RELN Mutations in Autism Spectrum Disorder
 Dawn B. Lammert
Dawn B. Lammert Brian W. Howell
Brian W. Howell- Department of Neuroscience and Physiology, SUNY Upstate Medical School, Syracuse, NY, USA
RELN encodes a large, secreted glycoprotein integral to proper neuronal positioning during development and regulation of synaptic function postnatally. Rare, homozygous, null mutations lead to lissencephaly with cerebellar hypoplasia (LCH), accompanied by developmental delay and epilepsy. Until recently, little was known about the frequency or consequences of heterozygous mutations. Several lines of evidence from multiple studies now implicate heterozygous mutations in RELN in autism spectrum disorders (ASD). RELN maps to the AUTS1 locus on 7q22, and at this time over 40 distinct mutations have been identified that would alter the protein sequence, four of which are de novo. The RELN mutations that are most clearly consequential are those that are predicted to inactivate the signaling function of the encoded protein and those that fall in a highly conserved RXR motif found at the core of the 16 Reelin subrepeats. Despite the growing evidence of RELN dysfunction in ASD, it appears that these mutations in isolation are insufficient and that secondary genetic or environmental factors are likely required for a diagnosis.
Autism spectrum disorder (ASD) currently affects as many as 1 in 45 children in the United States (Zablotsky et al., 2015). ASD incorporates Asperger, autism, and pervasive developmental disorder-not otherwise specified (PDD-NOS), and is characterized by social, behavioral, and language deficits. A small percentage (<20%) of ASD known as “syndromic autism” is attributable to monogenetic diseases, the two most common being fragile X syndrome and tuberous sclerosis (Miles, 2011; Persico and Napolioni, 2013). Other monogenetic disorders that have a high frequency of ASD but are less prevalent in the general population include Prader-Willi/Angelman, 15q microduplication, Rett, Smith-Lemli-Opitz, and Timothy syndromes.
The remaining 80% of ASD cases are considered “non-syndromic autism” and are the focus of high throughput sequencing efforts. A better understanding of how candidate genes contribute to ASD at the molecular level is key to understanding how so many variants converge on a common phenotype. RELN, encoding a large secreted glycoprotein, expressed in the brain and critical for proper brain development and synapse function, is consistently cited as a candidate gene for ASD (Persico and Napolioni, 2013).
In 2001 the International Molecular Genetic Study of Autism Consortium (IMGSAC) described a region on chromosome 7q as the peak region of linkage and first autism susceptibility locus (AUTS1; IMGSAC, 1998). Subsequent linkage studies supported this finding (IMGSAC, 2001a,b; Lamb et al., 2005). Given the role of RELN in neurodevelopment and its location at chromosome 7q22, RELN quickly emerged as a candidate gene for autism and numerous studies (>15) have investigated the occurrence of ASD risk-associated single nucleotide polymorphism (SNPs) in RELN (DeSilva et al., 1997; Persico et al., 2001; Krebs et al., 2002; Zhang et al., 2002). These and other studies had mixed results, possibly due to varying study designs, ethnic populations, and mathematical interpretations. A recent meta-analysis considered three known SNPs in RELN, and concluded that one rs362691 was significantly associated with an increased risk of ASD (Wang et al., 2014a).
While SNP analysis supports heterozygous mutations in RELN, it cannot explain how they contribute to ASD. Many genes have been proposed as candidates for ASD on the basis of sequencing analysis, but like RELN, their pathological mechanism remains speculative. Instead of perseverating on a particular individual SNP, researchers are now considering candidate genes on a much broader scale. The sum of coding and non-coding variants from genome-wide screens coupled with network analyses, gene and protein expression, and epigenetic modifications provide evidence that helps understand functionally how a gene contributes to ASD (Neale and Sham, 2004). From these types of analyses emerges an approach for deciphering the role of RELN in ASD at the molecular level, beyond association.
RELN expression is both spatially and temporally consistent with ASD, which is thought to originate as a neurodevelopmental disorder that persists into postnatal life. Homozygous loss of RELN leads to severe neuronal dysplasia in several brain regions including the neocortex, hippocampus, and cerebellum. Patients homozygous null for RELN suffer from lissencephaly with cerebellar hypoplasia (LCH), a profoundly developmentally debilitating disease (Hong et al., 2000; Chang et al., 2007). Patients with LCH also suffer from epilepsy, but no autistic behavior has been reported in the patients or their parents.
RELN is first expressed by Cajal Retzius (CR) cells, and other less well defined marginal zone neurons, that act as pioneer neurons by regulating the positioning of projection neurons into discrete layers in the neocortex (D’Arcangelo et al., 1995; Hirotsune et al., 1995; Ogawa et al., 1995; Ikeda and Terashima, 1997; Meyer et al., 2004). CR eventually degenerate and a population of GABA-ergic interneurons expresses RELN postnatally (Pesold et al., 1998). In the developing cerebellum, RELN is first expressed by cells of the rhombic lip that migrate to populate the external granule layer and regulate the position of Purkinje neurons (D’Arcangelo et al., 1995; Miyata et al., 1997). Postnatally, cerebellar granule cell neurons (GCNs) now positioned in the internal granule layer continue to secrete Reelin, although its postnatal role is not clear (Sinagra et al., 2008). The brain size and architecture are relatively normal in the heterozygous reeler mouse (HRM); however, male HRM, thought to model ASD, have decreased numbers of Purkinje cells (Hadj-Sahraoui et al., 1996; Biamonte et al., 2009).
Traditionally, the cerebellum is considered responsible for fine-tuning movement, but its role in cognitive and emotional functions is now appreciated (Buckner, 2013). Acute adult injury results in the classical cerebellar signs early, such as asynergy, followed by subtle, often overlooked cognitive and communication impairments. In contrast, damage to the cerebellum during development leads to cognitive and communication defects. Interestingly, these early injuries have also been associated with the occurrence of ASD, which highlights a role for the cerebellum in its etiology (Becker and Stoodley, 2013; Wang et al., 2014b).
One of the most consistent anatomic findings in ASD is a decrease in cerebellar Purkinje cells and decreased volume of the vermis (Fatemi et al., 2012; D’Mello et al., 2015; Hampson and Blatt, 2015). In the most recent stereologic study, Skefos et al. (2014) found that Purkinje cells were decreased in ASD individuals. Compared to previous studies, this group included patients with cognitive delay and epilepsy, showing that this defect is widespread across ASD. The role of Purkinje cells and their ability to drive ASD behaviors as seen in tuberous sclerosis, has been recently demonstrated in a mouse model (Tsai et al., 2012a). They used a conditional knock-out of TSC1 in Purkinje cells to show that dysfunction in these cells was sufficient to decrease interest in novel mouse social interaction, increase grooming, and increase ultrasonic vocalizations—behaviors consistent with other ASD mouse models (Silverman et al., 2010).
Not only is RELN expression consistent with ASD, but the Reelin signaling pathways intersect prominent ASD protein networks. To understand how so many disparate genes can converge on a similar phenotype, grouping candidate genes into networks has helped to uncover cellular processes that might be driving ASD. Network analysis continually implicates synaptic function and dysregulated protein translation, particularly at the synapse (Gilman et al., 2011; Sanders et al., 2012; Ebert and Greenberg, 2013; De Rubeis et al., 2014).
Canonical Reelin signaling is initiated by Reelin binding its receptors very-low-density-lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2; LRP8; D’Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999). Disabled-1 (Dab1) is recruited to the receptors, which then activates Src family kinases and leads to reciprocal activation through tyrosine phosphorylation of Dab1 (Howell et al., 1997, 1999; Rice et al., 1998; Arnaud et al., 2003; Bock and Herz, 2003). This signaling initiates a number of signaling cascades, which have been extensively reviewed (Tissir and Goffinet, 2003; D’Arcangelo, 2014; Sekine et al., 2014). Dab1 and VLDLR have been suggested to be associated with ASD risk, but overall there is little evidence directly implicating Dab1, VLDLR, ApoER2, SRC, or FYN in ASD (Fatemi et al., 2005; Iwata et al., 2012; Li et al., 2013; Shen et al., 2015).
Particularly relevant to ASD network analyses are recent studies of the integral role of Reelin as a modulator of the postnatal synapse and the ability of Reelin to enhance LTP in the hippocampus (Weeber et al., 2002; Beffert et al., 2005; Chen et al., 2005). Secretion of Reelin, however, is constitutive and independent of synaptic activity (Lacor et al., 2000). Canonical Reelin-ApoER2/VLDLR-Dab1 signaling leads to phosphorylation of the NMDA receptor (NMDAR), increased calcium flux with glutamate stimulation, as well as altered intermembrane mobility of NR2B and NR2A subunit-containing receptors (Beffert et al., 2005; Chen et al., 2005; Groc et al., 2007; Campo et al., 2009; Ventruti et al., 2011). Reelin is also capable of modulating presynaptic neurotransmitter release by regulating the VAMP7 and SNAP-25 interaction (Bal et al., 2013).
Structurally, ApoER2, NMDAR, PTEN, and PSD-95 form a complex at the post-synaptic density in a Reelin-dependent manner (Ventruti et al., 2011; Figure 1). Neurexins and neuroligins, pre- and post-synaptic cell adhesion molecules respectively, organize the synapse, and each has been implicated in ASD (Dean and Dresbach, 2006). Neuroligins interact with PSD-95, which in turn is anchored to the cytoskeleton through SHANK proteins (Ebert and Greenberg, 2013). SHANK3 mutations often lead to Phelan-McDermid syndrome, which frequently presents with ASD (Grabrucker et al., 2011). PSD-95 expression is also regulated by fragile X mental retardation protein (FMRP), the protein implicated in fragile X syndrome (Tsai et al., 2012b). Hypermethylation of a trinucleotide expansion leads to decreased expression of FMRP and subsequent augmented synaptic mRNA translation.
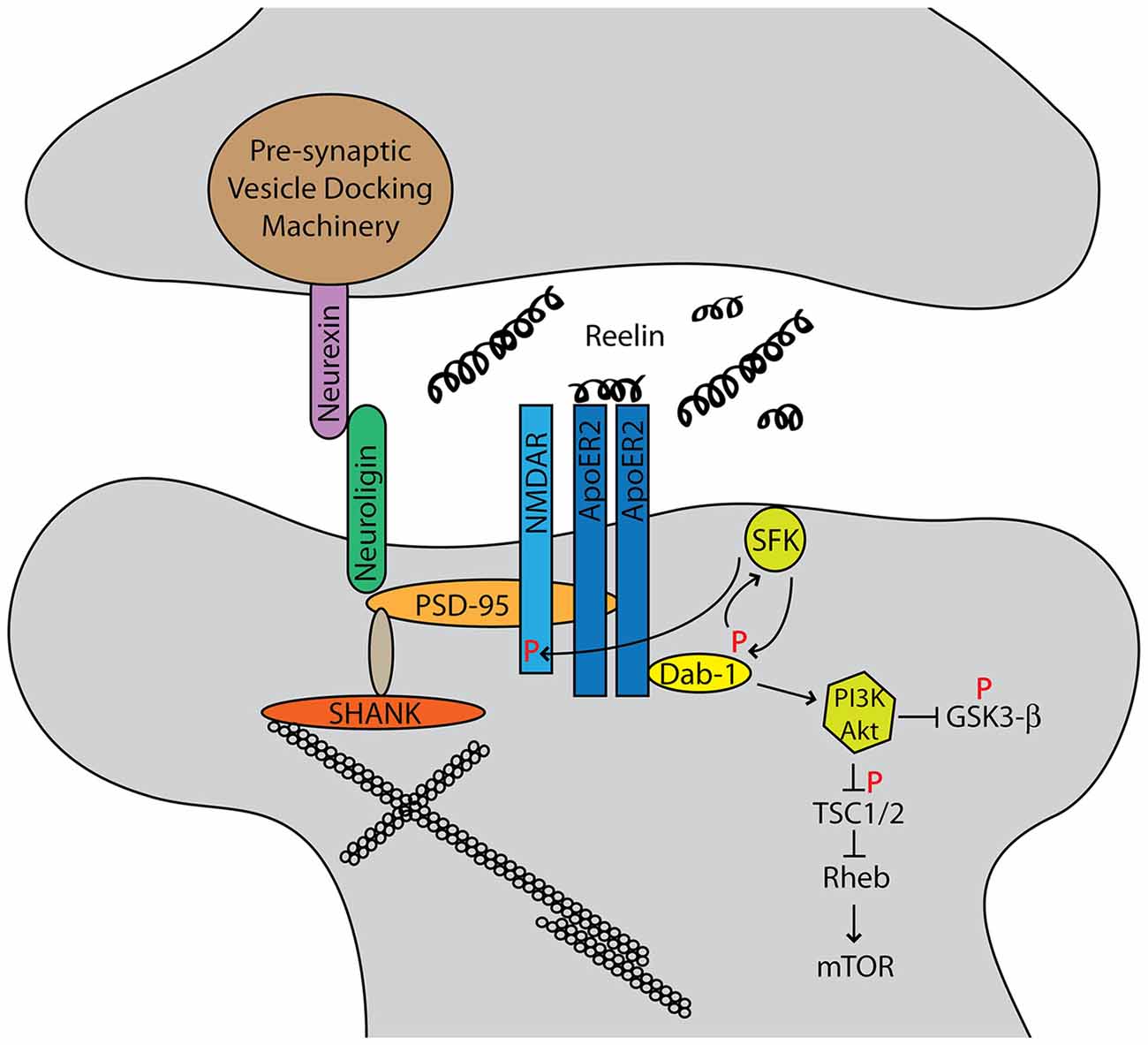
Figure 1. Autism spectrum disorders (ASD) candidate genes cluster into functional networks, and the two most prominent are synapse structure/function and protein translational control. The Reelin-signaling pathway intersects both of these networks. Reelin binds its receptors ApoER2 and very-low-density-lipoprotein receptor (VLDLR). The adapter protein Dab1 binds the cytoplasmic NPXY motif of the receptors and is phosphorylated by Src family kinases. This reciprocally activates Src, which leads to phosphorylation of the NMDA receptor (NMDAR) as well as downstream AKT/PI3K signaling that intersects the mTOR pathway.
Reelin also directly intersects protein translation control, the second major candidate gene network and hallmark of Fragile X and tuberous sclerosis syndromes. Tuberous sclerosis is caused by mutations in either TSC1 or TSC2 genes, which leads to hyperactivation of mTORC1 and subsequent increases in protein translation (Crino, 2011). Canonical Reelin signaling activates Akt, which phosphorylates TSC1/2 and leads to dendrite growth and branching (Jossin and Goffinet, 2007). Recently, Reelin and Dab1 protein expression were shown to be increased in TSC2 conditional knock-out mice as well as human cortical tubers (Moon et al., 2015). Here, activation of mTOR signaling may impair Cul5-mediated Dab1 degradation. Although Reelin signaling through mTOR is still incompletely understood, it is clear that it plays a significant role.
Further evidence for RELN involvement in ASD is the observation of decreased expression of RELN transcript and encoded protein in ASD patients. Decreased Reelin was detected in the cerebellum of ASD subjects as compared to controls (Fatemi et al., 2001, 2005) and in the superior frontal cortex (Fatemi et al., 2005). RELN mRNA in these areas was decreased, as was the dab1 transcript. VLDLR mRNA levels were increased.
Part of the elusiveness of ASD etiology is the likelihood of gene-environment interactions. Maternal stressors during gestation have been shown to alter RELN expression through promoter methylation (Giovanoli et al., 2014). MeCP2, the gene implicated in Rett and MeCP2 duplication syndromes, which share features of ASD, shows increased binding to the RELN promoter in human ASD cerebella (Zhubi et al., 2014). This binding corresponds with decreased RELN mRNA expression, consistent with the aforementioned reduced RELN expression in ASD tissue samples.
With the advent of more efficient and affordable sequencing technologies, whole-exome sequencing (WES) has become a new, popular approach for identifying candidate genes. WES identifies probable disease-contributing mutations that disrupt protein function. The average rate of mutation for the human genome is 1.2 × 10−8 per nucleotide. Over the entire genome, Kong et al. (2012) detected 63.2 de novo mutations per trio studied, but only 2% of the human genome is actually coding sequence (Kong et al., 2012). Therefore, in agreement with these findings, each exome has approximately only a single de novo protein changing allele (Gratten et al., 2013). Focusing then on only detected de novo events is a way to streamline candidate gene discovery.
Initial expectations were that individuals with yet unsolved complex disorders would have increased indels, CNVs, and frameshift, nonsense, and missense mutations compared to controls. While findings support that nonsense mutations may be more frequent in ASD than controls, the general finding is that there is not a dramatic overall increase in de novo mutation rates in ASD (Neale et al., 2012; Sanders et al., 2012; Samocha et al., 2014). Furthermore, de novo mutations do not make up a large enough proportion of cases to explain the elusive genetics of ASD, and likely represent less than 5% of the overall ASD risk (Neale et al., 2012).
Currently more than two de novo mutations in a gene support its candidacy, although this threshold will increase with increasing patients to control for multiple testing. RELN currently has four unique documented de novo ASD-associated mutations, three of which are likely pathological (Neale et al., 2012; De Rubeis et al., 2014; Iossifov et al., 2014; Yuen et al., 2015). Furthermore, RELN was 1 of 22 genes with a false discovery rate of < 0.05 in a study of nearly 4000 ASD patients (De Rubeis et al., 2014). De novo mutations, while directly explaining very few cases, are likely to contribute, at least in part, to disease in the proband in whom they were discovered. Given the repeated implication of particular gene signaling networks in ASD, understanding how a single de novo mutation influences this system at the molecular level will help explain a much larger number of ASD cases (Gratten et al., 2013).
Large and small scale WES studies of ASD individuals consistently identify missense and nonsense mutations in RELN, leading researchers to emphasize its importance in ASD (De Rubeis et al., 2014). There are currently over 40 unique RELN variants identified in ASD probands that are absent in controls (Bonora et al., 2003; Neale et al., 2012; Koshimizu et al., 2013; De Rubeis et al., 2014; Iossifov et al., 2014; Yuen et al., 2015; Zhang et al., 2015; Figure 2). These mutations have not been functionally characterized; however, strong predictions regarding their consequences can be deduced based on Reelin structure and function.

Figure 2. The structure of Reelin is diagrammed with conserved domain boundaries mapped (NCBI Conserved Domain Database). (A) Missense, nonsense, and frameshift mutations that are absent in controls and thus more likely to contribute to ASD are indicated (*Bonora et al., 2003; @Neale et al., 2012; +Koshimizu et al., 2013; #De Rubeis et al., 2014; ∧Iossifov et al., 2014; ~Yuen et al., 2015; =Zhang et al., 2015). Specifically, mutations identified by Bonora et al. (2003) and De Rubeis et al. (2014) as occurring in isolated controls from case-control studies, even if they overlap with mutations identified in other studies, are not pictured. This process did not remove any RXR mutations identified in ASD probands; however, three RXR mutations were identified in controls: R1198H, R2104H, and R2292H. Missense mutations are colored based on PolyPhen2 predictions (green, benign; orange, possibly damaging; red, probably damaging). Nonsense and frameshift mutations are indicated in black. (B) Clustal Omega alignment of the sub-repeat domains is annotated with the corresponding missense mutations from (A). The RXR consensus sequence is indicated below the aligned repeat sequence. Note: corrections to annotations for missense mutations from supplementary file S4 of De Rubeis et al. (2014) are as follows: R3439Q to R3441Q, G254V to G2737V, R156H to R2639H.
Reelin, a large 410 kDa protein, comprises eight Reelin repeat domains (D’Arcangelo et al., 1995; Nogi et al., 2006; Panteri et al., 2006; Yasui et al., 2007). Each Reelin repeat domain is composed of two subrepeat domains (A and B) linked by an EGF-like domain that share highly conserved sequences and are structurally similar. Reelin binds its receptors ApoER2 and VLDLR through two lysine residues on subrepeat 6A (Yasui et al., 2010).
The first prediction from structure-function analysis is that any nonsense mutation that truncates Reelin before the receptor-binding domain will be loss-of-function. In this instance, the transcript would either be degraded by nonsense-mediated decay or it would produce a protein product unable to initiate canonical signaling. Two mutations with this characteristic have been identified—a de novo mutation Q417X and a frameshift mutation that disrupts Reelin after E221 (De Rubeis et al., 2014). Both of these mutations occur before the receptor binding residues K2359 and K2466 (mouse equivalents K2360, K2467; Yasui et al., 2010). Whether these mutations could also contribute to a possible gain-of-function, perhaps through a non-canonical Reelin pathway is unclear, since the receptor-ligand domain is unknown (Lee et al., 2014).
Alternatively, one may predict that mutations may interfere with conserved domains, altering Reelin function in a way that contributes to the ASD phenotype. Aligning the subrepeat sequences of Reelin (Clustal Omega) and plotting the mutations identified in ASD genetic studies, we have found that Reelin is enriched in mutations that lie within an RXR consensus sequence that occurs once in each subrepeat (Bonora et al., 2003; De Rubeis et al., 2014; Iossifov et al., 2014; Figure 2). Of the identified variants, seven unique mutations fall within the RXR consensus sequence—a much larger percentage than would be expected by chance (R1742W, R1742Q, R2290C, R2290H, R2292C, R2639H, R2833S). R2290C, a mutation falling within the RXR consensus sequence, was discovered as a de novo variant originating on the paternal chromosome (Iossifov et al., 2014). This RXR consensus region is highly conserved across evolution, suggesting a particular functional relevance for this region that is linked to ASD pathogenesis.
Each subrepeat is composed of an 11-stranded beta-jelly roll fold, and the RXR consensus sequence is found at the beginning of the 10th beta sheet (Nogi et al., 2006). Arginine is important structurally for hydrogen bonding with the protein backbone (Borders et al., 1994). Disruption at this position could compromise protein folding, exposing the hydrophilic pore and enabling novel interactions. Alternatively, considering that Reelin may serve as an extracellular matrix (ECM) molecule, these closely spaced arginines and their neighboring tryptophan residues may be important for glycosaminoglycan binding (Panteri et al., 2006).
Clustering of mutations within this RXR consensus sequence argues against random mutations leading to complete loss-of-function. Presumably many mutations throughout the 3460 amino acids of Reelin could disrupt function. Therefore, a mutational hotspot might suggest a particular mode of loss-of- or gain-of-function, the details of which will need to be determined experimentally.
Animal models of RELN mutations may ultimately be necessary to parse out the link between RELN and ASD. Thus far, simple loss-of-function alleles have not provided overwhelming evidence that heterozygous RELN mutations in the mouse produce overt or consistent behavioral phenotypes reminiscent of ASD (Moy and Nadler, 2008). Similarly, the human genetics of RELN mutations suggests that a second hit, either environmental or genetic, may be necessary for ASD. Parents of patients with LCH are heterozygous for RELN loss-of-function alleles but do not have ASD (Hong et al., 2000; Chang et al., 2007). Approximately half of the ASD-associated mutations identified in RELN, including truncating and RXR mutations, are inherited from normal parents. In addition, following the same method of characterizing mutations in controls from ASD studies, here too there are examples of a nonsense mutation truncating Reelin before the receptor binding domain (Q849X) and RXR consensus mutations (R1198H, R2104H, and R2292H; Bonora et al., 2003; De Rubeis et al., 2014).
Since RELN is particularly susceptible to environment-driven epigenetic changes, one can hypothesize that perhaps a single mutation, which decreases its expression, combined with environmental down-regulation of Reelin production, could drive Reelin protein levels below a critical threshold in the brain. Or, perhaps another modifying gene allele in trans provides this added susceptibility. One likely contributing factor is sex. RELN mutations occur in approximately four times as many male as female probands. And indeed, testosterone and estrogen have differing effects on RELN expression and HRM phenotypes (Hadj-Sahraoui et al., 1996; Absil et al., 2003; Biamonte et al., 2009; Macri et al., 2010).
Adding to the excitement and promise of deciphering the role of RELN in ASD is evidence that Reelin supplementation or increased production could potentially reverse behavioral consequences of decreased Reelin signaling (Rogers et al., 2011, 2013; Hethorn et al., 2015). As mutations in RELN continue to be identified in genetic studies and the molecular mechanisms of these mutations are elucidated, we will better understand the role of Reelin in neuronal signaling, development, and ASD.
In the same way that science stresses transitioning from the bench to the bedside, we must also start to move gene candidates from the computer to the bench. RELN is now well positioned to make such a transition in ASD research. This approach will not be without its challenges, since it is predicted that for RELN and many of the other candidate genes, a single mutation may not be sufficient to cause overt ASD phenotypes, and gene-gene or gene-environment interactions will need to be considered.
Author Contributions
DBL wrote the manuscript and BWH suggested the topics to be covered and edited its content.
Funding
This work was supported by R01 NS073662 (BWH) and F31 NS086731 (DBL). National Institute of Neurological Disorders and Stroke/National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Eric Olson for discussions, Eric Larsen for explaining the processes for filtering and listing mutations on SFARI Gene-Autism Database, and Bonnie Lee Howell for editing the manuscript.
References
Absil, P., Pinxten, R., Balthazart, J., and Eens, M. (2003). Effects of testosterone on Reelin expression in the brain of male European starlings. Cell Tissue Res. 312, 81–93. doi: 10.1007/s00441-003-0701-9
Arnaud, L., Ballif, B. A., Förster, E., and Cooper, J. A. (2003). Fyn tyrosine kinase is a critical regulator of Disabled-1 during brain development. Curr. Biol. 13, 9–17. doi: 10.1016/s0960-9822(02)01397-0
Bal, M., Leitz, J., Reese, A. L., Ramirez, D. M., Durakoglugil, M., Herz, J., et al. (2013). Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 80, 934–946. doi: 10.1016/j.neuron.2013.08.024
Becker, E. B., and Stoodley, C. J. (2013). Autism spectrum disorder and the cerebellum. Int. Rev. Neurobiol. 113, 1–34. doi: 10.1016/b978-0-12-418700-9.00001-0
Beffert, U., Weeber, E. J., Durudas, A., Qiu, S., Masiulis, I., Sweatt, J. D., et al. (2005). Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 47, 567–579. doi: 10.1016/j.neuron.2005.07.007
Biamonte, F., Assenza, G., Marino, R., D’Amelio, M., Panteri, R., Caruso, D., et al. (2009). Interactions between neuroactive steroids and reelin haploinsufficiency in Purkinje cell survival. Neurobiol. Dis. 36, 103–115. doi: 10.1016/j.nbd.2009.07.001
Bock, H. H., and Herz, J. (2003). Reelin activates SRC family tyrosine kinases in neurons. Curr. Biol. 13, 18–26. doi: 10.1016/s0960-9822(02)01403-3
Bonora, E., Beyer, K. S., Lamb, J. A., Parr, J. R., Klauck, S. M., Benner, A., et al. (2003). Analysis of reelin as a candidate gene for autism. Mol. Psychiatry 8, 885–892. doi: 10.1038/sj.mp.4001310
Borders, C. L., Broadwater, J. A., Bekeny, P. A., Salmon, J. E., Lee, A. S., Eldridge, A. M., et al. (1994). A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 3, 541–548. doi: 10.1002/pro.5560030402
Buckner, R. L. (2013). The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron 80, 807–815. doi: 10.1016/j.neuron.2013.10.044
Campo, C. G., Sinagra, M., Verrier, D., Manzoni, O. J., and Chavis, P. (2009). Reelin secreted by GABAergic neurons regulates glutamate receptor homeostasis. PLoS One 4:e5505. doi: 10.1371/journal.pone.0005505
Chang, B. S., Duzcan, F., Kim, S., Cinbis, M., Aggarwal, A., Apse, K. A., et al. (2007). The role of RELN in lissencephaly and neuropsychiatric disease. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 144B, 58–63. doi: 10.1002/ajmg.b.30392
Chen, Y., Beffert, U., Ertunc, M., Tang, T. S., Kavalali, E. T., Bezprozvanny, I., et al. (2005). Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 25, 8209–8216. doi: 10.1523/jneurosci.1951-05.2005
Crino, P. B. (2011). mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 17, 734–742. doi: 10.1016/j.molmed.2011.07.008
D’Arcangelo, G. (2014). Reelin in the years: controlling neuronal migration and maturation in the mammalian brain. Adv. Neurosci. 2014:597395. doi: 10.1155/2014/597395
D’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D. S., Sheldon, M., and Curran, T. (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24, 471–479. doi: 10.1016/s0896-6273(00)80860-0
D’Arcangelo, G., Miao, G. G., Chen, S. C., Soares, H. D., Morgan, J. I., and Curran, T. (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723. doi: 10.1038/374719a0
Dean, C., and Dresbach, T. (2006). Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 29, 21–29. doi: 10.1016/j.tins.2005.11.003
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
DeSilva, U., D’Arcangelo, G., Braden, V. V., Chen, J., Miao, G. G., Curran, T., et al. (1997). The human reelin gene: isolation, sequencing and mapping on chromosome 7. Genome Res. 7, 157–164. doi: 10.1101/gr.7.2.157
D’Mello, A. M., Crocetti, D., Mostofsky, S. H., and Stoodley, C. J. (2015). Cerebellar gray matter and lobular volumes correlate with core autism symptoms. Neuroimage Clin. 7, 631–639. doi: 10.1016/j.nicl.2015.02.007
Ebert, D. H., and Greenberg, M. E. (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. doi: 10.1038/nature11860
Fatemi, S. H., Aldinger, K. A., Ashwood, P., Bauman, M. L., Blaha, C. D., Blatt, G. J., et al. (2012). Consensus paper: pathological role of the cerebellum in autism. Cerebellum 11, 777–807. doi: 10.1007/s12311-012-0355-9
Fatemi, S. H., Snow, A. V., Stary, J. M., Araghi-Niknam, M., Reutiman, T. J., Lee, S., et al. (2005). Reelin signaling is impaired in autism. Biol. Psychiatry 57, 777–787. doi: 10.1016/j.biopsych.2004.12.018
Fatemi, S. H., Stary, J. M., Halt, A. R., and Realmuto, G. R. (2001). Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J. Autism Dev. Disord. 31, 529–535. doi: 10.1023/A:1013234708757
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi: 10.1016/j.neuron.2011.05.021
Giovanoli, S., Weber, L., and Meyer, U. (2014). Single and combined effects of prenatal immune activation and peripubertal stress on parvalbumin and reelin expression in the hippocampal formation. Brain Behav. Immun. 40, 48–54. doi: 10.1016/j.bbi.2014.04.005
Grabrucker, A. M., Schmeisser, M. J., Schoen, M., and Boeckers, T. M. (2011). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603. doi: 10.1016/j.tcb.2011.07.003
Gratten, J., Visscher, P. M., Mowry, B. J., and Wray, N. R. (2013). Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat. Genet. 45, 234–238. doi: 10.1038/ng.2555
Groc, L., Choquet, D., Stephenson, F. A., Verrier, D., Manzoni, O. J., and Chavis, P. (2007). NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J. Neurosci. 27, 10165–10175. doi: 10.1523/jneurosci.1772-07.2007
Hadj-Sahraoui, N., Frederic, F., Delhaye-Bouchaud, N., and Mariani, J. (1996). Gender effect on Purkinje cell loss in the cerebellum of the heterozygous reeler mouse. J. Neurogenet. 11, 45–58. doi: 10.3109/01677069609107062
Hampson, D. R., and Blatt, G. J. (2015). Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 9:420. doi: 10.3389/fnins.2015.00420
Hethorn, W. R., Ciarlone, S. L., Filonova, I., Rogers, J. T., Aguirre, D., Ramirez, R. A., et al. (2015). Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur. J. Neurosci. 41, 1372–1380. doi: 10.1111/ejn.12893
Hiesberger, T., Trommsdorff, M., Howell, B. W., Goffinet, A., Mumby, M. C., Cooper, J. A., et al. (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489. doi: 10.1016/s0896-6273(00)80861-2
Hirotsune, S., Takahara, T., Sasaki, N., Hirose, K., Yoshiki, A., Ohashi, T., et al. (1995). The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat. Genet. 10, 77–83. doi: 10.1016/0168-9525(95)90549-9
Hong, S. E., Shugart, Y. Y., Huang, D. T., Shahwan, S. A., Grant, P. E., Hourihane, J. O., et al. (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96. doi: 10.1038/79246
Howell, B. W., Hawkes, R., Soriano, P., and Cooper, J. A. (1997). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389, 733–737.
Howell, B. W., Herrick, T. M., and Cooper, J. A. (1999). Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 13, 643–648. doi: 10.1101/gad.13.6.643
Ikeda, Y., and Terashima, T. (1997). Expression of reelin, the gene responsible for the reeler mutation, in embryonic development and adulthood in the mouse. Dev. Dyn. 210, 157–172. doi: 10.1002/(sici)1097-0177(199710)210:2<157::aidaja8>3.0.co;2-f
IMGSAC. (1998). A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Hum. Mol. Genet. 7, 571–578. doi: 10.1093/hmg/7.3.571
IMGSAC. (2001a). Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum. Mol. Genet. 10, 973–982. doi: 10.1093/hmg/10.9.973
IMGSAC. (2001b). A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q and 16p. Am. J. Hum. Genet. 69, 570–581. doi: 10.1086/323264
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iwata, K., Izumo, N., Matsuzaki, H., Manabe, T., Ishibashi, Y., Ichitani, Y., et al. (2012). Vldlr overexpression causes hyperactivity in rats. Mol. Autism 3:11. doi: 10.1186/2040-2392-3-11
Jossin, Y., and Goffinet, A. M. (2007). Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell Biol. 27, 7113–7124. doi: 10.1128/mcb.00928-07
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. doi: 10.1038/nature11396
Koshimizu, E., Miyatake, S., Okamoto, N., Nakashima, M., Tsurusaki, Y., Miyake, N., et al. (2013). Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PLoS One 8:e74167. doi: 10.1371/journal.pone.0074167
Krebs, M. O., Betancur, C., Leroy, S., Bourdel, M. C., Gillberg, C., Leboyer, M., et al. (2002). Absence of association between a polymorphic GGC repeat in the 5’ untranslated region of the reelin gene and autism. Mol. Psychiatry 7, 801–804. doi: 10.1038/sj.mp.4001071
Lacor, P. N., Grayson, D. R., Auta, J., Sugaya, I., Costa, E., and Guidotti, A. (2000). Reelin secretion from glutamatergic neurons in culture is independent from neurotransmitter regulation. Proc. Natl. Acad. Sci. U S A. 97, 3556–3561. doi: 10.1073/pnas.97.7.3556
Lamb, J. A., Barnby, G., Bonora, E., Sykes, N., Bacchelli, E., Blasi, F., et al. (2005). Analysis of IMGSAC autism susceptibility loci: evidence for sex limited and parent of origin specific effects. J. Med. Genet. 42, 132–137. doi: 10.1136/jmg.2004.025668
Lee, G. H., Chhangawala, Z., von Daake, S., Savas, J. N., Yates, J. R., Comoletti, D., et al. (2014). Reelin induces Erk1/2 signaling in cortical neurons through a non-canonical pathway. J. Biol. Chem. 289, 20307–20317. doi: 10.1074/jbc.m114.576249
Li, J., Liu, J., Zhao, L., Ma, Y., Jia, M., Lu, T., et al. (2013). Association study between genes in Reelin signaling pathway and autism identifies DAB1 as a susceptibility gene in a Chinese Han population. Prog. Neuropsychopharmacol. Biol. Psychiatry 44, 226–232. doi: 10.1016/j.pnpbp.2013.01.004
Macri, S., Biamonte, F., Romano, E., Marino, R., Keller, F., and Laviola, G. (2010). Perseverative responding and neuroanatomical alterations in adult heterozygous reeler mice are mitigated by neonatal estrogen administration. Psychoneuroendocrinology 35, 1374–1387. doi: 10.1016/j.psyneuen.2010.03.012
Meyer, G., Cabrera Socorro, A., Perez Garcia, C. G., Martinez Millan, L., Walker, N., and Caput, D. (2004). Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J. Neurosci. 24, 9878–9887. doi: 10.1523/jneurosci.3060-04.2004
Miles, J. H. (2011). Autism spectrum disorders–a genetics review. Genet. Med. 13, 278–294. doi: 10.1097/gim.0b013e3181ff67ba
Miyata, T., Nakajima, K., Mikoshiba, K., and Ogawa, M. (1997). Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J. Neurosci. 17, 3599–3609.
Moon, U. Y., Park, J. Y., Park, R., Cho, J. Y., Hughes, L. J., McKenna, J., et al. (2015). Impaired Reelin-Dab1 Signaling Contributes to Neuronal Migration Deficits of Tuberous Sclerosis Complex. Cell Rep. 12, 965–978. doi: 10.1016/j.celrep.2015.07.013
Moy, S. S., and Nadler, J. J. (2008). Advances in behavioral genetics: mouse models of autism. Mol. Psychiatry 13, 4–26. doi: 10.1038/sj.mp.4002082
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Neale, B. M., and Sham, P. C. (2004). The future of association studies: gene-based analysis and replication. Am. J. Hum. Genet. 75, 353–362. doi: 10.1086/423901
Nogi, T., Yasui, N., Hattori, M., Iwasaki, K., and Takagi, J. (2006). Structure of a signaling-competent reelin fragment revealed by X-ray crystallography and electron tomography. EMBO J. 25, 3675–3683. doi: 10.1038/sj.emboj.7601240
Ogawa, M., Miyata, T., Nakajima, K., Yagyu, K., Seike, M., Ikenaka, K., et al. (1995). The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 14, 899–912. doi: 10.1016/0168-9525(95)90534-0
Panteri, R., Paiardini, A., and Keller, F. (2006). A 3D model of Reelin subrepeat regions predicts Reelin binding to carbohydrates. Brain Res. 1116, 222–230. doi: 10.1016/j.brainres.2006.07.128
Persico, A. M., D’Agruma, L., Maiorano, N., Totaro, A., Militerni, R., Bravaccio, C., et al. (2001). Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 6, 150–159. doi: 10.1038/sj.mp.4000850
Persico, A. M., and Napolioni, V. (2013). Autism genetics. Behav. Brain Res. 251, 95–112. doi: 10.1016/j.bbr.2013.06.012
Pesold, C., Impagnatiello, F., Pisu, M. G., Uzunov, D. P., Costa, E., Guidotti, A., et al. (1998). Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. U S A. 95, 3221–3226. doi: 10.1073/pnas.95.6.3221
Rice, D. S., Sheldon, M., D’Arcangelo, G., Nakajima, K., Goldowitz, D., and Curran, T. (1998). Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125, 3719–3729.
Rogers, J. T., Rusiana, I., Trotter, J., Zhao, L., Donaldson, E., Pak, D. T., et al. (2011). Reelin supplementation enhances cognitive ability, synaptic plasticity and dendritic spine density. Learn. Mem. 18, 558–564. doi: 10.1101/lm.2153511
Rogers, J. T., Zhao, L., Trotter, J. H., Rusiana, I., Peters, M. M., Li, Q., et al. (2013). Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 27, 386–395. doi: 10.1177/0269881112463468
Samocha, K. E., Robinson, E. B., Sanders, S. J., Stevens, C., Sabo, A., McGrath, L. M., et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950. doi: 10.1038/ng.3050
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Sekine, K., Kubo, K., and Nakajima, K. (2014). How does Reelin control neuronal migration and layer formation in the developing mammalian neocortex? Neurosci. Res. 86, 50–58. doi: 10.1016/j.neures.2014.06.004
Shen, Y., Xun, G., Guo, H., He, Y., Ou, J., Dong, H., et al. (2015). Association and gene-gene interactions study of reelin signaling pathway related genes with autism in the Han Chinese population. Autism Res. doi: 10.1002/aur.1540 [Epub ahead of print].
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502. doi: 10.1038/nrn2851
Sinagra, M., Gonzalez Campo, C., Verrier, D., Moustie, O., Manzoni, O. J., and Chavis, P. (2008). Glutamatergic cerebellar granule neurons synthesize and secrete reelin in vitro. Neuron Glia Biol. 4, 189–196. doi: 10.1017/s1740925x09990214
Skefos, J., Cummings, C., Enzer, K., Holiday, J., Weed, K., Levy, E., et al. (2014). Regional alterations in purkinje cell density in patients with autism. PLoS One 9:e81255. doi: 10.1371/journal.pone.0081255
Tissir, F., and Goffinet, A. M. (2003). Reelin and brain development. Nat. Rev. Neurosci. 4, 496–505. doi: 10.1038/nrn1113
Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., et al. (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701. doi: 10.1016/s0092-8674(00)80782-5
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012a). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsai, N. P., Wilkerson, J. R., Guo, W., Maksimova, M. A., DeMartino, G. N., Cowan, C. W., et al. (2012b). Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594. doi: 10.1016/j.cell.2012.11.040
Ventruti, A., Kazdoba, T. M., Niu, S., and D’Arcangelo, G. (2011). Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 189, 32–42. doi: 10.1016/j.neuroscience.2011.05.050
Wang, Z., Hong, Y., Zou, L., Zhong, R., Zhu, B., Shen, N., et al. (2014a). Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 165B, 192–200. doi: 10.1002/ajmg.b.32222
Wang, S. S., Kloth, A. D., and Badura, A. (2014b). The cerebellum, sensitive periods and autism. Neuron 83, 518–532. doi: 10.1016/j.neuron.2014.07.016
Weeber, E. J., Beffert, U., Jones, C., Christian, J. M., Forster, E., Sweatt, J. D., et al. (2002). Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 277, 39944–39952. doi: 10.1074/jbc.m205147200
Yasui, N., Nogi, T., Kitao, T., Nakano, Y., Hattori, M., and Takagi, J. (2007). Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc. Natl. Acad. Sci. U S A. 104, 9988–9993. doi: 10.1073/pnas.0700438104
Yasui, N., Nogi, T., and Takagi, J. (2010). Structural basis for specific recognition of reelin by its receptors. Structure 18, 320–331. doi: 10.1016/j.str.2010.01.010
Yuen, R. K., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191. doi: 10.1038/nm.3792
Zablotsky, B., Black, L. I., Maenner, M. J., Schieve, L. A., and Blumberg, S. J. (2015). Estimated prevalence of autism and other developmental disabilities following questionnaire changes in the 2014 National Health Interview Survey. Natl. Health Stat. Report 1–20.
Zhang, Y., Kong, W., Gao, Y., Liu, X., Gao, K., Xie, H., et al. (2015). Gene mutation analysis in 253 chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS One 10:e0141782. doi: 10.1371/journal.pone.0141782
Zhang, H., Liu, X., Zhang, C., Mundo, E., Macciardi, F., Grayson, D. R., et al. (2002). Reelin gene alleles and susceptibility to autism spectrum disorders. Mol. Psychiatry 7, 1012–1017. doi: 10.1038/sj.mp.4001124
Keywords: RELN, Dab1, autism, autism spectrum disorder, brain development, cerebellum, neocortex
Citation: Lammert DB and Howell BW (2016) RELN Mutations in Autism Spectrum Disorder. Front. Cell. Neurosci. 10:84. doi: 10.3389/fncel.2016.00084
Received: 29 December 2015; Accepted: 18 March 2016;
Published: 31 March 2016.
Edited by:
Gabriella D’Arcangelo, Rutgers, The State Univesity of New Jersey, USAReviewed by:
Ivan Iossifov, Cold Spring Harbor Laboratory, USACarlos Gustavo Perez-Garcia, The Salk Institute, USA
Copyright © 2016 Lammert and Howell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian W. Howell, howellb@upstate.edu