liver despite being generally considered as an anti-inflammatory hormone (28). However, it has been shown that glucocorticoids preferentially stimulate the transcription of SAA1 but not SAA2 because only SAA1 promoter harbors a glucocorticoid response element (GRE) (29). The mouse SAA genes were mapped to chromosome 7, a region which is analogous to the region of human chromosome 11 (13). Likewise, the expression of Saa1 and Saa2 is inducible, while Saa4 is constitutively expressed in the mouse (13). By contrast, the mouse Saa3 is not a pseudogene but encodes an inducible SAA3 protein which is believed to be expressed mainly in extrahepatic tissues (13). Instead, Saa5 is a pseudogene in the mouse (13).
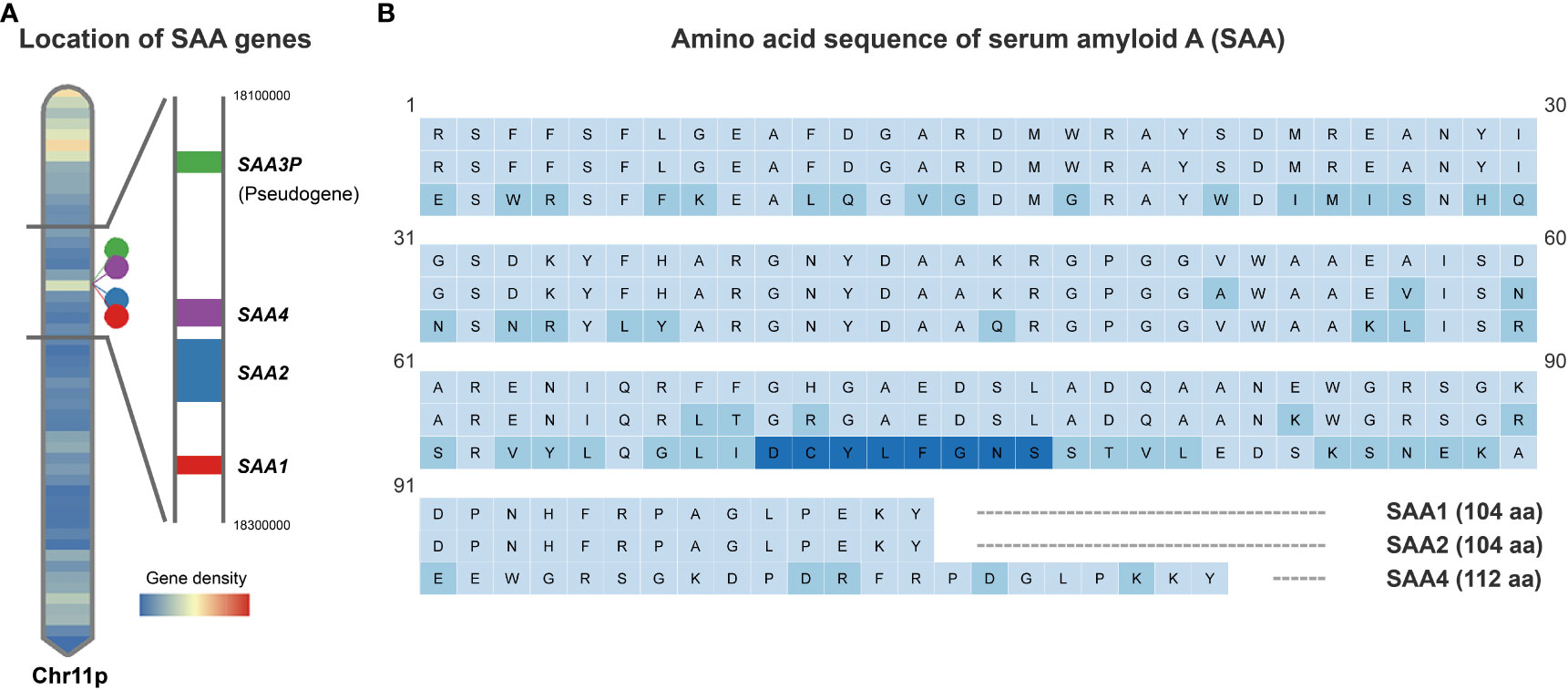
Figure 1 Schematic depiction of human SAA gene locations and protein sequences. (A). Human SAA genes including SAA1, SAA2, SAA4 and pseudogene SAA3P clustered in a 150-kb region on the short arm of chromosome 11. (B). Comparison of the amino acid sequences encoded by the human SAA1, SAA2 and SAA4 genes based on UniProt Knowledgebase entries P0DJI8, P0DJI9 and P35542. Amino acid residue variations compared to SAA1 are marked in light blue and an additional 8 amino acids inserted between amino acid residues 69 and 70 of the SAA1/2 in SAA4 are marked in dark blue.
The human SAA1 and 2 are small proteins consisting of 104 amino acids and share 92% of amino acids, while SAA4 carries an additional 8 amino acids inserted between amino acids 69 and 70 of the inducible SAAs (13, 20) (Figure 1B). The N-terminal of SAA harbors a hydrophobic α-helical domain, while the C-terminal is essential for the maintenance of SAA structure (13, 17). Proteolytic cleavage of the C-terminal results in the formation of a 76 amino acid N-terminal fragment, which is subject to aggregation and formation of highly-ordered β-sheets, a structure commonly seen in microfibrils of the amyloid deposit (13, 17, 30).
2.2 Functions of SAA in APR
The SAA molecule is amphipathic, but mostly lipophilic in circulation (31). Like the transportation of other lipophilic hormones such as the steroid and thyroid hormones, SAA is also transported by a carrier in the blood (31). The water-insoluble N-terminal α-helical domain of SAA carries a binding site for the high-density lipoprotein (HDL). It has been estimated that 95% of SAA released from the liver is transported in HDL despite that a small fraction of lipid-free form (about 50 μg/mL) also exists in the circulation, which may be in equilibrium with the fraction partitioned in HDL (31–33). Since HDL binding sequesters SAA from its biological activities, HDL is considered as a natural inhibitor of SAA biological activities (13, 34, 35). In addition, capture of SAA displaces the original apolipoproteins (Apo-AI) and cholesterol in HDL, and this alteration in HDL composition will increase the affinity of HDL for macrophages (36). It is known that macrophages are engaged in the clearance of dead cells at the injury site, which may overload the macrophages with cholesterol from the engulfed cell debris. Studies have shown that capture of SAA not only increases the affinity of HDL for macrophages, but also facilitates cholesterol efflux from the cholesterol-loaded macrophages to HDL. By this way, the phagocytic activity of macrophages can be restored, and the evacuated cholesterol can be recycled to be used by newly regenerated cells for the repairment damaged tissues (16, 37, 38). Therefore, it is suggested that increased proportion of SAA-containing HDL in the circulation in APR is to ensure individual survival at the early stage.
In addition to the liver-derived SAA, cells in the inflamed tissue including immune cells, fibroblasts, endothelial cells, epithelial cells, adipocytes, and smooth muscle cells are also capable of producing SAA (22, 39–44), which may contribute to a pool of lipid-free SAA together with the liver-derived SAA in the local tissue. This pool of free SAA has been shown to participate in immunomodulation, immunosurveillance and tissue remodeling in the inflamed tissue. SAA has been shown to act as a humoral DAMP molecule to enhance the chemotaxis of neutrophils, monocytes, mast cells, and T lymphocytes, as well as the production of a wide array of inflammatory mediators, including pro-inflammatory cytokines, prostaglandins and matrix metalloproteinases (MMPs) (13, 40, 41, 45–54). In addition, SAA has also been shown to induce macrophage polarization toward both pro-inflammatory M1 phenotype and anti-inflammatory M2 phenotype (55–57). The M2 phenotype helps resolve inflammation by secreting anti-inflammatory cytokines and clearing dead cell debris. Given all these properties of SAA in the inflamed tissue, it is likely that SAA helps the body survive the traumatic insults through reversible homeostatic inflammatory reactions by functioning as an early responsive host-derived DAMP molecule.
2.3 SAA receptors
The apparent importance of SAA in APR has aroused great interest in the identification of its mediating receptors, but the delineation develops slowly. To date, several cell surface receptors have been identified, including the G protein-coupled FPRL1 (formyl peptide receptor like–1) receptor, the pattern recognition receptors RAGE (receptor for advanced glycation end products) and TLR2/4 (toll-like receptor 2/4), and the scavenger receptors CD36 (cluster of differentiation 36) and CLA-1 (CD36 and LIMPII Analogous-1) (35, 54, 58–64). Activation of FPRL1, RAGE and TLR2/4 has been shown to be responsible for SAA-induced inflammatory reactions including macrophage activation, chemotaxis, cytokine expression, MMP9 and nitric oxide production with the activation of ERK and p38 MAPK and NF-κB pathways (35, 54, 58–61, 63). The scavenger receptor CLA-1 and CD36 have been demonstrated to mediate the effect of HDL-SAA on cholesterol efflux from cells (64, 65) and SAA-induced pro-inflammatory cytokine secretion (62) respectively. Although multiple SAA receptors have been identified, it is unclear whether these receptors are expressed in a cell-specific manner or multiple SAA receptors are jointly expressed in the same cell.
In summary, there are three SAA isoforms in humans, and the predominant role of the inducible SAAs in APR may help the body overcome traumatic insults by launching reversible homeostatic inflammatory reactions as an early host-derived DAMP molecule. For a comprehensive description of SAA in APR, the reader is referred to a wealth of elegant reviews in this area (13, 16, 20, 22, 52). Here, in the subsequent sections, we will focus on SAA in pregnancy, a subject that has not received adequate attention.
3 SAA in pregnancy
Inflammation is implicated in both normal and abnormal pregnancies (1–4). While optimal inflammation in gestational tissues is an indispensable event of implantation and parturition processes, aberrant inflammation is associated with pregnancy disorders such as abortion, premature preterm rupture of membranes (PPROM), preterm birth, gestational diabetes (GDM) and pre-eclampsia (66–69). In view of the role of SAA in inflammation in APR and the inflammatory states of normal and abnormal pregnancies, investigators have been trying to disclose the role of SAA in pregnancy for some time. Evidence gathered so far indicates that SAA in maternal circulation may function as a non-specific DAMP molecule in response to tissue inflammation and damage incurred in parturition and gestational disorders. However, SAA synthesized locally in gestational tissues may function as an early host-derived DAMP molecule exerting more specific effects in placentation, membrane rupture and parturition, thereby helping the embryo implant into the endometrium and aiding the fetus to escape from the endangered in-utero environment.
3.1 Changes of maternal plasma SAA in normal pregnancy
In view of the inflammatory effects of SAA and the dramatic hormonal changes in pregnancy, earlier studies assumed that maternal plasma SAA levels in normal pregnancy should differ from that in non-pregnancy. Ostensen et al. measured SAA levels in the serial plasma samples collected from the same pregnant woman, and found that maternal plasma SAA levels remained unaltered in the first, second and third trimesters, but manifested a dramatic increase immediately after parturition, which resumed normalization within the first 2 weeks postpartum (70). Subsequent studies confirmed the findings by Ostensen et al. (71, 72). These later studies established that maternal plasma SAA levels stayed unaltered during pregnancy but began to rise immediately after parturition and reached a plateau at 24 hours postpartum (71, 72). Of interest, these studies also revealed that SAA concentrations in the maternal blood were much higher than that in umbilical cord blood (71, 72), indicating that there is a lack of transplacental transfer of SAA, and any increases in SAA levels in cord blood may reflect increased fetal production in conditions such as fetal infection or trauma. However, only a few studies addressed whether SAA has already manifested a change at the onset of labor. Cicarelli et al. found that some but not all of the cases they examined displayed a rise of SAA levels in maternal circulation at the moment of delivery (72). By measuring SAA levels in the serial maternal venous blood samples collected before (1–2 days before labor onset) and after onset of labor as well as 24 hours after delivery, we found that there was a significant increase in SAA1 levels in maternal circulation after onset of labor, but this increase was much less than the increase 24 hours after delivery (40). It remains unsettled whether this increase after onset of labor is part of the initiating mechanism of parturition or a mere non-specific reflection of the inflammatory state of gestational tissues at parturition. Nonetheless, all these studies including ours showed a marked increase in maternal blood SAA levels at postpartum. It is very likely that this postpartum SAA elevation in maternal circulation is part of the non-specific acute phase response of the body to tissue inflammation and damage experienced in parturition (70), which may help the mother enhance host defense and repair the tissue damage.
3.2 Changes of maternal plasma SAA in gestational disorders
3.2.1 PPROM and chorioamnionitis
Fetal membranes are consisted of amnion and chorion layers, which enclose the amniotic fluid, and function as a barrier to ascending infections for the protection of the fetus. Preterm premature rupture of membranes (PPROM) occurs in 8–10% of all pregnancies (4, 73). Ruptured membranes are exposed to ascending infections with a high likelihood of the development of chorioamnionitis. Chorioamnionitis is a major cause of preterm birth, and unrestrained chorioamnionitis can spread infection to both mother and fetus leading to deadly outcomes (4, 74). It is estimated that PPROM accounts for approximately one-third of preterm birth and 18–20% of perinatal deaths (5, 6, 9). Therefore, understanding the cause of PPROM and chorioamnionitis is of utmost importance for the prevention of preterm birth as well as for the safety of both mother and fetus. In view of the tissue damage incurred and the high risk of chorioamnionitis in PPROM, SAA was expected to rise in PPROM, particularly those with infection. Yang et al. found that SAA 1 and 2 were among the proteins which were significantly elevated in maternal blood in lipopolysaccharide (LPS)-induced preterm birth in the mouse (75). In human studies, Koseoglu et al. found that SAA levels in maternal blood were significantly elevated in the PPROM group compared to the control group (76), and Kayabas et al. demonstrated that SAA levels in maternal circulation were further increased in patients with clinical chorioamnionitis (107 µg/mL) when compared with patients with only PPROM but not chorioamnionitis (21 µg/mL) (77). We also found that SAA1 levels were markedly increased in maternal circulation in preterm birth with chorioamnionitis when compared with gestational age-matched iatrogenic preterm birth (40). In addition to the rise of SAA levels in maternal circulation, SAA levels in cord blood were also significantly higher in patients with PPROM (115 µg/mL) than in patients without PPROM (26 µg/mL) (77). These results suggest that both mother and fetus may respond to PPROM, and the response may be particularly strong when chorioamnionitis is present. However, the exact role of increased SAA levels in PPROM and chorioamnionitis is not well understood. It is likely that increased SAA levels in maternal circulation in PPROM and chorioamnionitis are the acute phase response of the body to the mediators released from damaged and infected membranes.
3.2.2 Pre-eclampsia and placenta accreta
Pre-eclampsia is a hypertensive disease that develops during pregnancy, which accounts for 2% to 8% of pregnancy-related complications (78). The exact cause for pre-eclampsia remains elusive. Apart from majorly insufficient blood flow to the placenta due to shallow invasion of extravillous trophoblasts (EVT) into the endometrium (79), vascular endothelial dysfunction associated with maternal systemic inflammation has also been suggested to account for the pathogenesis of pre-eclampsia (68, 69, 80). It has been shown that maternal plasma SAA levels were significantly increased in pre-eclampsia, and were further increased in eclampsia and HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome (81–83). However, there is also a study that failed to show any changes of SAA levels in patients with pre-eclampsia irrespective of severity (84). It is not known what causes the disparity. The early or late onset of the disease may affect the results. Although these studies described that the recruited patients were diagnosed pre-eclampsia after 20 weeks’ gestation, they did not mention when patients develop hypertension.
In contrast to the shallow invasiveness of EVTs in pre-eclampsia, excessive invasion of EVTs can result in placenta accreta, which may lead to deadly bleeding at parturition. Zakaria et al. reported that SAA levels in maternal blood were significantly increased in patients with placenta accreta (85). As SAA levels were elevated in situations of both shallow and excessive invasiveness of EVTs, it is likely that the alteration of SAA levels in maternal blood in pre-eclampsia and placenta accreta are the non-specific reflection of the inflammatory and injury states of the diseases.
3.2.3 Gestational diabetes
The prevalence of gestational diabetes mellitus (GDM) is about 3 to 15% of pregnancies, which is thought to arise from adaptive failures to pregnancy with maternal obesity as an important contributor (66, 86). Pregnancies complicated with GDM are more likely to develop maternal and fetal complications, such as hypertensive disorders, pre-eclampsia, preterm birth, and increased risk of developing diabetes in later lives. Accumulating evidence indicates that both obesity and GDM may be associated with a state of low-grade chronic inflammation (66, 67). As a sensitive marker of inflammatory diseases, SAA is hypothesized to contribute to the inflammatory state of patients with GDM. Erin et al. found that maternal circulating SAA levels were significantly higher in women with GDM when compared with healthy pregnant controls (87). Their correlation analysis showed that SAA levels were significantly correlated with age, BMI, mean arterial blood pressure, glucose tolerance and carotid intima-media thickness (CIMT) (87), a valid predictor of atherosclerosis. Thus, they suggested that increased maternal plasma SAA levels in GDM might be an indicator of an increased risk of subclinical atherosclerosis and future atherosclerotic heart disease (87). In contrast to the findings by Erin et al., Pöyhönen-Alho et al. showed that both SAA and CRP levels (SAA: 1.24 ± 0.27 mg/L, CRP: 0.60 ± 0.37 mg/L) were reluctant to change in pregnant women with GDM when compared with normal pregnant women (SAA: 1.32 ± 0.38 mg/L, CRP: 0.59 ± 0.61 mg/L) (88). Notably, their CRP data were also in contradiction with previous studies which showed elevated CRP levels in GDM (89, 90). Further investigations may be required to affirm the change of maternal blood SAA levels and its significance in GDM.
3.2.4 Recurrent spontaneous abortion
Recurrent spontaneous abortion (RSA) is defined as three or more consecutive spontaneous pregnancy losses (91), which occurs in approximately 1% of pregnancies (92). However, about half of the RSA cases are unexplained pregnancy losses (93, 94). Ibrahim et al. demonstrated that plasma SAA levels were significantly increased in women with unexplained RSA (Median 50.0 μg/mL, interquartile range 26.0-69.0) when compared with the control group (Median 11.6 μg/mL, interquartile range 6.2-15.5; P<0.001) (95). These findings were confirmed by an independent group (96). However, the significance of SAA elevation in RSA was not investigated, but it was suggested that SAA might be a biomarker of RSA (95).
The reported changes of SAA levels in the circulation in different gestational disorders are summarized in Table 1. It appears that SAA levels in maternal circulation may not be an ideal biomarker for specific pregnancy complications, but the extent of SAA elevation may reflect the severity of gestational disorders.
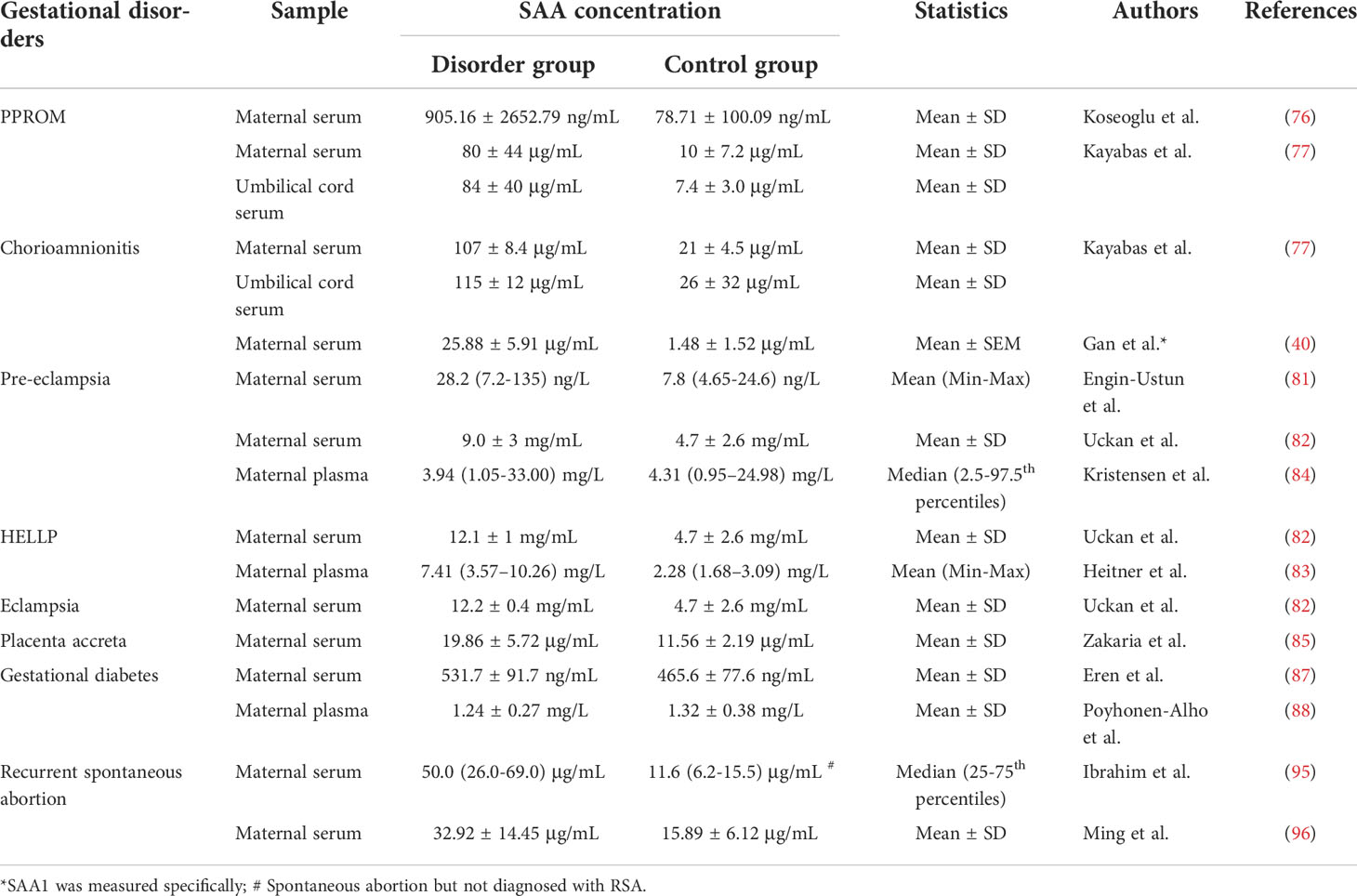
Table 1 SAA levels in the circulation in gestational disorders.
4 SAA synthesized within gestational tissues
It is widely accepted that the liver is the major source of acute phase SAA in the circulation. However, accumulating evidence indicates that a wide range of cells in extrahepatic tissues including immune cells, fibroblasts, endothelial cells, epithelial cells, adipocytes, and smooth muscle cells are capable of de novo synthesis of SAA (22, 39–44). SAA produced locally in extrahepatic tissues may be more efficient than the liver-derived SAA in launching inflammatory reactions within the tissue. In pregnancy, almost all gestational tissues including the placenta, fetal membranes and uterus are capable of SAA synthesis, and SAA synthesized in these gestational tissues has been shown to be implicated in the initiation of inflammatory processes as well as specific actions involved in placentation, membrane rupture and parturition.
4.1 Placenta
The placenta is a temporary fetal organ in pregnancy, which plays critical roles in nutrient, gas, and waste exchange between maternal and fetal circulations (97, 98). The placental cytotrophoblasts can either differentiate into villous syncytiotrophoblasts or EVTs. The villous syncytiotrophoblasts serve as an important endocrine organ producing critical hormones which not only maintain pregnancy, but also promote parturition. In contrast to the villous trophoblasts, EVTs invade into the endometrium whereby they remodel uterine spiral arteries for the establishment of implantation (99). Sandri et al. demonstrated that SAA was expressed in EVTs and decidual cells at the maternal-fetal interface in humans (100), where SAA promoted the invasion of EVTs into the endometrium through toll-like receptor 4 (TLR4)-mediated induction of MMP2 and 9 (100). This function of SAA in EVTs mimics very much the situation of tumor cells in metastasis. Both circulatory and local SAA levels have been shown to be increased in many tumors (101, 102), and SAA has been shown to aid in tumor cell invasion and metastasis by enhancing ECM degradation through induction of MMPs (49, 58, 103).
In addition to EVTs, we and others demonstrated that SAA was also synthesized in human placental villous trophoblasts (40, 100, 104, 105), and its expression was significantly increased upon syncytialization of trophoblasts (40). However, it appeared that SAA played no role in the syncytialization of villous trophoblasts. SAA treatment of BeWo cells (a trophoblast cell line) did not significantly increase the number of multinucleated syncytiotrophoblasts as observed with F-actin staining, and instead, SAA inhibited the secretion of hCG, a hormone synthesized primarily by syncytiotrophoblasts, in BeWo cells (100). However, SAA may participate in parturition by launching inflammatory reactions through sensing the inflammatory stimuli as well as by executing specific actions within gestational tissues.
As previously described, inflammation of gestational tissues is indispensable in parturition (1, 3, 106). Depending on the presence or absence of infection, inflammation of the gestational tissues can be classified into infectious and sterile inflammation (1, 3, 7, 106, 107). While infectious inflammation plays an important role in infection-induced preterm birth, sterile inflammation is more crucial in normal parturition. Although oxidative stress and cell senescence have been suggested to play a role in the initiation of sterile inflammation in parturition (7, 108), it remains elusive whether there exists any humoral factor involved in the initiation of sterile inflammation in gestational tissues. We found that SAA1 expression was significantly increased in the placenta villous tissue in normal spontaneous deliveries without infection at term compared with elective c section without labor at term (40). SAA1 in maternal blood was also significantly increased in infection-induced preterm birth compared with iatrogenic preterm birth without labor and histologic chorioamnionitis (40). Further investigation showed that inflammatory stimuli such as LPS and pro-inflammatory cytokines could stimulate the expression of SAA1 in placenta villous trophoblasts, and in turn, SAA1 increased the expression of inflammatory mediators including IL-1β, IL-8, TNF-α and cyclooxygenase 2 (COX-2) with concomitantly increased prostaglandin F2α (PGF2α) production (40), a major prostaglandin isoform synthesized by the placenta (109). Prostaglandins E2 (PGE2) and PGF2α are regarded as the final common mediators of labor onset in virtually all species, and they are not only potent stimulators of myometrial contraction, but also strong inducers of cervical ripening (109). Given the crucial role of prostaglandins and inflammation in parturition, we believe that SAA1 in the placenta is implicated in the creation of an inflammatory environment resulting in the increased prostaglandin production for parturition. Studies in the mouse support such a notion (40). SAA1 was found in the junctional zone of the mouse placenta, which was significantly increased by LPS administration. Moreover, intraperitoneal injection of SAA1 (8 μg/kg body weight) on gestational day 16.5 induced preterm birth with concurrently increased abundance of IL-1β, TNF-α, and COX-2 in the mouse placenta (40). These data gathered in the placenta suggest that SAA may be a humoral factor involved in the initiation of both sterile and infectious inflammation in gestational tissues in parturition.
In addition to inflammatory mediators, the stress hormone glucocorticoids also stimulated SAA1 expression in placental villous trophoblasts (40). The findings in the placenta are in line with the findings in the liver where glucocorticoids have been shown to stimulate SAA1 expression either on their own or in synergy with pro-inflammatory cytokines (24, 28). It is widely accepted that glucocorticoids play an important role in the initiation of parturition across different species including humans (110–114). Despite the differences in their origins and target issues, induction of COX-2 expression with concomitantly increased PGE2 and PGF2α production comprises one of the key effects of glucocorticoids in parturition in virtually all species (115–119). The finding that glucocorticoids stimulated the expression of SAA1 in human placental villous trophoblasts indicates that induction of SAA1 expression in gestational tissues may also play a role in glucocorticoid-induced parturition. In addition to the induction of SAA expression in the placenta, glucocorticoids also induce SAA expression either on their own or in synergy with SAA in the fetal membranes, which will be described in the following section.
4.2 Fetal membranes
As previously described, the fetal membranes make up the amniotic sac which provides protection for the fetus (120). Of the amnion and chorion layers, the amnion is known to be more tensile (114, 120–122). Toward the end of gestation, the amnion layer normally undergoes extensive extracellular matrix (ECM) remodeling as well as cell apoptosis and senescence in order to rupture in parturition (7, 108, 121, 122). However, premature preterm rupture of membranes may predispose the membranes to ascending infection resulting in the development of chorioamnionitis and preterm birth. In addition to the protective role described above, the fetal membranes are also known as a rich source of vital biochemicals which plays a pivotal role in parturition (108, 109, 114). Among these biochemicals, PGE2 and cortisol should be emphasized particularly (109). The fetal membranes not only produce the most PGE2 in pregnancy (109, 123, 124) but also possess the highest capacity of cortisol regeneration through 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) among fetal tissues (114, 125–127). Moreover, both PGE2 and cortisol can stimulate the expression of COX-2 and 11β-HSD1, the enzymes responsible for their respective production in the fetal membranes (115, 117–119, 128–131), thus forming a feedforward loop of mutual enhancement between cortisol and PGE2 production, which is believed to be a crucial feedforward mechanism underlying membrane activation at parturition (114, 125).
In addition to the identified biochemicals, we discovered that almost all cell types including amnion fibroblasts, epithelial cells and chorion trophoblasts in human fetal membranes are capable of de novo synthesis of SAA1 (41). Interestingly, by using cultured human amnion fibroblasts, a major site of cortisol regeneration and PGE2 production, we demonstrated that cortisol and SAA1 synergistically induced the expression of not only 11β-HSD1 but also SAA1 per se (132, 133), and in turn, SAA1 produced by fibroblasts might induce the expression of a number of inflammatory mediators including IL-1β, IL-6 as well as COX-2 with increased PGE2 production in fibroblasts in an autocrine or paracrine manner (41). Our data suggest that SAA1 may be part of the feedforward loop described above in membrane activation at parturition. Further investigation revealed that the synergistic induction of 11β-HSD1 and SAA1 expression by cortisol and SAA1 involved the participation of the transcription factor STAT3 in amnion fibroblasts (132). Of interest, STAT3 has been shown to mediate the transcription of several acute phase genes including SAA1 in the liver (134, 135). Previously, we also demonstrated that STAT3 was a key transcription factor mediating the induction of COX-2 expression by glucocorticoids in amnion fibroblasts (119). Given all these transcriptive effects in amnion fibroblasts, STAT3 appears to be a central transcription factor orchestrating the expression of multiple biochemicals of the feedforward loop in the activation of membranes at parturition (Figure 2).
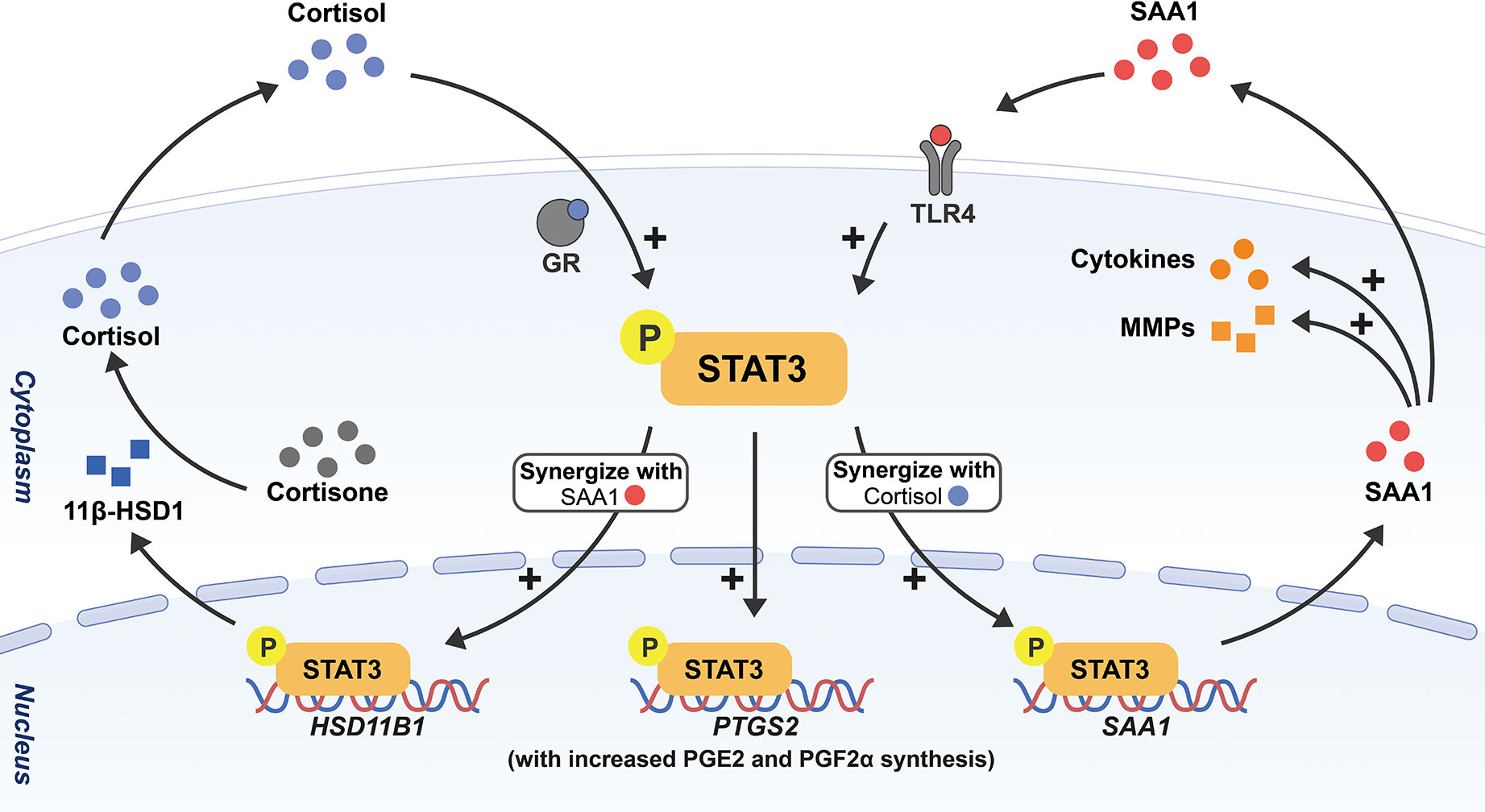
Figure 2 Schematic drawing depicting the feedforward loop among SAA, cortisol and prostaglandins in the activation of fetal membranes with STAT3 as a central transcription factor. Through phosphorylation of STAT3, cortisol and SAA1 can induce the expression of not only 11β-HSD1 but also SAA1 per se synergistically in amnion fibroblasts, and in turn, 11β-HSD1 converts biologically inactive cortisone to cortisol. Cortisol and SAA1 further stimulate the expression of COX-2 (encoded by PTGS2) with subsequent increased PGE2 and PGF2α synthesis. In addition, SAA1 also increases the expression of a number of pro-inflammatory cytokines and MMPs in amnion fibroblasts. All these effects will lead to membrane activation and ECM remodeling in parturition.
In preparation for membrane rupture, the amnion undergoes profound ECM remodeling. Interestingly, SAA synthesized locally in synovial cells was found to be a stimulator of the expression of collagenase and other MMPs, which might be responsible for joint destruction in rheumatism (51, 136, 137). We found that SAA1 was also involved in ECM remodeling in the amnion by inducing the expression of several MMPs including MMP-1, MMP-2, MMP-8, MMP-9 and MMP-13 (138, 139), and decreasing the expression of lysyl oxidase‐like 1 (LOXL1) (139), a collagen cross-linking enzyme, in amnion fibroblasts. In addition to the enhancement of enzymatic cleavage of collagens by MMPs, we also found that SAA1 induced autophagy-mediated collagen breakdown in amnion fibroblasts (138). These findings highlight that upon sensing danger signals, increased SAA synthesis locally in the amnion may participate in parturition by launching the feedforward loop of membrane activation as well as membrane rupture through multiple ECM remodeling approaches.
4.3 Uterus
It is within the uterus that the conception is established and the fetus develops. A successful implantation of the fertilized egg requires the proper timing of decidualization of the endometrium (140). Impairment of this process can lead to a series of pregnancy disorders (140–142). Recently, Goolam et al. demonstrated that SAA3 is robustly upregulated (272-fold) in the decidua of a mouse model with defective formation of primary decidual zone (143). In view of the role of SAA in the invasiveness of EVTs and in the inflammatory reactions of gestational tissues (40, 41, 100, 132, 139), it is possible that SAA synthesized locally at the fetal-maternal interface plays a crucial role in the fine regulation of inflammatory reactions and tissue remodeling for the establishment of pregnancy.
In addition to the endometrium, the myometrium also undergoes dramatic changes during pregnancy. The myometrium is distended and quiet during gestation, but becomes highly contractile in parturition. It has been suggested that inflammation may be involved in the myometrium transition from a quiescent to a contractile phenotype (6, 144). Jiang et al. demonstrated that SAA1 expression was significantly increased in human myometrium at term laboring compared to non-laboring (145), and they found that SAA1 stimulated the expression of pro-inflammatory cytokines (IL-8, IL-6), chemokines (CXCL5, CCL2), adhesion molecules (ICAM1, ICAM5) and PGE2 via activating the Yes-associated protein (YAP) pathway in human primary myometrial cells (145). These results suggest that SAA1 synthesized in the myometrium may help create an inflammatory microenvironment in the myometrium to assist its transition to the contractile phenotype in parturition.
Notably, almost all the inflammatory, endocrine and ECM remodeling effects of SAA1 in gestational tissues were mediated by the toll-like receptor TLR4 (40, 41, 100, 132, 138, 139). The toll-like receptors function as pattern recognition receptors (PRRs) in the detection of danger signals including a wide range of microbial products and host-derived DAMP proteins (146, 147). High-mobility group box 1 (HMGB1) is such a host-derived protein that interacts with multiple TLRs (148). Given the inflammatory properties of SAA depicted above, we believe that SAA may be another host-derived DAMP molecule which can be sensed by TLR4 in gestational tissues to initiate tissue-controlled inflammation and tissue remodeling at parturition. The idea is reinforced by the findings that the abundance of SAA1 was significantly increased in gestational tissues in parturition with or without infection, and upon stimulation by bacterial products, pro-inflammatory cytokines and stress hormone glucocorticoids (40, 41, 132), and in turn, SAA stimulated the production of pro-inflammatory mediators including SAA per se, tissue remodeling proteases MMPs as well as prostaglandins PGE2 and PGF2α, the common mediators of labor onset, in gestational tissues (40, 41, 100, 132, 138, 139).
5 Conclusions and perspectives
SAA is emerging as a host-derived DAMP molecule in the detection of either sterile or infectious signals in the initiation of inflammation in gestational tissues. SAA undergoes a modest rise during parturition but a dramatic rise in postpartum in maternal circulation, which may be a non-specific host innate immune response to the inflammation and trauma experienced during parturition. In gestational disorders such as PPROM, infection-induced preterm birth, pre-eclampsia, gestational diabetes and recurrent spontaneous abortion, maternal plasma SAA levels may increase non-specifically during pregnancy possibly due to the inflammatory nature of these diseases. Thus, SAA levels in maternal circulation may not be an ideal biomarker for specific pregnancy complications, but the extent of SAA elevation may reflect the severity of gestational disorders. Although the liver has been known to be a major source of plasma SAA in APR, cells in gestational tissues including villous and extravillous trophoblasts of the placenta, amnion epithelial and fibroblast cells, and chorion trophoblasts of the fetal membranes, decidual and myometrial cells of the uterus are all capable of SAA1 synthesis. SAA produced locally in gestational tissues may function as a TLR4-sensed DAMP molecule to be involved in the inflammatory reactions as well as specific actions associated with placentation, membrane rupture and initiation of parturition (Figure 3). However, the role of SAA in pregnancy is just starting to emerge. Several unresolved issues await further investigation. It is intriguing why our body needs two inducible SAAs and a constituent SAA4. It is necessary to decipher whether these isoforms play any differential roles in pregnancy in the future. Moreover, thorough investigations are required to understand the exact role of SAA in normal pregnancy, particularly in postpartum and gestational disorders. We believe that in-depth investigation of SAA in pregnancy may help better understand how inflammation is initiated in gestational tissues in both normal and abnormal pregnancies.
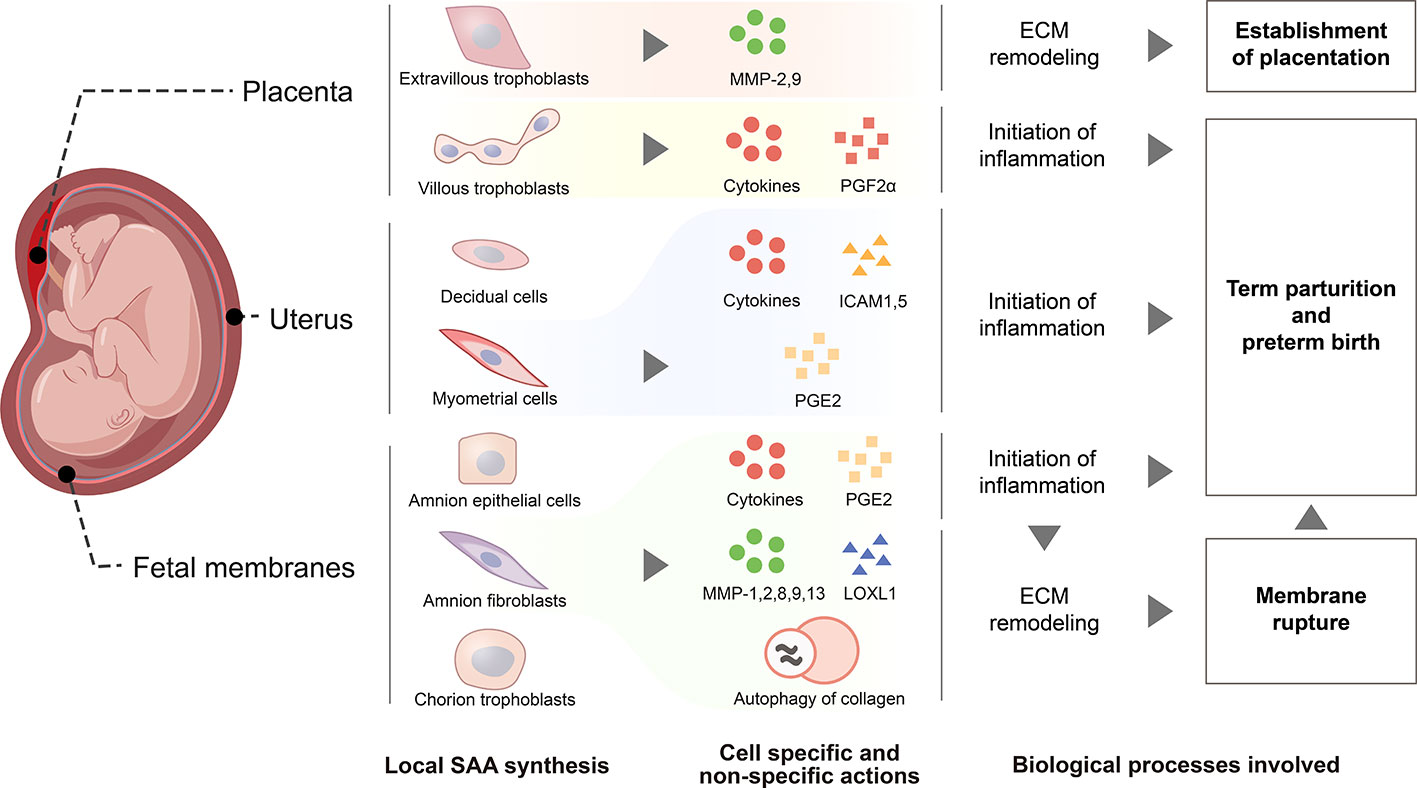
Figure 3 Schematic drawing depicting functions of SAA synthesized within gestational tissues. De novo SAA synthesis is detected in extravillous trophoblasts and syncytiotrophoblasts of the placenta, myometrial and decidual cells of the uterus, epithelial and fibroblast cells of the amnion, cytotrophoblasts of the chorionic membrane. In extravillous trophoblasts, SAA may promote their invasion into the endometrium through induction of metalloproteases 2 and 9 for the establishment of placentation. SAA synthesized within myometrium, placenta and fetal membranes may promote parturition via enhancing inflammatory reactions through induction of pro-inflammatory cytokines and other inflammatory mediators including prostaglandins PGF2α and PGE2, ICAM1 and ICAM5, the adhesion molecules for the infiltrated leukocytes. Moreover, SAA may also facilitate fetal membrane rupture in parturition through induction of extracellular matrix remodeling via increasing the expression of metalloproteases 1, 2, 8, 9 and 13, decreasing the expression of lysyl oxidase‐like 1 and inducing autophagy-mediated collagen breakdown.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by National Natural Science Foundation of China (81830042 and 82071677), National Key R & D Program of China (2020YFA0803900) and Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZLCX20210200).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Challis JR, Lockwood CJ, Myatt L, Norman JE, Strauss JF 3rd, Petraglia F. Inflammation and pregnancy. Reprod Sci (2009) 16(2):206–15. doi: 10.1177/1933719108329095
2. Jabbour HN, Sales KJ, Catalano RD, Norman JE. Inflammatory pathways in female reproductive health and disease. Reproduction (2009) 138(6):903–19. doi: 10.1530/REP-09-0247
3. Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel LA, Nien JK. Inflammation in preterm and term labour and delivery. Semin Fetal Neonatal Med (2006) 11(5):317–26. doi: 10.1016/j.siny.2006.05.001
4. Romero R, Espinoza J, Kusanovic JP, Gotsch F, Hassan S, Erez O, et al. The preterm parturition syndrome. BJOG (2006) 113 Suppl 3:17–42. doi: 10.1111/j.1471-0528.2006.01120.x
5. Keelan JA, Blumenstein M, Helliwell RJ, Sato TA, Marvin KW, Mitchell MD. Cytokines, prostaglandins and parturition–a review. Placenta (2003) 24 Suppl A:S33–46. doi: 10.1053/plac.2002.0948
6. Osman I, Young A, Ledingham MA, Thomson AJ, Jordan F, Greer IA, et al. Leukocyte density and pro-inflammatory cytokine expression in human fetal membranes, decidua, cervix and myometrium before and during labour at term. Mol Hum Reprod (2003) 9(1):41–5. doi: 10.1093/molehr/gag001
7. Menon R, Behnia F, Polettini J, Richardson LS. Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes. Semin Immunopathol (2020) 42(4):431–50. doi: 10.1007/s00281-020-00808-x
8. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw (2018) 18(4):e27. doi: 10.4110/in.2018.18.e27
10. Baumann H, Gauldie J. The acute phase response. Immunol Today (1994) 15(2):74–80. doi: 10.1016/0167-5699(94)90137-6
11. Jain S, Gautam V, Naseem S. Acute-phase proteins: As diagnostic tool. J Pharm Bioallied Sci (2011) 3(1):118–27. doi: 10.4103/0975-7406.76489
12. Herpers BL, Endeman H, de Jong BA, de Jongh BM, Grutters JC, Biesma DH, et al. Acute-phase responsiveness of mannose-binding lectin in community-acquired pneumonia is highly dependent upon MBL2 genotypes. Clin Exp Immunol (2009) 156(3):488–94. doi: 10.1111/j.1365-2249.2009.03929.x
15. Clyne B, Olshaker JS. The c-reactive protein. J Emerg Med (1999) 17(6):1019–25. doi: 10.1016/S0736-4679(99)00135-3
16. Kisilevsky R, Manley PN. Acute-phase serum amyloid a: perspectives on its physiological and pathological roles. Amyloid (2012) 19(1):5–14. doi: 10.3109/13506129.2011.654294
17. Lu J, Yu Y, Zhu I, Cheng Y, Sun PD. Structural mechanism of serum amyloid a-mediated inflammatory amyloidosis. Proc Natl Acad Sci U S A (2014) 111(14):5189–94. doi: 10.1073/pnas.1322357111
18. Rosenthal CJ, Franklin EC. Variation with age and disease of an amyloid a protein-related serum component. J Clin Invest (1975) 55(4):746–53. doi: 10.1172/JCI107985
19. Watson G, See CG, Woo P. Use of somatic cell hybrids and fluorescence in situ hybridization to localize the functional serum amyloid a (SAA) genes to chromosome 11p15.4-p15.1 and the entire SAA superfamily to chromosome 11p15. Genomics (1994) 23(3):694–6. doi: 10.1006/geno.1994.1559
20. Sun L, Ye RD. Serum amyloid A1: Structure, function and gene polymorphism. Gene (2016) 583(1):48–57. doi: 10.1016/j.gene.2016.02.044
21. Kluve-Beckerman B, Drumm ML, Benson MD. Nonexpression of the human serum amyloid a three (SAA3) gene. DNA Cell Biol (1991) 10(9):651–61. doi: 10.1089/dna.1991.10.651
22. Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid a, a major acute-phase protein, in normal and disease states. Curr Opin Hematol (2000) 7(1):64–9. doi: 10.1097/00062752-200001000-00012
23. Edbrooke MR, Burt DW, Cheshire JK, Woo P. Identification of cis-acting sequences responsible for phorbol ester induction of human serum amyloid a gene expression via a nuclear factor kappaB-like transcription factor. Mol Cell Biol (1989) 9(5):1908–16. doi: 10.1128/mcb.9.5.1908-1916.1989
24. Thorn CF, Lu ZY, Whitehead AS. Regulation of the human acute phase serum amyloid a genes by tumour necrosis factor-alpha, interleukin-6 and glucocorticoids in hepatic and epithelial cell lines. Scand J Immunol (2004) 59(2):152–8. doi: 10.1111/j.0300-9475.2004.01369.x
25. Betts JC, Cheshire JK, Akira S, Kishimoto T, Woo P. The role of NF-kappa b and NF-IL6 transactivating factors in the synergistic activation of human serum amyloid a gene expression by interleukin-1 and interleukin-6. J Biol Chem (1993) 268(34):25624–31. doi: 10.1016/S0021-9258(19)74435-4
26. Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, Yoshizaki K. IL-6 plays a critical role in the synergistic induction of human serum amyloid a (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem Biophys Res Commun (2004) 314(2):363–9. doi: 10.1016/j.bbrc.2003.12.096
27. Maury CP, Enholm E, Teppo AM. Is interferon an "inducer" of serum amyloid a? N Engl J Med (1983) 309(17):1060–1. doi: 10.1056/NEJM198310273091715
28. Thorn CF, Lu ZY, Whitehead AS. Tissue-specific regulation of the human acute-phase serum amyloid a genes, SAA1 and SAA2, by glucocorticoids in hepatic and epithelial cells. Eur J Immunol (2003) 33(9):2630–9. doi: 10.1002/eji.200323985
29. Thorn CF, Whitehead AS. Differential glucocorticoid enhancement of the cytokine-driven transcriptional activation of the human acute phase serum amyloid a genes, SAA1 and SAA2. J Immunol (2002) 169(1):399–406. doi: 10.4049/jimmunol.169.1.399
30. Husby G, Marhaug G, Sletten K. Amyloid a in systemic amyloidosis associated with cancer. Cancer Res (1982) 42(4):1600–3.
31. Benditt EP, Eriksen N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc Natl Acad Sci U S A (1977) 74(9):4025–8. doi: 10.1073/pnas.74.9.4025
32. Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid a-containing human high density lipoprotein 3. density, size, and apolipoprotein composition. J Biol Chem (1986) 261(21):9644–51.
33. Van der Westhuyzen D, Coetzee G, De Beer F. Serum amyloid a protein in plasma: characteristics of acute phase HDL. In: Marrink J, van Rijswijk MH, editors. Amyloidosis, vol. p . Dordrecht: Springer (1986). p. 115–25.
34. Zhu S, Wang Y, Chen W, Li W, Wang A, Wong S, et al. High-density lipoprotein (HDL) counter-regulates serum amyloid a (SAA)-induced sPLA2-IIE and sPLA2-V expression in macrophages. PloS One (2016) 11(11):e0167468. doi: 10.1371/journal.pone.0167468
35. Cai H, Song C, Endoh I, Goyette J, Jessup W, Freedman SB, et al. Serum amyloid a induces monocyte tissue factor. J Immunol (2007) 178(3):1852–60. doi: 10.4049/jimmunol.178.3.1852
36. Getz GS, Reardon CA. SAA. HDL biogenesis, and inflammation. J Lipid Res (2008) 49(2):269–70. doi: 10.1194/jlr.E700012-JLR200
37. Kisilevsky R, Tam SP. Macrophage cholesterol efflux and the active domains of serum amyloid a 2.1. J Lipid Res (2003) 44(12):2257–69. doi: 10.1194/jlr.M300133-JLR200
38. Kisilevsky R, Subrahmanyan L. Serum amyloid a changes high density lipoprotein's cellular affinity. a clue to serum amyloid a's principal function. Lab Invest (1992) 66(6):778–85.
39. Urieli-Shoval S, Cohen P, Eisenberg S, Matzner Y. Widespread expression of serum amyloid a in histologically normal human tissues. predominant localization to the epithelium. J Histochem Cytochem (1998) 46(12):1377–84. doi: 10.1177/002215549804601206
40. Gan XW, Wang WS, Lu JW, Ling LJ, Zhou Q, Zhang HJ, et al. De novo synthesis of SAA1 in the placenta participates in parturition. Front Immunol (2020) 11:1038. doi: 10.3389/fimmu.2020.01038
41. Li W, Wang W, Zuo R, Liu C, Shu Q, Ying H, et al. Induction of pro-inflammatory genes by serum amyloid A1 in human amnion fibroblasts. Sci Rep (2017) 7(1):693. doi: 10.1038/s41598-017-00782-9
42. Cunnane G, Whitehead AS. Amyloid precursors and amyloidosis in rheumatoid arthritis. Baillieres Best Pract Res Clin Rheumatol (1999) 13(4):615–28. doi: 10.1053/berh.1999.0049
43. Fyfe AI, Rothenberg LS, DeBeer FC, Cantor RM, Rotter JI, Lusis AJ. Association between serum amyloid a proteins and coronary artery disease: evidence from two distinct arteriosclerotic processes. Circulation (1997) 96(9):2914–9. doi: 10.1161/01.CIR.96.9.2914
44. Duff GW. Cytokines and acute phase proteins in rheumatoid arthritis. Scand J Rheumatol Suppl. (1994) 100:9–19. doi: 10.3109/03009749409095197
45. Smole U, Gour N, Phelan J, Hofer G, Kohler C, Kratzer B, et al. Serum amyloid a is a soluble pattern recognition receptor that drives type 2 immunity. Nat Immunol (2020) 21(7):756–65. doi: 10.1038/s41590-020-0698-1
46. Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, et al. Serum amyloid a is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med (1994) 180(1):203–9. doi: 10.1084/jem.180.1.203
47. Xu L, Badolato R, Murphy WJ, Longo DL, Anver M, Hale S, et al. A novel biologic function of serum amyloid a. induction of T lymphocyte migration and adhesion. J Immunol (1995) 155(3):1184–90.
48. Olsson N, Siegbahn A, Nilsson G. Serum amyloid a induces chemotaxis of human mast cells by activating a pertussis toxin-sensitive signal transduction pathway. Biochem Biophys Res Commun (1999) 254(1):143–6. doi: 10.1006/bbrc.1998.9911
49. Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B, et al. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene (2015) 34(4):424–35. doi: 10.1038/onc.2013.568
50. Vallon R, Freuler F, Desta-Tsedu N, Robeva A, Dawson J, Wenner P, et al. (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol (2001) 166(4):2801–7. doi: 10.4049/jimmunol.166.4.2801
51. Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid a protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest (1998) 78(5):535–9.
52. Ye RD, Sun L. Emerging functions of serum amyloid a in inflammation. J Leukoc Biol (2015) 98(6):923–9. doi: 10.1189/jlb.3VMR0315-080R
53. Li H, Zhao Y, Zhou S, Heng CK. Serum amyloid a activates peroxisome proliferator-activated receptor gamma through extracellularly regulated kinase 1/2 and COX-2 expression in hepatocytes. Biochemistry (2010) 49(44):9508–17. doi: 10.1021/bi100645m
54. Shah C, Hari-Dass R, Raynes JG. Serum amyloid a is an innate immune opsonin for gram-negative bacteria. Blood (2006) 108(5):1751–7. doi: 10.1182/blood-2005-11-011932
55. Gaiser AK, Bauer S, Ruez S, Holzmann K, Fandrich M, Syrovets T, et al. Serum amyloid A1 induces classically activated macrophages: A role for enhanced fibril formation. Front Immunol (2021) 12:691155. doi: 10.3389/fimmu.2021.691155
56. Sun L, Zhou H, Zhu Z, Yan Q, Wang L, Liang Q, et al. Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J Immunol (2015) 194(10):4891–900. doi: 10.4049/jimmunol.1402164
57. Wang Y, Huang H, Sun R, Chen B, Han F, Li Q, et al. Serum amyloid a induces M2b-like macrophage polarization during liver inflammation. Oncotarget (2017) 8(65):109238–46. doi: 10.18632/oncotarget.22652
58. Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JI, et al. Serum amyloid a stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem Biophys Res Commun (2005) 330(3):989–98. doi: 10.1016/j.bbrc.2005.03.069
59. He R, Sang H, Ye RD. Serum amyloid a induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood (2003) 101(4):1572–81. doi: 10.1182/blood-2002-05-1431
60. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid a. J Immunol (2008) 181(1):22–6. doi: 10.4049/jimmunol.181.1.22
61. Chen M, Zhou H, Cheng N, Qian F, Ye RD. Serum amyloid A1 isoforms display different efficacy at toll-like receptor 2 and formyl peptide receptor 2. Immunobiology (2014) 219(12):916–23. doi: 10.1016/j.imbio.2014.08.002
62. Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, et al. CD36 is a novel serum amyloid a (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem (2010) 285(11):8492–506. doi: 10.1074/jbc.M109.007526
63. Ebert R, Benisch P, Krug M, Zeck S, Meissner-Weigl J, Steinert A, et al. Acute phase serum amyloid a induces proinflammatory cytokines and mineralization via toll-like receptor 4 in mesenchymal stem cells. Stem Cell Res (2015) 15(1):231–9. doi: 10.1016/j.scr.2015.06.008
64. Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, et al. Serum amyloid a binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid a protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem (2005) 280(9):8031–40. doi: 10.1074/jbc.M405009200
65. Calvo D, Gomez-Coronado D, Lasuncion MA, Vega MA. CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler Thromb Vasc Biol (1997) 17(11):2341–9. doi: 10.1161/01.ATV.17.11.2341
66. Pantham P, Aye IL, Powell TL. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta (2015) 36(7):709–15. doi: 10.1016/j.placenta.2015.04.006
67. de Mendonca E, Fragoso MBT, de Oliveira JM, Xavier JA, Goulart MOF, de Oliveira ACM. Gestational diabetes mellitus: The crosslink among inflammation, nitroxidative stress, intestinal microbiota and alternative therapies. Antioxidants (Basel) (2022) 11(1):129. doi: 10.3390/antiox11010129
68. Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol (1998) 179(1):80–6. doi: 10.1016/S0002-9378(98)70254-6
69. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol (1999) 180(2 Pt 1):499–506. doi: 10.1016/S0002-9378(99)70239-5
70. Ostensen M, Marhaug G, Husby G. Amyloid-related serum protein (SAA) during and after pregnancy in healthy women and women with rheumatic disease. Acta Pathol Microbiol Immunol Scand C (1985) 93(1):1–5. doi: 10.1111/j.1699-0463.1985.tb02913.x
71. de Villiers WJ, Louw JP, Strachan AF, Etsebeth SM, Shephard EG, de Beer FC. C-reactive protein and serum amyloid a protein in pregnancy and labour. Br J Obstet Gynaecol (1990) 97(8):725–30. doi: 10.1111/j.1471-0528.1990.tb16247.x
72. Cicarelli LM, Perroni AG, Zugaib M, de Albuquerque PB, Campa A. Maternal and cord blood levels of serum amyloid a, c-reactive protein, tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 during and after delivery. Mediators Inflamm (2005) 2005(2):96–100. doi: 10.1155/MI.2005.96
73. Ohlsson A. Treatments of preterm premature rupture of the membranes: a meta-analysis. Am J Obstet Gynecol (1989) 160(4):890–906. doi: 10.1016/0002-9378(89)90306-2
74. Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am J Obstet Gynecol (2015) 213(4 Suppl):S29–52. doi: 10.1016/j.ajog.2015.08.040
75. Yang Q, Whitin JC, Ling XB, Nayak NR, Cohen HJ, Jin J, et al. Plasma biomarkers in a mouse model of preterm labor. Pediatr Res (2009) 66(1):11–6. doi: 10.1203/PDR.0b013e3181a207e3
76. Koseoglu SB, Guzel AI, Deveer R, Tokmak A, Engin-Ustun Y, Ozdas S, et al. Maternal serum amyloid a levels in pregnancies complicated with preterm prelabour rupture of membranes. Ginekol Pol (2014) 85(7):516–20. doi: 10.17772/gp/1763
77. Kayabas H, Bildircin FD, Avci B, Karli P, Ozdemir AZ, Alper T. Serum amyloid-a may be an early marker in diagnosis of preterm premature rupture of membrane and chorioamnionitis. Ann Med Res (2019) 26(12):2802–6. doi: 10.5455/annalsmedres.2019.11.751
79. Lim KH, Zhou Y, Janatpour M, McMaster M, Bass K, Chun SH, et al. Human cytotrophoblast differentiation/invasion is abnormal in pre-eclampsia. Am J Pathol (1997) 151(6):1809–18.
80. Roberts JM, Taylor RN, Musci TJ, Rodgers GM, Hubel CA, McLaughlin MK. Preeclampsia: an endothelial cell disorder. Am J Obstet Gynecol (1989) 161(5):1200–4. doi: 10.1016/0002-9378(89)90665-0
81. Engin-Ustun Y, Ustun Y, Karabulut AB, Ozkaplan E, Meydanli MM, Kafkasli A. Serum amyloid a levels are increased in pre-eclampsia. Gynecol Obstet Invest (2007) 64(2):117–20. doi: 10.1159/000100329
82. Uckan K, Sahin HG. Serum amyloid a, procalcitonin, highly sensitive c reactive protein and tumor necrosis factor alpha levels and acute inflammatory response in patients with hemolysis, elevated liver enzymes, low platelet count (HELLP) and eclampsia. J Obstet Gynaecol Res (2018) 44(3):440–7. doi: 10.1111/jog.13532
83. Heitner JC, Koy C, Kreutzer M, Gerber B, Reimer T, Glocker MO. Differentiation of HELLP patients from healthy pregnant women by proteome analysis–on the way towards a clinical marker set. J Chromatogr B Analyt Technol BioMed Life Sci (2006) 840(1):10–9. doi: 10.1016/j.jchromb.2006.06.002
84. Kristensen K, Wide-Swensson D, Lindstrom V, Schmidt C, Grubb A, Strevens H. Serum amyloid a protein and c-reactive protein in normal pregnancy and preeclampsia. Gynecol Obstet Invest (2009) 67(4):275–80. doi: 10.1159/000214081
85. Zakaria El-Sheikha K, Abd El-Hamed Sedek AE-M, Abdul-Hameid Abo Zeid A, Hassan Mohamed El-Shayeb A. Serum amyloid a as a novel marker to morbidly adherent placenta. Al Azhar Med J (2020) 49(2):437–46. doi: 10.21608/amj.2020.68555
86. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol (2012) 8(11):639–49. doi: 10.1038/nrendo.2012.96
87. Eren MA, Vural M, Cece H, Camuzcuoglu H, Yildiz S, Toy H, et al. Association of serum amyloid a with subclinical atherosclerosis in women with gestational diabetes. Gynecol Endocrinol (2012) 28(12):1010–3. doi: 10.3109/09513590.2012.705371
88. Poyhonen-Alho M, Ebeling P, Saarinen A, Kaaja R. Decreased variation of inflammatory markers in gestational diabetes. Diabetes Metab Res Rev (2011) 27(3):269–76. doi: 10.1002/dmrr.1170
89. Rota S, Yildirim B, Kaleli B, Aybek H, Duman K, Kaptanoglu B. C-reactive protein levels in non-obese pregnant women with gestational diabetes. Tohoku J Exp Med (2005) 206(4):341–5. doi: 10.1620/tjem.206.341
90. Leipold H, Worda C, Gruber CJ, Prikoszovich T, Wagner O, Kautzky-Willer A. Gestational diabetes mellitus is associated with increased c-reactive protein concentrations in the third but not second trimester. Eur J Clin Invest (2005) 35(12):752–7. doi: 10.1111/j.1365-2362.2005.01574.x
91. Stirrat GM. Recurrent miscarriage I: definition and epidemiology. Lancet (1990) 336(8716):673–5. doi: 10.1016/0140-6736(90)92159-F
92. Quenby S, Gallos ID, Dhillon-Smith RK, Podesek M, Stephenson MD, Fisher J, et al. Miscarriage matters: the epidemiological, physical, psychological, and economic costs of early pregnancy loss. Lancet (2021) 397(10285):1658–67. doi: 10.1016/S0140-6736(21)00682-6
93. Jaslow CR, Carney JL, Kutteh WH. Diagnostic factors identified in 1020 women with two versus three or more recurrent pregnancy losses. Fertil Steril (2010) 93(4):1234–43. doi: 10.1016/j.fertnstert.2009.01.166
94. Stephenson MD. Frequency of factors associated with habitual abortion in 197 couples. Fertil Steril (1996) 66(1):24–9.
95. Ibrahim MI, Ramy AR, Abdelhamid AS, Ellaithy MI, Omar A, Harara RM, et al. Maternal serum amyloid a level as a novel marker of primary unexplained recurrent early pregnancy loss. Int J Gynaecol Obstet (2017) 136(3):298–303. doi: 10.1002/ijgo.12076
96. Ming N, Liu Z, Wu H, Zhang G, Liu L. Expression and significance of serum interferon-γ, serum amyloid a protein and soluble fms-like tyrosine kinase-1 in patients with unexplained recurrent spontaneous abortion. J Clin Med Pract (2022) 26(9):25–8. doi: 10.7619/jcmp.20220861
97. Jansson T. Placenta plays a critical role in maternal-fetal resource allocation. Proc Natl Acad Sci USA (2016) 113(40):11066–8. doi: 10.1073/pnas.1613437113
98. Nelson DM, Kay H, Wang Y. The placenta: From development to disease. Hoboken (NJ): John Wiley & Sons (2011).
99. Menkhorst E, Winship A, Van Sinderen M, Dimitriadis E. Human extravillous trophoblast invasion: intrinsic and extrinsic regulation. Reprod Fertil Dev (2016) 28(4):406–15. doi: 10.1071/RD14208
100. Sandri S, Urban Borbely A, Fernandes I, de Oliveira EM, Knebel FH, Ruano R, et al. Serum amyloid a in the placenta and its role in trophoblast invasion. PloS One (2014) 9(3):e90881. doi: 10.1371/journal.pone.0090881
101. Rosenthal CJ, Sullivan LM. Serum amyloid a to monitor cancer dissemination. Ann Intern Med (1979) 91(3):383–90. doi: 10.7326/0003-4819-91-3-383
102. Biran H, Friedman N, Neumann L, Pras M, Shainkin-Kestenbaum R. Serum amyloid a (SAA) variations in patients with cancer: correlation with disease activity, stage, primary site, and prognosis. J Clin Pathol (1986) 39(7):794–7. doi: 10.1136/jcp.39.7.794
103. Paret C, Schon Z, Szponar A, Kovacs G. Inflammatory protein serum amyloid A1 marks a subset of conventional renal cell carcinomas with fatal outcome. Eur Urol (2010) 57(5):859–66. doi: 10.1016/j.eururo.2009.08.014
104. Kovacevic A, Hammer A, Sundl M, Pfister B, Hrzenjak A, Ray A, et al. Expression of serum amyloid a transcripts in human trophoblast and fetal-derived trophoblast-like choriocarcinoma cells. FEBS Lett (2006) 580(1):161–7. doi: 10.1016/j.febslet.2005.11.067
105. Rossmann C, Hammer A, Koyani CN, Kovacevic A, Siwetz M, Desoye G, et al. Expression of serum amyloid A4 in human trophoblast-like choriocarcinoma cell lines and human first trimester/term trophoblast cells. Placenta (2014) 35(8):661–4. doi: 10.1016/j.placenta.2014.05.012
106. Bukowski R, Sadovsky Y, Goodarzi H, Zhang H, Biggio JR, Varner M, et al. Onset of human preterm and term birth is related to unique inflammatory transcriptome profiles at the maternal fetal interface. PeerJ (2017) 5:e3685. doi: 10.7717/peerj.3685
107. Nadeau-Vallee M, Obari D, Palacios J, Brien ME, Duval C, Chemtob S, et al. Sterile inflammation and pregnancy complications: a review. Reproduction (2016) 152(6):R277–R92. doi: 10.1530/REP-16-0453
108. Menon R. Human fetal membranes at term: Dead tissue or signalers of parturition? Placenta (2016) 44:1–5. doi: 10.1016/j.placenta.2016.05.013
109. Li WJ, Lu JW, Zhang CY, Wang WS, Ying H, Myatt L, et al. PGE2 vs PGF2alpha in human parturition. Placenta (2021) 104:208–19. doi: 10.1016/j.placenta.2020.12.012
110. Jenkin G, Young IR. Mechanisms responsible for parturition; the use of experimental models. Anim Reprod Sci (2004) 82-83:567–81. doi: 10.1016/j.anireprosci.2004.05.010
111. Liggins GC, Forster CS, Grieves SA, Schwartz AL. Control of parturition in man. Biol Reprod (1977) 16(1):39–56. doi: 10.1095/biolreprod16.1.39
112. Lanman JT. Parturition in nonhuman primates. Biol Reprod (1977) 16(1):28–38. doi: 10.1093/biolreprod/16.1.28
113. Thorburn GD, Challis JR, Currie WB. Control of parturition in domestic animals. Biol Reprod (1977) 16(1):18–27. doi: 10.1095/biolreprod16.1.18
114. Wang W, Chen ZJ, Myatt L, Sun K. 11beta-HSD1 in human fetal membranes as a potential therapeutic target for preterm birth. Endocr Rev (2018) 39(3):241–60. doi: 10.1210/er.2017-00188
115. Casey ML, MacDonald PC, Mitchell MD. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis. a paradox? J Clin Invest (1985) 75(6):1852–7. doi: 10.1172/JCI111899
116. Whittle WL, Patel FA, Alfaidy N, Holloway AC, Fraser M, Gyomorey S, et al. Glucocorticoid regulation of human and ovine parturition: the relationship between fetal hypothalamic-pituitary-adrenal axis activation and intrauterine prostaglandin production. Biol Reprod (2001) 64(4):1019–32. doi: 10.1095/biolreprod64.4.1019
117. Sun K, Ma R, Cui X, Campos B, Webster R, Brockman D, et al. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin h synthase type 2 but not microsomal prostaglandin e synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J Clin Endocrinol Metab (2003) 88(11):5564–71. doi: 10.1210/jc.2003-030875
118. Lu J, Wang W, Mi Y, Zhang C, Ying H, Wang L, et al. AKAP95-mediated nuclear anchoring of PKA mediates cortisol-induced PTGS2 expression in human amnion fibroblasts. Sci Signal (2017) 10(506):eaac6160. doi: 10.1126/scisignal.aac6160
119. Wang W, Guo C, Zhu P, Lu J, Li W, Liu C, et al. Phosphorylation of STAT3 mediates the induction of cyclooxygenase-2 by cortisol in the human amnion at parturition. Sci Signal (2015) 8(400):ra106. doi: 10.1126/scisignal.aac6151
120. Myatt L, Sun K. Role of fetal membranes in signaling of fetal maturation and parturition. Int J Dev Biol (2010) 54(2-3):545–53. doi: 10.1387/ijdb.082771lm
121. Parry S, Strauss JF 3rd. Premature rupture of the fetal membranes. N Engl J Med (1998) 338(10):663–70. doi: 10.1056/NEJM199803053381006
122. Oyen ML, Calvin SE, Landers DV. Premature rupture of the fetal membranes: is the amnion the major determinant? Am J Obstet Gynecol (2006) 195(2):510–5. doi: 10.1016/j.ajog.2006.02.010
123. Duchesne MJ, Thaler-Dao H, de Paulet AC. Prostaglandin synthesis in human placenta and fetal membranes. Prostaglandins (1978) 15(1):19–42. doi: 10.1016/S0090-6980(78)80003-3
124. Mitchell MD, Bibby J, Hicks BR, Turnbull AC. Specific production of prostaglandin e by human amnion in vitro. Prostaglandins (1978) 15(2):377–82. doi: 10.1016/0090-6980(78)90177-6
125. Wang WS, Guo CM, Sun K. Cortisol regeneration in the fetal membranes, a coincidental or requisite event in human parturition? Front Physiol (2020) 11:462. doi: 10.3389/fphys.2020.00462
126. Murphy BE. Ontogeny of cortisol-cortisone interconversion in human tissues: a role for cortisone in human fetal development. J Steroid Biochem (1981) 14(9):811–7. doi: 10.1016/0022-4731(81)90226-0
127. Murphy BE. Chorionic membrane as an extra-adrenal source of foetal cortisol in human amniotic fluid. Nature (1977) 266(5598):179–81. doi: 10.1038/266179a0
128. Lu JW, Wang WS, Zhou Q, Ling LJ, Ying H, Sun Y, et al. C/EBPdelta drives key endocrine signals in the human amnion at parturition. Clin Transl Med (2021) 11(6):e416. doi: 10.1002/ctm2.416
129. Sun K, Myatt L. Enhancement of glucocorticoid-induced 11beta-hydroxysteroid dehydrogenase type 1 expression by proinflammatory cytokines in cultured human amnion fibroblasts. Endocrinology (2003) 144(12):5568–77. doi: 10.1210/en.2003-0780
130. Lu JW, Wang WS, Zhou Q, Gan XW, Myatt L, Sun K. Activation of prostaglandin EP4 receptor attenuates the induction of cyclooxygenase-2 expression by EP2 receptor activation in human amnion fibroblasts: implications for parturition. FASEB J (2019) 33(7):8148–60. doi: 10.1096/fj.201802642R
131. Li W, Gao L, Wang Y, Duan T, Myatt L, Sun K. Enhancement of cortisol-induced 11beta-hydroxysteroid dehydrogenase type 1 expression by interleukin 1beta in cultured human chorionic trophoblast cells. Endocrinology (2006) 147(5):2490–5. doi: 10.1210/en.2005-1626
132. Lu Y, Wang WS, Lin YK, Lu JW, Li WJ, Zhang CY, et al. Enhancement of cortisol-induced SAA1 transcription by SAA1 in the human amnion. J Mol Endocrinol (2019) 62(4):149–58. doi: 10.1530/JME-18-0263
133. Lu Y, Zhou Q, Lu JW, Wang WS, Sun K. Involvement of STAT3 in the synergistic induction of 11beta-HSD1 by SAA1 and cortisol in human amnion fibroblasts. Am J Reprod Immunol (2019) 82(2):e13150. doi: 10.1111/aji.13150
134. Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Poli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol (2001) 21(5):1621–32. doi: 10.1128/MCB.21.5.1621-1632.2001
135. Ochrietor JD, Harrison KA, Zahedi K, Mortensen RF. Role of STAT3 and C/EBP in cytokine-dependent expression of the mouse serum amyloid p-component (SAP) and c-reactive protein (CRP) genes. Cytokine (2000) 12(7):888–99. doi: 10.1006/cyto.2000.0668
136. Kumon Y, Suehiro T, Hashimoto K, Nakatani K, Sipe JD. Local expression of acute phase serum amyloid a mRNA in rheumatoid arthritis synovial tissue and cells. J Rheumatol (1999) 26(4):785–90.
137. Brinckerhoff CE, Mitchell TI, Karmilowicz MJ, Kluve-Beckerman B, Benson MD. Autocrine induction of collagenase by serum amyloid a-like and beta 2-microglobulin-like proteins. Science (1989) 243(4891):655–7. doi: 10.1126/science.2536953
138. Wang WS, Li WJ, Wang YW, Wang LY, Mi YB, Lu JW, et al. Involvement of serum amyloid A1 in the rupture of fetal membranes through induction of collagen I degradation. Clin Sci (Lond) (2019) 133(3):515–30. doi: 10.1042/CS20180950
139. Wang YW, Wang WS, Wang LY, Bao YR, Lu JW, Lu Y, et al. Extracellular matrix remodeling effects of serum amyloid A1 in the human amnion: Implications for fetal membrane rupture. Am J Reprod Immunol (2019) 81(1):e13073. doi: 10.1111/aji.13073
140. Evans J, Salamonsen LA, Winship A, Menkhorst E, Nie G, Gargett CE, et al. Fertile ground: human endometrial programming and lessons in health and disease. Nat Rev Endocrinol (2016) 12(11):654–67. doi: 10.1038/nrendo.2016.116
141. Garrido-Gomez T, Dominguez F, Quinonero A, Diaz-Gimeno P, Kapidzic M, Gormley M, et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc Natl Acad Sci U S A (2017) 114(40):E8468–E77. doi: 10.1073/pnas.1706546114
142. Wu D, Kimura F, Zheng L, Ishida M, Niwa Y, Hirata K, et al. Chronic endometritis modifies decidualization in human endometrial stromal cells. Reprod Biol Endocrinol (2017) 15(1):16. doi: 10.1186/s12958-017-0233-x
143. Goolam M, Xypolita ME, Costello I, Lydon JP, DeMayo FJ, Bikoff EK, et al. The transcriptional repressor Blimp1/PRDM1 regulates the maternal decidual response in mice. Nat Commun (2020) 11(1):2782. doi: 10.1038/s41467-020-16603-z
144. Thomson AJ, Telfer JF, Young A, Campbell S, Stewart CJ, Cameron IT, et al. Leukocytes infiltrate the myometrium during human parturition: further evidence that labour is an inflammatory process. Hum Reprod (1999) 14(1):229–36. doi: 10.1093/humrep/14.1.229
145. Jiang Y, Pin L, Shi W, Huang Q, Wang L, Liu H. SAA1 regulates pro-labour mediators in term labour by activating YAP pathway. Mol Cell Biochem (2021) 476(7):2791–801. doi: 10.1007/s11010-021-04125-1
146. Arpaia N, Barton GM. Toll-like receptors: key players in antiviral immunity. Curr Opin Virol (2011) 1(6):447–54. doi: 10.1016/j.coviro.2011.10.006
147. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol (2005) 17(1):1–14. doi: 10.1093/intimm/dxh186
Keywords: acute phase protein, inflammation, parturition, pregnancy, SAA, DAMP, gestation
Citation: Lin Y-k, Zhu P, Wang W-s and Sun K (2022) Serum amyloid A, a host-derived DAMP in pregnancy? Front. Immunol. 13:978929. doi: 10.3389/fimmu.2022.978929
Received: 27 June 2022; Accepted: 18 July 2022;
Published: 05 August 2022.
Edited by:
Alessandro Rolfo, University of Turin, ItalyReviewed by:
Olesya Plazyo, Michigan Medicine, University of Michigan, United StatesRamkumar Menon, University of Texas Medical Branch at Galveston, United States
Copyright © 2022 Lin, Zhu, Wang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kang Sun, sungangrenji@hotmail.com
†These authors have contributed equally to this work