Dysregulation of GABAergic Signaling in Neurodevelomental Disorders: Targeting Cation-Chloride Co-transporters to Re-establish a Proper E/I Balance
 Enrico Cherubini
Enrico Cherubini Graziella Di Cristo
Graziella Di Cristo Massimo Avoli
Massimo Avoli- 1European Brain Research Institute (EBRI)-Rita Levi-Montalcini, Roma, Italy
- 2Neurosciences Department, Université de Montréal and CHU Sainte-Justine Research Center, Montreal, QC, Canada
- 3Montreal Neurological Institute-Hospital and Departments of Neurology and Neurosurgery and of Physiology, McGill University, Montreal, QC, Canada
The construction of the brain relies on a series of well-defined genetically and experience- or activity -dependent mechanisms which allow to adapt to the external environment. Disruption of these processes leads to neurological and psychiatric disorders, which in many cases are manifest already early in postnatal life. GABA, the main inhibitory neurotransmitter in the adult brain is one of the major players in the early assembly and formation of neuronal circuits. In the prenatal and immediate postnatal period GABA, acting on GABAA receptors, depolarizes and excites targeted cells via an outwardly directed flux of chloride. In this way it activates NMDA receptors and voltage-dependent calcium channels contributing, through intracellular calcium rise, to shape neuronal activity and to establish, through the formation of new synapses and elimination of others, adult neuronal circuits. The direction of GABAA-mediated neurotransmission (depolarizing or hyperpolarizing) depends on the intracellular levels of chloride [Cl−]i, which in turn are maintained by the activity of the cation-chloride importer and exporter KCC2 and NKCC1, respectively. Thus, the premature hyperpolarizing action of GABA or its persistent depolarizing effect beyond the postnatal period, leads to behavioral deficits associated with morphological alterations and an excitatory (E)/inhibitory (I) imbalance in selective brain areas. The aim of this review is to summarize recent data concerning the functional role of GABAergic transmission in building up and refining neuronal circuits early in development and its dysfunction in neurodevelopmental disorders such as Autism Spectrum Disorders (ASDs), schizophrenia and epilepsy. In particular, we focus on novel information concerning the mechanisms by which alterations in cation-chloride co-transporters (CCC) generate behavioral and cognitive impairment in these diseases. We discuss also the possibility to re-establish a proper GABAA-mediated neurotransmission and excitatory (E)/inhibitory (I) balance within selective brain areas acting on CCC.
Introduction
In the adult mammalian central nervous system (CNS), γ-aminobutyric acid (GABA) inhibits neuronal firing by activating two different classes of receptors: GABAA and GABAB. While GABAA receptors are integral ion channels, GABAB receptors are coupled to ion channels via guanine nucleotide-binding proteins and second messengers. The opening of GABAA receptor channels by GABA leads to an inwardly directed flux of Cl− that, by hyperpolarizing the membrane, inhibits neuronal firing. Early in postnatal life, instead, GABA, via GABAA receptors, depolarizes and excites targeted cells by an outwardly directed flux of Cl− (Ben-Ari et al., 1989). This phenomenon is due to the high levels of intracellular Cl− ([Cl−]i) that result from the differential temporal expression of the cation-chloride co-transporters NKCC1 and KCC2, which are involved in Cl− uptake and extrusion, respectively. The low expression of the KCC2 extruder at birth leads to Cl− accumulation inside the neuron via NKCC1. The developmentally up-regulated expression of KCC2, which in rodents occurs toward the end of the first postnatal week, results in the extrusion of Cl−, causing the shift of GABA from depolarizing to hyperpolarizing direction (Rivera et al., 1999) (Figure 1).
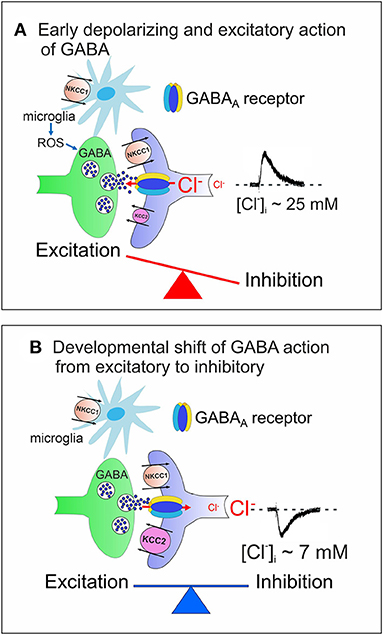
Figure 1. Cation chloride co-transporters contribute to maintain a proper E/I balance in neuronal circuits. (A) Early in postnatal life, in rodent's hippocampus, GABA, released from GABAergic interneurons, exerts via GABAA receptors a depolarizing, excitatory action in targeted cells by an outwardly directed flux of Cl−. High [Cl−]I results from the differential temporal expression of Cl− importer and exporter NKCC1 and KCC2, respectively, and accumulation of Cl− inside via NKCC1. Early in postnatal life, GABAergic transmission is controlled also by microglia via ROS. The early depolarizing and excitatory action of GABA, which leads to E/I unbalance, is instrumental in stimulating synaptogenesis and in shaping early neuronal circuits. (B) In juvenile and adult animals, the upregulated expression of KCC2 contributes to maintain a low [Cl−]I, responsible for the hyperpolarizing action of GABA and for preserving a proper E/I balance in neuronal circuits. In (A,B) both pre- (GABAergic interneurons, green) and post-synaptic (targeted cells, violet) elements are represented. Red arrows indicate the direction of Cl− flux through GABAA receptor channels.
At the network level, the interplay between the depolarizing action of GABA and glutamate generates a primordial form of synchrony which varies in its specific patterns among different brain regions (Buzsáki and Draguhn, 2004; Griguoli and Cherubini, 2017). In the hippocampus, the so-called Giant Depolarizing Potentials (GDPs), are crucial for synaptic wiring and refinement of local neuronal circuits (Ben-Ari et al., 2012). Principal neurons are driven by GABAergic interneurons (Mohajerani and Cherubini, 2005), which act as functional hubs to synchronize large neuronal ensembles (Bonifazi et al., 2009). Early Ca2+ signals associated with GDPs act as coincident detectors for enhancing synaptic efficacy at emerging GABAergic (Kasyanov et al., 2004) and glutamatergic synapses (Mohajerani et al., 2007). GDPs are indeed instrumental in converting silent synapses into active ones (Kasyanov et al., 2004), a key mechanism for persistently increasing synaptic efficacy (Voronin and Cherubini, 2004). Immediately after birth, at least in the rodent CA3 hippocampal region, GDPs are associated to intrinsic bursts, driven by a persistent Na+ current (Sipilä et al., 2006) and are facilitated by the low expression of Kv7.2/Kv7.3 channels, responsible for the non-inactivating, low-threshold M current (IM) (Safiulina et al., 2008). GDPs disappear toward the end of the first postnatal week, when the polarity of GABA shifts from depolarizing to hyperpolarizing.
It is therefore not surprising that the depolarizing action of GABA at early stages of postnatal development coincides with the period of maximal synaptogenesis (Huttenlocher, 1979; De Felipe et al., 1997; Virtanen et al., 2018). Interestingly, GABAergic signals operate before glutamatergic ones, which appear later in concomitance with the development of dendritic arborization (Tyzio et al., 1999; Khazipov et al., 2001; Ben-Ari et al., 2007). The late switch of GABA polarity at the axon initial segment of principal cells favors more organized forms of network oscillations such as those occurring in the gamma range (20–80 Hz), as demonstrated in the somatosensory (Khirug et al., 2008) and in the prefrontal cortex (Rinetti-Vargas et al., 2017). The early depolarizing action of GABA and its developmental shift, mainly documented in in vitro studies, have been challenged because of the lack of direct in vivo demonstrations (Ben-Ari et al., 2012). Using a combined electrophysiological and imaging techniques from anesthetized neonatal mice, it has been shown that, in spite of its depolarizing action, GABA inhibits cortical activity via a shunting inhibitory action (Kirmse et al., 2015).
Similarly, an inhibitory effect of GABA on spontaneous glutamatergic events has been reported in the hippocampus of anesthetized animals during the first postnatal week, following photo-stimulation of GABAergic interneurons expressing channelrhodopsin (Valeeva et al., 2016).
Evidence for an early depolarizing and excitatory action of GABA in vivo has been provided by Oh et al. (2016) who have demonstrated that, in the developing mouse cortex, synapses formation requires GABA-mediated activation of T-type voltage dependent Ca2+ channels. The early depolarizing and excitatory action of GABA in in vivo conditions has been further confirmed by Sulis Sato et al. (2017) and Murata and Colonnese (2020). Using a particular probe formed by the fusion of a Cl− and pH-sensitive GFP mutant with an ion-insensitive red fluorescent protein, which allows the combined measurement of [Cl−]I and pH, Sulis Sato et al. (2017) have demonstrated, by means of two-photon in vivo imaging from individual pyramidal cells in the mouse cortex, the developmental shift of GABAergic signaling; this effect could be mimicked by the selective NKCC1 antagonist bumetanide. In addition, using chemogenetic and optogenetic approaches, Murata and Colonnese (2020) have proved that, at postnatal day 3 in non-anesthetized mice, GABA released from GABAergic interneurons increases the firing of CA1 principal cells. However, according to these authors, the shift of GABA polarity is region-specific, since at the same age GABAergic interneurons exert an inhibitory action on visual cortex principal cells.
Our review will focus on evidence concerning the functional role of GABAergic signaling in brain maturation and its alterations in neurodevelopmental disorders, highlighting the contribution of CCC to these effects. CCC are intrinsic membrane proteins that transport Cl− ions, together with Na+ and/or K+ ions, in an electroneutral manner due to the stoichiometric coupling and directionality of translocated ions. Therefore, members of this family are prime regulators of [Cl−]i. After the initial discovery of the depolarizing action of GABA (Ben-Ari et al., 1989; Cherubini et al., 1991), a fundamental step forward to understanding why in immature neurons the equilibrium potential for chloride () is positive relative to the resting membrane potential (Vm) has been made by Rivera et al. (1999) who demonstrated that the developmental regulated expression of the cation-chloride exporter KCC2 leads to Cl− accumulation inside young neurons with consequent shift of GABA action from hyperpolarizing to depolarizing. It is worth mentioning that GABAA receptor channels are permeable not only to Cl− but also to and therefore, the equilibrium potential for Cl− () does not correspond precisely to the equilibrium potential of GABA (EGABA), which usually shifts toward more positive values, being EHCO3− less negative than (Kaila, 1994). Taking into account a HCO3−/Cl− permeability ratio of 0.2–0.4, the quantitative influence of on EGABA can be estimated using the Goldman-Hodgkin-Katz equation, which shows that the []i (~15 mM at a pH of 7.1–7.2) influences EGABA in the same way as 3–5 mM of [Cl−]i. Thus, as compared to adult neurons, in the immature CNS, in which [Cl−]i is relatively high, the depolarizing influence of on EGABA is negligible. In addition to controlling EGABA, the direction of GABA action and network excitability, CCCs regulate many physiological processes including cell volume, water transport and intracellular pH (Delpire and Gagnon, 2018).
Distribution of NKCC1 and KCC2
Encoded by the SLC12 gene's family, CCCs are glycoproteins that are widely distributed in all organ systems, including the brain (Kaila et al., 2014). Among CCCs, the main chloride extruder KCC2 has been found almost exclusively in the CNS. Consistent with a developmental gradient, at birth KCC2 is already present in the spinal cord and in the brainstem in rodents, while it starts to be upregulated later in most rostral regions of the brain (Kaila et al., 2014; Virtanen et al., 2020). In the human cortex, upregulation of KCC2 starts prenatally from the 25th postconceptional week and peaks at birth (Sedmak et al., 2016). In preterm infants, the relatively low expression of KCC2 is associated with a discontinuous type of EEG—organized in intermittent bursts of activity, separated by silent periods reminiscent of GDPs—that disappears at birth (Khazipov and Luhmann, 2006). It is worth noting that KCC2 has been also detected in pancreatic β-cells where it plays a crucial role in modulating insulin secretion (Kursan et al., 2017).
In the hippocampus, KCC2 is involved in regulating GDPs, which are driven by the synergistic depolarizing action of glutamate and GABA (Bolea et al., 1999; Ben-Ari et al., 2012) and by the intrinsic pacemaker properties of CA3 pyramidal neurons (Strata et al., 1997; Safiulina et al., 2008; Griguoli and Cherubini, 2017). Interestingly, during the first week of postnatal life, using whole-cell Cl− loading experiments, Spoljaric et al. (2019) have reported that the selective KCC2 antagonist VU0463271 can increase the firing rate of CA3 principal cells as well as their synchrony during the rising phase of GDPs, suggesting that these neurons are able to actively extrude Cl− in a KCC2-dependent way, particularly when GABA is applied at the dendritic level. In addition to its canonical function of transporting Cl− outside the neuron, KCC2 plays a key role in controlling actin cytoskeleton's dynamics and spinogenesis at early developmental stages (Blaesse et al., 2009; Virtanen et al., 2020). Of note, during the first postnatal week, KCC2 promotes spinogenesis independent of KCC2 Cl− transport function in the somatosensory cortex (Li et al., 2007; Fiumelli et al., 2013), but, at the same age, it constrains spine density in hippocampal CA1 neurons, an effect that is instead dependent on the transporter function (Awad et al., 2018). This difference might be explained by distinct membrane localizations of KCC2 in different neuron types or/and brain region during early postnatal development. Nevertheless, it is safe to state that KCC2 plays multiple roles in brain development, contributing to shape neuronal circuits shortly after birth and network plasticity at later stages of postnatal development (Virtanen et al., 2021).
Unlike KCC2, the main Cl− importer NKCC1 is expressed in the brain already at birth, where it plays a key role in maintaining high [Cl−]i in immature neurons. NKCC1 is widely distributed not only in CNS and in the peripheral nervous system (PNS) but also in a variety of different tissues including the inner ear, in skeletal and smooth muscles, exocrine glands, epithelial cells and kidneys, where it contributes to regulate major physiological functions (Delpire and Gagnon, 2018; Virtanen et al., 2020). While in the CNS, the majority of neurons express low levels of NKCC1, in the PNS sensory neurons such as dorsal root or trigeminal ganglion cells, exhibit high amounts of the protein. When GABA from local interneurons is released at the terminals of sensory afferent fibers, it causes a membrane depolarization that leads to a reduction of glutamatergic transmission and to an inhibitory response. This effect is reduced in NKCC1 knock-out mice, indicating that the depolarizing value of EGABA is maintained via NKCC1. Such mechanism may have a strong implication for nociception, as demonstrated by deficits in thermal nociceptive threshold in NKCC1 knock-out mice (Sung et al., 2000). Primary sensory afferents (i.e., dorsal root ganglion fibers) contain GABA receptors, whose activation by GABA, released from interneurons localized in lamina I/II of the dorsal horn, causes a depolarization (primary afferent depolarization or PAD). The depolarization, maintained by the expression of NKCC1 and the lack of KCC2 (Alvarez-Leefmans et al., 2001) leads to suppression of nociceptive signals mainly via inactivation of voltage-gated Na+ channels and consequent reduction of transmitter release (Price et al., 2009) An inflammatory insult, after peripheral nerve injury, may cause an upregulation of NKCC1 activity in nociceptive afferent fibers leading to an increased [Cl−]i and an excessive GABAA-mediated depolarization that would facilitate cross-excitation between low and high threshold nociceptive afferent fibers and nociception (Price et al., 2005).
In the CNS, the selective deletion from hippocampal CA1 pyramidal cells of the SLC12a2 gene, leads to an attenuation of the depolarizing action of GABA, due to a reduction of [Cl−]i and to a severe impairment of GDPs activity, in a region-specific way (Graf et al., 2021). However, such deletion only slightly affects the in vivo network dynamics or hippocampal-dependent behavioral tasks, suggesting that most of the effects observed in NKCC1 knock-out mice or after pharmacological blockade of the transporter, may be attributed to the loss of the protein from non-neuronal cells or from cells localized outside the brain (Graf et al., 2021). In the brain, NKCC1 has been found to be expressed in several non-neuronal cell types such as choroid plexus epithelial cells, astrocytes, oligodendrocytes and microglia (Tóth et al., 2021). In particular NKCC1 is highly expressed in microglia (DePaula-Silva et al., 2019) (Figure 1) where it plays a fundamental role in neuro-inflammation. Thus, the selective deletion of NKCC1 on microglia affects their cell volume and baseline morphology and boosts cytokines production in response to inflammatory stimuli (Tóth et al., 2021). Interestingly, it has been recently reported that long-term potentiation triggers in potentiated synapses withdrawal of perisynaptic astrocytic processes, which involves the NKCC1 transporter and the actin-controlling protein cofilin. This favors glutamate spillover and NMDA-mediated inter synaptic cross-talk, crucial for LTP and memory formation (Henneberger et al., 2020).
Transcriptional and Post-translational Regulation of NKCC1 and KCC2
Among transcriptional regulators of CCCs, a key factor is represented by Brain Derived Neurotrophic Factor (BDNF) and its tropomyosin kinase B (TrkB) receptor. Employing transgenic embryos that overexpress BDNF under the control of the nestin promoter, Aguado et al. (2003) demonstrated that, in embryonic hippocampal slices, BDNF powerfully controls the developmental switch of GABAergic transmission. At the network level, by upregulating KCC2 expression, BDNF reduced [Cl−]i and GABAA-activated Ca2+ transients. Furthermore, in immature cultured hippocampal neurons, BDNF enhanced KCC2 mRNA and protein expression levels via ERK1/2-dependent upregulation of Egr4 transcription factor (Ludwig et al., 2011a). BDNF can further increase KCC2 activation, by promoting the localization at the membrane of already synthetized KCC2 in the developing brain (Khirug et al., 2010; Puskarjov et al., 2015; Awad et al., 2018). Egr4 mRNA expression can be also triggered by the trophic factor neurturin which leads to the developmental upregulation of KCC2 in an ERK1/2-dependent way (Ludwig et al., 2011b). Furthermore, TrkB-deficient mice exhibit a reduced number of GABAergic synapses associated with decreased expression levels of KCC2, further indicating that BDNF is determinant for its expression (Carmona et al., 2006). These data are inconsistent with those reported by Puskarjov et al. (2015) on BDNF-deficient mice in which no developmental changes in GABA shift were detected. Although the lack of the developmental upregulation of KCC2 in BDNF null mice may be related to compensatory processes, the reason for this discrepancy is still unclear.
While the impact of BDNF/TrkB signaling on KCC2 expression at early stages of postnatal development has been well-documented, the role of this neurotrophin on NKCC1 expression is still debated. A recent study by Badurek et al. (2020) has however unveiled that the selective deletion of TrkB from immature dentate granule cells (DGC), when these cells integrate the hippocampal circuit, induces a premature shift of GABA from the depolarizing to the hyperpolarizing direction at mossy fibers-CA3 synapses, which at birth are GABAergic (Safiulina et al., 2006a). A dysfunction in BDNF/TrkB signaling leads due to downregulation of NKCC1 expression and low [Cl−]i, in the absence of any effect on KCC2. In agreement with a previous study on immature neocortical neurons (Cancedda et al., 2007), the premature hyperpolarizing shift of GABA prevents the establishment of proper synaptic connectivity in targeted neurons, an effect that persists in adulthood (Badurek et al., 2020). However, how in immature DGCs, BDNF/TrkB signaling regulates the expression of the Cl− importer NKCC1 remains to be elucidated. Another trophic factor that controls KCC2 expression and the developmental GABA switch is the insulin growth factor 1 (IGF-1), which presumably requires protein tyrosine kinase c-Src (Kelsch et al., 2001). It is worth noting that, independently on its action on CCCs, the BDNF/TrkB signaling pathway is instrumental in tuning hippocampal wiring at emerging GABAergic (Sivakumaran et al., 2009) and glutamatergic (Mohajerani et al., 2007) synapses, during spike time dependent plasticity, an Hebbian form of learning.
Interestingly, neuroligin 2 (NLG2) a cell adhesion molecule involved in regulating GABAergic synaptogenesis has recently emerged as a key modulator of the developmental GABAergic switch. It was unexpectedly discovered that, knocking down NLG2, leads to a reduced expression of KCC2, which is in turn associated with a delayed switch of GABA from the depolarizing to the hyperpolarizing direction. The down-regulation of KCC2 was accompanied by a reduced number of dendritic spines and glutamatergic synaptic events, suggesting that, in neural networks, NLG2 may serve as a master regulator of the delicate balance between glutamatergic and GABAergic functions (Sun et al., 2013).
Among post-translational mechanisms controlling the activity and stabilization of CCC, protein phosphorylation represents the main functional substrate. KCC2 and NKCC1 are regulated in a reciprocal fashion by threonine-dependent phosphorylation/de-phosphorylation residues, targeted by WNK (with-No-Lysine) kinases that are responsible for increasing or decreasing [Cl−]i levels, respectively (Ben-Ari et al., 2012; Kahle et al., 2013; Kaila et al., 2014). The reciprocal activation is probably determined by similar four amino acid phosphorylation motifs that are present on C and N terminus domains of KCC2 and NKCC1, respectively (Rinehart et al., 2009). Interestingly, early in postnatal life, oxytocin, a hypothalamic hormone known to promote parturition and lactation and to be involved in social behavior, regulates GABA switch by upregulating the activity of KCC2, through the promotion of its phosphorylation at Ser940 and its insertion at the plasma membrane without impairing NKCC1 (Leonzino et al., 2016).
Involvement of NKCC1 and KCC2 in Maintaining a Proper Ratio Between Excitation and Inhibition Within Neuronal Circuits
By regulating the direction of GABA action and therefore the efficacy of inhibition, NKCC1 and KCC2 contribute to set a proper E/I balance within selective neuronal circuits (Figure 1B). A proper ratio between excitation (E) and inhibition (I), the so-called E/I balance is thought to be critical for controlling spike rate and information processing. It requires precise connections through dynamic processes involving neurotransmitter receptors, transporters, scaffolding proteins, and the cytoskeleton. Using an open source software to map the distribution and morphology of excitatory and inhibitory synapses along the dendritic tree of layer 2/3 mouse cortical pyramidal neurons with computational modeling, Iascone et al. (2020) unveiled that E/I synapses are highly regulated by molecular mechanisms operating locally to generate a relative invariant E/I ratio across dendritic segments.
Failure to maintain a proper E/I balance within key neuronal circuits is thought to account for behavioral deficits observed in several neurological diseases (Yizhar et al., 2011; Lee et al., 2017; Ghatak et al., 2021). A reduced inhibition or an excessive excitation may cause an increased signal to noise ratio with consequent neuronal hyper-excitability and seizures. Conversely, an enhanced inhibition may lead to a reduced signal to noise ratio and to a lower level of activity (Sohal and Rubenstein, 2019). Both conditions would affect information processing. Changes occurring at synaptic and circuit levels would influence the interplay between GABAergic interneurons and targeted pyramidal cells leading to altered temporal integration and abnormal rhythmogenesis. In cortical circuits, the E/I balance plays a critical role in regulating the responses of neuronal circuits to sensory stimuli. Thus, in juvenile mice carrying the human R451C mutation of the gene encoding for neuroligin 3 (an adhesion molecule essential for synaptic stabilization) found in some families with children affected by Autism Spectrum Disorders (ASDs), the impairment of GABA release from parvalbumin (PV)+ basket cells was found to severely alter the E/I balance in layer IV neuronal microcircuit of the somatosensory cortex (Cellot and Cherubini, 2014a). This represents a critical issue, since PV+ cells, which are innervated by the same thalamic afferents to excitatory layer IV spiny neurons, play a crucial role in sensory information, acting as an inhibitory gate for incoming thalamic inputs via feed-forward disynaptic inhibition (Cellot and Cherubini, 2014a). Changes in the inhibitory gate may alter sensory processing in ASD patients leading to misleading sensory representations with difficulties to combine pieces of information into a unified perceptual whole.
Although an E/I imbalance has been implicated in various brain disorders, this concept is rather broad and oversimplified. It should be used indeed with caution, particularly in view of our progress in understanding, thanks also to the development of optogenetics, the functional role of selective neuronal circuits in behavior. Both excitation and inhibition are not unidimensional entities but originate from multiple sources, which are dynamically regulated in space and time (He and Cline, 2019). Differences in local circuits connectivity, can produce various levels of inhibition or disinhibition in different pathways. Distinct classes of cortical GABAergic interneurons may differently contribute to seizures, by suppressing or prolonging them (Khoshkhoo et al., 2017). Therefore, in these cases a therapeutic intervention directed against a particular type of interneuron will be more effective than one aimed at inhibition in general.
CCC dynamically regulate in an activity-dependent way GABAA-mediated synaptic strength (Woodin et al., 2003; Fiumelli and Woodin, 2007). Hence, brief synaptic stimulations or pairing pre- and postsynaptic activity induce long-term synaptic plasticity changes at GABAergic synapses, exhibiting a positive shift in (EGABA), mediated by a decrease in KCC2's function and increase of [Cl−]i, with consequent decline of synaptic inhibition (Balena and Woodin, 2008). Activity-dependent changes in EGABA requires Ca2+ influx through voltage-gated calcium channels and NMDA receptors (Balena and Woodin, 2008). CCC also very labile, and they can be disrupted in several neuropsychiatric disorders particularly in those originating early in developmental such as ASDs, schizophrenia and epilepsy (Fiumelli and Woodin, 2007). In the following sections, an outline of the involvement of GABAergic signaling in these disorders will be discussed in line with possible therapeutic interventions aimed at targeting CCC to re-establish a proper E/I balance at synaptic level and/or neuronal connectivity at the circuit level.
Autism Spectrum Disorders
ASDs comprise a heterogeneous group of neurodevelopmental disorders characterized by impaired social interactions, deficits in verbal and non-verbal communication, restricted interests and stereotyped behaviors with high incidence (~1/70 children) and a significant economic and social burden for families and society. Impaired chloride homeostasis with consequent changes in the direction of GABA shift during time-sensitive windows, may account for behavioral alterations found in some animal models of ASDs, reminiscent of those observed in autistic patients (Pizzarelli and Cherubini, 2011; Cellot and Cherubini, 2014b). GABA-mediated enhancement in network excitability may account for the high co-morbidity of ASDs with epilepsy (Frye et al., 2013; Bozzi et al., 2018; Sierra-Arregui et al., 2020) and for the paradoxical action exerted by benzodiazepines in some ASD patients (Marrosu et al., 1987). The E/I imbalance in selective brain areas may result either from the persistent depolarizing and excitatory action of GABA beyond the critical period (Tyzio et al., 2014; Corradini et al., 2018; Fernandez et al., 2019) (Figure 2A), or from the early hyperpolarizing action of this neurotransmitter, following downregulation of the chloride importer NKCC1 (Figure 2B).
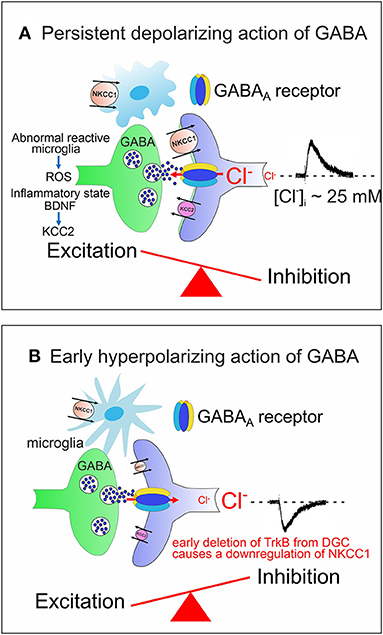
Figure 2. Alterations in developmental GABA shift lead to neuropsychiatric disorders. (A) The persistent depolarizing action of GABA beyond the critical period, impairs network excitability and the E/I balance. Abnormal reactive microglia, produces high levels of ROS, often associated with an inflammatory state caused by a dysfunction of the immune system. BDNF, released from microglia contributes to downregulate KCC2 with consequent enhancement of network excitability. (B) The early hyperpolarizing action of GABA at birth, caused by the reduced expression of NKCC1, following deletion of TrkB from immature DGCs, at the time when these cells integrate the classical tri-synaptic pathway, severely alters the morphology and circuitry downstream of DGCs. In (A,B) both pre (GABAergic interneurons, green) and post synaptic (targeted cells, violet) elements are represented. Red arrows indicate the direction of Cl− flux through GABAA receptor channels.
As already mentioned, the early depolarizing and excitatory action of GABA is essential for shaping neuronal networks, as demonstrated by the morphological impairment of cortical pyramidal neurons (Cancedda et al., 2007) and the premature termination of interneuron migration (Bortone and Polleux, 2009) following precocious KCC2 expression by KCC2 electroporation. A reduced expression of NKCC1 at birth has been recently demonstrated in a novel genetic mouse model (Ntrk2/Trkb) in which the selective deletion of TrkB from immature dentate granule cells (DGCs) leads to a disruption of downstream circuits, associated with a severe impairment of synaptic plasticity and cognitive processes (Badurek et al., 2020). BDNF, via TrkB signaling pathway, is known to play a crucial role in the maturation of inhibition as demonstrated in cortical and hippocampal neurons (Huang et al., 1999; Yamada et al., 2002). Interestingly, as shown in Figures 1A, 2A, at early stages of brain development, GABAergic signaling is controlled by Reactive Oxygen Species (ROS, Safiulina et al., 2006b), which in ASDs are produced at high levels by abnormal reactive microglia, often associated with a dysfunction of the immune system sustained by a strong inflammatory state (Pangrazzi et al., 2020). The exact mechanisms by which microglia alters the strength of inhibition is still unclear. One possibility is that microglia interacts with CCC via the BDNF-TrkB signaling pathway. Thus, BDNF, released from microglia, would cause disinhibition via downregulation of KCC2 (Rivera et al., 2002; Coull et al., 2005). This effect may involve KCC2 de-phosphorylation with consequent reduction of surface protein expression and increased protein turnover (Wake et al., 2007). In addition, neuroinflammation associated ROS and inflammatory cytokines would activate NKCC1, thereby enhancing neuronal excitation (Alahmari et al., 2015) (Figure 2A). Therefore, it is reasonable to hypothesize that targeting CCC may allow, at least in some cases, to improve cognitive deficits in ASDs by re-establishing a correct GABAergic signaling in neuronal circuits.
One simple approach to restore physiological levels of [Cl−]i is to reduce NKCC1 activity with the selective high affinity antagonist bumetanide. Therefore, this drug has been extensively tested as a potential treatment for a variety of neuropsychiatric disorders (Ben-Ari, 2017). In both syndromic (Fragile X) and non-syndromic (valproic acid and maternal immune activation) animal models of ASDs, which are known to be associated with a dysfunction of GABAergic signaling (Tyzio et al., 2014; Corradini et al., 2018; Fernandez et al., 2019), bumetanide, via maternal administration, is able to reduce chloride accumulation and to rescue behavioral deficits by re-establishing an appropriate E/I balance in the brain of offspring (Tyzio et al., 2014). Similarly, bumetanide (0.5–2 mg twice daily for 3 months) has been demonstrated to ameliorate cognitive functions in autistic children (Ben-Ari, 2017). Following a pilot study by Lemonnier and Ben-Ari (2010), this drug has been tested by the same group in two placebo-controlled randomized studies from 60 and 88 children, respectively (Lemonnier et al., 2012, 2017). These and other studies on autistic children from different countries (Bruining et al., 2015; Hajri et al., 2019; Zhang et al., 2020; Fernell et al., 2021), have demonstrated beneficial effects of bumetanide on cognitive functions, as assessed by Child Autistic Rating Scale (CARS), Clinical Global Impressions Improvement Scale (CGI-I), Social Responsiveness Scale (SRS), and Aberrant Behavior Checklist (ABC), with only few minor side effects (such as mild hypokalaemia; loss of appetite, diuresis, dehydration, asthenia). In one clinical trial, bumetanide was associated with the Applied Behavior Analysis (ABA) training. In this case, more positive results were obtained in children treated with bumetanide and ABA respect to those treated with the ABA alone (Du et al., 2015). In adolescents with ASDs, chronic treatment with bumetanide has been shown to significantly improve visual recognition of emotive figures and to reduce amygdala activation in response to eye contacts (Hadjikhani et al., 2015). This would allow increasing the time spontaneously spent looking in the eyes to acquire the necessary information for social processing (Hadjikhani et al., 2018). However, in a recent double blind randomized study from 92 participants (Sprengers et al., 2021), bumetanide did not differ from placebo on sociability effects (assessed by SRS) but, unlike placebo, exerted clear positive effects on repetitive behavior, a core symptom of ASDs (measured with the Repetitive Behavior Scale-Revised). Although, the reason for the discrepant results between these studies is unclear, the possibility that the drug may be effective only in some forms of autism whose symptoms are more related to a GABAergic dysfunction, cannot be excluded. The high heterogeneity among ASD patients may explain why a large phase 3 clinical trial performed in 50 centers from 14 different countries failed to reach significant differences between bumetanide- and placebo-treated children as recently announced by a press release from Servier and Neurochlore (France). Bumetanide has also been reported to attenuate autism's traits but not seizures in patients with Tuberous Sclerosis (Van Andel et al., 2020). Overall, bumetanide has been shown to exert a symptomatic action mitigating, at least in a subpopulation of autistic children, the severity of symptoms, which reappear after interruption of the treatment. A limitation however in using bumetanide for treating Neurodevelopmental Disorders lies on the fact that this drug has poor pharmacokinetic properties and low capability to cross the blood brain barrier (BBB) to reach either neuronal or non-neuronal targets (Puskarjov et al., 2014a; Virtanen et al., 2020; Löscher and Kaila, 2021; Tóth et al., 2021). Moreover, an active efflux of bumetanide from the brain to the blood, which involves several transporters expressed at the BBB, including organic anion ones, would contribute to maintain a very low concentration of the drug in the brain (Römermann et al., 2017). This raises the possibility that the observed beneficial effects of bumetanide are related to some still unknown peripheral-central type of communication.
Another alternative approach to attenuate E/I imbalance in ASDs is to use KCC2 activators to selectively enhance the activity of KCC2, which is a key player in Cl− homeostasis and, unlike NKCC1, it is expressed mainly by neurons (Schulte et al., 2018; Virtanen et al., 2021). Targeting KCC2 specificity may prevent adverse side effects occurring in non-neuronal tissues or in other organs as in the case of NKCC1. Among these, the KCC2 analog CLP257 was able, by lowering [Cl−]i, to restore chloride transport in neurons with reduced KCC2 activity and to alleviate hypersensitivity in a rat model of neuropathic pain (Gagnon et al., 2013). KCC2 expression is known to be upregulated by phosphatases, insulin growth factor I (IGF-1) and 5-hydroxytryptamine type 2A (5-HT2A) receptors (Kelsch et al., 2001; Bos et al., 2013; Baroncelli et al., 2017). More recently, by generating a robust high-throughput drug screening platform that allows for the rapid assessment of KCC2 gene expression in genome-edited human reporter neurons, Tang et al. (2019) identified a group of small molecules that are able to increase KCC2 expression (KCC2 Expression-Enhancing Compounds or KEECs). In an animal model of Rett syndrome (MeCP2 mutant mouse), exhibiting reduced KCC2 activity, these molecules were able, by enhancing KCC2 expression levels, to restore a proper E/I balance and to ameliorate disease-associated respiratory and locomotion phenotypes. This study did not address whether KEECs can ameliorate other deficits in the mutant mice, In particular social behavioral deficits. Nevertheless, these very promising results pave the way to test whether, at preclinical and clinical levels, these drugs may possibly restore cognitive functions in other neurodevelopmental disorders associated with [Cl−]i imbalance.
Schizophrenia
Schizophrenia is a debilitating psychiatric illness affecting 0.5–1% of the global population, which is characterized by positive (hallucinations and delusions), negative (lack of communication, social interaction, motivation), and cognitive symptoms (Lewis and Lieberman, 2000). Although positive symptoms are the most notable feature of this illness, cognitive disturbances are typically present before the onset of psychosis and are the best predictor of long-term functional outcome (Kahn and Keefe, 2013). The affected domains of cognition include working memory, executive function, learning and long-term memory, visual/auditory perception, and attention (Carter et al., 2008). In particular, working memory impairment is thought to be a core feature of schizophrenia, because it can influence all other observed cognitive deficits (Silver et al., 2003).
Working memory function is associated with oscillatory activity in the gamma frequency range (30–80 Hz) in the prefrontal cortex. The power of gamma oscillations in the prefrontal cortex normally increases in proportion to working memory load (Howard et al., 2003; Jensen et al., 2007), but in individuals with schizophrenia this increase is reduced (Uhlhaas and Singer, 2010). Moreover, these deficits have been detected in both individuals with chronic illness (Cho et al., 2006) and in subjects with the first-episode of psychosis (Minzenberg et al., 2010), suggesting that working memory impairments and disrupted gamma power reflect the disease process of schizophrenia and are not due to chronic illness or the use of antipsychotic medications.
Gamma oscillations refer to the synchronous firing of large ensembles of excitatory glutamatergic pyramidal neurons within and across brain regions, which is paced by GABAergic interneurons (Gonzalez-Burgos and Lewis, 2008). In particular, fast spiking PV+ GABAergic interneurons, connected by gap junctions, shape via feed-forward inhibition, the spatial and temporal profile of pyramidal cells firing, to functionally impact the information processing (Hu et al., 2014). The coordinated activity of glutamatergic and GABAergic neurons in triggering gamma activity, is commonly referred as the pyramidal interneuron network gamma (PING) (Gonzalez-Burgos and Lewis, 2008). The role of GABAergic cells in the generation and modulation of gamma oscillation is strongly supported by numerous studies using pharmacology-, and more recently optogenetics-, based manipulations both in vitro and in vivo (Whittington et al., 1995; Traub et al., 1996; Fuchs et al., 2007; Cardin et al., 2009; Sohal et al., 2009). It is thus reasonable to hypothesize that the altered gamma oscillations dynamics observed in schizophrenic individuals may be caused, at least in part, by functional abnormalities in GABA neurotransmission in the prefrontal cortex.
Another observation supporting a putative role for altered GABAergic transmission in schizophrenia is the frequency of co-morbidity of schizophrenia and epilepsy. In fact, both individual and family history of epileptic disorders appear to be associated with schizophrenia (Qin et al., 2005). A Danish population-based cohort study found that a history of juvenile febrile seizures was associated with a 44% increased risk of schizophrenia, while a history of both febrile seizures plus epilepsy was associated with 204% increased risk of schizophrenia (Vestergaard et al., 2005). Overall, it has been estimated that 2–9% of epileptic patients are also diagnosed with schizophrenia compared with an estimated 1% prevalence in the general population (Clarke et al., 2012; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). This comorbidity may be explained by a common genetic architecture between the two disorders; however, this hypothesis needs to be more extensively explored by systematic genome sequencing of patients affected by both epilepsy and schizophrenia. Another potential cause of comorbidity between epilepsy and schizophrenia might be environmental (or non-genetic). For example, neonatal seizures due to global cerebral hypoxia or intracranial hemorrhage are a strong risk factor for later development of both epilepsy and cognitive impairment (Tekgul et al., 2006). Independently of the underlying causes, and as it is the case of ASD, it has been suggested that alterations in GABAergic transmission may account for the co-morbidity of schizophrenia with epilepsy (Kalkman, 2011).
Overall, numerous in vivo imaging and postmortem studies have consistently revealed alterations in GABA levels and components of GABAergic circuits in the prefrontal cortex of schizophrenic subjects compared to control cohorts, in particular regarding PV+ basket and chandelier GABAergic cells (extensively reviewed by Dienel and Lewis, 2019). As described above, one of the factors regulating the function of GABA neurotransmission is its nature, which can be hyperpolarizing, depolarizing, or shunting depending on the flow of Cl− ions through GABA-A receptors channels once these are activated. This has prompted researchers to look at the reversal potential of GABA and/or the expression of NKCC1, KCC2 cation-chloride co-transporters and their associated regulatory pathways in mouse models of schizophrenia and post-mortem human tissues.
In the subchronic phencyclidine (scPCP)-treated mice, a well-studied animal model mimicking the cognitive impairment symptoms associated with schizophrenia (Jentsch and Roth, 1999; Steeds et al., 2015; Kim et al., 2021) found that the reversal potential of GABA (EGABA) recorded from pyramidal neurons in the infralimbic cortex (the more ventral portion of the prefrontal cortex) was more positive in scPCP-treated mice as compared to those treated with vehicle. This effect was highly specific since it was not found in the prelimbic cortex, which is also part of the prefrontal cortex and sits just above the infralimbic cortex. Changes in EGABA in scPCP mice led to a depolarizing and excitatory action of the neurotransmitter with consequent increase in firing of infralimbic cortex pyramidal neurons that, following a 20 Hz stimulation, generated twice as many action potentials as those from vehicle-treated mice. Using RNAscope in situ hybridization, they further reported that NKCC1 mRNAs were increased in infralimbic, but not in prelimbic neurons, of 5 postnatal weeks-old scPCP mice, while KCC2 mRNAs were not altered. Based on this observation, Kim et al. (2021) hypothesized that limiting NKCC1 function may rescue the electrophysiological and behavioral phenotypes. In keeping with this hypothesis, their data showed that intraperitoneal or focal intracortical injection of the selective NKCC1 blocker bumetanide, before initiating behavioral testing, ameliorated scPCP mice performance on different behavioral tasks assessing declarative memory, working memory, and executive function, more specifically in the novel object recognition, Y-maze spontaneous alternation and operant reversal learning tests. Since bumetanide exhibits low brain penetration (Römermann et al., 2017), it has been frequently questioned whether its effects on behavior in different disease animal models was dependent on its specific inhibitory effects on NKCC1 in the brain or rather due to secondary unspecific effects. In the scPCP mouse model, short hairpin RNAi-mediated downregulation of endogenous NKCC1 mimicked the effects of bumetanide on behaviors, therefore supporting the hypothesis that more depolarized GABA reversal potential in the infralimbic cortex pyramidal neurons plays a role in the cognitive deficits observed in this mouse model.
Another evidence supporting the occurrence of dysregulated Cl− balance in schizophrenia-like animal models comes from the study of the sandy mouse, a dysbindin null mutation. Dysbindin is encoded by DTNBP1. DTNBP1 polymorphisms are considered risk factors for schizophrenia onset (Straub et al., 2002; Van Den Bogaert et al., 2003), even if a consensus has not been reached yet (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Farrell et al., 2015). Recent transcriptome studies showed that, in addition to alterations associated with PV+ GABAergic cells, the dysbindin mutant mice showed reduction of both NKCC1 and KCC2 mRNA levels at the late embryonic stage (Larimore et al., 2017).
In humans, clinical genetic studies support an association between NKCC1 (coded by the gene SLC12A2) and schizophrenia. By correlating blood oxygen level-dependent (BOLD) signals in the prefrontal cortex from schizophrenia patients and controls during a working memory task and genotyping data from a genome wide single nucleotide polymorphism (SNP)-array, Potkin et al. (2009) found that the BOLD signal significantly correlated with the presence of two SNPs within the SLC12A2 gene. Kim et al. (2012) analyzed two independent case control datasets of patients with schizophrenia and healthy controls and found that SNPs in Disrupted-in-Schizophrenia 1 (DISC1) and SLC12A2 interact epistatically to affect risk for schizophrenia. Finally, one recent study identified a novel heterozygous NKCC1 missense variant in a French-Canadian cohort of schizophrenic patients. Functional studies showed that this variant lead to a gain of function of the transporter (Merner et al., 2016).
Conversely, post-mortem studies in human tissue produced conflicting results. Hyde et al. (2011) found a reduction of KCC2-codying mRNA in the hippocampus, but not dorsolateral prefrontal cortex, of schizophrenic patients. The same study did not find any significant difference in full length NKCC1-codying transcripts. On the other hand Sullivan et al. (2015) reported decreased KCC2 protein expression, by western blot, in the dorsal lateral prefrontal cortex, but not the anterior cingulate cortex, in subjects with schizophrenia. Of note, this group also reported that individuals with schizophrenia off antipsychotic medication at the time of death had decreased KCC2 protein expression compared to both normal controls and subjects with schizophrenia on antipsychotics, suggesting that antipsychotic may affect KCC2 expression levels. Other studies found illness-associated alterations in the expression of NKCC1 transcripts. In particular, Dean et al. (2007) reported an upregulation of NKCC1 mRNA in prefrontal cortex tissue from schizophrenia cases as compared to controls. On the other hand, Morita et al. (2014) analyzed the expression of different NKCC1-codying transcripts produced by alternative splicing and reported reduced expression of the shorter variants of NKCC1 transcripts (NKCC1b and 1-2a) in the dorsolateral prefrontal cortex of subject with schizophrenia compared to a control cohort. Consistently, Zhang et al. (2021) reported reduced NKCC1 mRNA levels in peripheral blood mononuclear cells of patients presenting with the first episode of schizophrenia, and thus not under the effects of antipsychotic medications. Finally, Arion and Lewis (2011) analyzed mRNA expression levels of NKCC1 and KCC2 and their associated regulatory kinases STK39, OXSR1, WNK1, WNK3, and WNK4 in the dorsolateral prefrontal cortex of subjects with schizophrenia compared to non-psychiatric subjects and reported no differences in the expression of either KCC2 or NKCC1 transcripts. However, they found overexpression of OXSR1 and WNK3 transcripts in schizophrenia. These alterations of transcript levels were consistent across subjects with the illness. In addition, they were not altered in monkeys chronically exposed to antipsychotic medications. Together, these findings suggest that the observed difference in OXSR1 and WNK3 levels in schizophrenic individuals were not caused by other factors (medications, substance abuse, etc.). WNK3 is a potent activator of NKCC1, while it can inhibit KCC2 activity (Kahle et al., 2005; de Los Heros et al., 2006). OXSR1 binds to and phosphorylates NKCC1, resulting in an increase of NKCC1 activity (Vitari et al., 2006). Therefore, if increased OXSRI and WNK3 mRNA levels translate into increased protein and thus kinase activity, then this would lead to both enhanced NKCC1 and decreased KCC2 activity, respectively, thus steering neurons toward a predicted higher intracellular Cl− concentration (Arion and Lewis, 2011).
The discrepancy in the findings reported by these studies could be explained in part by differences in the technical approaches used to analyse NKCC1/KCC2 levels and in the medication history of the subjects, by the relative small sample size of each study and heterogeneity of the disease. It is possible that Cl− imbalance may be restricted to specific brain regions, or neuronal circuits, and thus, that only same types of behavioral abnormalities may be affected by targeting Cl− imbalance. More likely, due to the heterogeneity of the diseases, Cl− imbalance mediated by dysregulation of chloride transporters may not be a common pathogenic mechanism in all patients, but it may affect only a subset. If the latter hypothesis is correct, then it is essential to develop and characterize reliable in vivo biomarkers for measuring excitation/inhibition imbalance with high spatial and temporal resolution during cognitive tasks. These biomarkers would help stratifying the patients in different populations and identifying those with higher probability of responding to Cl− balance-targeting medications.
Epilepsy
Epilepsy is a neurological condition characterized by recurrent spontaneous seizures that can be classified, perhaps in an oversimplified manner, as primary generalized (e.g., those occurring in absence epilepsy), and focal (with possible secondary generalization), such as those observed in patients presenting with mesial temporal lobe epilepsy (MTLE) or focal cortical dysplasia (Avoli and Gloor, 1987). Here, we will mainly address the role of GABAA receptor signaling in the epileptiform synchronization of limbic networks. Accordingly, focal seizures recorded in MTLE patients arise from limbic structures such as the hippocampus, the amygdala and the rhinal cortices (Gloor, 1997). To note that an epileptic brain with focal abnormalities generates interictal spikes (i.e., short electrographic events <2 s in duration, which are not accompanied by any detectable clinical symptom) (arrows and asterisks in Figures 3Aa,B) and ictal discharges or seizures (i.e., abnormal, hypersynchronous activity lasting up to several tens of seconds, which disrupt normal brain function) (continuous lines in Figures 3Ab, B) (Avoli and Gloor, 1987).
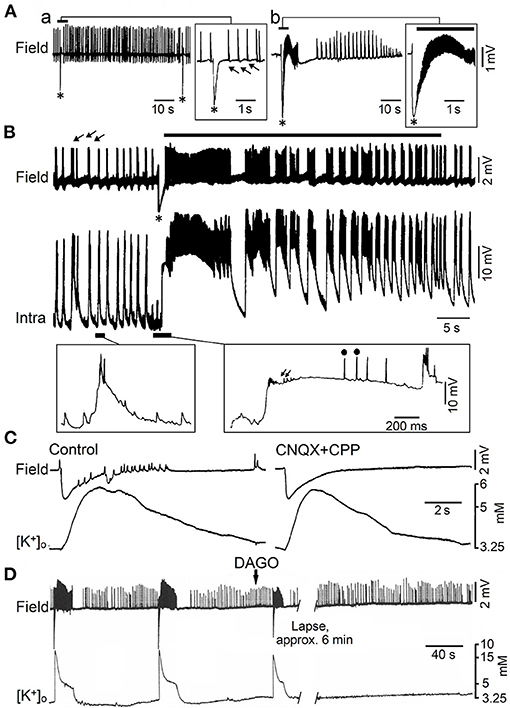
Figure 3. Epileptiform activity induced by 4AP in juvenile rat hippocampal slices maintained in vitro. (A) Field potential recordings obtained during perfusion with medium containing 4AP from the CA3 subfield of adult (a) and young (14 day-old) brain slices (b). Note that only fast (arrows) and slow interictal discharges (asterisk) are recorded in adult brain slices while ictal (continuous line) and interictal epileptiform discharges occur in the young hippocampus; note also in (b) that the slow (asterisk) interictal discharge leads to the onset of a prolonged ictal event. (B) Field and intracellular (Intra) recordings obtained during perfusion with medium containing 4AP from the CA3 area of a 22-day-old rat hippocampal slice; arrows point to the fast interictal spikes while asterisk identifies the slow interictal that initiates the ictal discharge (thick line). Selected portions of the intracellular recording are illustrated at an expanded time base in the panels below; there, small arrows and dots identify depolarizing potentials and fractionated spikes, respectively. (C) Effects induced by application of non-NMDA (CNQX) and NMDA (CPP) receptor antagonists on the 4AP-induced epileptiform activity recorded with field and K+ selective microelectrodes from the CA3 subfield of a 19-day-old rat hippocampal slice. Note that this pharmacological procedure blocks the fast interictal events as well as the short-lasting ictal discharge; however the elevation in extracellular [K+] that correlates with the negative-going slow field potential is not modified. (D) Effects of the mu-opioid receptor agonist DAGO on the synchronous activity induced by 4AP in the CA3 subfield of a 15-day-old rat; note that DAGO abolishes the negative-going slow field potential along with the subsequent ictal discharge while fast interictal spikes continue to occur. [(A) Is modified from Fueta and Avoli, 1992; (B) is modified from Avoli et al. (1993); (C,D) are modified from Avoli et al. (1996b)].
Epilepsy is believed to result from a pathological shift of the E/I balance toward excitation. Accordingly, in the 1950s, clinical studies reported that interfering with GABA synthesis leads to convulsions (Coursin, 1954). In the following decades, in vitro experiments have demonstrated that several convulsive drugs are GABAA receptor antagonists (Schwartzkroin and Prince, 1980). In addition, in vivo studies have highlighted a decrease of GABAergic inhibition shortly before the onset of electrographic seizures recorded from the hippocampus (Ben-Ari et al., 1979) or neocortex (Kostopoulos et al., 1983). These findings were in line with those indicating that the functional disconnection of interneurons from excitatory inputs impairs inhibition in animal models of focal seizures (Sloviter, 1987), and that alterations in GABA transporter function or in GABAA receptor subunits composition occur in patients affected by focal epileptic disorders and in animal models mimicking them (McDonald et al., 1991; Johnson et al., 1992; Williamson et al., 1995; Brooks-Kayal et al., 1998). Therefore, in the 1990s, weakening of inhibition was considered a sine qua non conditio for the occurrence of focal seizures and thus for the establishment of epilepsy.
However, successive studies have challenged this view since epileptiform activity can occur in hippocampal slices maintained in vitro during pharmacological manipulations that do not interfere with GABAA receptor mediated inhibition and even enhance it. These experimental procedures include the application of Mg2+ free-medium (Mody et al., 1987; Derchansky et al., 2008) or of K+ channel blockers such as 4-aminopyridine (4AP) (Voskuyl and Albus, 1985; Rutecki et al., 1987; Perreault and Avoli, 1991, 1992). As illustrated in Figure 3Aa, field potential recordings obtained from the CA3 subfield of adult rat hippocampal slices treated with 4AP, have revealed the occurrence of two distinct types of interictal activity. The first type (arrows in Figure 3Aa), also identified as fast, occurs frequently, is driven by CA3 network, and is abolished by non-NMDA glutamatergic receptor antagonists. The second type (asterisks in Figure 3Aa) has a lower rate of occurrence, can initiate in any hippocampal areas, continues to occur when ionotropic glutamatergic excitatory transmission is blocked, and it is abolished by GABAA receptor antagonists (Perreault and Avoli, 1991, 1992) suggesting its GABAergic origin; hence, these 4AP-induced, interictal events have been often referred to as slow GABAergic spikes (Perreault and Avoli, 1991, 1992). It has been also found that, in hippocampal slices obtained from young (10–24 day old) rats, 4AP induces ictal discharges (Chesnut and Swann, 1988; Avoli, 1990), which are preceded by a slow GABAergic spike (Figure 3Ab, continuous line and asterisk, respectively). Shortly later, intracellular recordings from CA3 pyramidal cells in young hippocampal slices demonstrated that this initial GABAergic spike is characterized by a prolonged depolarization, associated to sparse, fractionated (presumably ectopic) action potentials, leading to the generation of repetitive bursts of action potentials once the ictal activity is established (Avoli et al., 1993) (Figure 3B).
The mechanistic link between such initial synchronous GABAergic event and the onset of electrographic seizure activity in the young rodent CA3 area was supported by findings obtained in successive studies. As illustrated in Figure 3C, the initial GABAergic spikes were mirrored by sizeable elevations in extracellular [K+] that were not influenced by application of ionotropic glutamatergic receptor antagonists (Avoli et al., 1996b; see also Figure 5). In addition, GABAA receptor antagonists or pharmacological procedures interfering with GABA signaling, such as the application of a μ-type opioid receptor agonist (Capogna et al., 1993), abolished both the initial GABAergic spike and the subsequent ictal activity, that was replaced by on-going, short-lasting interictal spikes (Figure 3D) (Avoli et al., 1996b).
Overall, these studies have demonstrated that during application of 4AP, the slow GABAergic spikes mostly reflect the synchronous activation of postsynaptic GABAA receptors that leads to increases in extracellular [K+] sufficiently large to trigger ictal activity (Morris et al., 1996; Lamsa and Kaila, 1997). Originally, it was proposed that such mechanism only occurs in the juvenile hippocampus because of a decreased homeostasis of extracellular [K+] at this early stage of brain maturation (Avoli et al., 1993, 1996b). This hypothesis was, however, not confirmed in successive experiments performed in adult rodent brain slices that could include the entorhinal, perirhinal, piriform, insular cortices, or the amygdala (see for review, Avoli and de Curtis, 2011). In all these cortical structures, 4AP was able to induce slow interictal spikes along with ictal discharges that were often preceded by a single (also termed “sentinel”) slow GABAergic event (Figure 4A) (see also data obtained from the guinea pig whole brain preparation, Carriero et al., 2010). Seizures with a similar electrographic onset have been recorded with intracranial electrodes in the adult human's brain of patients affected by focal epileptic disorders, including MTLE (Perucca et al., 2014) as well as in in vivo animal models (Lévesque et al., 2012).
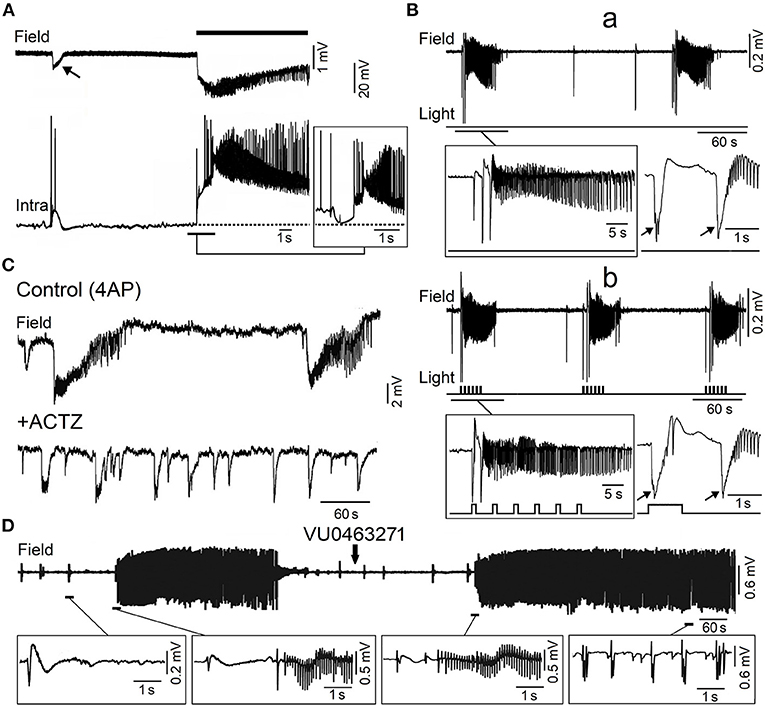
Figure 4. Epileptiform activity induced by 4AP in the adult entorhinal cortex in in vitro brain slices. (A) Simultaneous field (Field) and intracellular (Intra) recordings obtained from adult rat entorhinal cortex in vitro during bath application of 4AP; the recordings shown in the insert were obtained from a different neuron that was depolarized by intracellular current injection to a steady membrane potential at ~62 mV. (B) Low voltage fast onset ictal discharges occur spontaneously during bath application of 4AP in the mouse entorhinal cortex (a) but are also triggered by optogenetic stimulation of parvalbumin positive-interneuron (b). The onset of the ictal discharges occurring under each condition is further expanded in the inserts below; note that the ictal discharges are preceded by one or two slow interictal spikes (arrows) under both spontaneous and stimulated conditions. (C) Field potential recordings obtained from the rat entorhinal cortex under control conditions (4AP) and 20 min after bath application of carbonic anhydrase inhibitor acetazolamide (ACTZ, 10 μM); note that this pharmacological procedure greatly reduces the duration of the ictal discharges. (D) Effects of the KCC2 blocker VU0463271 on the 4AP-induced field potential activity recorded from the rat entorhinal cortex; note that addition of VU0463271 induces a pattern of continuous interictal spikes in the field potential recording. Enlarged examples of the field potential recordings are shown below. [(A) Is modified from Lopantsev and Avoli (1998); (B) is modified from Shiri et al. (2016); (C) is modified from Hamidi and Avoli (2015a); (D) is modified from Chen et al. (2019)].
These ictal discharges have been termed low-voltage fast onset seizures since they initiate with a pattern of low voltage oscillatory activity in the beta-gamma range (see for review Avoli et al., 2016).
As illustrated in Figure 4A, the onset of an ictal discharge recorded in vitro in the adult rat entorhinal cortex in the presence of 4AP coincides with principal cell depolarization that, as observed in young rat CA3 pyramidal neurons (cf. with insert in Figure 3B), is associated with few “fractionated” (and thus presumptive ectopic) action potentials. Moreover, this initial depolarizing event becomes hyperpolarizing when the neuronal membrane potential is depolarized to values less negative than −60 mV by injecting a steady depolarizing current (Figure 4A; Lopantsev and Avoli, 1998). Indeed, it was known at that time that activation of GABAA receptors can depolarize cortical neurons since these receptors are permeable not only to Cl− but also to , which has an equilibrium potential more positive than Cl− (Grover et al., 1993; Kaila, 1994). Similar intracellular patterns were also recorded at the onset of 4AP-induced ictal discharges in principal cells of the amygdala in an in vitro slice preparation (Benini et al., 2003).
These results, therefore, demonstrate that, paradoxically, the initiation of electrographic seizures occurring in vitro coincides with, and thus may be caused by, a robust synchronous inhibitory event. In line with this hypothesis, studies performed by several laboratories have shown that interneurons fire at the onset of these ictal discharges as well as that GABAA receptor antagonists can abolish them (see for review, Avoli and de Curtis, 2011; Avoli et al., 2016). Intense interneuronal firing leading to GABA release and subsequent, excessive activation of GABAA receptors, as identified at the onset of 4AP-induced ictal discharges, coincides also in the adult entorhinal cortex with elevations of extracellular [K+] (Avoli et al., 1996a; Librizzi et al., 2017). In turn, these transient increases in extracellular [K+] depolarize, and thus recruit neighboring neurons into the synchronous firing that is associated with the ongoing ictal activity (see for review: Avoli and de Curtis, 2011; Avoli et al., 2016). The essential role of interneurons in initiating 4AP-induced ictal discharges was later confirmed by optogenetic studies performed in several laboratories in the entorhinal (Shiri et al., 2015, 2016; Yekhlef et al., 2015) and somatosensory cortex (Chang et al., 2018). As shown in Figure 4B, optogenetic activation of parvalbumin- or somatostatin-positive interneurons initiates ictal discharges with an onset pattern similar to what identified in spontaneously occurring events. Specifically, the onset of both spontaneous and optogenetic-induced ictal discharges is characterized by one-two interictal-like spikes that are followed by fast, beta-gamma oscillations, i.e., the hallmark of low-voltage fast ictal discharges. In line with the participation of inhibitory cells in the initiation of this type of seizure activity, single unit recordings from the seizure onset zone in epileptic patients have shown that GABAergic interneurons increase their firing rate, during the onset of low-voltage fast seizures, earlier than excitatory cells (see for review, Weiss et al., 2019). To note, as well, that 4AP-induced electrographic seizures associated with an increase of extracellular [K+], presumably due to the high firing rate of interneurons resulting in the activation of postsynaptic GABAA receptors by the massive release of GABA, occur in neocortical slices obtained from epileptic patients with Taylor's type focal cortical dysplasia (D'Antuono et al., 2004; Gigout et al., 2006) but not in brain slices from epileptic patients with no obvious structural abnormalities (Louvel et al., 2001).
As mentioned above, findings obtained from the in vitro 4AP model point at the synchronous postsynaptic activation of GABAA receptors, leading to membrane depolarizations—which are contributed by efflux (Figure 5A; Grover et al., 1993; Kaila, 1994)—and to sizable increases in extracellular [K+] (Figure 5C; Avoli et al., 1996a,b; Morris et al., 1996; Lamsa and Kaila, 1997), as the fundamental mechanism for triggering low-voltage fast ictal activity (Avoli and de Curtis, 2011; Avoli et al., 2016). In line with this hypothesis, application of the carbonic anhydrase inhibitor acetazolamide reduced the duration and the interval of occurrence of ictal discharges induced by 4AP in the piriform and entorhinal cortices (Hamidi and Avoli, 2015a) (Figure 4C). Also to note that Zuckermann and Glaser (1968) discovered over 50 years ago that elevations of [K+]o induce neuronal hyperexcitability (Figure 5B) and seizures. Successive studies have demonstrated that the increased extracellular [K+] weakens inhibition by causing a positive shift of the reversal of GABAA receptor mediated inhibitory currents (Jensen et al., 1993), and leads to neuronal network resonance, which generates oscillatory patterns in the beta-gamma range (Bartos et al., 2007). It has also been established that activation of GABAA receptors leads to accumulation of intracellular [Cl−], which in turn activates KCC2 which extrudes both Cl− and K+ from the intraneuronal compartment (Figure 5A; Viitanen et al., 2010). Therefore, the activity of KCC2 may play a role in seizure generation and epileptogenesis (see for review: Di Cristo et al., 2018).
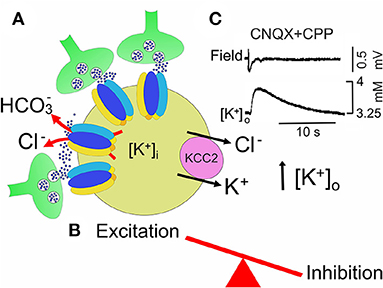
Figure 5. In the 4-AP model of MTLE, neuronal synchronization preceeding ictal dischages, may occur in the absence of glutamatergic transmission. (A) GABA, abundantly released from GABAergic interneurons (green) which make synapses with a principal cell (yellow), binds to GABAA receptor-channels, permeable to chloride and . permeability would allow to shift the equilibrium potential for GABA (EGABA) toward more positive values respect to . The high firing rate of GABAergic interneurons, electrically coupled via gap junctions, leads to accumulation of K+ in the extracellular space (high [K+]o), which is instrumental in triggering ictal activity. By producing a further shift of EGABA toward more positive potentials, high [K+]o contributes to weaken inhibition. In addition, activation of GABAA receptors would cause an inward flux of Cl− that would activate the cation-chloride exporter KCC2. By extruding both Cl− and K+, this would further enhance neuronal excitability by further rising [K+]o (B). (C) Field potential recording of a slow GABAergic event due to the synchronous activation of GABAA receptors (slow GABAergic spike) preceeding elevation of extracellular K+ measured with ion-sensitive electrodes in the presence of AMPA and NMDA receptor antagonists, CNQX and CPP, respectively (modified from Avoli et al. (1996a)].
In line with this view, 4AP-induced electrographic ictal discharges recorded from the entorhinal and piriform cortices in vitro can be abolished or facilitated by inhibiting or enhancing the activity of KCC2, respectively (Hamidi and Avoli, 2015b; Chen et al., 2019). As illustrated in Figure 4D, the KCC2 antagonist VU0463271 transformed the dynamic pattern of 4AP-induced interictal and ictal activity into a continuous pattern of interictal-like epileptiform events. Similar effects (i.e., the induction of recurrent, robust spiking in both 0-Mg2+ and 4AP in vitro models) have been reported by Moss' laboratory in the presence of KCC2 antagonists (Sivakumaran et al., 2015; Kelley et al., 2016; Moore et al., 2018). Furthermore, these investigators have found that in vivo microinfusion of VU0463271 into the mouse dorsal hippocampus induces recurrent epileptiform discharges thus confirming that KCC2 function is essential for making GABAA receptor mediated inhibition operative (Sivakumaran et al., 2015). More recently, Dzhala and Staley (2021) have identified in an organotypic hippocampal slice model that the KCC2 antagonist VU0463271, by decreasing Cl− extrusion from the intracellular compartment, increases its intracellular elevations during ictal events and discloses a pattern of continuous interictal-like discharges resembling status epilepticus. However, in one in vitro study performed by Moss' group, KCC2 antagonism could prolong the ictal events induced by 4AP in the entorhinal cortex (Kelley et al., 2016). To note also that Silayeva et al. (2015) have reported that KCC2 activity is essential for controlling status epilepticus induced by systemic injections of kainic acid in vivo.
In vitro electrophysiological recordings from human resected “epileptogenic” brain tissue have demonstrated that the shift of GABAA receptor signaling from hyperpolarizing to depolarizing direction can contribute to network hyperexcitability and neuronal synchronization. Thus, in hippocampal slices from brain tissue removed from patients affected by drug-resistant forms of MTLE, the depolarizing action of GABA in a subset of pyramidal cells (~30%) is present during interictal spikes recorded in the subiculum, the output structure of the hippocampus that projects to the temporal lobe, suggesting a perturbed homeostasis of intracellular [Cl−] Cohen et al. (2002). This hypothesis has been confirmed by Huberfeld et al. (2007) who, using combined intracellular recordings and KCC2 immunochemistry, found that subicular cells generating hyperpolarizing responses to GABA were immunopositive for KCC2, whereas cells generating depolarizing responses were immunonegative. Thus, the depolarizing responses to GABA presumably result from excessively high intracellular [Cl−] since they were abolished (together with spontaneously occurring field potential events) by bumetanide, a selective blocker of NKCC1; this pharmacological procedure caused a shift of EGABA toward more hyperpolarized values, reinstating GABAA-mediated inhibition (Huberfeld et al., 2015). A depolarizing action of GABA, resulting from altered intracellular [Cl−] homeostasis, has been also detected in cortical tissue samples obtained from pediatric patients undergoing surgical resection for the treatment of pharmaco-resistant forms of focal epilepsy due to cortical dysplasia (Abdijadid et al., 2015).
An immature depolarizing GABAergic signaling, resulting from a down-regulation of KCC2, has been found also in Scn1b−/− and Scn1a+/− mouse models as well as in brain extracts from patients affected by the Dravet Syndrome, a devastating developmental form of epileptic encephalopathy (Yuan et al., 2019). These data point to KCC2, the major Cl− extruder, as a key determinant for maintaining intracellular [Cl−] homeostasis and for regulating neuronal excitability. A rise of intracellular [Cl−] following KCC2 dysfunctions impairs GABAA-mediated inhibition, with consequent changes in the E/I balance toward excitation, making the brain more prone to seizures. Interestingly, several mutations of the SLC12A5 gene encoding for KCC2 have been identified in epileptic patients (Duy et al., 2019). Puskarjov et al. (2014b) have reported the R952H mutation of the SLC12A5 gene in an Australian family with early childhood onset of febrile seizures. In rodent neurons, this mutation led to deficits in neuronal Cl− extrusion associated to impaired formation of cortical dendritic spines. Febrile seizures, which might later lead to idiopathic generalized seizures, have been reported to occur in a French-Canadian cohort carrying both the R952H and the R1049C KCC2 mutations, located in conserved residues of the KCC2 cytoplasmic C-terminus, an important regulatory region of transport function (Kahle et al., 2014).
The role played by the activation of GABAA receptors in promoting epileptiform activity is also supported by the ability of hippocampal networks maintained in vitro to generate ictal discharges during pharmacological blockade of both GABAB and ionotropic glutamatergic receptors (Uusisaari et al., 2002). Such unexpected role of GABAA signaling may explain the limited therapeutic efficacy of some antiepileptic drugs that were developed to potentiate GABAA receptor function; these compounds include progabide, γ-vinyl-GABA and tiagabine (Rogawski and Löscher, 2004). Moreover, benzodiazepines—which increase GABAA receptor function in the brain by acting on the allosteric “benzodiazepine site” (Costa et al., 1975; Choi et al., 1977)—can halt seizure activity and status epilepticus (Pang and Hirsch, 2005) but do not represent first choice drugs for treating chronic epileptic conditions. It is also worth mentioning that compatible with an excitatory action of GABA, benzodiazepines may paradoxically worsen seizures (Perucca et al., 1998). Bumetanide, by inhibiting NKCC1, may have a beneficial effect, restoring neuronal intracellular [Cl−] as demonstrated in the case of a girl affected by epilepsy, cortical dysplasia and ASD (Bruining et al., 2015). Beneficial effects of bumetanide were also detected in a double-blind pilot study on 43 randomized babies affected by neonatal seizures caused by hypoxic-ischemic encephalopathy. This diuretic, added to phenobarbital in dose-escalation design, was able to reduce, in a statistically significant way, seizure burden in the group of subjects treated with phenobarbital and bumetanide (n = 27), respect to the control group treated with phenobarbital alone (n = 16) (Soul et al., 2021). However, more work is required to establish bumetanide exposure response and safety. Although in the hypoxic-ischemic encephalopathy, the BBB may be compromised (Römermann et al., 2017), usually bumetanide's effectiveness is limited by its poor brain penetration, an effect that can be overcome with new derivatives, currently under development, capable of better permeate the BBB (Savardi et al., 2021).
Conclusions and Future Perspectives
The discovery of CCC as key regulators of neuronal Cl− concentration has allowed to better understand how GABA, acting on GABAA receptors, influences via its depolarizing and excitatory action, several developmental processes whose alterations lead to pathological conditions including ASD, schizophrenia and epilepsy. However, in spite of such progress many questions are still open.
Although bumetanide has shown to have positive effects in a wide range of pathological conditions, its exact mechanisms of action are still poorly understood. In addition, in humans, its beneficial effects are symptomatic and it is unclear whether they are linked to its ability to shift GABA action from the depolarizing to the hyperpolarizing direction by restoring low [Cl−]i and a proper E/I balance in neuronal ensembles. The high expression levels of NKCC1 on microglia opens, however new perspectives on the mechanisms of action of this transporter, especially in relation to the role played by activated glial cells in brain inflammation and oxidative stress, which are key features of many neuropsychiatric diseases. Furthermore, to better understand the mechanisms controlling the expression of CCC in the brain and their contribution in shaping, via GABAergic signaling, neuronal circuits in both physiological and pathological conditions, it will be crucial to clarify how BDNF/TrkB signaling pathway regulates NKCC1 neuronal expression early in postnatal life (Badurek et al., 2020). An alternative way to re-establish in neurodevelopmental disorders a proper E/I balance by acting on GABAA-mediated neurotransmission may be the development of new therapeutic tools selectively targeting the chloride exporters KCC2 which in the CNS are exclusively expressed on neurons (Gagnon et al., 2013; Puskarjov et al., 2014a). By acting either on KCC2 membrane trafficking or on their intrinsic transport kinetics, these compounds will allow attenuating neuronal [Cl−]i, reinstating an appropriate Cl− homeostasis in selective brain regions. However, new KCC2 activators should not interfere with the well-known KCC2 structural function on dendritic spines formation and dynamics (Blaesse et al., 2009). These new molecules may be particularly useful for treating drug-resistant forms of epilepsy, such as cortical focal dysplasia, occurring in the pediatric age. It is worth mentioning that the use of KCC2 analogs may be limited by their possible proconvulsant effects triggered by accumulation of K+ in the extracellular space.
Studies from animal models have allowed identifying early changes in GABAergic signaling as a contributing cause of cognitive deficits observed in Neurodevelopmental Disorders. These are often associated with a loss of particular subtypes of inhibitory interneurons which, by pacing principal cells, give rise to coherent network oscillations, thought to support different behavioral states of the animals and high cognitive tasks. However, with the exception of epilepsy, a direct proof that these processes may occur also in individual affected by neurodevelopmental disorders remains to be demonstrated, making difficult to translate data obtained in preclinical studies from animal models to humans. In the latter case, evidence in favor of an altered GABAA-mediated neurotransmission indirectly relies on: (i) genetic studies; (ii) immunohistochemistry from postmortem brain samples showing reduction in GABAergic markers or particular subtypes of GABAergic interneurons; (iii) high incidence of epileptic activity as a comorbidity effect; (iv) alterations in oscillatory activity particularly in the gamma power, detected on the EEG or MEG; and (v) in some cases the paradoxical action of benzodiazepines (for a review see Cellot and Cherubini, 2014b).
An innovative approach consisting in reprogramming human dermal fibroblast obtained through skin biopsy from patients (at progressive stages of the diseases) into induced pluripotent stem cells (iPSCs) and 3D cerebral organoids will provide an innovative platform to uncover disease mechanisms directly in humans. These processes will allow identifying disease mechanisms in a personalized type of manner and to test novel strategies for prevention, diagnosis, patient stratification, therapy and/or rehabilitation.
Author Contributions
EC, GDC, and MA designed, revised the literature, and wrote the manuscript. EC and MA prepared the Figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Telethon (GGP 16083) and from Fondo Ordinario Enti (FOE D.M 865/2019) funds in the framework of a collaboration agreement between the Italian National Research Council and EBRI (2019–2021) to EC; from Canadian Institutes for Health Research and Natural Sciences and Engineering Research Council of Canada to GDC; from Canadian Institutes of Health Research (Grants 8109, 74609 and 130328) and the Savoy Foundation to MA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank our colleagues who contributed to original works reported in this review. We are also grateful to the members of our labs for useful discussions and suggestions.
References
Abdijadid, S., Mathern, G. W., Levine, M. S., and Cepeda, C. (2015). Basic mechanisms of epileptogenesis in pediatric cortical dysplasia. CNS Neurosci. Ther. 21, 92–103. doi: 10.1111/cns.12345
Aguado, F., Carmona, M. A., Pozas, E., Aguiló, A., Martínez-Guijarro, F. J., Alcantara, S., et al. (2003). BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development 130, 1267–1280. doi: 10.1242/dev.00351
Alahmari, K. A., Prabhakaran, H., Prabhakaran, K., Chandramoorthy, H. C., and Ramugounder, R. (2015). Antioxidants and NOS inhibitors selectively targets manganese-induced cell volume via Na-K-Cl cotransporter-1 in astrocytes. Brain Res. 1610, 69–79. doi: 10.1016/j.brainres.2015.03.035
Alvarez-Leefmans, F. J., León-Olea, M., Mendoza-Sotelo, J., Alvarez, F. J., Antón, B., and Garduño, R. (2001). Immunolocalization of the Na(+)-K(+)-2Cl(-) cotransporter in peripheral nervous tissue of vertebrates. Neuroscience 104, 569–582. doi: 10.1016/S0306-4522(01)00091-4
Arion, D., and Lewis, D. A. (2011). Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch. Gen. Psychiatry 68, 21–31. doi: 10.1001/archgenpsychiatry.2010.114
Avoli, M. (1990). Epileptiform discharges and a synchronous GABAergic potential induced by 4-aminopyridine in the rat immature hippocampus. Neurosci. Lett. 117, 93–98. doi: 10.1016/0304-3940(90)90125-S
Avoli, M., Barbarosie, M., Lücke, A., Nagao, T., Lopantsev, V., and Köhling, R. (1996a). Synchronous GABA-mediated pote,ntials and epileptiform discharges in the rat limbic system in vitro. J. Neurosci. 16, 3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996
Avoli, M., and de Curtis, M. (2011). GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol 95, 104–132. doi: 10.1016/j.pneurobio.2011.07.003
Avoli, M., de Curtis, M., Gnatkovsky, V., Gotman, J., Köhling, R., Lévesque, M., et al. (2016). Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J. Neurophysiol. 115, 3229–3237. doi: 10.1152/jn.01128.2015
Avoli, M., and Gloor, P. (1987). “Epilepsy” in Encyclopedia of Neuroscience, Vol I. ed G. Adelman (Basel; Stuttgart: Birkhäuser Boston), 400–403.
Avoli, M., Louvel, J., Kurcewicz, I., Pumain, R., and Barbarosie, M. (1996b). Extracellular free potassium and calcium during synchrono,us activity induced by 4,-aminopyridine in the juvenile rat hippocampus. J. Physiol. 493, 707–717. doi: 10.1113/jphysiol.1996.sp021416
Avoli, M., Psarropoulou, C., Tancredi, V., and Fueta, Y. (1993). On the synchronous activity induced by 4-aminopyridine in the CA3 subfield of juvenile rat hippocampus. J. Neurophysiol. 70, 1018–1029. doi: 10.1152/jn.1993.70.3.1018
Awad, P. N., Amegandjin, C. A., Szczurkowska, J., Carriço, J. N., Fernandes do Nascimento, A. S., Baho, E., et al. (2018). KCC2 regulates dendritic spine formation in a brain-region specific and BDNF dependent manner. Cereb. Cortex 28, 4049–4062. doi: 10.1093/cercor/bhy198
Badurek, S., Griguoli, M., Asif-Malik, A., Zonta, B., Guo, F., Middei, S., et al. (2020). Immature Dentate Granule Cells Require Ntrk2/Trkb for the Formation of Functional Hippocampal Circuitry. iScience May 22;23:101078. doi: 10.1016/j.isci.2020.101078
Balena, T., and Woodin, M. A. (2008). Coincident pre- and postsynaptic activity downregulates NKCC1 to hyperpolarize E(Cl) during development. Eur. J. Neurosci. 27, 2402–2412. doi: 10.1111/j.1460-9568.2008.06194.x
Baroncelli, L., Cenni, M. C., Melani, R., Deidda, G., Landi, S., Narducci, R., et al. (2017). Early IGF-1 primes visual cortex maturation and accelerates developmental switch between NKCC1 and KCC2 chloride transporters in enriched animals. Neuropharmacology 113, 167–177. doi: 10.1016/j.neuropharm.2016.02.034
Bartos, M., Vida, I., and Jonas, P. (2007). Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 8, 45–56. doi: 10.1038/nrn2044
Ben-Ari, Y. (2017). NKCC1 chloride importer antagonists attenuate many neurological and psychiatric disorders. Trends Neurosci. 40, 536–554. doi: 10.1016/j.tins.2017.07.001
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284. doi: 10.1152/physrev.00017.2006
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. doi: 10.1177/1073858412438697
Ben-Ari, Y., Krnjevi,ć, K., and Reinhardt, W. (1979). Hippocampal seizures and failure of inhibition. Can. J. Physiol. Pharmacol. 57, 1462–1466. doi: 10.1139/y79-218
Benini, R., D'Antuono, M., Pralong, E., and Avoli, M. (2003). Involvement of amygdala networks in epileptiform synchronization in vitro. Neuroscience 120, 75–84. doi: 10.1016/S0306-4522(03)00262-8
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838. doi: 10.1016/j.neuron.2009.03.003
Bolea, S., Avignone, E., Berretta, N., Sanchez-Andres, J. V., and Cherubini, E. (1999). Glutamate controls the induction of GABA-mediated, depolarizing potentials through AMPA receptors in neonatal rat hippo.mpal slices. J. Neurophysiol. 81, 2095–2102. doi: 10.1152/jn.1999.81.5.2095
Bonifazi, P., Goldin, M., Picardo, M. A., Jorquera, I., Cattani, A., Bianconi, G., et al. (2009). GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science 326, 1419–1424. doi: 10.1126/science.1175509
Bortone, D., and Polleux, F. (2009). KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71. doi: 10.1016/j.neuron.2009.01.034
Bos, R., Sadlaoud, K., Boulenguez, P., Buttigieg, D., Liabeuf, S., Brocar, C., et al. (2013). Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 110, 348–353. doi: 10.1073/pnas.1213680110
Bozzi, Y., Provenzano, G., and Casarosa, S. (2018). Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/inhibition imbalance. Eur. J. Neurosci. 47, 534–548. doi: 10.1111/ejn.13595
Brooks-Kayal, A. R., Shumate, M., Jin, H., Rikhter, T. Y., and Coulter, D. A. (1998). Selective changes in single cell GABAA receptor sub,unit expression and function in temporal lobe epilepsy. Nat. Med. 4, 1166–1172. doi: 10.1038/2661
Bruining, H., Passtoors, L., Goriounova, N., Jansen, F., Hakvoort, B., de Jonge, M., et al. (2015). Paradoxical benzodiazepine response: a ration,ale for bumetanide in neurodevelopmental disorders? Pediatrics 136, 539–543. doi: 10.1542/peds.2014-4133
Buzsáki, G., and Draguhn, A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929. doi: 10.1126/science.1099745
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007
Capogna, M., Gähwiler, B. H., and Thompson, S. M. (1993). Mechanism of mu-opioid receptor-mediated presynaptic inhibition in the rat hippocampus in vitro. J. Physiol. 470, 539–558. doi: 10.1113/jphysiol.1993.sp019874
Cardin, J. A., Carlén, M., Meletis, K., Knoblich, U., Zhang, F., Deisseroth, K., et al. (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. doi: 10.1038/nature08002
Carmona, M. A., Pozas, E., Martínez, A., Espinosa-Parrilla, J. F., Soriano, E., and Aguado, F. (2006). Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb. Cortex 16, 47–63. doi: 10.1093/cercor/bhi083
Carriero, G., Uva, L., Gnatkovsky, V., Avoli, M., and de Curtis, M. (2010). Independent epileptiform discharge patterns in the olfactory and limbic areas of the in vitro isolated Guinea pig brain during 4-aminopyridine treatment. J. Neurophysiol. 103, 2728–2736. doi: 10.1152/jn.00862.2009
Carter, C. S., Barch, D. M., Buchanan, R. W., Bullmore, E., Krystal, J. H., Cohen, J., et al. (2008). Identifying cognitive mechanisms targeted for treatment development in schizophrenia: an overview of the first meeting of the Cognitive Neuroscience Treatment Research to Improve Cognition in Schizophrenia Initiative. Biol. Psychiatry 64, 4–10. doi: 10.1016/j.biopsych.2008.03.020
Cellot, G., and Cherubini, E. (2014a). Reduced inhibitory gate in the barrel cortex of Neuroligin3R451C knock-in mice, an animal model of autism spectrum disorders. Physiol. Rep. 16:e12077. doi: 10.14814/phy2.12077
Cellot, G., and Cherubini, E. (2014b). GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. (2014) 2:70. doi: 10.3389/fped.2014.00070
Chang, M., Dian, J. A., Dufour, S., Wang, L., Moradi Chameh, H., Ramani, M., et al. (2018). Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol. Dis. 109, 102–116. doi: 10.1016/j.nbd.2017.10.007
Chen, L. Y., Lévesque, M., and Avoli, M. (2019). KCC2 antagonism increases neuronal network excitability but disrupts ictogenesis in vitro. J. Neurophysiol. 122. 1163–1173. doi: 10.1152/jn.00266.2019
Cherubini, E., Gaiarsa, J. L., and Ben-Ari, Y. (1991). GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 14, 515–519. doi: 10.1016/0166-2236(91)90003-D
Chesnut, T. J., and Swann, J. W. (1988). Epileptiform activity induced by 4-aminopyridine in immature hippocampus. Epilepsy Res. 2, 187–195. doi: 10.1016/0920-1211(88)90056-3
Cho, R. Y., Konecky, R. O., and Carter, C. S. (2006). Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 103, 19878–19883. doi: 10.1073/pnas.0609440103
Choi, D. W., Farb, D. H., and Fischbach, G. D. (1977). Chlordiazepoxide selectively augments GABA action in spinal cord cell cultures. Nature 269, 342–344. doi: 10.1038/269342a0
Clarke, M. C., Tanskanen, A., Huttunen, M. O., Clancy, M., Cotter, D. R., and Cannon, M. (2012). Evidence for shared susceptibility to epilepsy and psychosis: a population-based family study. Biol. Psychiatry 71, 836–839. doi: 10.1016/j.biopsych.2012.01.011
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421. doi: 10.1126/science.1076510
Corradini, I., Focchi, E., Rasile, M., Morini, R., Desiato, G., Tomasoni, R., et al. (2018). Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691. doi: 10.1016/j.biopsych.2017.09.030
Costa, E., Guidotti, A., and Mao, C. C. (1975). Evidence for involvement of GABA in the action of benzodiazepines: studies on rat cerebellum. Adv. Biochem. Psychopharmacol. 14, 113–130.
Coull, J. A., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi: 10.1038/nature04223
Coursin, D. B. (1954). Convulsive seizures in infants with pyridoxine-deficient diet. J. Am. Med. Assoc. 154, 406–408. doi: 10.1001/jama.1954.02940390030009
D'Antuono, M., Louvel, J., Köhling, R., Mattia, D., Bernasconi, A., Olivier, A., et al. (2004). GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex. Brain 127, 1626–1640. doi: 10.1093/brain/awh181
De Felipe, J., Marco, P., Fairén, A., and Jones, E. G. (1997). Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb. Cortex 7, 619–634. doi: 10.1093/cercor/7.7.619
de Los Heros, P., Kahle, K. T., Rinehart, J., Bobadilla, N. A., Vázquez, N., San Cristobal, P., et al. (2006). WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 1976–1981. doi: 10.1073/pnas.0510947103
Dean, B., Keriakous, D., Scarr, E., and Thomas, E. A. (2007). Gene expression profiling in Brodmann's area 46 from subjects with schizophrenia. Aust. N. Z. J. Psychiatry 41, 308–320. doi: 10.1080/00048670701213245
Delpire, E., and Gagnon, K. B. (2018). Na+ -K+−2Cl− Cotransporter (NKCC) physiological function in nonpolarized cells and transporting epithelia. Compr. Physiol. 8, 871–901. doi: 10.1002/cphy.c170018
DePaula-Silva, A. B., Gorbea, C., Doty, D. J., Libbey, J. E., Sanchez, J. M. S., Hanak, T. J., et al. (2019). Differential transcriptional profiles identify microglial- and macrophage-specific gene markers expressed during virus-induced neuroinflammation. J. Neuroinflammation 16:152. doi: 10.1186/s12974-019-1545-x
Derchansky, M., Jahromi, S. S., Mamani, M., Shin, D. S., Sik, A., and Carlen, P. L. (2008). Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J. Physiol. 586, 477–494. doi: 10.1113/jphysiol.2007.143065
Di Cristo, G., Awad, P. N., Hamidi, S., and Avoli, M. (2018). KCC2, epileptiform synchronization, and epileptic disorders. Prog. Neurobiol. 162, 1–16. doi: 10.1016/j.pneurobio.2017.11.002
Dienel, S. J., and Lewis, D. A. (2019). Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 131:104208. doi: 10.1016/j.nbd.2018.06.020
Du, L., Shan, L., Wang, B., Li, H., Xu, Z., Staal, W. G., et al. (2015). A pilot study on the combination of applied behavior analysis and bumetanide treatment for children with autism. J. Child Adolesc. Psychopharmacol. 25, 585–588. doi: 10.1089/cap.2015.0045
Duy, P. Q., David, W. B., and Kahle, K. T. (2019). Identification of KCC2 mutations in human epilepsy suggests strategies for therapeutic transporter modulation. Front. Cell. Neurosci. 12:515. doi: 10.3389/fncel.2019.00515
Dzhala, V. I., and Staley, K. J. (2021). KCC2 chloride transport contributes to the termination of Ictal epileptiform activity. eNeuro 8: ENEURO.0208-20.2020, 2021. doi: 10.1523/ENEURO.0208-20.2020
Farrell, M. S., Werge, T., Sklar, P., Owen, M. J., Ophoff, R. A., O'Donovan, M. C., et al. (2015). Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 20, 555–562. doi: 10.1038/mp.2015.16
Fernandez, A., Dumon, C., Guimond, D., Tyzio, R., Bonifazi, P., Lozovaya, N., et al. (2019). The GABA developmental shift is abolished by maternal immune activation already at birth. Cereb. Cortex 29, 3982–3992. doi: 10.1093/cercor/bhy279
Fernell, E., Gustafsson, P., and Gillberg, C. (2021). Bumetanide for autism: open-label trial in six children. Acta Paediatr. 110, 1548–1553. doi: 10.1111/apa.15723
Fiumelli, H., Briner, A., Puskarjov, M., Blaesse, P., Belem, B. J., Dayer, A. G., et al. (2013). An ion transportindependent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb. Cortex 23, 378–388. doi: 10.1093/cercor/bhs027
Fiumelli, H., and Woodin, M. A. (2007). Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr. Opin. Neurobiol. 17, 81–86. doi: 10.1016/j.conb.2007.01.002
Frye, R. E., Rossignol, D., Casanova, M. F., Brown, G. L., Martin, V., Edelson, S., et al. (2013). A review of traditional and novel treatments for seizures in autism spectrum disorder: findings from a systematic review and expert panel. Front Public Health 1:31. doi: 10.3389/fpubh.2013.00031
Fuchs, E. C., Zivkovic, A. R., Cunningham, M. O., Middleton, S., Lebeau, F. E. N., Bannerman, D. M., et al. (2007). Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron 53, 591–604. doi: 10.1016/j.neuron.2007.01.031
Fueta, Y., and Avoli, M. (1992). Effects of antiepileptic drugs on 4-aminopyridine-induced epileptiform activity in young and adult rat hippocampus. Epilepsy Res. 12, 207–215. doi: 10.1016/0920-1211(92)90075-5
Gagnon, M., Bergern, M. J., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R. P., et al. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528. doi: 10.1038/nm.3356
Ghatak, S., Talantova, M., McKercher, S. R., and Lipton, S. A. (2021). Novel therapeutic approach for excitatory/inhibitory imbalance in neurodevelopmental and neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 61, 701–721. doi: 10.1146/annurev-pharmtox-032320-015420
Gigout, S. 1., Louvel, J., Kawasaki, H., D'Antuono, M., Armand, V., Kurcewicz, I., et al. (2006). Effects of gap junction blockers on human neocortical synchronization. Neurobiol. Dis. 22, 496–508. doi: 10.1016/j.nbd.2005.12.011
Gloor, P. (1997). The Temporal Lobe and Limbic System. New York, NY: Oxford University Press (1997).
Gonzalez-Burgos, G., and Lewis, D. A. (2008). GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 34, 944–961. doi: 10.1093/schbul/sbn070
Graf, J., Zhang, C., Marguet, S. L., Herrmann, T., Flossmann, T., Hinsch, R., et al. (2021). A limited role of NKCC1 in telencephalic glutamatergic neurons for developing hippocampal network dynamics and behavior. Proc. Natl. Acad. Sci. U.S.A. 118:e2014784118. doi: 10.1073/pnas.2014784118
Griguoli, M., and Cherubini, E. (2017). Early correlated network activity in the hippocampus: its putative role in shaping neuronal circuits. Front Cell Neurosci. 11:255. doi: 10.3389/fncel.2017.00255
Grover, L. M., Lambert, N. A., Schwartzkroin, P. A., and Teyler, T. J. (1993). Role of ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J. Neurophysiol. 69, 1541–1555. doi: 10.1152/jn.1993.69.5.1541
Hadjikhani, N., Åsberg Johnels, J., Lassalle, A., Zürcher, N. R., Hippolyte, L., Gillberg, C., et al. (2018). Bumetanide for autism: more eye contact, less amygdala activation. Sci. Rep. (2018) 8:3602. doi: 10.1038/s41598-018-21958-x
Hadjikhani, N., Zürcher, N. R., Rogier, O., Ruest, T., Hippolyte, L., Ben-Ari, Y., et al. (2015). Improving emotional face perception in autism with diuretic bumetanide: a proof-of-concept behavioral and functional brain imaging pilot study. Autism 19, 149–157. doi: 10.1177/1362361313514141
Hajri, M., Ben Amor, A., Abbes, Z., Dhouib, S., Ouanes, S., Mrabet, A., et al. (2019). Bumetanide in the management of autism. Tunisian experience in Razi Hospital. Tunis. Med. 97, 971–977.
Hamidi, S., and Avoli, M. (2015a). Carbonic anhydrase inhibition by acetazolamide reduces in vitro epileptiform synchronization. Neuropharmacology 95, 377–387. doi: 10.1016/j.neuropharm.2015.04.015
Hamidi, S., and Avoli, M. (2015b). KCC2 function modulates in vitro ictogenesis. Neurobiol. Dis. 79, 51–58. doi: 10.1016/j.nbd.2015.04.006
He, H. Y., and Cline, H. T. (2019). What is excitation/inhibition and how is it regulated? A case of the elephant and the wisemen. J. Exp. Neurosci. 13:1179069519859371. doi: 10.1177/1179069519859371
Henneberger, C., Bard, L., Panatier, A., Reynolds, J. P., Kopach, O., Medvedev, N. I., et al. (2020). LTP induction boosts glutamate spillover by driving withdrawal of perisynaptic astroglia. Neuron 108, 919–936. doi: 10.1016/j.neuron.2020.08.030
Howard, M. W., Rizzuto, D. S., Caplan, J. B., Madsen, J. R., Lisman, J., Aschenbrenner-Scheibe, R., et al. (2003). Gamma oscillations correlate with working memory load in humans. Cereb. Cortex 13, 1369–1374. doi: 10.1093/cercor/bhg084
Hu, H., Gan, J., and Jonas, P. (2014). Interneurons. Fast-spiking, parvalbumin? GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263. doi: 10.1126/science.1255263
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., et al. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. doi: 10.1016/S0092-8674(00)81509-3
Huberfeld, G., Blauwblomme, T., and Miles, R. (2015). Hippocampus and epilepsy: Findings from human tissues. Rev. Neurol. 171, 236–251. doi: 10.1016/j.neurol.2015.01.563
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007
Huttenlocher, P. R. (1979). Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res. 163, 195–205. doi: 10.1016/0006-8993(79)90349-4
Hyde, T. M., Lipska, B. K., Ali, T., Mathew, S. V., Law, A. J., Metitiri, O. E., et al. (2011). Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011
Iascone, D. M., Li, Y., Sümbül, U., Doron, M., Chen, H., Andreu, V., et al. (2020). Whole-neuron synaptic mapping reveals spatially precise excitatory/inhibitory balance limiting dendritic and somatic spiking. Neuron 106, 566–578. doi: 10.1016/j.neuron.2020.02.015
Jensen, M. S., Cherubini, E., and Yaari, Y. (1993). Opponent effects of potassium on GABAA-mediated postsynaptic inhibition in the rat hippocampus. J. Neurophysiol. 69, 764–771. doi: 10.1152/jn.1993.69.3.764
Jensen, O., Kaiser, J., and Lachaux, J. P. (2007). Human gamma-frequency oscillations associated with attention and memory. Trends Neurosci. 30, 317–324. doi: 10.1016/j.tins.2007.05.001
Jentsch, J. D., and Roth, R. H. (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20, 201–225. doi: 10.1016/S0893-133X(98)00060-8
Johnson, E. W., de Lanerolle, N. C., Kim, J. H., Sundaresan, S., Spencer, D. D., Mattson, R. H., et al. (1992). “Central” and “peripheral” benzodiazepine receptors: opposite changes in human epileptogenic tissue. Neurology 42, 811–815. doi: 10.1212/WNL.42.4.811
Kahle, K. T., Deeb, T. Z., Puskarjov, M., Silayeva, L., Liang, B., Kaila, K., et al. (2013). Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737. doi: 10.1016/j.tins.2013.08.006
Kahle, K. T., Merner, N. D., Friedel, P., Silayeva, L., Liang, B., Khanna, A., et al. (2014). Rouleau GA. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774. doi: 10.15252/embr.201438840
Kahle, K. T., Rinehart, J., de Los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl- in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788. doi: 10.1073/pnas.0508307102
Kahn, R. S., and Keefe, R. S. (2013). Schizophrenia is a cognitive illness: time for a change in focus. JAMA Psychiatry 70, 1107–1112. doi: 10.1001/jamapsychiatry.2013.155
Kaila, K. (1994). Ionic basis of GABA A receptor channel function in the nervous system. Prog. Neurobiol. 42, 489–537. doi: 10.1016/0301-0082(94)90049-3
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M., and Voipio, J. (2014). Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654. doi: 10.1038/nrn3819
Kalkman, H. O. (2011). Alterations in the expression of neuronal chloride transporters may contribute to schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 410–414. doi: 10.1016/j.pnpbp.2011.01.004
Kasyanov, A. M., Safiulina, V. F., Voronin, L. L., and Cherubini, E. (2004). GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc. Natl. Acad. Sci. U.S.A. 101, 3967–3972. doi: 10.1073/pnas.0305974101
Kelley, M. R., Deeb, T. Z., Brandon, N. J., Dunlop, J., Davies, P. A., and Moss, S. J. (2016). Compromising KCC2 transporter activity enhances the development of continuous seizure activity. Neuropharmacology 108, 103–110. doi: 10.1016/j.neuropharm.2016.04.029
Kelsch, W., Hormuzdi, S., Straube, E., Lewen, A., Monyer, H., and Misgeld, U. (2001). Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J. Neurosci. 21, 8339–8347. doi: 10.1523/JNEUROSCI.21-21-08339.2001
Khazipov, R., Esclapez, M., Caillard, O., Bernard, C., Khalilov, I., Tyzio, R., et al. (2001). Early development of neuronal activity in the primate hippocampus in utero. J. Neurosci. 21, 9770–9781. doi: 10.1523/JNEUROSCI.21-24-09770.2001
Khazipov, R., and Luhmann, H. J. (2006). Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 29, 414–418. doi: 10.1016/j.tins.2006.05.007
Khirug, S., Ahmad, F., Puskarjov, M., Afzalov, R., Kaila, K., and Blaesse, P. A. (2010). Single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J. Neurosci. 30, 12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008
Khoshkhoo, S., Vogt, D., and Sohal, V. S. (2017). Dynamic, cell-type-specific roles for GABAergic interneurons in a mouse model of optogenetically inducible seizures. Neuron 93, 291–298. doi: 10.1016/j.neuron.2016.11.043
Kim, H. R., Rajagopal, L., Meltzer, H. Y., and Martina, M. (2021). Depolarizing GABAA current in the prefrontal cortex is linked with cognitive impairment in a mouse model relevant for schizophrenia. Sci. Adv. 7:eaba5032. doi: 10.1126/sciadv.aba5032
Kim, J. Y., Liu, C. Y., Zhang, F., Duan, X., Wen, Z., Song, J., et al. (2012). Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148, 1051–1064. doi: 10.1016/j.cell.2011.12.037
Kirmse, K., Kummer, M., Kovalchuk, Y., Witte, O. W., Garaschuk, O., and Holthoff, K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6:7750. doi: 10.1038/ncomms8750
Kostopoulos, G., Avoli, M., and Gloor, P. (1983). Participation of cortical recurrent inhibition in the genesis of spike and wave discharges in feline generalized penicillin epilepsy. Brain Res. 267, 101–112. doi: 10.1016/0006-8993(83)91043-0
Kursan, S., McMillen, T. S., Beesetty, P., Dias-Junior, E., Almutairi, M. M., Sajib, A. A., et al. (2017). The neuronal K+Cl− co-transporter 2 (Slc12a5) modulates insulin secretion. Sci. Rep. 7:1732. doi: 10.1038/s41598-017-01814-0
Lamsa, K., and Kaila, K. (1997). Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4-aminopyridine. J. Neurophysiol. 78, 2582–2591. doi: 10.1152/jn.1997.78.5.2582
Larimore, J., Zlatic, S. A., Arnold, M., Singleton, K. S., Cross, R., Rudolph, H., et al. (2017). Dysbindin deficiency modifies the expression of GABA neuron and ion permeation transcripts in the developing hippocampus. Front. Genet. 8:28. doi: 10.3389/fgene.2017.00028
Lee, E., Lee, J., and Kim, E. (2017). Excitation/Inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847. doi: 10.1016/j.biopsych.2016.05.011
Lemonnier, E., and Ben-Ari, Y. (2010). The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 99, 1885–1888. doi: 10.1111/j.1651-2227.2010.01933.x
Lemonnier, E., Degrez, C., Phelep, M., Tyzio, R., Josse, F., Grandgeorge, M., et al. (2012). A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. Psychiatry 2:e202. doi: 10.1038/tp.2012.124
Lemonnier, E., Villeneuve, N., Sonie, S., Serret, S., Rosier, A., Roue, M., et al. (2017). Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 7:e1056. doi: 10.1038/tp.2017.10
Leonzino, M., Busnelli, M., Antonucci, F., Verderio, C., Mazzanti, M., and Chini, B. (2016). The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 15, 96–103. doi: 10.1016/j.celrep.2016.03.013
Lévesque, M., Salami, P., Gotman, J., and Avoli, M. (2012). Two seizure-onset types reveal specific patterns of high-frequency oscillations in a model of temporal lobe epilepsy. J. Neurosci. 32, 13264–13272. doi: 10.1523/JNEUROSCI.5086-11.2012
Lewis, D. A., and Lieberman, J. A. (2000). Catching up on schizophrenia: natural history and neurobiology. Neuron 28, 325–334. doi: 10.1016/S0896-6273(00)00111-2
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., et al. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033. doi: 10.1016/j.neuron.2007.10.039
Librizzi, L., Losi, G., Marcon, I., Sessolo, M., Scalmani, P., Carmignoto, G., et al. (2017). Interneuronal network activity at the onset of seizure-like events in entorhinal cortex slices. J. Neurosci. 37, 10398–10407. doi: 10.1523/JNEUROSCI.3906-16.2017
Lopantsev, V., and Avoli, M. (1998). Participation of GABAA-mediated inhibition in ictallike discharges in the rat entorhinal cortex. J. Neurophysiol. 79, 352–360. doi: 10.1152/jn.1998.79.1.352
Löscher, W., and Kaila, K. (2021). Reply to the commentary by Ben-Ari and Delpire: Bumetanide and neonatal seizures: Fiction versus reality. Epilepsia 62, 941–946. doi: 10.1111/epi.16866
Louvel, J., Papatheodoropoulos, C., Siniscalchi, A., Kurcewicz, I., Pumain, R., Devaux, B., et al. (2001). GABA-mediated synchronization in the human neocortex: elevations in extracellular potassium and presynaptic mechanisms. Neuroscience 105, 803–813. doi: 10.1016/S0306-4522(01)00247-0
Ludwig, A., Uvarov, P., Pellegrino, C., Thomas-Crusells, J., Schuchmann, S., Saarma, M., et al. (2011a). Neurturin evokes MAPK-dependent upregulation of Egr4 and KCC2 in developing neurons. Neural Plast. 2011, 1–8. doi: 10.1155/2011/641248
Ludwig, A., Uvarov, P., Soni, S., Thomas-Crusells, J., Airaksinen, M. S., and Rivera, C. (2011b). Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. (2011b). J. Neurosci. 31, 644–649. doi: 10.1523/JNEUROSCI.2006-10.2011
Marrosu, F., Marrosu, G., Rachel, M., and Biggio, G. (1987). Paradoxical reactions elicited by diazepam in children with classic autism. Funct. Neurol. 2, 355–361.
McDonald, J. W., Garofalo, E. A., Hood, T., Sackellares, J. C., Gilman, S., McKeever, P. E., et al. (1991). Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 29, 529–541. doi: 10.1002/ana.410290513
Merner, N. D., Mercado, A., Khanna, A. R., Hodgkinson, A., Bruat, V., Awadalla, P., et al. (2016). Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psychiatr. Res. 77, 22–26. doi: 10.1016/j.jpsychires.2016.02.016
Minzenberg, M. J., Firl, A. J., Yoon, J. H., Gomes, G. C., Reinking, C., and Carter, C. S. (2010). Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology 35, 2590–2599. doi: 10.1038/npp.2010.150
Mody, I., Lambert, J. D., and Heinemann, U. (1987). Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol. 57, 869–888. doi: 10.1152/jn.1987.57.3.869
Mohajerani, M. H., and Cherubini, E. (2005). Spontaneous recurrent network activity in organotypic rat hippocampal slices. Eur. J. Neurosci. 22, 107–118. doi: 10.1111/j.1460-9568.2005.04198.x
Mohajerani, M. H., Sivakumaran, S., Zacchi, P., Aguilera, P., and Cherubini, E. (2007). Correlated network activity enhances synaptic efficacy via BD,NF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 104, 13176–13181. doi: 10.1073/pnas.0704533104
Moore, Y. E., Deeb, T. Z., Chadchankar, H., Brandon, N. J., and Moss, S. J. (2018). Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. U.S.A. 115, 10166–10171. doi: 10.1073/pnas.1810134115
Morita, Y., Callicott, J. H., Testa, L. R., Mighdoll, M. I., Dickinson, D., Chen, Q., et al. (2014). Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J. Neurosci. 34, 4929–4940. doi: 10.1523/JNEUROSCI.1423-13.2014
Morris, M. E., Obrocea, G. V., and Avoli, M. (1996). Extracellular K+ accumulations and synchronous GABA-mediated potentials evoked by 4-aminopyridine in the adult rat hippocampus. Exp. Brain Res. 109, 71–82. doi: 10.1007/BF00228628
Murata, Y., and Colonnese, M. T. (2020). GABAergic interneurons excite neonatal hippocampus in vivo. Sci Adv. 6:eaba1430. doi: 10.1126/sciadv.aba1430
Oh, W. C., Lutzu, S., Castillo, P. E., and Kwon, H. B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353, 1037–1040. doi: 10.1126/science.aaf5206
Pang, T., and Hirsch, L. J. (2005). Treatment of convulsive and nonconvulsive status epilepticus. Curr. Treat. Options Neurol. 7, 247–259. doi: 10.1007/s11940-005-0035-x
Pangrazzi, L., Balasco, L., and Bozzi, Y. (2020). Oxidative stress and immune system dysfunction in autism spectrum disorders. Int. J. Mol. Sci. 21:3293. doi: 10.3390/ijms21093293
Perreault, P., and Avoli, M. (1991). Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J. Neurophysiol. 65, 771–785. doi: 10.1152/jn.1991.65.4.771
Perreault, P., and Avoli, M. (1992). 4-Aminopyridine-induced epileptiform activity and a GABA-mediated long- lasting depolarization in the rat hippocampus. J. Neurosci. 12, 104–115. doi: 10.1523/JNEUROSCI.12-01-00104.1992
Perucca, E., Gram, L., Avanzini, G., and Dulac, O. (1998). Antiepileptic drugs as a cause of worsening seizures. Epilepsia 39, 5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x
Perucca, P., Dubeau, F., and Gotman, J. (2014). Intracranial electroencephalographic seizure-onset patterns: effect of underly.ing pathology. Brain 137, 183–196. doi: 10.1093/brain/awt299
Pizzarelli, R., and Cherubini, E. (2011). Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast. 2011:297153. doi: 10.1155/2011/297153
Potkin, S. G., Turner, J. A., Guffanti, G., Lakatos, A., Fallon, J. H., Nguyen, D. D., et al. (2009). A genome-wide association study of schizophrenia using brain activation as a quantitative phenotype. Schizophr. Bull. 35, 96–108. doi: 10.1093/schbul/sbn155
Price, T. J., Cervero, F., and de Koninck, Y. (2005). Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr. Top. Med. Chem. 5, 547–555. doi: 10.2174/1568026054367629
Price, T. J., Cervero, F., Gold, M. S., Hammond, D. L., and Prescott, S. A. (2009). Chloride regulation in the pain pathway. Brain Res. Rev. 60, 149–170. doi: 10.1016/j.brainresrev.2008.12.015
Puskarjov, M., Ahmad, F., Khirug, S., Sivakumaran, S., Kaila, K., and Blaesse, P. (2015). BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology 88, 103–109. doi: 10.1016/j.neuropharm.2014.09.005
Puskarjov, M., Kahle, K. T., Ruusuvuori, E., and Kaila, K. (2014a). Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 55, 806–818. doi: 10.1111/epi.12620
Puskarjov, M., Seja, P., Heron, S. E., Williams, T. C., Ahmad, F., Iona, X., et al. (2014b). A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. 15, 723–729. doi: 10.1002/embr.201438749
Qin, P., Xu, H., Laursen, T. M., Vestergaard, M., and Mortensen, P. B. (2005). Risk for schizophrenia and schizophrenia-like psychosis among patients with epilepsy: population based cohort study. BMJ 331:23. doi: 10.1136/bmj.38488.462037.8F
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Hodson, C. A., Zhang, J., et al. (2009). Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536. doi: 10.1016/j.cell.2009.05.031
Rinetti-Vargas, G., Phamluong, K., Ron, D., and Bender, K. J. (2017). Periadolescent maturation of GABAergic hyperpolarization at the axon initial segment. Cell Rep. 20, 21–29. doi: 10.1016/j.celrep.2017.06.030
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., et al. (2002). BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and imairs neuronal Cl- extrusion. J. Cell Biol. 159, 747–752. doi: 10.1083/jcb.200209011
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Rogawski, M. A., and Löscher, W. (2004). The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 5, 553–564. doi: 10.1038/nrn1430
Römermann, K., Fedrowitz, M., Hampel, P., Kaczmarek, E., Töllner, K., Erker, T., et al. (2017). Multiple blood-brain barrier transport mechanisms limit bumetanide accumulation, and therapeutic potential, in the mammalian brain. Neuropharmacology 117, 182–194. doi: 10.1016/j.neuropharm.2017.02.006
Rutecki, P. A., Lebeda, F. J., and Johnston, D. (1987). 4-Aminopyridine produces epileptiform activity in hippocampus and enhances synaptic excitation and inhibition. J. Neurophysiol. 57, 1911–1924. doi: 10.1152/jn.1987.57.6.1911
Safiulina, V. F., Afzalov, R., Khiroug, L., Cherubini, E., and Giniatullin, R. (2006a). Reactive oxygen species mediate the potentiating effects of ATP on GABAergic synaptic transmission in the immature hippocampus. J. Biol. Chem. 281, 23464–23470. doi: 10.1074/jbc.M601627200
Safiulina, V. F., Fattorini, G., Conti, F., and Cherubini, E. (2006b). GABAergic signaling at mossy fiber synapses in neonatal rat hippocampus. J. Neurosci. 26, 597–608. doi: 10.1523/JNEUROSCI.4493-05.2006
Safiulina, V. F., Zacchi, P., Taglialatela, M., Yaari, Y., and Cherubini, E. (2008). Low expression of Kv7/M channels facilitates intrinsic and network bursting in the developing rat hippocampus. J. Physiol. 586, 5437–5453. doi: 10.1113/jphysiol.2008.156257
Savardi, A., Borgogno, M., De Vivo, M., and Cancedda, L. (2021). Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 42, P1009–P1034. doi: 10.1016/j.tips.2021.09.005
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schulte, J. T., Wierenga, C. J., and Bruining, H. (2018). Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 90, 260–271. doi: 10.1016/j.neubiorev.2018.05.001
Schwartzkroin, P. A., and Prince, D. A. (1980). Changes in excitatory and inhibitory synaptic potentials leading to epileptogenic activity. Brain Res. 183, 61–76. doi: 10.1016/0006-8993(80)90119-5
Sedmak, G., Jovanov-Milošević, N., Puskarjov, M., Ulamec, M., Krušlin, B., Kaila, K., et al. (2016). Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb. Cortex 26, 4574–4589. doi: 10.1093/cercor/bhv218
Shiri, Z., Manseau, F., Lévesque, M., Williams, S., and Avoli, M. (2015). Interneuron activity leads to initiation of low-voltage fast-onset seizures. Ann. Neurol. 77, 541–546. doi: 10.1002/ana.24342
Shiri, Z., Manseau, F., Lévesque, M., Williams, S., and Avoli, M. (2016). Activation of specific neuronal networks leads to different seizure onset types. Ann. Neurol. 79, 354–365. doi: 10.1002/ana.24570
Sierra-Arregui, T., Llorente, J., Giménez Minguez, P., Tønnesen, J., and Peñagarikano, O. (2020). Neurobiological mechanisms of autism,spectrum disorder and epilepsy, insights from animal models. Neuroscience 445, 69–82. doi: 10.1016/j.neuroscience.2020.02.043
Silayeva, L., Deeb, T. Z., Hines, R. M., Kelley, M. R., Munoz, M. B., Lee, H. H., et al. (2015). KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U.S.A. 112, 3523–3528. doi: 10.1073/pnas.1415126112
Silver, H., Feldman, P., Bilker, W., and Gur, R. C. (2003). Working memory deficit as a core neuropsychological dysfunction in schizophrenia. Am. J. Psychiatry 160, 1809–1816. doi: 10.1176/appi.ajp.160.10.1809
Sipilä, S. T., Huttu, K., Voipio, J., and Kaila, K. (2006). Intrinsic bursting of immature CA3 pyramidal neurons and consequent giant depolarizing potentials are driven by a persistent Na+ current and terminated by a slow Ca2+ -activated K+ current. Eur. J. Neurosci. 23, 2330–2338. doi: 10.1111/j.1460-9568.2006.04757.x
Sivakumaran, S., Cardarelli, R. A., Maguire, J., Kelley, M. R., Silayeva, L., Morrow, D. H., et al. (2015). Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J. Neurosci. 35, 8291–8296. doi: 10.1523/JNEUROSCI.5205-14.2015
Sivakumaran, S., Mohajerani, M. H., and Cherubini, E. (2009). At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J. Neurosci. 29, 2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009
Sloviter, R. S. (1987). Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science 235, 73–76. doi: 10.1126/science.2879352
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. doi: 10.1038/nature07991
Soul, J. S., Bergin, A. M., Stopp, C., Hayes, B., Singh, A., Fortuno, C. R., et al. (2021). A pilot randomized, controlled, double-blind trial of bumetanide to treat neonatal seizures. Ann. Neurol. 89, 327–340. doi: 10.1002/ana.25959
Spoljaric, I., Spoljaric, A., Mavrovic, M., Seja, P., Puskarjov, M., and Kaila, K. (2019). KCC2-Mediated Cl− extrusion modulates spontaneous hippocampal network events in perinatal rats and mice. Cell Rep. 26, 1073–1081. doi: 10.1016/j.celrep.2019.01.011
Sprengers, J. J., van Andel, D. M., Zuithoff, N. P. A., Keijzer-Veen, M. G., Schulp, A. J. A., Scheepers, F.E., et al. (2021). Bumetanide for core symptoms of autism spectrum disorder (BAMBI): a single center, double-blinded, participant-randomized, placebo-controlled, phase-2 superiority trial. J. Am. Acad. Child Adolesc. Psychiatry 60, 865–876. doi: 10.1016/j.jaac.2020.07.888
Steeds, H., Carhart-Harris, R. L., and Stone, J. M. (2015). Drug models of schizophrenia. Ther. Adv. Psychopharmacol. 5, 43–58. doi: 10.1177/2045125314557797
Strata, F., Atzori, M., Molnar, M., Ugolini, G., Tempia, F., and Cherubini, E. (1997). A pacemaker current in dye-coupled hilar interneurons contributes to the generation of giant GABAergic potentials in developing hippocampus. J. Neurosci. 17, 1435–1446. doi: 10.1523/JNEUROSCI.17-04-01435.1997
Straub, R. E., Jiang, Y., MacLean, C. J., Ma, Y., Webb, B. T., Myakishev, M. V., et al. (2002). Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 71, 337–348. doi: 10.1086/341750
Sulis Sato, S., Artoni, P., Landi, S., Cozzolino, O., Parra, R., Pracucci, E., et al. (2017). Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. U.S.A. 114, E8770–E8779. doi: 10.1073/pnas.1702861114
Sullivan, C. R., Funk, A. J., Shan, D., Haroutunian, V., and McCullumsmith, R. E. (2015). Decreased chloride channel expression in the dorsolateral prefrontal cortex in schizophrenia. PLoS ONE 10:e0123158. doi: 10.1371/journal.pone.0123158
Sun, C., Zhang, L., and Chen, G. (2013). An unexpected role of neuroligin-2 in regulating KCC2 and GABA functional switch. Mol. Brain 6 :23. doi: 10.1186/1756-6606-6-23
Sung, K. W., Kirby, M., McDonald, M. P., Lovinger, D. M., and Delpire, E. (2000). Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter null mice. J. Neurosci. 20, 7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000
Tang, X., Drotar, J., Li, K., Clairmont, C. D., Brumm, A. S., Sullins, A. J., et al. (2019). Pha,rmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 11:eaau0164. doi: 10.1126/scitranslmed.aau0164
Tekgul, H., Gauvreau, K., Murphy, L., Robertson, R., Stewart, J., Volpe, J., et al. (2006). The current etiological profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics 117, 1270–1280. doi: 10.1542/peds.2005-1178
Tóth, K., Lénárt, N., Szabadits, E., Pósfai, B., Fekete, R., Cserép, C., et al. (2021). Microglial NKCC1 shapes microglial phenotype, cerebral inflammatory responses and brain injury. bioRxiv 2021.01.21.427597. doi: 10.1101/2021.01.21.427597
Traub, R. D., Whittington, M. A., Colling, S. B., Buzsáki, G., and Jefferys, J. G. (1996). Analysis of gamma rhythms in the rat hippocampus in vitro and in vivo. J. Physiol. 493, 471–484. doi: 10.1113/jphysiol.1996.sp021397
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-Mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Aniksztejn, L. (1999). The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 19, 10372–10382. doi: 10.1523/JNEUROSCI.19-23-10372.1999
Uhlhaas, P. J., and Singer, W. (2010). Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 11, 100–113. doi: 10.1038/nrn2774
Uusisaari, M., Smirnov, S., Voipio, J., and Kaila, K. (2002). Spontaneous epileptiform activity mediated by GABA(A) receptors and gap junctions in the rat hippocampal slice following long-term exposure to GABA(B) antagonists. Neuropharmacology 43, 563–572. doi: 10.1016/S0028-3908(02)00156-9
Valeeva, G., Tressard, T., Mukhtarov, M., Baude, A., and Khazipov, R. (2016). An optogenetic approach for investigation of excitatory and inhibitory network GABA actions in mice expressing Channelrhodopsin-2 in GABAergic neurons. J. Neurosci. 36, 5961–5973. doi: 10.1523/JNEUROSCI.3482-15.2016
Van Andel, D. M., Sprengers, J. J., Oranje, B., Scheepers, F. E., Jansen, F. E., and Bruining, H. (2020). Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: an open-label pilot study. Mol. Autism 11:30. doi: 10.1186/s13229-020-00335-4
Van Den Bogaert, A., Schumacher, J., Schulze, T. G., Otte, A. C., Ohlraun, S., Kovalenko, S., et al. (2003). The DTNBP1 (dysbindin) gene contributes to schizophrenia, depending on family history of the disease. Am. J. Hum. Genet. 73, 1438–1443. doi: 10.1086/379928
Vestergaard, M., Pedersen, C. B., Christensen, J., Madsen, K. M., Olsen, J., and Mortensen, P. B. (2005). Febrile seizures and risk of schizophrenia. Schizophr. Res. 73, 343–349. doi: 10.1016/j.schres.2004.07.004
Viitanen, T., Ruusuvuori, E., Kaila, K., and Voipio, J. (2010). The K+-Cl− cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J. Physiol. 588, 1527–1540. doi: 10.1113/jphysiol.2009.181826
Virtanen, M. A., Lacoh, C. M., Fiumelli, H., Kosel, M., Tyagarajan, S., de Roo, M., et al. (2018). Development of inhibitory synaptic inputs on layer 2/3 pyramidal neurons in the rat medial prefrontal cortex. Brain Struct. Funct. 223, 1999–2012. doi: 10.1007/s00429-017-1602-0
Virtanen, M. A., Uvarov, P., Hübner, C. A., and Kaila, K. (2020). NKCC1, an elusive molecular target in brain development: making sense of the existing data. Cells 9:2607. doi: 10.3390/cells9122607
Virtanen, M. A., Uvarov, P., Mavrovic, M., Poncer, J. C., and Kaila, K. (2021). The multifaceted roles of KCC2 in cortical development. Trends Neurosci. 44, 378–392. doi: 10.1016/j.tins.2021.01.004
Vitari, A. C., Thastrup, J., Rafiqi, F. H., Deak, M., Morrice, N. A., Karlsson, H. K., et al. (2006). Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem. J. 397, 223–231. doi: 10.1042/BJ20060220
Voronin, L. L., and Cherubini, E. (2004). ‘Deaf, mute and whispering’ silent synapses: their role in synaptic plasticity. J. Physiol. 557, 3–12. doi: 10.1113/jphysiol.2003.058966
Voskuyl, R. A., and Albus, H. (1985). Spontaneous epileptiform discharges in hippocampal slices induced by 4-aminopyridine. Brain Res. 342, 54–66. doi: 10.1016/0006-8993(85)91352-6
Wake, H., Watanabe, M., Moorhouse, A. J., Kanematsu, T., Horibe, S., Matsukawa, N., et al. (2007). Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 27, 1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007
Weiss, S. A., Staba, R., Bragin, A., Moxon, K., Sperling, M., Avoli, M. et al. (2019). Interneurons and principal cell firing in human limbic areas at focal seizure onset". Neurobiol. Dis. 124, 183–188. doi: 10.1016/j.nbd.2018.11.014
Whittington, M. A., Traub, R. D., and Jefferys, J. G. R. (1995). Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 373, 612–615. doi: 10.1038/373612a0
Williamson, A., Telfeian, A. E., and Spencer, D. D. (1995). Prolonged GABA responses in dentate granule cells in slices isolated from patients with temporal lobe sclerosis. J. Neurophysiol. 74, 378–387. doi: 10.1152/jn.1995.74.1.378
Woodin, M. A., Ganguly, K., and Poo, M. M. (2003). Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl– transporter activity. Neuron 39, 807–820 doi: 10.1016/S0896-6273(03)00507-5
Yamada, M. K., Nakanishi, K., Ohba, S., Nakamura, T., Ikegaya, Y., Nishiyama, N., et al. (2002). Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J. Neurosci. 22, 7580–7585. doi: 10.1523/JNEUROSCI.22-17-07580.2002
Yekhlef, L., Breschi, G. L., Lagostena, L., Russo, G., and Taverna, S. (2015). Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizure-like activity in mouse medial entorhinal cortex. J. Neurophysiol. 113, 1616–1630. doi: 10.1152/jn.00841.2014
Yizhar, O., Fenno, L. E., Prigge, M., Schneider, F., Davidson, T. J., O'Shea, D. J., et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. doi: 10.1038/nature10360
Yuan, Y., O'Malley, H. A., Smaldino, M. A., Bouza, A. A., Hull, J. M., and Isom, L. L. (2019). Delayed maturation of GABAergic signaling in the Scn1a and Scn1b mouse models of Dravet Syndrome. Sci. Rep. 9:6210. doi: 10.1038/s41598-019-42191-0
Zhang, L., Huang, C. C., Dai, Y., Luo, Q., Ji, Y., Wang, K., et al. (2020). Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 10:9. doi: 10.1038/s41398-020-0692-2
Zhang, T., Tang, Y., Yang, X., Wang, X., Ding, S., Huang, K., et al. (2021). Expression of GSK3β, PICK1, NEFL, C4, NKCC1 and Synaptophysin in peripheral blood mononuclear cells of the first-episode schizophrenia patients. Asian J. Psychiatr. 61:102683. doi: 10.1016/j.ajp.2020.102520
Keywords: GABAergic signaling, cation-chloride co-transporters, neurodevelopmental disorders, neuronal oscillations, E/I balance, autism spectrum disorders, schizophrenia, epilepsy
Citation: Cherubini E, Di Cristo G and Avoli M (2022) Dysregulation of GABAergic Signaling in Neurodevelomental Disorders: Targeting Cation-Chloride Co-transporters to Re-establish a Proper E/I Balance. Front. Cell. Neurosci. 15:813441. doi: 10.3389/fncel.2021.813441
Received: 11 November 2021; Accepted: 30 November 2021;
Published: 05 January 2022.
Edited by:
Heiko J. Luhmann, Johannes Gutenberg University Mainz, GermanyReviewed by:
Melanie A. Woodin, University of Toronto, CanadaYuri Bozzi, University of Trento, Italy
Copyright © 2022 Cherubini, Di Cristo and Avoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico Cherubini, cher@sissa.it