 Daniele Guadagnolo1
Daniele Guadagnolo1 Maria Piane
Maria Piane Antonio Pizzuti
Antonio Pizzuti Simona Petrucci
Simona Petrucci- 1Department of Experimental Medicine, Policlinico Umberto i Hospital, Sapienza University of Rome, Rome, Italy
- 2Department of Clinical and Molecular Medicine, Sapienza University of Rome, Rome, Italy
- 3Medical Genetics and Advanced Cell Diagnostics Unit, S. Andrea University Hospital, Rome, Italy
Parkinson disease (PD) is a complex neurodegenerative disorder, usually with multifactorial etiology. It is characterized by prominent movement disorders and non-motor symptoms. Movement disorders commonly include bradykinesia, rigidity, and resting tremor. Non-motor symptoms can include behavior disorders, sleep disturbances, hyposmia, cognitive impairment, and depression. A fraction of PD cases instead is due to Parkinsonian conditions with Mendelian inheritance. The study of the genetic causes of these phenotypes has shed light onto common pathogenetic mechanisms underlying Parkinsonian conditions. Monogenic Parkinsonisms can present autosomal dominant, autosomal recessive, or even X-linked inheritance patterns. Clinical presentations vary from forms indistinguishable from idiopathic PD to severe childhood-onset conditions with other neurological signs. We provided a comprehensive description of each condition, discussing current knowledge on genotype-phenotype correlations. Despite the broad clinical spectrum and the many genes involved, the phenotype appears to be related to the disrupted cell function and inheritance pattern, and several assumptions about genotype-phenotype correlations can be made. The interest in these assumptions is not merely speculative, in the light of novel promising targeted therapies currently under development.
Introduction
Parkinson disease (PD) is a complex, progressive, neurodegenerative disorder with worldwide incidence of 5–35 in 100,000 cases per year and prevalence reaching 2–4% at the age of 85. PD prevalence is expected to double in the next two decades because of population aging. Mortality is higher in patients with a more than 10-year-long history of disease (1). Pathological hallmarks of the condition include Lewy bodies, Lewy neurites, and loss of dopaminergic neurons of the substantia nigra (SN) pars compacta (SNpc). However, PD neuropathology is pleomorphic, as Lewy bodies are absent in some monogenic forms of PD (2). Clinical manifestations include motor signs (resting tremor, rigidity, bradykinesia, and postural instability) and non-motor features such as hyposmia, constipation, mood disorders, and rapid eye movement sleep behavior disorder (RBD), often preceding the motor signs. In later stages, cognitive decline and autonomic dysfunction may appear. The mean age of onset is in the sixth decade of life, ranging from <40 to more than 80 years. Early-onset PD (EOPD) is commonly defined as an age of onset under 45 years, while juvenile Parkinsonism (JOPD) refers to those cases with onset within 21 years (3).
A PD case is defined as familial or sporadic, according to the presence or absence of a clear family history. Approximately 5–10% can be classified as familial (4).
Most PD cases have a multifactorial etiology, resulting from the combined effects of environmental and genetic factors, while about 5–10% are caused by pathogenic variants in single genes. Monogenic forms of PD are more frequent in EOPD patients, being more than 10% of cases with onset before 45 years and more than 40% in those with onset before 30 years (5). Familial and monogenic PD must not be regarded as synonymous because many familial cases do not have a Mendelian transmission model. To date, more than 20 genes whose mutations cause autosomal dominant (AD), autosomal recessive (AR), and X-linked Parkinsonisms are known, with related phenotypes ranging from idiopathic PD-like (iPD-like) to young-onset Parkinsonisms, either pure or complicated by atypical motor and non-motor features (6). This review will focus on monogenic conditions with Parkinsonian signs as the prominent feature. Parkinsonisms presenting as an iPD-like condition will be classified as “typical PD.” Cases presenting complex phenotypes, featuring prominent additional neurological signs, such as dementia, spasticity, dystonia, and/or abnormal ocular movements, will be classified as “atypical PD.” We will discuss the clinical presentation and genetic cause of each condition, with insights into underlying molecular pathology, to provide genotype–phenotype correlations. Genetic variants will be identified with the most widely used traditional literature nomenclature. The phenotypes will be presented and discussed individually, based on transmission patterns and clinical features. A comprehensive overview of the discussed conditions is provided in Table 1.
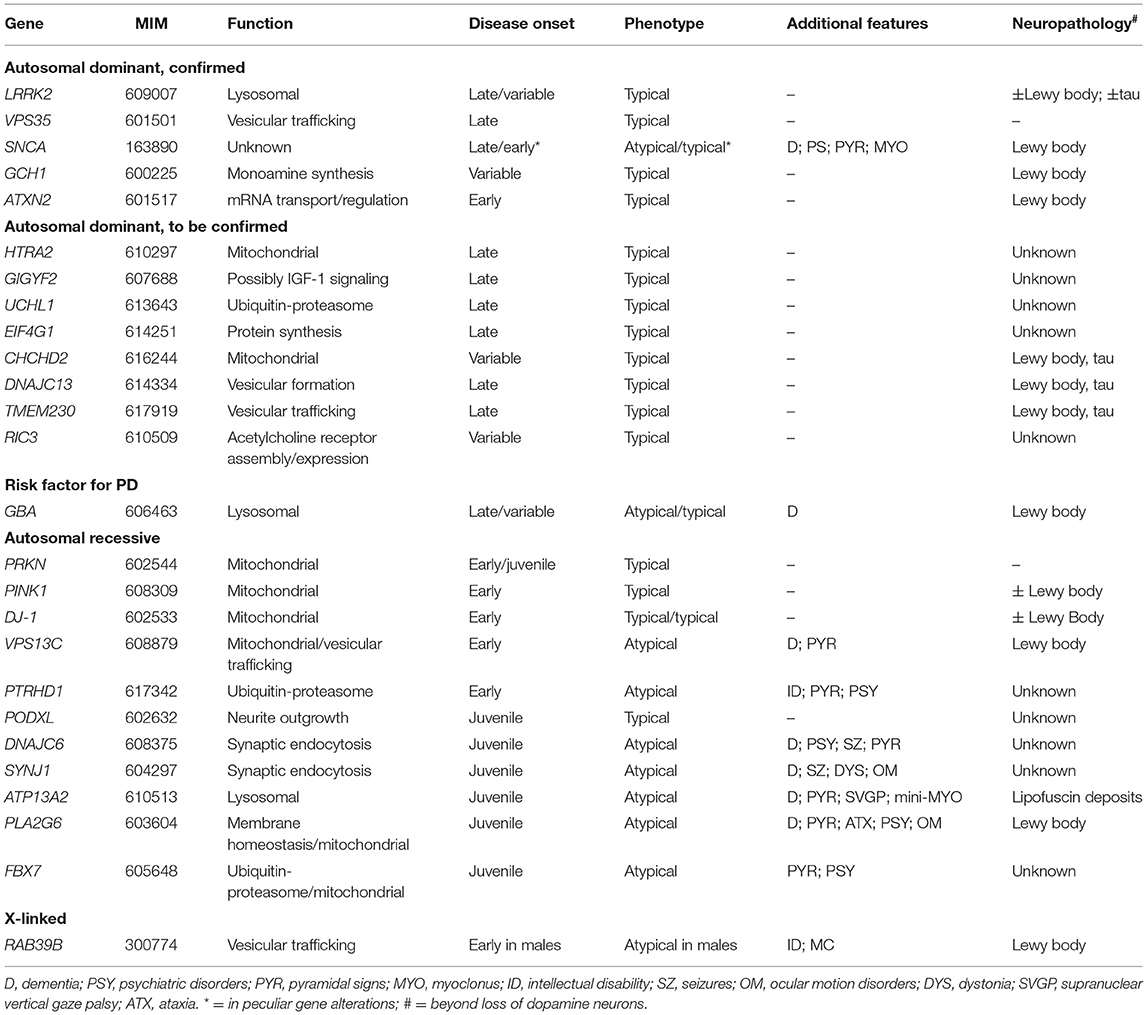
Table 1. Genes causing Parkinsonian conditions and related phenotypes.
Autosomal Dominant PD
Autosomal dominant PD (AD PD) includes different forms of Parkinsonisms that share many peculiarities such as incomplete penetrance, a mean age of onset during the fifth decade or later, and a good response to dopaminergic treatment. However, additional neurological signs, when present, distinguish iPD-like conditions from atypical Parkinsonism. Pathogenic variants in LRRK2 and VPS35 are usually related to Parkinsonisms resembling typical PD, while alterations in SNCA are more frequently found in atypical forms. However, phenotypes may overlap.
Idiopathic PD-Like
LRRK2
Pathogenic variants in the LRRK2 gene (leucine-rich repeat kinase 2, MIM*609007), also known as Dardarin, are the most commonly known causes of AD PD, accounting for 5% of familial and 1% of sporadic cases (PARK8, MIM#607060) (7). They were identified more than 15 years ago by two independent groups in two unrelated families, with late-onset Parkinsonism resembling iPD (8, 9). Many rare LRRK2 variants have been detected over the years but only six of them (p.R1441G/C/H, p.G2019S, p.Y1699C, and p.I2020T) are considered disease causing (8–13). Three additional variants (p.A211V, p.K544E, and p.T1410M), recently demonstrated to cause neurotoxicity, are waiting for confirmation (14). Two coding substitutions, p.G2385R and p.R1628P, mostly identified in Asian populations, act instead as genetic risk factors, each conferring a 2-fold risk of developing PD (15, 16). The p.G2019S variant is by far the most common, being detected in 4% of familial and 1% of sporadic PD cases worldwide (17). Its frequency is even higher among Mediterranean populations and some ethnic groups, including Ashkenazi Jews and North Africans, in which it is found in 23 and 37% of patients with familial PD, respectively (18). Conversely, p.G2019S is extremely rare in East Asia (17). The other known pathogenic LRRK2 variants are very rare, with the exception of p.R1441G and p.R1441C, which are founder mutations in Basque and south Italian ethnicities, respectively (19, 20). The penetrance of the p.G2019S variant is incomplete and age-dependent, peaking at 42.5–74% at the age of ~80 years (17, 21), but varies among different ethnicities (22). Additional genetic variants, acting as single or cumulative risk factors, have been demonstrated to contribute to such variability (23, 24). Age-related penetrance has also been reported for the p.R1441G mutation, ranging from 13% at age 65 to 83% at age 80 (25). Less is known about the penetrance of the other pathogenic variants.
The LRRK2-related phenotype, closely resembling iPD, is characterized by a late-onset progressive Parkinsonism, with resting tremor as a common presenting feature, good response to levodopa therapy, and, usually, positive outcomes with deep brain stimulation (DBS) (17, 26). However, the mean age of onset is slightly lower in LRRK2-related cases than iPD, as patients with PD onset before 40 years are more common among the p.G2019S carriers (7), and differs among different populations, for example, being 10 years earlier among Tunisian carriers compared to Norwegian ones (22). Further features differentiating LRRK2 p.G2019S-related Parkinsonism from iPD are the absence of gender differences (27), the slower progression for motor signs and cognitive impairment (28), and the more frequent occurrence of postural-instability-gait-difficulty (29). Typical iPD non-motor features, such as hyposmia, sleep, and cognitive and dysautonomic alterations, occur in LRRK2 PD cases, but with less frequency (30). Dementia is also rarer in LRRK2-related Parkinsonism (17). When present, cognitive deterioration is usually milder and more slowly progressive than in iPD (31) and is characterized by better attention, executive function, and language (32). Pathogenic variants in LRRK2 were found to be extremely rare in multiple system atrophy (MSA), progressive supranuclear palsy, and corticobasal degeneration (33–36).
Prodromal and premotor symptoms in LRRK2 carriers are still poorly known. In pre-diagnostic PD phases, asymptomatic cases may present subtle motor alterations or isolated Parkinsonian signs, including decreased arm swing, gait asymmetry, and voice changes (37). Non-motor symptoms, such as constipation and urinary urgency, anxiety, daytime sleepiness or poorer performances in executive functioning, and subtle gait changes, are more frequent in asymptomatic LRRK2 variant carriers than in heathy controls (7).
Brain imaging alterations have been found in LRRK2 mutation carriers. Increased gray matter volume of different anatomical structures associated with motor loops has been documented in symptomatic and asymptomatic LRRK2 carriers compared to controls. In contrast, a decreased basal ganglia gray matter volume has been found in LRRK2 PD cases (38). Inversion recovery MRI sequences, assessing brain iron content, showed excessive iron deposition in the SN of brains from LRRK2 carriers (39). Abnormal DAT-SPECT scans have been found in all LRRK2 patients manifesting PD, as well as in some carriers showing prodromal signs and in a subgroup of non-manifesting carriers (40). Moreover, PET studies showed increased dopaminergic and cholinergic activity in LRRK2 non-manifesting carriers compared to sporadic PD cases, possibly reflecting compensatory changes preceding the motor onset of PD (41, 42).
The neuropathology of LRRK2-related disease, mainly investigated in p.G201S carriers, is characterized by neuronal loss in the SNpc and locus coeruleus in all cases, with or without protein aggregation. Typical Lewy-type pathology and alpha-synuclein (α-syn) aggregates are present in 65–80% of LRRK2 manifesting carriers, at a lower frequency than iPD cases (43). Tau inclusions, with variable distribution and severity, are also common, being found in about half the brains from patients with LRRK2- PD (43). a-syn aggregates prevail in p.G2019S carriers, while pure nigral degeneration has been described in about half of p.I2020T patients. Nevertheless, neuropathology can differ among relatives carrying the same pathogenic variant (44). Neuropathological and clinical features are strictly correlated, as Lewy-type pathology has been associated with the occurrence of non-motor symptoms, while pure neurodegeneration has been found in brains from patients with PD with only motor signs (43, 44).
More recently, genome-wide association studies (GWAS) confirmed the linkage of LRRK2 locus variants to sporadic PD (45–47). Contrary to rare pathogenic mutations, these common variants confer modest risk for PD, suggesting a possible role of LRRK2 in influencing iPD susceptibility (37).
LRRK2 is a large and multifunctional protein with multiple domains for various protein–protein interactions and enzymatic serine–threonine kinase and GTPase activities. All the clearly pathogenic mutations cause a toxic gain-of-function. The increase in the LRRK2 kinase activity mainly compromises neuronal vesicular trafficking, through an aberrant excessive phosphorylation of Rab GTPases (48). These findings have suggested that LRRK2 kinase inhibitors might be a therapeutic target in LRRK2-related PD not only for monogenic LRRK2-related Parkinsonisms but also for the more common iPD (49).
VPS35
Late-onset PD with autosomal dominant inheritance (PARK17, MIM#614203) is also the phenotypic picture related to pathogenic variants in the VPS35 gene (vacuolar protein sorting 35, MIM*601501). The phenotype is quite similar to iPD, mostly homogeneous and with a benign course. It may differ for an earlier age of onset (mean age at onset 50 years) (50). No atypical signs have been described in the VPS35 p.D620N variant carriers since its discovery in two unrelated families with Swiss and Austrian origin, respectively (51, 52). Scarce cognitive and neuropsychiatric features, hyposmia in about 50% of patients, and excellent response to levodopa are reported (52, 53). Among these kindreds, the penetrance was high but incomplete (53). Beyond the p.D620N, identified in a few other familial or sporadic PD cases worldwide, no other proved pathogenic variants have been reported (54). The only patient who underwent neuropathology after death did not show signs of α-syn (55).
The VPS35 protein is part of the retromer complex, involved in the neuronal vesicular recycling from endosomes to the trans-Golgi network. Altered VPS35 is supposed to compromise the intracellular localization and stability of these organelles (56).
Atypical AD PD
SNCA
Pathogenic variants in SNCA (alpha-synuclein, MIM*163890), encoding α-syn, were identified 20 years ago as the first genetic cause of AD PD (PARK1, MIM#168601; PARK4, MIM#605543). In the large “Contursi kindred” the SNCA p.A53T mutation segregated with a PD and dementia with Lewy bodies phenotype (DLB, MIM#127750) (57, 58). Many other point mutations and whole gene multiplications have been detected in hundreds of patients with hereditary forms of autosomal dominant forms of PD, DLB, and other neurodegenerative conditions thus far (14). The identification of α-syn as a major component of Lewy bodies (LBs) and Lewy neurites (LNs), the neuropathological pathognomonic hallmarks of PD and DLB, confirmed the role of SNCA in the pathogenesis of PD. The detection of Lewy-type pathology in sporadic PD supported the involvement of α-syn in iPD, too (44, 59). Damaging mutations and whole gene multiplications favor α-syn aggregation with potential deleterious consequences at both the synaptic level and lysosomal/endosomal compartments, by a gain-of-function mechanism (60).
Duplication and triplications of an otherwise normal SNCA gene have been described in more than 100 patients (61). SNCA missense mutations (p.A53T, p.A30P, p.E46K, p.H50Q, and p.G51D) are instead very rare worldwide.
Many different PD phenotypes have been related to SNCA mutations, ranging from the more common late-onset Parkinsonism, either with or without non-motor symptoms, to the rarer early-onset aggressive diseases with atypical signs.
In whole-gene multiplications, the number of SNCA copies clearly correlates with the disease severity, supporting the notion of a “dosage effect” (62). Indeed, patients with four copies of SNCA (heterozygous triplication or homozygous duplication carriers) have a 10-year earlier age of onset, a more rapid progression and a more severe phenotype, often complicated by myoclonus, severe cognitive impairment, and psychiatric features, compared to heterozygous duplication carriers (61). Marked weight loss, dysautonomia, and fatigue can precede motor symptoms onset with death occurring within 7 years. Brain imaging reveals frontoparietal atrophy and a severe striatal dopaminergic deficit (63, 64). SNCA duplications cause a more variable phenotype, even within the same family, ranging from asymptomatic carriers to iPD-like or, more rarely, to severe forms resembling SNCA triplication carrier phenotypes (61, 65). The mean age of onset for SNCA duplication-related PD is 50 years. Non-motor symptoms are inconstantly present, and death occurs after 15 years from onset (66, 67). Atypical phenotypes have been described, including fronto-temporal dementia (FTD) with marked anxiety and obsessive–compulsive disorder, and a singular head shaking movement disorder (68, 69).
Compared to copy-gain variants, missense mutations cause more complex phenotypes with mutation-specific trends in clinical presentations (70). In most cases, despite similar ages of onset (average age 47.6 + 12.9 years), motor and non-motor features differ among patients with different specific mutations.
The p.A53T PD is characterized by marked intra-familial and inter-familial variability (67, 71). Penetrance is incomplete but high (80–90%). Parkinsonism resembles iPD, with a more aggressive and rapid course. Tremor is not common. Motor signs are initially L-dopa responsive but worsen early because of the occurrence of motor complications. Non-motor features, including hyposmia, orthostatic hypotension, RBD, and depression are inconstantly present. Myoclonic jerks and central hypoventilation have been reported. Cognitive impairment may vary, but dementia usually occurs within 5–7 years from disease onset (67, 72, 73). Parkinson dementia disease (PDD) and DLB have been described (74). More rarely, prominent language dysfunction resembling primary progressive aphasia and frontotemporal dementia with behavioral dysregulation and speech-related problems have been reported (75). Olfactory dysfunction, RBD, and dopaminergic deficit at DATSCAN have been proposed as possible premotor signs after their identification in otherwise asymptomatic p.A53T carriers (71, 72).
Conversely, a late-onset Parkinsonism with tremor as a rather constant motor sign and cognitive impairment ranging from mild cognitive decline to frank dementia have been reported in p.H50Q carriers, all with English ancestry (76, 77).
Patients with the p.A30P mutation, found in a single German family, present a condition similar to iPD, with onset around 60 years, incomplete penetrance, and a benign course of disease. Cognitive impairment and hallucination occur, although inconsistently (78, 79).
More severe phenotypes are related to the p. E46K and p.G51D variants. The first one, identified in a Basque family, causes a high penetrant and severe Parkinsonism, presenting at 50–65 years (80). Dementia with LB phenotype and autonomic dysregulation occur a few years after the onset of motor signs. However, disease severity may vary among families. Marked cardiac denervation has been found in patients and in p.E46K asymptomatic carriers (81).
Conversely, the Parkinsonian condition related to p.G51D, found in a few European and Asian cases, strongly differs from the late-onset PD caused by the other SNCA damaging missense mutations. The age of onset is very early, before 20 years in one case, and pyramidal signs, myoclonus and seizures coexist, inconstantly complicated by psychiatric symptoms, dementia, and autonomic dysfunction (82, 83).
Another early onset SNCA-related Parkinsonism, with no atypical signs, has been described in a Finnish family with the p.A53E SNCA mutation (84).
A proper definition of phenotypes related to SNCA pathogenic missense variants is limited by the extreme rarity of such mutations and by the lack of thorough clinical evaluations for each individual case. A comprehensive international database considering complete and standardized information from individual patients would help to overcome these limits.
Pathogenic SNCA variants are localized in exons 2 and 3, which encode for an α-helical domain with lipid binding activity and for a hydrophobic domain (85). These mutations probably prolong α-syn half-life by interfering with lysosomal degradation (86). The tendency of some of these variants to accelerate α-syn aggregation and to recruit tau proteins into inclusions has been demonstrated with in vitro and in vivo studies. Further investigation will clarify if phenotype differences for distinct missense mutations depend on toxic gain-of-function mechanisms or another prolonged mutated protein half-life (87).
Soon after the discovery of the role of SNCA in AD PD, common variants at this locus were investigated for association with sporadic PD. A positive association emerged between iPD and expanded alleles at the NACP-Rep1 repeat, located 10 kb upstream of the transcriptional start site of SNCA (88, 89). Many additional GWAS analyses since then have supported a statistically significant association between the risk for PD and several single-nucleotide polymorphisms (SNP) located both at the 5′ end and the 3′ end of the SNCA gene (46). Although the effect of each SNP is individually low (odd ratio 1.3), the cumulative risk can be substantial. The NACP-Rep1 alleles and the rs356168 SNP increase α-syn expression both in vitro and in vivo, supporting the hypothesis that common SNCA variants increasing α-syn expression also increase the risk for apparently sporadic PD (87).
AD Genes Awaiting Confirmation
In addition to these AD PD genes, many others have been proposed as monogenic causes of hereditary iPD-like conditions. However, their pathogenicity is still debated or waiting for confirmation. GIGYF2 (GRB10-interacting GYF protein 2, MIM*612003; PARK11, MIM#607688), HTRA2 (HTRA serine peptidase 2, MIM*610297; PARK13, MIM#606441), UCHL1 (ubiquitin carboxyl-terminal esterase L1, MIM*191342; PARK15, MIM#613643), and EIF4G1 (eukaryotic translation initiation factor 4-G, MIM*614251; PARK18, MIM#614251) variants were detected in families with late-onset Parkinsonism resembling iPD and segregating with autosomal dominant fashion. However, many studies denied their pathogenic role in PD and none of them is still considered as a PD gene (90, 91). With the advent of NGS and the increasing availability of whole exome/genome sequencing (WES/WGS) many genes such as DNAJC13, CHCHD2, TMEM230, LRP10, and RIC3 have been identified in AD PD families. Variants in CHCHD2 (coiled-coil-helix domain containing protein 2, MIM*616244), a gene involved in the mitochondrial respiratory function, were identified in a few families with AD PD (PARK22, MIM#616710). Patients presented variable disease onset (mean age 52 years), good response to levodopa, depression, and the absence of cognitive impairment. A brain autopsy of a CHCHD2 PD patient revealed widespread LB pathology with amyloid plaques and neurofibrillary tangles in the brainstem, limbic regions, and cortex (92–94). Despite these results, large-scale studies did not support the causative role of CHCHD2 in PD [reviewed in (95)]. A definitive confirmation of its pathogenicity is still lacking.
Evidence is less robust for the remaining four genes. The c.2564A>G mutation in DNAJC13 (DNAJ/HSP40 homolog subfamily C, member 13, MIM*614334) gene was identified in patients with late onset (mean age 63 years) and slow progressive PD from a large Mennonite kindred. Brain pathology of three mutation carriers showed LB with cell loss in Meynert nucleus and SN, as well as tau pathology. However, no other PD cases with pathogenic variants in DNAJC13 have been detected to date and its role in PD etiology has been recently reconsidered (91). An independent study in the same Mennonite kindred identified the c.422G>T variant in TMEM230 (Transmembrane protein 230, MIM*617019) as the cause of the Parkinsonism segregating in that family. The same variant was also detected in a few sporadic PD cases (96). As no other rare pathogenic mutation in TMEM230 has been detected in familial and sporadic PD patients, pathogenicity for this gene still needs confirmation (97).
Recently, the c.169C>A mutation in RIC3 (resistance to inhibitors of cholinesterase 3, MIM*610509) was found to segregate with a variable onset (30–68 years) Parkinsonism in a large family from South India. Nine mutated patients across three consecutive generations presented typical PD with RBD, depression, and restless leg syndrome (98). RIC3, not detected in other PD cases, is also waiting for confirmation (99). Heterozygous mutations in LRP10 (low-density lipoprotein receptor-related protein 10, MIM*609921) were detected in Italian kindred with late-onset PD and in other unrelated patients with Parkinsonism and dementia PDD and DLB (100). However, inconsistent findings emerged by replications studies worldwide, with no differences in LRP10 variant frequency between patients and controls, and for the incongruity of segregation analysis (101). Other population and functional studies are required to elucidate the role of this gene in PD.
Other Disease-Causing Genes Associated With PD
Other Movement Disorder Genes Possibly Manifesting as AD PD
An iPD phenotype can be related to genes known to cause other movement disorders. In particular, pathogenic variants in GCH1 (GTP cyclohydrolase I, MIM*600225), the most common cause of levodopa-responsive dystonia (MIM#128230), have been found in patients and families with AD PD characterized by variable disease onset (mean age 43 years), long-term motor complications, and non-motor signs such as cognitive impairment, sleep disorders, hyposmia, and autonomic dysfunction, without dystonia. Nigrostriatal degeneration in affected patients was also proved by a reduced tracer uptake in SPECT studies (102). The association between PD phenotype and GCH1 pathogenic variants was confirmed by further studies (103, 104). Also, triplet expansion variants in ATXN2 (MIM*601517), responsible for autosomal dominant cerebellar ataxia type 2 (SCA2, MIM#183090), may cause a form of typical PD with onset after 40 years, good response to levodopa therapy without cognitive impairment, and cerebellar signs. LB pathology and neuronal loss have been documented, in the absence of cerebellar atrophy. A CAG repeat expansion exceeding 33 is considered pathogenic. However, while in SCA2 phenotypes the mean of repeats is 43; in PD cases, repeats are lower in number (mean 36 ± 1) and with CAA interruptions (105, 106).
GBA
Heterozygous variants in the GBA (acid beta glucosidase, MIM*606463) gene are the most common genetic risk factor for PD (MIM#168600) worldwide. GBA encodes the β-glucocerebrosidase (GCase), a lysosomal enzyme that cleaves the glucosylceramide sphingolipid into glucose and ceramide. Biallelic mutations in GBA cause Gaucher's disease (GD), the most common AR lysosomal storage disease with a variable involvement of the central nervous system. About 300 different pathogenic GBA variants have been found, many of them resulting in a significant loss of GCase activity (107). Different mutations can lead to different phenotypes of GD. Variants are overall classified according to the GD subtype they are related to. Mutations causing the non-neuronopathic GD type 1 are defined as mild (e.g., the c.1226A>G, also known as N370S in the traditional nomenclature); those causing neuronopathic GD type 2 and 3 are classified as severe (e.g., c.1448T > C, L444P) (108).
The association between GBA variants and increased risk of Parkinsonism arose after clinical observations of higher incidence of PD among GD type 1 patients and their heterozygous parents. This suggestion was confirmed by a large, worldwide, multicenter association study, and by many other papers which demonstrated higher frequencies of the GBA variants in PD patients compared to healthy controls (109). The N370S and the L444P mutations are the two most common alleles worldwide, accounting for 70–80% of GBA variants associated with PD in some populations. In particular, the N370S is the most frequent among Ashkenazi Jews from eastern Europe whereas L444P is more common among non-Jew European descendants (110). Nevertheless, GBA mutations represent only a risk factor for PD. Population studies showed that a little more than 9% of GBA mutation carriers develop PD (111). Heterozygotes for a GBA mutation have a 5-fold increased risk of developing PD compared to the age-matched general population. For homozygotes, the risk increase is 10- to 20-fold (108). Pathogenic variants in GBA are detected in 8.5% of PD patients (112). However, carrier frequency varies across different ethnic groups, ranging from 10 to 31% in Ashkenazi Jews, 2.9–12% in non-Jewish Europeans, and 2.3% in Norwegians (113). PD cases with GBA mutations are usually similar to iPD. However, some peculiarities are emerging by large population studies comparing carrier and non-carrier PD phenotypes and clinical features related to mild and severe mutations. In heterozygous GBA PD, the first symptoms manifest 3–6 years earlier than in iPD, a rigid akinetic motor phenotype is more common, and the response to levodopa is good. However, at least in a subset of patients, the progression of the disease can be faster and therapeutic outcomes are often limited by earlier development of motor fluctuations and dyskinesias, as well as cognitive decline after DBS (114, 115). GBA mutation carriers are also more likely to manifest non-motor symptoms. Cognitive involvement is more frequent, and the risk of developing dementia is at least 3-fold higher than in iPD (116, 117). Non-motor symptoms, such as hallucinations, depression and anxiety, impulse control disorders, RBD, and autonomic dysfunctions, are also more common among GBA patients, and more frequent in severe mutation carriers. Similarly, motor complications, including dysphagia, dysarthria, and freezing of gait, are more frequent in GBA carriers (118).
Compared to milder variants, severe GBA mutations are associated with a higher risk of developing PD, younger onset, worse motor progression, more frequent cognitive involvement, and more complex non-motor phenotype (27, 119).
GBA PD patients have a distributed pattern of white matter abnormalities involving the interhemispheric, frontal cortico-cortical, and parahippocampal tracts, and no gray matter atrophy at structural MRI (120). Spectroscopic MRI shows a neurodegeneration pattern more pronounced in the putamen than in the midbrain (121). GBA mutation carriers also differ from iPD cases in PET studies, showing reduced cerebral blood flow in the parieto-occipital cortex and reduced nigrostriatal function. This resembles the pattern typically seen in DLB, especially in severe mutation carriers (119).
Mutations in GBA are also a significant risk factor for DLB, conferring a more than 8-fold increased risk of developing this condition compared to controls (122). The association between GBA pathogenic variants and other Parkinsonian conditions is instead less consistent. Correlations with PSP are still weak (123, 124), but recent studies demonstrated an association between GBA variants and MSA (125, 126).
The exact mechanism by which GBA mutations lead to PD is still unclear. It is well-known that GCase is part of the endo-lysosomal pathway, particularly crucial in many pathogenetic pathways leading to PD. Mutated GCase is not able to fold properly and accumulates in different cellular compartments of the dopaminergic neurons, causing a cell stress response, damage, and neuronal death. In addition, the entrapment of the beta GCase in the endoplasmic reticulum reduces enzyme levels in the cell, triggering α-syn accumulation (127). Intriguingly, LB pathology has been found in cortical areas of brains from 10 PD patients with Parkinsonism and from almost all GBA-related PD cases who underwent autopsy. Less is known about the distribution of neuronal loss or additional neuropathology (128).
At present, clinical trials assessing the safety and the efficacy of molecules aiding proper GCase folding, improving enzymatic activity, or reducing the GCase substrates accumulation are ongoing (108).
Autosomal Recessive Early-Onset Parkinsonisms
Among AR parkinsonisms, forms caused by biallelic pathogenic variants in the PRKN, PINK1, and DJ-1 genes are thus far considered pure forms of EOPD. Together, they account for ~13% of EOPD cases (73). VPS13C and PTRHD1 are mutated in families with EOPD complicated by pyramidal signs and cognitive involvement. Other genes such as APT13A2, PLA2G6, FBXO7, and the more recently identified DNAJC6, SYNJ1, and VPSC13 are usually related to younger onset Parkinsonism, complicated by atypical motor and non-motor phenotypes. Biallelic mutations in another gene, PODXL, recently have been detected in siblings with a juvenile form of pure PD. However, this gene is still waiting for confirmation.
Pure EOPD Forms
The PRKN, PINK1, and DJ1 genes share similar PD phenotypes and the same cellular pathway. They are involved in mitochondrial homeostasis and mitophagy. Variants in these genes can impair mitochondrial function, leading to cellular stress and neurotoxicity. PRKN encodes the E3-ubiquitin ligase, involved in the proteasome pathway for damaged proteins degradation and in mitochondrial homeostasis. PINK1 encodes a mitochondrial kinase involved in mitophagy, acting upstream of Parkin. DJ-1 encodes for a protein involved in the antioxidant response that shares biochemical pathways with PINK1 and Parkin (129–131).
PRKN
Biallelic pathogenic variants in PRKN (Parkin, MIM*602544), are the most common genetic cause of early-onset pure Parkinsonism (PARK2, MIM#602544) (132). First identified in a Japanese family with AR PD, PRKN variants currently account for 10–20% of PD with onset within 40 years (133). The occurrence of biallelic mutation is inversely related to the age of the disease onset: the younger the onset, the higher the probability to detect homozygous or compound heterozygous PRKN carriers. The onset is usually before 40 years, with a median age of 31 years for the first motor signs. Juvenile onset, with the first symptoms within 20 years, is described in 16% of PD patients with biallelic PRKN mutations (73). Cardinal signs of PRKN-related PD are a more common bradykinetic motor phenotype, benign and slow progression, and good response to levodopa therapy, although frequently complicated by iatrogenic dyskinesia, to anticholinergic medication, and to DBS (26, 73, 134). Hyperreflexia and/or dystonia may occur, and lower-limb dystonia can be a presenting sign. Cognitive decline is uncommon, being as frequent as in the general population, and dementia is extremely rare. Sense of smell is usually well-preserved and additional features, such as psychiatric manifestations and dysautonomia, are also rare (134, 135). Disease duration can reach 50 years. Later disease stages may be complicated by freezing of gait, postural deformities, and motor fluctuations (136).
Unlike iPD, women and men are equally affected and the loss of dopaminergic striatal innervation, revealed by 18F-DOPA PET/SPECT, is rather symmetric and slowly progressive (134).
Neuronal loss in pigmented nuclei of the brainstem is the prominent feature found in brains of PRKN PD patients. Neurodegeneration is more prominent in the SNpc than in the locus coeruleus. Typical LBs containing α-syn have been identified in very few affected individuals (136, 137). Pathogenic variants are highly diverse, including exonic deletions or multiplications and missense, nonsense, and frameshift variants, described in homozygous or compound heterozygous states. Exonic rearrangements are the most common anomalies. Functional studies demonstrated protein loss-of-function or absence of protein due to nonsense mRNA decay for most of them (138, 139).
PINK1
The phenotype related to biallelic pathogenic variants in PINK1 (PTEN-induced putative kinase 1, MIM*608309; PARK6 MIM#605909) is similar to PRKN PD, with early onset, good response to levodopa, and rare cognitive compromission. However, hyperreflexia is less common and hyposmia, dysautonomic features, and psychiatric symptoms including anxiety, psychosis, and affective disorders may occur (73). Neuronal loss is prevalent in SN and, contrary to PRKN-related disease, LBs have been found on neuropathological examinations (140).
PINK1 biallelic mutations are the second most common cause of EOPD worldwide, accounting for 1–5% of cases, but with variations among ethnic groups, being more frequent in north Africa (90). More than 60 variants, including deletions and missense and nonsense variations, have been reported (73).
DJ-1
Biallelic variants in DJ-1 (oncogene Dj1, MIM*602533) cause a rare Parkinsonism (PARK7, MIM#606324), the third most common AR PD after the PRKN- and PINK1-related forms, accounting for the 0.4 and 1% of EOPD cases, respectively (1). Patients usually share the same phenotype of both PRKN and PINK1 cases, presenting early onset slowly progressive Parkinsonism (mean age 27 years), good response to dopaminergic therapy, frequent focal dystonia, motor complications upon treatment, and psychiatric symptoms, in particular anxiety that often presents as the first symptom (73, 132). Unlike PRKN and PINK1, additional features, including depression, cognitive decline, motor neuron disease, and bulbar signs are seldom described (141, 142). The only reported neuropathological brain examination showed loss of neurons in the substantia nigra and locus coeruleus with widespread α-syn, akin to sporadic PD brains (142).
Heterozygous Variants in PRKN, PINK1, and DJ-1
While biallelic pathogenic variants in PRKN, PINK1, and DJ-1 are clearly causative for EOPD, the role of single heterozygous mutations in these genes is debated. PRKN and PINK1 heterozygous mutations have been detected in a substantial number of PD patients but also in healthy controls, raising the question of whether they may contribute to the disease or are incidental findings. The results of many case-controlled studies suggest that they may represent minor susceptibility factors that mildly contribute to the risk of sporadic PD (Parkin odds ratio 2:53; PINK1 odds ratio 1:65) (143, 144). Interestingly, multimodal neuroimaging and electrophysiologic studies disclosed a latent nigrostriatal impairment in otherwise healthy subjects carrying heterozygous Parkin or PINK1 mutations (145–147). However, the role of heterozygous variants in these AR genes cannot be conclusively established as prospective studies of healthy heterozygous carriers are still lacking (144).
Atypical EOPD Forms
VPS13C
VPS13C (vacuolar protein sorting 13 homolog C, MIM* 608879) is the most recently identified gene causing a rare, atypical form of early-onset Parkinsonism (PARK 23, MIM#616840). To date, only three families have been described. All affected patients presented asymmetric akinetic-rigid Parkinsonism starting from age 25 to 45, with initially good response to dopaminergic treatment. The disease progression is severe and rapid with dramatic early cognitive involvement, dysautonomia, limb dystonia, hyperreflexia, and pyramidal signs leading to tetraplegia. The patients are bedridden after about 10 years of disease. MRI documented asymmetric brain atrophy. Neuropathology of a single case showed widespread and abundant α-syn positive LBs and neuritis, together with tau pathology with neurofibrillary tangles (148). VPS13C encodes the vacuolar protein sorting 13 protein involved in mitochondrial activity and vesicular trafficking. It has been shown that VPS13C mutations alter mitochondrial function and PINK1-Parkin-dependent mitophagy (149).
PTRHD1
Homozygous missense mutations and 28-nucleotid frameshift deletion in PTRHD1 (peptidyl-tRNA hydrolase domain-containing 1, MIM*617342) have been recently identified in two unrelated consanguineous Iranian families and in a sub-Saharan African kindred with early onset Parkinsonism and intellectual disability (150, 151). Motor signs of Parkinsonism appeared, at 20–30 years of age. Phenotypes were variably complicated by muscle stiffness, postural tremor, pyramidal signs, sensory-motor polyneuropathy, behavioral disorders, and hypersomnia. No other PTRHD1 cases have been identified to date (152). Chromosomal microdeletions encompassing the PTRHD1 gene have been previously related to many syndromes with intellectual disability. PDRHT1 encodes a protein involved in the ubiquitin proteasome pathway. Intriguingly, the pathogenic variants causing Parkinsonism are in the ubiquitin-like (UBL) domain-binding site of the protein. The suppression of the ubiquitin protein degradation is a well-known mechanism of PD. Despite these findings, further population and functional studies are needed to confirm the role of this rare gene in determining PD.
Juvenile Parkinsonism
Juvenile Parkinsonism includes those forms of PD with onset within 21 years, often combined with other hyperkinetic movement disorders and neurological and imaging abnormalities (153). With the exclusion of PRKN mutations, responsible for 77% of juvenile PD (see section Other Movement Disorder Genes Possibly Manifesting as AD PD) and the extremely rare PODXL-related cases, more than 90% of patients have a complex or atypical presentation, with dystonia, pyramidal signs, neuropsychiatric disorders, abnormal eye movements, and brain imaging. Many genes have been associated with AR young JOPD. This section will focus on those forms in which Parkinsonism is the predominant sign.
Pure JOPD
PODXL
A homozygous frameshift variant in the PODXL gene (podocalyxin-like, MIM*602632) has been described in three siblings from an Indian consanguineous family, who developed a pure levodopa-responsive Parkinsonism manifested at 13–17 years of age, later complicated by dyskinesia and off-dystonia, with no additional signs. PODXL encodes a glycoprotein involved in the regulation of neurite outgrowth. The frameshift mutation (c.89_90insGTCGCCCC) resulted in a complete loss of protein function (154). Replications of these findings and confirmation of PODXL as a causative PD gene are still awaited.
Atypical
JOPD
DNAJC6 Biallelic damaging variants in DNAJC6 (DNAJ/HSP40 homolog, subfamily c, member 6, MIM* 608375) are associated with a form of juvenile Parkinsonism (PARK19, MIM#615528), with a mean age of onset at 11 years (range 7–42 years). The wide phenotypic spectrum ranges from typical pallido-pyramidal syndrome to pure EOPD. Motor signs at onset vary from tremor to bradykinesia but the disease later manifests with Parkinsonism, postural instability, and usually good response to levodopa, often limited by treatment-induced dyskinesia, psychosis, and hallucinations (155, 156). In complex forms, Parkinsonism is often complicated by atypical features such as cognitive decline, spasticity/pyramidal signs, dysarthria/anarthria, seizures, and hallucinations. Disease progression is severe, especially in cases with younger onset, who need a wheelchair after 2–10 years of disease (155, 157, 158). Brain atrophy has been described in all but one case (158). Conversely, those cases with pure Parkinsonism show variable age of onset, slow disease progression, and good response to dopaminergic therapies (156). DNAJC6 encodes for Auxilin, a protein involved in calthrin-mediated endocytosis. Mutations in DNAJC6 cause an impairment in synaptic vesicle recycling, compromising endocytosis (155). A clear genotype-phenotype correlation has been defined: nonsense mutations have been related to juvenile complex Parkinsonism, while patients with missense mutations or variants resulting in reduced protein production have been found in pure Parkinsonism cases (156, 157).
SYNJ1
SYNJ1 (synaptojanin, MIM*604297) mutations cause another AR form of JOPD (PARK20, MIM#615530) characterized by motor PD features presenting at a median age of 21 years (range 12–31 years), poor response to levodopa treatment, early induced dyskinesias, gait disorders, and dysarthria/anarthria (159, 160). In most cases, dystonia, cognitive decline, seizures, and oculomotor abnormalities were described as well (33). Cerebral cortical atrophy, quadrigeminal plate thinning, and hippocampal T2-hyperintensity at MRI have been inconsistently reported (95). Recently, a good response to clonazepam therapy, especially for trunk dystonia, has been reported in two SYNJ1 compound heterozygous siblings with diplopia, dystonia, and Parkinsonism (161). At present, 12 families with atypical Parkinsonism and biallelic SYNJ1 variants have been described (33, 161). SYNJ1 encodes for a poly-phosphoinositide phosphatase, which contains two consecutive phosphatase domains. Mutations affecting the SAC1-like domain are responsible for Parkinsonism while variants impairing the dual phosphatase domain cause a recessive infantile epilepsy syndrome with severe and progressive neurodegeneration (MIM# 617389) (162). No pathology from SYNJ1 patients with Parkinsonism is available today. However, brain neuropathology on a child with biallelic SYNJ1 pathogenic variants affected by refractory epilepsy and severe neurodegeneration showed white matter atrophy and prominent cell loss, tau pathology, and neurofibrillary tangles in SN and, although less represented, in basal ganglia (163).
ATP13A2
ATP13A2 (ATPase 13A2, MIM*610513) causes Kufor Rakeb syndrome (MIM#606693), an AR atypical JOPD with onset before 20 years (164, 165). The disease is characterized by a combination of partially levodopa-responsive Parkinsonism and pyramidal signs, complicated in most cases by dementia, hallucinations, dystonia, impaired saccadic movements, vertical gaze palsy, and mini-myoclonus. Symptoms at onset are variable, encompassing akineto-rigid syndrome, learning disability, cognitive deterioration during school years, or behavioral dysfunctions. Progression of the disease is slow, sometimes associated with cerebellar dysfunction (33). Brain MRI shows diffuse atrophy and, in many cases, iron accumulation in the putamen and caudate (165, 166). ATP13A2 encodes for a lysosomal P5-type ATPase protein that transports inorganic cations and regulates endo-lysosomal cargo sorting and neuronal integrity. Homozygous or compound heterozygous variants in ATP13A2 also have been associated with complex hereditary spastic paraplegia (SPG78, MIM# 617225), with no Parkinsonism in most reported cases and to neuronal ceroid lipofuscinosis. In brains from Kufor Rakeb patients, neuronal and glial lipofuscin deposits at the cortex, cerebellum, and basal ganglia have been described (167, 168).
PLA2G6
Biallelic mutations in PLA2G6 (phospholipase A2, group 6, MIM*603604) cause an early-onset dystonia-Parkinsonism (PARK14, MIM#612953) with variable age at onset (10–30 years) (169, 170). The disease is also characterized by rapid cognitive decline, pyramidal signs, eye movement abnormalities, psychiatric and behavioral problems, cerebellar ataxia, and autonomic dysfunction. The response to levodopa therapy is good but compromised by early-onset treatment-induced dyskinesias. The progression of the disease is rapid and severe, leading to loss of autonomy (171). MRI may show brain iron accumulations and frontal lobe and general white matter atrophy (172). Beyond atypical Parkinsonism, biallelic pathogenic variants in PLA2G6 are also the cause of other neurodegenerative diseases, including infantile neuroaxonal dystrophy (INAD, MIM#256600) and idiopathic neurodegeneration with brain iron accumulation, type 2 (NBIA2, MIM# 610217), all sharing many pathological and clinical features. They are characterized by spheroid axonal inclusions in the brain and motor regression, progressive cognitive decline, axial hypotonia, spasticity, bulbar and ophthalmic dysfunctions, dystonia, and cerebellar atrophy, starting in the first year of life (171). PLA2G6 encodes for calcium-independent phospholipase A2 beta enzyme (iPLA2β), which participates in cell membrane homeostasis, mitochondrial function, fatty acid oxidation, and calcium signaling. Defects of this protein lead to membrane fluidity alteration and neuronal function impairment. a-syn with LBs, Lewy neurites, and neuroaxonal dystrophy are documented in patients with PARK14 and in INAD cases. Differences in phenotypes are related to the effects of the mutations: variants causing loss of PLA2G6 catalytic activity leads to INAD/NBIA2, whereas mutations altering substrate specificity or regulatory domains are responsible for PARK14 (173).
FBXO7
A form of juvenile pallido-pyramidal syndrome (PARK15, MIM#260300) is caused by biallelic mutations in FBXO7 (F-box only protein, MIM*605648). Parkinsonism is usually the first manifestation with rigidity, bradykinesia, postural instability, and, less frequently, tremor, around the age of 17 (range 10–52) years. Pyramidal signs are common and cognitive decline, vertical gaze palsy, and autonomic dysfunctions also occur, although less frequently. Response to levodopa is good but often limited by psychiatric and dyskinetic complications. No abnormalities have been detected in brain MRI (174, 175).
FBXO7 encodes an adaptor protein involved in substrate degradation and in mitochondrial maintenance interacting with PINK1 and Parkin.
X-Linked PD
Pathogenic whole gene deletion, missense, and splicing mutations in RAB39B (RAS-associated protein RAB39B, MIM* 300774) were identified in a few families with non-progressive intellectual disability, macrocephaly, and early-onset Parkinsonism in males (Waisman syndrome, MIM# 311510). Tremor was the presenting motor sign, followed by a levodopa-responsive akinetic-rigid Parkinsonism. Seizures were inconsistently present. Brain atrophy of one affected patient documented a clear α-syn with LBs in the SN and in the cortex, together with tau positive neurofibrillary tangles in the SN and axonal spheroid in the white matter of basal ganglia (176–178). Other mutations of RAB39B were detected in patients with intellectual disability of variable degree, autism, seizure, and macrocephaly without PD (95), as well as in male and female patients with early- or later-onset Parkinsonism, respectively, without intellectual disability (179). RAB39B encodes a neuronal protein involved in vesicular recycling trafficking and in the maintenance of α-syn homeostasis. Parkinsonian phenotypes have been mainly related to loss of function mutations (179). To date, RAB39B is a proven but rare cause of Parkinsonism and intellectual disability in males. Many studies failed to identify pathogenic alterations of this gene in large cohorts of PD patients (180).
Discussion
The advent of next generation sequencing (NGS) increased the number of genes known to cause Mendelian forms of Parkinsonism. They are involved in many specific biological pathways, suggesting that several cellular functions are critical in the pathogenesis of PD. SNCA is directly responsible for altering the expression of α-syn, the main component of LBs (181, 182). PRKN, PINK1, and DJ-1 are related to mitochondrial function and mitophagy (183–185), as well as other genes linked to classical and atypical PD such as FBXO7, PLA2G6, VPS13C, and CHCHD2, which are involved in the mitochondrial quality control system (186). The impairment of lysosomal function has also been linked to the pathogenesis of PD. Altered LRRK2 compromises cell autophagy, reducing α-syn degradation (187, 188), ATP13A2 mutations cause lysosomal dysfunctions (189), and reduced enzymatic activity of GCAse determinates α-syn accumulation, endoplasmic reticulum stress, and mitochondrial dysfunction (190). Recently, a novel disease mechanism involving vesicular trafficking and synaptic endocytosis has been proposed after the identification of many other PD-related genes, such as VPS35, DNAJC6, SYNJ1, and LRP10 (49).
A precise genetic diagnosis enables proper genetic counseling according to the mode of inheritance and penetrance of the mutation and may help define the disease prognosis and influence therapeutic choices.
Some genes and some mutations are associated with specific phenotypes. The relationship between clinical phenotypes and their molecular bases is depicted in Figure 1. The LRRK2 p.G2019S mutation usually causes a slow progressive iPD-like condition with variable age of onset. SNCA pathogenic variants are responsible for a more aggressive Parkinsonism with cognitive decline and other non-motor features. Instead, GBA variant carriers can present more heterogeneous phenotypes, ranging from absence of disease to severe Parkinsonian conditions. In affected carriers, the disease usually manifests as classical late-onset levodopa-responsive PD. However, in a subgroup of patients, the condition can be more severe, sometimes presenting with cognitive decline as PDD or DLB (122, 191). Among EOPD cases, PRKN, PINK1, and DJ-1, sharing mitochondria and mitophagy related functions, are usually responsible for pure PD forms with only motor signs, slow progression, and good response to dopaminergic therapy. Complex EOPD forms with rapid progression, early cognitive deterioration, and additional movement disorders instead are usually related to new AR genes. Except for PODXL, mutations in genes causing juvenile Parkinsonisms are always related to complex phenotypes in which pallido-pyramidal signs, oculomotor abnormalities, cognitive impairment, and seizures variably occur. Intellectual disability in males is a red flag for RAB39B mutations. However, phenotypes rarely overlap, hindering the achievement of the correct diagnostic assumptions. Early onset LRRK2-related PD as well as milder forms of DNAJC6-related Parkinsonism may present with a phenotype resembling pure PRKN/PINK1 recessive cases (132, 156). Clinical features in SNCA triplication carriers are similar to the VPS13C-related phenotype (63, 148), while the complex phenotype of SNCA p.G51D patients, characterized by precocious disease onset, pyramidal signs, myoclonus, seizures, and, inconstantly, cognitive decline, significantly overlaps with DNAJC6- and SYNJ1-related phenotypes (82, 155, 157–160). Differential diagnosis among complex juvenile Parkinsonisms is more tangled and only partially helped by ancillary tests (33). The easier availability of NGS in clinical practice has notably unraveled the emerging genotype and phenotype heterogeneity in Parkinsonian conditions. However, the clinical reasoning is too often overshadowed by excessive reliance on the diagnostic power of this technique. Exonic, multiexonic, and/or whole gene rearrangements, frequently implied in many PD genes, including PRKN, PINK1, DJ-1, and SNCA, and even pathogenic repeat expansions of ATXN2, are not generally identified by NGS techniques. The request of the proper molecular analysis, and thus the possibility to thereby reach the correct diagnosis still requires a scrupulous clinical observation and, whenever possible, a focused clinical diagnostic suspicion, within the frame of accurate genetic counseling.
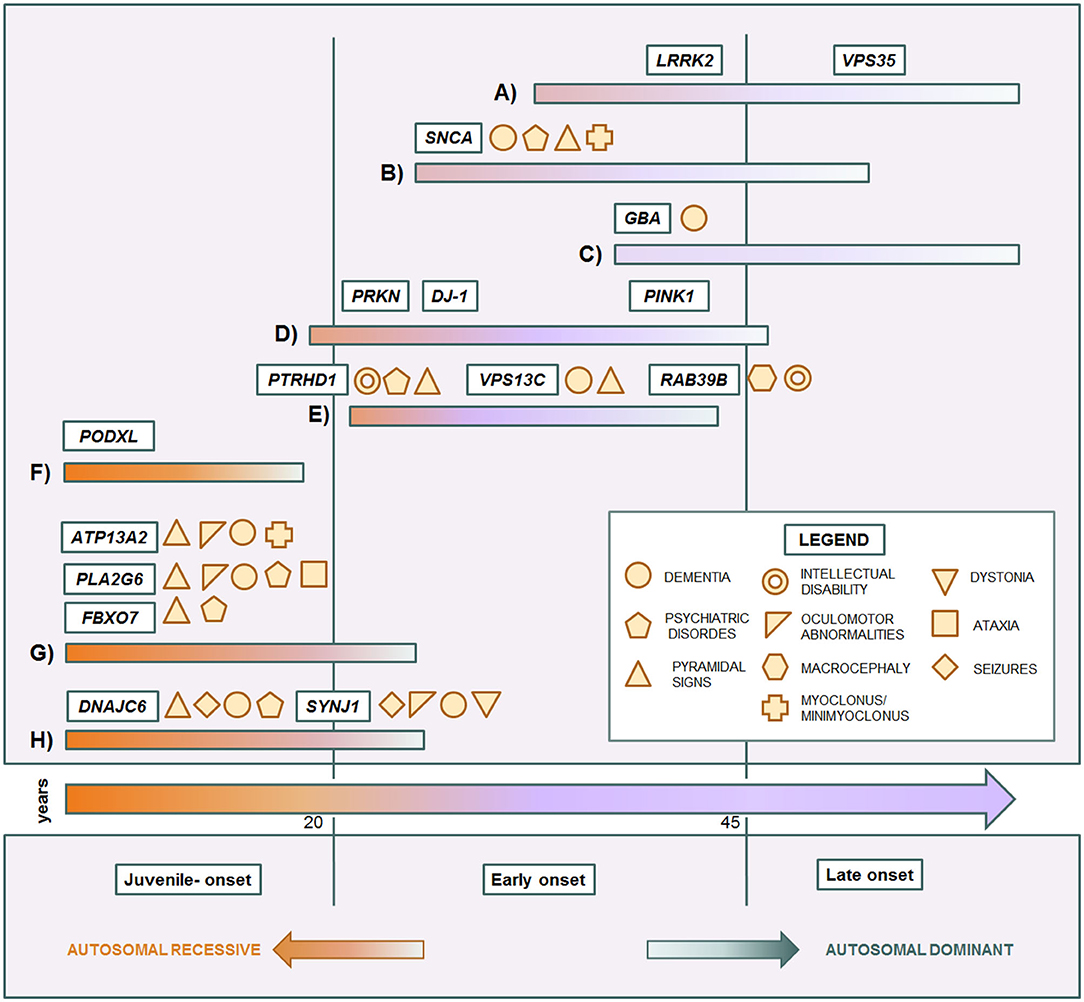
Figure 1. Genotype-phenotype correlation in monogenic Parkinsonian conditions. Central arrow: time of onset. Each colored bar represents a subset of conditions. (A) iPD-like, late-onset autosomal dominant Parkinsonisms. (B) Complicated autosomal dominant Parkinsonisms. (C) Genetic risk factor causing late-onset Parkinsonism. (D) Typical autosomal recessive early-onset Parkinsonisms. (E) Complicated autosomal recessive early onset Parkinsonisms. (F) Juvenile uncomplicated Parkinsonism. (G) Juvenile pallido-pyramidal syndromes. (H) Juvenile atypical Parkinsonisms.
Precision medicine is becoming a reality as well for PD. The presence of defined pathogenic mechanisms, identifiable with genetic testing, is appealing for targeted therapies. Ongoing clinical trials are mainly recruiting participants carrying mutations in specific genes. However, some of these therapies might also be used for broader iPD cohorts. GCse, α-syn, LRRK2, and mitochondrial functions are currently seen as targets for personalized treatments.
The reduction of α-syn accumulation has been the first target of precision medicine. Many different therapeutic approaches have been proposed and even investigated in clinical trials.
One of them aims to reduce the synthesis of α-syn before its aggregation by neutralizing mRNA molecules with RNA interference (RNAi) technologies (192, 193), others by reducing SNCA transcription with molecules that interfere with histones acetylation (2-adrenergic receptor agonists) (194), or by using antisense oligonucleotide (ASO) therapy (49). A second therapeutic approach, aimed at preventing SNCA misfolding or aggregation, utilizes small antibody fragments (intrabodies) that prevent oligomerization by binding intracellular α-syn (195). Two such molecules are currently in early clinical trials (196, 197). An alternative strategy is the enhancement of α-syn clearance by immunotherapies or the activation of autophagy. The first method is based on passive immunization with α-syn specific antibodies or active immunization with injections of modified α-syn stimulating the production of endogenous antibodies. The second one is based on the administration of autophagy enhancers such as rapamycin, lithium, or the antineoplastic drug nilotinib (198). Clinical trials are ongoing for these compounds as well.
Another target of great interest is GCase. Clinical trials for GBA-targeted therapies are studying drugs able to increase GCase production or activity. The glucosylceramide synthase inhibitor GZ/SAR402671, which reduces glucocerebrosidase substrates, and ambroxol hydrochloride, a small chaperone molecule able to increase GCase activity, are present research targets. Other small molecular chaperones have been shown to reduce α-syn accumulation in GBA PD patients and for one of them a clinical trial is currently running (Netherlands Trial Register: NTR6960). Another way to restore GCase activity is to introduce wild-type GBA genes into the genome of GBA mutation carriers. Gene therapy, at present under investigation in other neuromuscular diseases, also will be tested in GBA-mutated patients (49, 199).
The evidence that LRRK2 kinase activity inhibition reverses the pathological features and reduces α-syn accumulation in cellular and animal models makes this protein another candidate for a target therapy. Preclinical research investigated many LRRK2 inhibitors that failed in brain penetration or in pharmacokinetic properties. Recently, two molecules with exceptional potency and selectivity in inhibiting LRRK2 kinase activity and a good safety profile have been identified (200–202). Based on the good results on animal models, the administration of this molecule started in healthy and affected human subjects, with and without LRRK2 mutations (ClinicalTrials.gov Identifier: NCT03710707; ClinicalTrials.gov Identifier: NCT04056689).
Finally, various therapeutic approaches focusing on mitochondrial dysfunctions have been proposed. Molecules improving mitochondrial functions, such as kinetin triphosphate, able to ameliorate the kinase activity of mutated PINK1, and selective MAO-B inhibitors, including selegilin and regasilin, are being studied. Good results of these treatments have been reported only after a proper stratification of patients, with the administration of these compounds in the “mitochondrial subtypes PDs” (203).
Currently, genetically determined PD offers the unique framework of a constant dialogue between cell biology, clinical acumen, medical genetics, and targeted therapy. Only appropriate genetic testing allows identification of the specific condition in patients, pointing which pathway, or pathways, is involved in the underlying pathogenesis for that case. Most of the ongoing trials about target therapy involve PD cases with molecular diagnosis as positive controls for the different forms of PD. In the era of precision medicine, the importance of genetics in PD is overstepping the boundaries of the research to play an increasingly pivotal role in clinical practice. Nevertheless, the identification of the compromised pathways and genes, driving the choice of the proper therapy, is still strictly dependent on clinical reasoning that, despite the availability of innovative diagnostic techniques, continues to be the irreplaceable key in reaching a genetic diagnosis.
Author Contributions
DG: conceptualization, investigation, data curation, original draft, and visualization. MP: data curation, methodology, supervision, review, and editing. MT: resources, methodology, supervision, review, and editing. AP: methodology, data curation, supervision, review, and editing. SP: conceptualization, investigation, data curation, original draft, methodology, and supervision. All authors contributed to the article and approved the submitted version.
Funding
This research received no external funding. AP was funded by Sapienza, University of Rome.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Simon DK, Tanner CM, Brundin P. Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med. (2020) 36:1–12. doi: 10.1016/j.cger.2019.08.002
2. Johansen KK, Torp SH, Farrer MJ, Gustavsson EK, Aasly JO. A case of Parkinson's disease with no lewy body pathology due to a homozygous exon deletion in Parkin. Case Rep Neurol Med. (2018) 2018:6838965. doi: 10.1155/2018/6838965
3. Schrag A, Schott JM. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. (2006) 5:355–63. doi: 10.1016/S1474-4422(06)70411-2
4. Kumar KR, Djarmati-Westenberger A, Grunewald A. Genetics of Parkinson's disease. Semin Neurol. (2011) 31:433–40. doi: 10.1055/s-0031-1299782
5. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:17013. doi: 10.1038/nrdp.2017.13
6. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol. (2020) 19:170–8. doi: 10.1016/S1474-4422(19)30287-X
7. Kestenbaum M, Alcalay RN. Clinical features of lrrk2 carriers with Parkinson's disease. Adv Neurobiol. (2017) 14:31–48. doi: 10.1007/978-3-319-49969-7_2
8. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. (2004) 44:595–600. doi: 10.1016/j.neuron.2004.10.023
9. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
10. Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, et al. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. (2005) 65:741–4. doi: 10.1212/01.WNL.0000172630.22804.73
11. Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. (2005) 365:412–5. doi: 10.1016/S0140-6736(05)17829-5
12. Hernandez DG, Paisan-Ruiz C, McInerney-Leo A, Jain S, Meyer-Lindenberg A, Evans EW, et al. Clinical and positron emission tomography of Parkinson's disease caused by LRRK2. Ann Neurol. (2005) 57:453–6. doi: 10.1002/ana.20401
13. Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. (2005) 57:918–21. doi: 10.1002/ana.20484
14. Koros C, Simitsi A, Stefanis L. Genetics of Parkinson's disease: genotype-phenotype correlations. Int Rev Neurobiol. (2017) 132:197–231. doi: 10.1016/bs.irn.2017.01.009
15. Li C, Ting Z, Qin X, Ying W, Li B, Guo Qiang L, et al. The prevalence of LRRK2 Gly2385Arg variant in Chinese Han population with Parkinson's disease. Mov Disord. (2007) 22:2439–43. doi: 10.1002/mds.21763
16. Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI, et al. Analysis of Lrrk2 R1628P as a risk factor for Parkinson's disease. Ann Neurol. (2008) 64:88–92. doi: 10.1002/ana.21405
17. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
18. Monfrini E, Di Fonzo A. Leucine-rich repeat kinase (LRRK2) genetics and Parkinson's Disease. Adv Neurobiol. (2017) 14:3–30. doi: 10.1007/978-3-319-49969-7_1
19. Simon-Sanchez J, Marti-Masso JF, Sanchez-Mut JV, Paisan-Ruiz C, Martinez-Gil A, Ruiz-Martinez J, et al. Parkinson's disease due to the R1441G mutation in Dardarin: a founder effect in the Basques. Mov Disord. (2006) 21:1954–9. doi: 10.1002/mds.21114
20. De Rosa A, De Michele G, Guacci A, Carbone R, Lieto M, Peluso S, et al. Genetic screening for the LRRK2 R1441C and G2019S mutations in Parkinsonian patients from Campania. J Parkinsons Dis. (2014) 4:123–8. doi: 10.3233/JPD-130312
21. Lee AJ, Wang Y, Alcalay RN, Mejia-Santana H, Saunders-Pullman R, Bressman S, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord. (2017) 32:1432–8. doi: 10.1002/mds.27059
22. Hentati F, Trinh J, Thompson C, Nosova E, Farrer MJ, Aasly JO. LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology. (2014) 83:568–9. doi: 10.1212/WNL.0000000000000675
23. Fernandez-Santiago R, Garrido A, Infante J, Gonzalez-Aramburu I, Sierra M, Fernandez M, et al. alpha-synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine-rich repeat kinase 2 (LRRK2) Parkinson's disease in Spain. Mov Disord. (2018) 33:637–41. doi: 10.1002/mds.27295
24. Iwaki H, Blauwendraat C, Makarious MB, Bandres-Ciga S, Leonard HL, Gibbs JR, et al. Penetrance of Parkinson's disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. Mov Disord. (2020) 35:774–80. doi: 10.1101/738260
25. Ruiz-Martinez J, Gorostidi A, Ibanez B, Alzualde A, Otaegui D, Moreno F, et al. Penetrance in Parkinson's disease related to the LRRK2 R1441G mutation in the Basque country (Spain). Mov Disord. (2010) 25:2340–5. doi: 10.1002/mds.23278
26. Rizzone MG, Martone T, Balestrino R, Lopiano L. Genetic background and outcome of deep brain stimulation in Parkinson's disease. Parkinsonism Relat Disord. (2019) 64:8–19. doi: 10.1016/j.parkreldis.2018.08.006
27. Gan-Or Z, Leblond CS, Mallett V, Orr-Urtreger A, Dion PA, Rouleau GA. LRRK2 mutations in Parkinson disease; a sex effect or lack thereof? A meta-analysis. Parkinsonism Relat Disord. (2015) 21:778–82. doi: 10.1016/j.parkreldis.2015.05.002
28. Saunders-Pullman R, Mirelman A, Alcalay RN, Wang C, Ortega RA, Raymond D, et al. Progression in the LRRK2-Asssociated Parkinson disease population. JAMA Neurol. (2018) 75:312–9. doi: 10.1001/jamaneurol.2017.4019
29. Mirelman A, Heman T, Yasinovsky K, Thaler A, Gurevich T, Marder K, et al. Fall risk and gait in Parkinson's disease: the role of the LRRK2 G2019S mutation. Mov Disord. (2013) 28:1683–90. doi: 10.1002/mds.25587
30. Marras C, Schule B, Munhoz RP, Rogaeva E, Langston JW, Kasten M, et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology. (2011) 77:325–33. doi: 10.1212/WNL.0b013e318227042d
31. Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, et al. Clinical features of LRRK2-associated Parkinson's disease in central Norway. Ann Neurol. (2005) 57:762–5. doi: 10.1002/ana.20456
32. Alcalay RN, Mejia-Santana H, Mirelman A, Saunders-Pullman R, Raymond D, Palmese C, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson's disease. Parkinsonism Relat Disord. (2015) 21:106–10. doi: 10.1016/j.parkreldis.2014.09.033
33. Weissbach A, Wittke C, Kasten M, Klein C. 'Atypical' Parkinson's disease - genetic. Int Rev Neurobiol. (2019) 149:207–35. doi: 10.1016/bs.irn.2019.10.011
34. Sanchez-Contreras M, Heckman MG, Tacik P, Diehl N, Brown PH, Soto-Ortolaza AI, et al. Study of LRRK2 variation in tauopathy: progressive supranuclear palsy and corticobasal degeneration. Mov Disord. (2017) 32:115–23. doi: 10.1002/mds.26815
35. Tan EK, Skipper L, Chua E, Wong MC, Pavanni R, Bonnard C, et al. Analysis of 14 LRRK2 mutations in Parkinson's plus syndromes and late-onset Parkinson's disease. Mov Disord. (2006) 21:997–1001. doi: 10.1002/mds.20875
36. Cho JW, Kim SY, Park SS, Jeon BS. The G2019S LRRK2 mutation is rare in Korean patients with Parkinson's disease and multiple system atrophy. J Clin Neurol. (2009) 5:29–32. doi: 10.3988/jcn.2009.5.1.29
37. Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. (2020) 16:97–107. doi: 10.1038/s41582-019-0301-2
38. Brockmann K, Groger A, Di Santo A, Liepelt I, Schulte C, Klose U, et al. Clinical and brain imaging characteristics in leucine-rich repeat kinase 2-associated PD and asymptomatic mutation carriers. Mov Disord. (2011) 26:2335–42. doi: 10.1002/mds.23991
39. Pyatigorskaya N, Sharman M, Corvol JC, Valabregue R, Yahia-Cherif L, Poupon F, et al. High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord. (2015) 30:1077–84. doi: 10.1002/mds.26218
40. Hustad E, Aasly JO. Clinical and imaging markers of prodromal Parkinson's disease. Front Neurol. (2020) 11:395. doi: 10.3389/fneur.2020.00395
41. Wile DJ, Agarwal PA, Schulzer M, Mak E, Dinelle K, Shahinfard E, et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol. (2017) 16:351–9. doi: 10.1016/S1474-4422(17)30056-X
42. Liu SY, Wile DJ, Fu J, Valerio FJ, Shahinfard E, McCormick S, et al. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson's disease: a cross-sectional PET study. Lancet Neurol. (2018) 17:309–16. doi: 10.1016/S1474-4422(18)30032-2
43. Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. (2015) 72:100–5. doi: 10.1001/jamaneurol.2014.2704
44. Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson's disease. Mov Disord. (2012) 27:831–42. doi: 10.1002/mds.24962
45. Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: mendelian versus non-Mendelian inheritance. J Neurochem. (2016) 139(Suppl 1):59–74. doi: 10.1111/jnc.13593
46. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. (2014) 46:989–93. doi: 10.1038/ng.3043
47. Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. (2017) 49:1511–6. doi: 10.1038/ng.3955
48. Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener. (2016) 11:73. doi: 10.1186/s13024-016-0140-1
49. Toffoli M, Vieira SRL, Schapira AHV. Genetic causes of PD: a pathway to disease modification. Neuropharmacology. (2020) 170:108022. doi: 10.1016/j.neuropharm.2020.108022
50. Struhal W, Presslauer S, Spielberger S, Zimprich A, Auff E, Bruecke T, et al. VPS35 Parkinson's disease phenotype resembles the sporadic disease. J Neural Transm. (2014) 121:755–9. doi: 10.1007/s00702-014-1179-1
51. Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. (2011) 89:168–75. doi: 10.1016/j.ajhg.2011.06.008
52. Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. (2011) 89:162–7. doi: 10.1016/j.ajhg.2011.06.001
53. Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Bertram L, et al. A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. (2012) 49:721–6. doi: 10.1136/jmedgenet-2012-101155
54. Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson's disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord. (2018) 33:1857–70. doi: 10.1002/mds.27527
55. Wider C, Wszolek ZK. Etiology and pathophysiology of frontotemporal dementia, Parkinson disease and Alzheimer disease: lessons from genetic studies. Neurodegener Dis. (2008) 5:122–5. doi: 10.1159/000113680
56. Mohan M, Mellick GD. Role of the VPS35 D620N mutation in Parkinson's disease. Parkinsonism Relat Disord. (2017) 36:10–18. doi: 10.1016/j.parkreldis.2016.12.001
57. Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. (1996) 274:1197–9. doi: 10.1126/science.274.5290.1197
58. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. (1997) 276:2045–7. doi: 10.1126/science.276.5321.2045
59. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
60. Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. (2011) 10:1015–25. doi: 10.1016/S1474-4422(11)70213-7
61. Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson's disease caused by SNCA duplications. Parkinsonism Relat Disord. (2016) 22 (Suppl 1):S1–6. doi: 10.1016/j.parkreldis.2015.09.007
62. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
63. Gwinn K, Devine MJ, Jin LW, Johnson J, Bird T, Muenter M, et al. Clinical features, with video documentation, of the original familial lewy body parkinsonism caused by alpha-synuclein triplication (Iowa kindred). Mov Disord. (2011) 26:2134–6. doi: 10.1002/mds.23776
64. Olgiati S, Thomas A, Quadri M, Breedveld GJ, Graafland J, Eussen H, et al. Early-onset parkinsonism caused by alpha-synuclein gene triplication: clinical and genetic findings in a novel family. Parkinsonism Relat Disord. (2015) 21:981–6. doi: 10.1016/j.parkreldis.2015.06.005
65. Elia AE, Petrucci S, Fasano A, Guidi M, Valbonesi S, Bernardini L, et al. Alpha-synuclein gene duplication: marked intrafamilial variability in two novel pedigrees. Mov Disord. (2013) 28:813–7. doi: 10.1002/mds.25518
66. Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, et al. Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord. (2009) 24:1811–9. doi: 10.1002/mds.22682
67. Kasten M, Klein C. The many faces of alpha-synuclein mutations. Mov Disord. (2013) 28:697–701. doi: 10.1002/mds.25499
68. Kara E, Kiely AP, Proukakis C, Giffin N, Love S, Hehir J, et al. A 6.4 Mb duplication of the alpha-synuclein locus causing frontotemporal dementia and Parkinsonism: phenotype-genotype correlations. JAMA Neurol. (2014) 71:1162–71. doi: 10.1001/jamaneurol.2014.994
69. Itokawa K, Sekine T, Funayama M, Tomiyama H, Fukui M, Yamamoto T, et al. A case of alpha-synuclein gene duplication presenting with head-shaking movements. Mov Disord. (2013) 28:384–7. doi: 10.1002/mds.25243
70. Tambasco N, Nigro P, Romoli M, Prontera P, Simoni S, Calabresi P. A53T in a parkinsonian family: a clinical update of the SNCA phenotypes. J Neural Transm. (2016) 123:1301–7. doi: 10.1007/s00702-016-1578-6
71. Ricciardi L, Petrucci S, Di Giuda D, Serra L, Spano B, Sensi M, et al. The contursi family 20 years later: intrafamilial phenotypic variability of the SNCA p.A53T mutation. Mov Disord. (2016) 31:257–8. doi: 10.1002/mds.26549
72. Papadimitriou D, Antonelou R, Miligkos M, Maniati M, Papagiannakis N, Bostantjopoulou S, et al. Motor and nonmotor features of carriers of the p.A53T alpha-synuclein mutation: a longitudinal study. Mov Disord. (2016) 31:1226–30. doi: 10.1002/mds.26615
73. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson's disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord. (2018) 33:730–41. doi: 10.1002/mds.27352
74. Morfis L, Cordato DJ. Dementia with Lewy bodies in an elderly Greek male due to alpha-synuclein gene mutation. J Clin Neurosci. (2006) 13:942–4. doi: 10.1016/j.jocn.2005.11.040
75. Bougea A, Koros C, Stamelou M, Simitsi A, Papagiannakis N, Antonelou R, et al. Frontotemporal dementia as the presenting phenotype of p.A53T mutation carriers in the alpha-synuclein gene. Parkinsonism Relat Disord. (2017) 35:82–7. doi: 10.1016/j.parkreldis.2016.12.002
76. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. (2013) 28:811–3. doi: 10.1002/mds.25421
77. Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. (2013) 80:1062–4. doi: 10.1212/WNL.0b013e31828727ba
78. Kruger R, Kuhn W, Leenders KL, Sprengelmeyer R, Muller T, Woitalla D, et al. Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology. (2001) 56:1355–62. doi: 10.1212/WNL.56.10.1355
79. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. doi: 10.1038/ng0298-106
80. Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. (2004) 55:164–73. doi: 10.1002/ana.10795
81. Tijero B, Gomez-Esteban JC, Lezcano E, Fernandez-Gonzalez C, Somme J, Llorens V, et al. Cardiac sympathetic denervation in symptomatic and asymptomatic carriers of the E46K mutation in the alpha synuclein gene. Parkinsonism Relat Disord. (2013) 19:95–100. doi: 10.1016/j.parkreldis.2012.08.001
82. Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. (2013) 73:459–71. doi: 10.1002/ana.23894
83. Tokutake T, Ishikawa A, Yoshimura N, Miyashita A, Kuwano R, Nishizawa M, et al. Clinical and neuroimaging features of patient with early-onset Parkinson's disease with dementia carrying SNCA p.G51D mutation. Parkinsonism Relat Disord. (2014) 20:262–4. doi: 10.1016/j.parkreldis.2013.11.008
84. Martikainen MH, Paivarinta M, Hietala M, Kaasinen V. Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet. (2015) 1:e27. doi: 10.1212/NXG.0000000000000027
85. George JM. The synucleins. Genome Biol. (2002) 3:REVIEWS3002. doi: 10.1186/gb-2001-3-1-reviews3002
86. Fraldi A, Klein AD, Medina DL, Settembre C. Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci. (2016) 39:277–95. doi: 10.1146/annurev-neuro-070815-014031
87. Nussbaum RL. Genetics of synucleinopathies. Cold Spring Harb Perspect Med. (2018) 8:a024109. doi: 10.1101/cshperspect.a024109
88. Tan EK, Matsuura T, Nagamitsu S, Khajavi M, Jankovic J., Ashizawa T. Polymorphism of NACP-Rep1 in Parkinson's disease: an etiologic link with essential tremor? Neurology. (2000) 54:1195–8. doi: 10.1212/WNL.54.5.1195
89. Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, et al. alpha-Synuclein gene haplotypes are associated with Parkinson's disease. Hum Mol Genet. (2001) 10:1847–51. doi: 10.1093/hmg/10.17.1847
90. Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson's disease. Rev Neurol. (2018) 174:628–43. doi: 10.1016/j.neurol.2018.08.004
91. Saini P, Rudakou U, Yu E, Ruskey JA, Asayesh F, Laurent SB, et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson's disease. Neurobiol Aging. (2021) 100:119.e7-119.e13. doi: 10.1016/j.neurobiolaging.2020.10.019
92. Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol. (2015) 14:274–82. doi: 10.1016/S1474-4422(14)70266-2
93. Ikeda A, Matsushima T, Daida K, Nakajima S, Conedera S, Li Y, et al. A novel mutation of CHCHD2 p.R8H in a sporadic case of Parkinson's disease. Parkinsonism Relat Disord. (2017) 34:66–8. doi: 10.1016/j.parkreldis.2016.10.018
94. Ikeda A, Nishioka K, Meng H, Takanashi M, Hasegawa I, Inoshita T, et al. Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum Mol Genet. (2019) 28:3895–911. doi: 10.1093/hmg/ddz241
95. Puschmann A. New genes causing hereditary Parkinson's disease or parkinsonism. Curr Neurol Neurosci Rep. (2017) 17:66. doi: 10.1007/s11910-017-0780-8
96. Deng HX, Shi Y, Yang Y, Ahmeti KB, Miller N, Huang C, et al. Identification of TMEM230 mutations in familial Parkinson's disease. Nat Genet. (2016) 48:733–9. doi: 10.1038/ng.3589
97. Wang X, Whelan E, Liu Z, Liu CF, Smith WW. Controversy of TMEM230 Associated with Parkinson's Disease. Neuroscience. (2021) 453:280–6. doi: 10.1016/j.neuroscience.2020.11.004
98. Sudhaman S, Muthane UB, Behari M, Govindappa ST, Juyal RC, Thelma BK. Evidence of mutations in RIC3 acetylcholine receptor chaperone as a novel cause of autosomal-dominant Parkinson's disease with non-motor phenotypes. J Med Genet. (2016) 53:559–66. doi: 10.1136/jmedgenet-2015-103616
99. Ross JP, Dupre N, Dauvilliers Y, Strong S, Dionne-Laporte A, Dion PA, et al. RIC3 variants are not associated with Parkinson's disease in French-Canadians and French. Neurobiol Aging. (2017) 53:194 e9–11. doi: 10.1016/j.neurobiolaging.2017.01.005
100. Quadri M, Mandemakers W, Grochowska MM, Masius R, Geut H, Fabrizio E, et al. LRP10 genetic variants in familial Parkinson's disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol. (2018) 17:597–608. doi: 10.1016/S1474-4422(18)30408-3
101. Chen Y, Cen Z, Zheng X, Pan Q, Chen X, Zhu, et al. LRP10 in autosomal-dominant Parkinson's disease. Mov Disord. (2019) 34:912–6. doi: 10.1002/mds.27693
102. Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain. (2014) 137(Pt 9):2480–92. doi: 10.1093/brain/awu179
103. Guella I, Sherman HE, Appel-Cresswell S, Rajput A, Rajput, Farrer AH, et al. Parkinsonism in GTP cyclohydrolase 1 mutation carriers. Brain. (2015) 138(Pt 5):e349. doi: 10.1093/brain/awu341
104. Pan HX, Zhao YW, Mei JP, Fang ZH, Wang Y, Zhou X, et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson's disease: a two-cohort case-control study. Transl Neurodegener. (2020) 9:31. doi: 10.1186/s40035-020-00212-3
105. Kim YE, Jeon B, Farrer MJ, Scott E, Guella I, Park SS, et al. SCA2 family presenting as typical Parkinson's disease: 34 year follow up. Parkinsonism Relat Disord. (2017) 40:69–72. doi: 10.1016/j.parkreldis.2017.04.003
106. Takao M, Aoyama M, Ishikawa K, Sakiyama Y, Yomono H, Saito Y, et al. Spinocerebellar ataxia type 2 is associated with Parkinsonism and Lewy body pathology. BMJ Case Rep. (2011) 2011:bcr0120113685. doi: 10.1136/bcr.01.2011.3685
107. Smith L, Mullin S, Schapira AHV. Insights into the structural biology of Gaucher disease. Exp Neurol. (2017) 298(Pt B):180–90. doi: 10.1016/j.expneurol.2017.09.010
108. Riboldi GM, Di Fonzo AB. GBA, Gaucher disease, and Parkinson's disease: from genetic to clinic to new therapeutic approaches. Cells. (2019) 8:364. doi: 10.3390/cells8040364
109. Sidransky E, Samaddar T, Tayebi N. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. (2009) 73:1424–5, author reply 1425–6. doi: 10.1212/WNL.0b013e3181b28601
110. Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet. (2011) 20:202–10. doi: 10.1093/hmg/ddq454
111. Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. (2012) 78:417–20. doi: 10.1212/WNL.0b013e318245f476
112. Skrahina V, Gaber H, Vollstedt EJ, Forster TM, Usnich T, Curado F, et al. The Rostock International Parkinson's Disease (ROPAD) study: protocol and initial findings. Mov Disord. (2021) 36:1005–10. doi: 10.1002/mds.28416
113. Neumann J, Bras J, Deas E, O'Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. (2009) 132(Pt 7):1783–94. doi: 10.1093/brain/awp044
114. Mangone G, Bekadar S, Cormier-Dequaire F, Tahiri K, Welaratne A, Czernecki V, et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson's disease patients with GBA mutations. Parkinsonism Relat Disord. (2020) 76:56–62. doi: 10.1016/j.parkreldis.2020.04.002
115. Pal G, Robertson E, O'Keefe J, Hall D. The neuropsychiatric and motor profile of GBA-Associated Parkinson's disease: a review. Mov Disord Clin Pract. (2016) 3:4–8. doi: 10.1002/mdc3.12229
116. Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson's disease phenotype. Parkinsonism Relat Disord. (2017) 36:47–51. doi: 10.1016/j.parkreldis.2016.12.014
117. Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. (2016) 73:1217–24. doi: 10.1001/jamaneurol.2016.2245
118. Petrucci S, Ginevrino M, Trezzi I, Monfrini E, Ricciardi L, Albanese A, et al. GBA-Related Parkinson's disease: dissection of genotype-phenotype correlates in a large Italian Cohort. Mov Disord. (2020) 35:2106–11. doi: 10.1002/mds.28200
119. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol. (2016) 80:662–73. doi: 10.1002/ana.24777
120. Agosta F, Kostic VS, Davidovic K, Kresojevic N, Sarro L, Svetel M, et al. White matter abnormalities in Parkinson's disease patients with glucocerebrosidase gene mutations. Mov Disord. (2013) 28:772–8. doi: 10.1002/mds.25397
121. Brockmann K, Hilker R, Pilatus U, Baudrexel S, Srulijes K, Magerkurth J, et al. GBA-associated PD. Neurodegeneration, altered membrane metabolism, and lack of energy failure. Neurology. (2012) 79:213–20. doi: 10.1212/WNL.0b013e31825dd369
122. Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. (2013) 70:727–35. doi: 10.1001/jamaneurol.2013.1925
123. Pilotto A, Schulte C, Hauser AK, Biskup S, Munz M, Brockmann K, et al. GBA-associated parkinsonism and dementia: beyond alpha-synucleinopathies? Eur J Neurol. (2016) 23:520–6. doi: 10.1111/ene.12894
124. Picillo M, Petrucci S, Valente EM, Pappata S, Squame F, Ginevrino M, et al. Progressive supranuclear palsy-like phenotype in a GBA E326K mutation carrier. Mov Disord Clin Pract. (2017) 4:444–6. doi: 10.1002/mdc3.12406
125. Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. (2015) 2:417–26. doi: 10.1002/acn3.185
126. Sklerov M, Kang UJ, Liong C, Clark L, Marder K, Pauciulo M, et al. Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract. (2017) 4:574–81. doi: 10.1002/mdc3.12481
127. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. (2014) 137(Pt 5):1481–95. doi: 10.1093/brain/awu020
128. Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord. (2017) 32:1504–23. doi: 10.1002/mds.27193
129. Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci. (2015) 40:200–10. doi: 10.1016/j.tibs.2015.02.003
130. van der Merwe C, Jalali Sefid Dashti Z, Christoffels A, Loos B, Bardien S. Evidence for a common biological pathway linking three Parkinson's disease-causing genes: Parkin, PINK1 and DJ-1. Eur J Neurosci. (2015) 41:1113–25. doi: 10.1111/ejn.12872
131. Voigt A, Berlemann LA, Winklhofer KF. The mitochondrial kinase PINK1: functions beyond mitophagy. J Neurochem. (2016) 139(Suppl 1):232–9. doi: 10.1111/jnc.13655
132. Kilarski LL, Pearson JP, Newsway V, Majounie E, Knipe MD, Misbahuddin A, et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson's disease. Mov Disord. (2012) 27:1522–9. doi: 10.1002/mds.25132
133. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
134. Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain. (2003) 126(Pt 6):1279–92. doi: 10.1093/brain/awg142
135. Alcalay RN, Siderowf A, Ottman R, Caccappolo E, Mejia-Santana H, Tang MX, et al. Olfaction in Parkin heterozygotes and compound heterozygotes: the CORE-PD study. Neurology. (2011) 76:319–26. doi: 10.1212/WNL.0b013e31820882aa
136. Doherty KM, Silveira-Moriyama L, Parkkinen L, Healy DG, Farrell M, Mencacci NE, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol. (2013) 70:571–9. doi: 10.1001/jamaneurol.2013.172
137. Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. (2001) 50:293–300. doi: 10.1002/ana.1132
138. Fiesel FC, Caulfield TR, Moussaud-Lamodiere EL, Ogaki K, Dourado DF, Flores SC, et al. Structural and functional impact of Parkinson disease-associated mutations in the E3 Ubiquitin Ligase Parkin. Hum Mutat. (2015) 36:774–86. doi: 10.1002/humu.22808
139. Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, et al. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. (2005) 14:2571–86. doi: 10.1093/hmg/ddi292
140. Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. (2010) 133(Pt 4):1128–42. doi: 10.1093/brain/awq051
141. Annesi G, Savettieri G, Pugliese P, D'Amelio M, Tarantino P, Ragonese P, et al. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. (2005) 58:803–7. doi: 10.1002/ana.20666
142. Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain. (2016) 139(Pt 6):1680–7. doi: 10.1093/brain/aww080
143. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. (2007) 6:652–62. doi: 10.1016/S1474-4422(07)70174-6
144. Marongiu R, Ferraris A, Ialongo T, Michiorri S, Soleti F, Ferrari F, et al. PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum. Hum Mutat. (2008) 29:565. doi: 10.1002/humu.20719
145. Bruggemann N, Mitterer M, Lanthaler AJ, Djarmati A, Hagenah J, Wiegers K, et al. Frequency of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers. Parkinsonism Relat Disord. (2009) 15:425–9. doi: 10.1016/j.parkreldis.2008.11.014
146. Pavese N, Khan NL, Scherfler C, Cohen L, Brooks DJ, Wood NW, et al. Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: an 18F-dopa PET progression study. Mov Disord. (2009) 24:2260–6. doi: 10.1002/mds.22817
147. van Nuenen BF, van Eimeren T, van der Vegt JP, Buhmann C, Klein C, Bloem BR, et al. Mapping preclinical compensation in Parkinson's disease: an imaging genomics approach. Mov Disord. (2009) 24(Suppl. 2):S703–10. doi: 10.1002/mds.22635
148. Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive Parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet. (2016) 98:500–13. doi: 10.1016/j.ajhg.2016.01.014
149. Schreglmann SR, Houlden H. VPS13C-another hint at mitochondrial dysfunction in familial Parkinson's disease. Mov Disord. (2016) 31:1340. doi: 10.1002/mds.26682
150. Khodadadi H, Azcona LJ, Aghamollaii V, Omrani MD, Garshasbi M, Taghavi S, et al. PTRHD1 (C2orf79) mutations lead to autosomal-recessive intellectual disability and parkinsonism. Mov Disord. (2017) 32:287–91. doi: 10.1002/mds.26824
151. Kuipers DJS, Carr J, Bardien S, Thomas P, Sebate B, Breedveld GJ, et al. PTRHD1 Loss-of-function mutation in an african family with juvenile-onset Parkinsonism and intellectual disability. Mov Disord. (2018) 33:1814–9. doi: 10.1002/mds.27501
152. Chen SJ, Ho CH, Lin HY, Lin CH, Wu RM. Lack of PTRHD1 mutation in patients with young-onset and familial Parkinson's disease in a Taiwanese population. Neurobiol Aging. (2021) 100:188e15–118e16. doi: 10.1016/j.neurobiolaging.2020.09.002
153. Niemann N, Jankovic J. Juvenile Parkinsonism: differential diagnosis, genetics, and treatment. Parkinsonism Relat Disord. (2019) 67:74–89. doi: 10.1016/j.parkreldis.2019.06.025
154. Sudhaman S, Prasad K, Behari M, Muthane UB, Juyal RC, Thelma BK. Discovery of a frameshift mutation in podocalyxin-like (PODXL) gene, coding for a neural adhesion molecule, as causal for autosomal-recessive juvenile Parkinsonism. J Med Genet. (2016) 53:450–6. doi: 10.1136/jmedgenet-2015-103459
155. Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE. (2012) 7:e36458. doi: 10.1371/journal.pone.0036458
156. Olgiati S, Quadri M, Fang M, Rood JP, Saute JA, Chien HF, et al. DNAJC6 mutations associated with early-onset Parkinson's disease. Ann Neurol. (2016) 79:244–56. doi: 10.1002/ana.24553
157. Elsayed LE, Drouet V, Usenko T, Mohammed IN, Hamed AA, Elseed MA, et al. A Novel nonsense mutation in DNAJC6 expands the phenotype of autosomal-recessive juvenile-onset Parkinson's disease. Ann Neurol. (2016) 79:335–7. doi: 10.1002/ana.24591
158. Koroglu C, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. (2013) 19:320–4. doi: 10.1016/j.parkreldis.2012.11.006
159. Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. (2013) 34:1208–15. doi: 10.1002/humu.22373
160. Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. (2013) 34:1200–7. doi: 10.1002/humu.22372
161. Hong D, Cong L, Zhong S, He Y, Xin L, Gao X, et al. Clonazepam improves the symptoms of two siblings with novel variants in the SYNJ1 gene. Parkinsonism Relat Disord. (2019) 62:221–25. doi: 10.1016/j.parkreldis.2018.11.020
162. Hardies K, Cai Y, Jardel C, Jansen AC, Cao M, May P, et al. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain. (2016) 139(Pt 9):2420–30. doi: 10.1093/brain/aww180
163. Dyment DA, Smith AC, Humphreys P, Schwartzentruber J, Beaulieu CL, Consortium FC, et al. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol Aging. (2015) 36:1222 e1–5. doi: 10.1016/j.neurobiolaging.2014.09.005
164. Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. (2006) 38:1184–91. doi: 10.1038/ng1884
165. Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. (2007) 68:1557–62. doi: 10.1212/01.wnl.0000260963.08711.08
166. Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord. (2010) 25:979–84. doi: 10.1002/mds.22947
167. Estiar MA, Leveille E, Spiegelman D, Dupre N, Trempe JF, Rouleau GA, et al. Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol Genet Genomic Med. (2020) 8:e1052. doi: 10.1002/mgg3.1052
168. Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. (2012) 21:2646–50. doi: 10.1093/hmg/dds089
169. Paisan-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. (2010) 25:1791–800. doi: 10.1002/mds.23221
170. Bohlega SA, Al-Mubarak BR, Alyemni EA, Abouelhoda M, Monies D, et al. Clinical heterogeneity of PLA2G6-related Parkinsonism: analysis of two Saudi families. BMC Res Notes. (2016) 9:295. doi: 10.1186/s13104-016-2102-7
171. Guo YP, Tang BS, Guo JF. PLA2G6-associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol. (2018) 9:1100. doi: 10.3389/fneur.2018.01100
172. Kurian MA, Morgan NV, MacPherson L, Foster K, Peake D, Gupts R, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology. (2008) 70:1623–9. doi: 10.1212/01.wnl.0000310986.48286.8e
173. Engel LA, Jing Z, O'Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia-parkinsonism. PLoS ONE. (2010) 5:e12897. doi: 10.1371/journal.pone.0012897
174. Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. (2009) 72:240–5. doi: 10.1212/01.wnl.0000338144.10967.2b
175. Wei L, Ding L, Li H, Lin Y, Dai Y, Xu X, et al. Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-Box only protein 7 gene. Parkinsonism Relat Disord. (2018) 47:76–9. doi: 10.1016/j.parkreldis.2017.11.332
176. Wilson GR, Sim JC, McLean C, Giannandrea M, Galea CA, Riseley JR, et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am J Hum Genet. (2014) 95:729–35. doi: 10.1016/j.ajhg.2014.10.015
177. Shi CH, Zhang SY, Yang ZH, Yang J, Shang DD, Mao CY, et al. A novel RAB39B gene mutation in X-linked juvenile parkinsonism with basal ganglia calcification. Mov Disord. (2016) 31:1905–9. doi: 10.1002/mds.26828
178. Lesage S, Bras J, Cormier-Dequaire F, Condroyer C, Nicolas A, Darwent L, et al. Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol Genet. (2015) 1:e9. doi: 10.1212/NXG.0000000000000009
179. Mata IF, Jang Y, Kim CH, Hanna DS, Dorschner MO, Samii A, et al. The RAB39B p.G192R mutation causes X-linked dominant Parkinson's disease. Mol Neurodegener. (2015) 10:50. doi: 10.1186/s13024-015-0045-4
180. Gao Y, Wilson GR, Salce N, Romano A, Mellick GD, Stephenson SEM, et al. Genetic analysis of RAB39B in an early-onset Parkinson's disease Cohort. Front Neurol. (2020) 11:523. doi: 10.3389/fneur.2020.00523
181. Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. (2017) 140:3191–203. doi: 10.1093/brain/awx285
182. Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, Billingsley K, Vitale D, Blauwendraat C, et al. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson's disease. Mov Disord. (2019) 34:460–8. doi: 10.1002/mds.27614
183. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. (2014) 205:143–53. doi: 10.1083/jcb.201402104
184. Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. (2006) 441:1162–6. doi: 10.1038/nature04779
185. Cookson MR. DJ-1, PINK1, and their effects on mitochondrial pathways. Mov Disord. (2010) 25(Suppl. 1):S44–8. doi: 10.1002/mds.22713
186. Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol Dis. (2020) 137:104782. doi: 10.1016/j.nbd.2020.104782
187. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. (2002) 51:296–301. doi: 10.1002/ana.10113
188. Yue Z, Yang XW. Dangerous duet: LRRK2 and alpha-synuclein jam at CMA. Nat Neurosci. (2013) 16:375–7. doi: 10.1038/nn.3361
189. Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA. (2012) 109:9611–6. doi: 10.1073/pnas.1112368109
190. Balestrino R, Schapira AHV. Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist. (2018) 24:540–59. doi: 10.1177/1073858417748875
191. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community-based incident cohort. Brain. (2013) 136(Pt 2):392–9. doi: 10.1093/brain/aws318
192. Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. (2008) 3:19. doi: 10.1186/1750-1326-3-19
193. McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, et al. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE. (2010) 5:e12122. doi: 10.1371/journal.pone.0012122
194. Gronich N, Abernethy DR, Auriel E, Lavi I, Rennert G, Saliba W. beta2-adrenoceptor agonists and antagonists and risk of Parkinson's disease. Mov Disord. (2018) 33:1465–71. doi: 10.1002/mds.108
195. Chatterjee D, Bhatt M, Butler D, De Genst E, Dobson CM, Messer A, et al. Proteasome-targeted nanobodies alleviate pathology and functional decline in an alpha-synuclein-based Parkinson's disease model. NPJ Parkinsons Dis. (2018) 4:25. doi: 10.1038/s41531-018-0062-4
196. McFarthing K, Simuni T. Clinical trial highlights: targetting alpha-synuclein. J Parkinsons Dis. (2019) 9:5–16. doi: 10.3233/JPD-189004
197. Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S, et al. A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies. J Mol Biol. (2014) 426:2500–19. doi: 10.1016/j.jmb.2014.04.015
198. Wang Z, Gao G, Duan C, Yang H. Progress of immunotherapy of anti-alpha-synuclein in Parkinson's disease. Biomed Pharmacother. (2019) 115:108843. doi: 10.1016/j.biopha.2019.108843
199. Dangouloff T, Servais L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag. (2019) 15:1153–61. doi: 10.2147/TCRM.S172291
200. Andersen MA, Wegener KM, Larsen S, Badolo L, Smith GP, Jeggo R, et al. PFE-360-induced LRRK2 inhibition induces reversible, non-adverse renal changes in rats. Toxicology. (2018) 395:15–22. doi: 10.1016/j.tox.2018.01.003
201. Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, et al. MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J Pharmacol Exp Ther. (2015) 355:397–409. doi: 10.1124/jpet.115.227587
202. Scott JD, DeMong DE, Greshock TJ, Basu K, Dai X, Harris J, et al. Discovery of a 3-(4-Pyrimidinyl) indazole (MLi-2), an orally available and selective leucine-rich repeat kinase 2 (LRRK2) inhibitor that reduces brain kinase activity. J Med Chem. (2017) 60:2983–92. doi: 10.1021/acs.jmedchem.7b00045
Keywords: Parkinson's disease, phenotype, monogenic, early onset parkinsonism, Juvenile parkinsonism
Citation: Guadagnolo D, Piane M, Torrisi MR, Pizzuti A and Petrucci S (2021) Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol. 12:648588. doi: 10.3389/fneur.2021.648588
Received: 31 December 2020; Accepted: 08 March 2021;
Published: 22 September 2021.
Edited by:
Tim Anderson, University of Otago, Christchurch, New ZealandReviewed by:
Avner Thaler, Tel Aviv Sourasky Medical Center, IsraelNorbert Brüggemann, University of Lübeck, Germany
Copyright © 2021 Guadagnolo, Piane, Torrisi, Pizzuti and Petrucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Petrucci, simona.petrucci@uniroma1.it