Visual Outcomes in Experimental Rodent Models of Blast-Mediated Traumatic Brain Injury
 Lucy P. Evans
Lucy P. Evans Ariel M. Roghair
Ariel M. Roghair Noah J. Gilkes
Noah J. Gilkes Alexander G. Bassuk1*
Alexander G. Bassuk1*- 1Department of Pediatrics, University of Iowa, Iowa City, IA, United States
- 2Medical Scientist Training Program, University of Iowa, Iowa City, IA, United States
Blast-mediated traumatic brain injuries (bTBI) cause long-lasting physical, cognitive, and psychological disorders, including persistent visual impairment. No known therapies are currently utilized in humans to lessen the lingering and often serious symptoms. With TBI mortality decreasing due to advancements in medical and protective technologies, there is growing interest in understanding the pathology of visual dysfunction after bTBI. However, this is complicated by numerous variables, e.g., injury location, severity, and head and body shielding. This review summarizes the visual outcomes observed by various, current experimental rodent models of bTBI, and identifies data showing that bTBI activates inflammatory and apoptotic signaling leading to visual dysfunction. Pharmacologic treatments blocking inflammation and cell death pathways reported to alleviate visual deficits in post-bTBI animal models are discussed. Notably, techniques for assessing bTBI outcomes across exposure paradigms differed widely, so we urge future studies to compare multiple models of blast injury, to allow data to be directly compared.
Introduction
Cases of traumatic brain injury (TBI) morbidity are increasing as people are more often surviving blast-mediated TBI (bTBI), an injury especially prevalent among military personnel. Over the past two decades, 417,503 U.S. service members sustained at least one TBI as active military (Defense and Veterans Brain Injury Center, 2020) with nearly 2/3 involving an explosive blast (McKee and Robinson, 2014). Unfortunately, bTBIs are typically classified as mild due to the lack of obvious acute macroscopic injury; consequently, affected service members often return to duty prematurely (McKee and Robinson, 2014; Bryden et al., 2019; Regasa et al., 2019). Many neuropathological processes—microvascular injury, axonal injury, and neuroinflammation—can appear in the days to weeks after blast and have long-term effects on physical, cognitive, and emotional health (Hernandez et al., 2018).
Visual impairments are reported by some 75% of TBI patients, including blurry/double vision, difficulties reading, light sensitivity, and decreased peripheral vision (Armstrong, 2018; Frick and Singman, 2019). These visual impairments can arise due to optic neuropathy, axonal injury, and the loss of retinal ganglion cells (RGCs), which transmit visual stimuli to higher-level processing centers in the brain (Sen, 2017). In animal studies, decreased RGC survival and axonal integrity are strongly implicated with the activation of microglia and macrophages, with unregulated oxidative stress further contributing to RGC loss and optic nerve degeneration (Wang et al., 2013; Gupta et al., 2019).
Technological advancements in protective body armor and headgear have improved survival in combat, producing survivors with an increased number of co-morbidities. Polycarbonate eye protection does reduce the number of penetrating eye injuries, but does not prevent closed-globe damage to the eyes after a blast (Cockerham et al., 2011). Additionally, this protective gear in particular is not always worn, as dust and sweat can accumulate and reduce visibility, leaving the eye susceptible to injury (Cockerham et al., 2009). Furthermore, while many types of TBI produce visual impairments in humans, the variations in injury mechanics and pathophysiology necessitate studying blast-induced visual damage as its own entity. Experimental models of bTBI are critical in studying the mechanisms driving visual pathologies and can be utilized to identify and test novel therapeutic targets to prevent long-term visual dysfunction.
In humans, bTBI can be caused by a wide range of severities applied to multiple organ systems, eliciting various reparative responses from the body. Injury severity can depend on the subject’s orientation to the blast wave, location and duration of impact, distance from the source, and protective equipment. It is further complicated by patient demographics such as sex, age, and coinciding co-morbidities (Cernak, 2017; Bryden et al., 2019). Mirroring the complexity of human injury, current murine experimental models vary in terms of injury location, level of protection from the blast wave, and the device and blast magnitude used to administer a bTBI. Many of these successfully model bTBI pathology, but lack of standardization makes direct comparison of data difficult. Here, we review the devices, exposure paradigms, and assessment criteria currently used in rodent models of bTBI-induced visual impairment.
Materials and Methods
Literature Search Process and Inclusion Criteria
To identify relevant literature, we used the default settings on PubMed Legacy edition, using three-part search terms: 1) the subject: mouse/rat/rodent; 2) the injury type: blast/TBI/traumatic brain injury/blast brain injury/brain injury; 3) the visual outcome: eye/vision. Different combinations yielded 28 search terms (e.g., “mouse traumatic brain injury vision”). The process was completed on June 9, 2020, and produced 1,152 results. A manual filtering process was used to exclude non-relevant results, ensuring that the included sources met three necessary inclusion criteria: 1) used a rodent TBI model, 2) evaluated visual outcomes associated with TBI, and 3) employed a blast-TBI model (Figure 1). 35 original literature sources were selected for the review, three from manual cross-referencing.
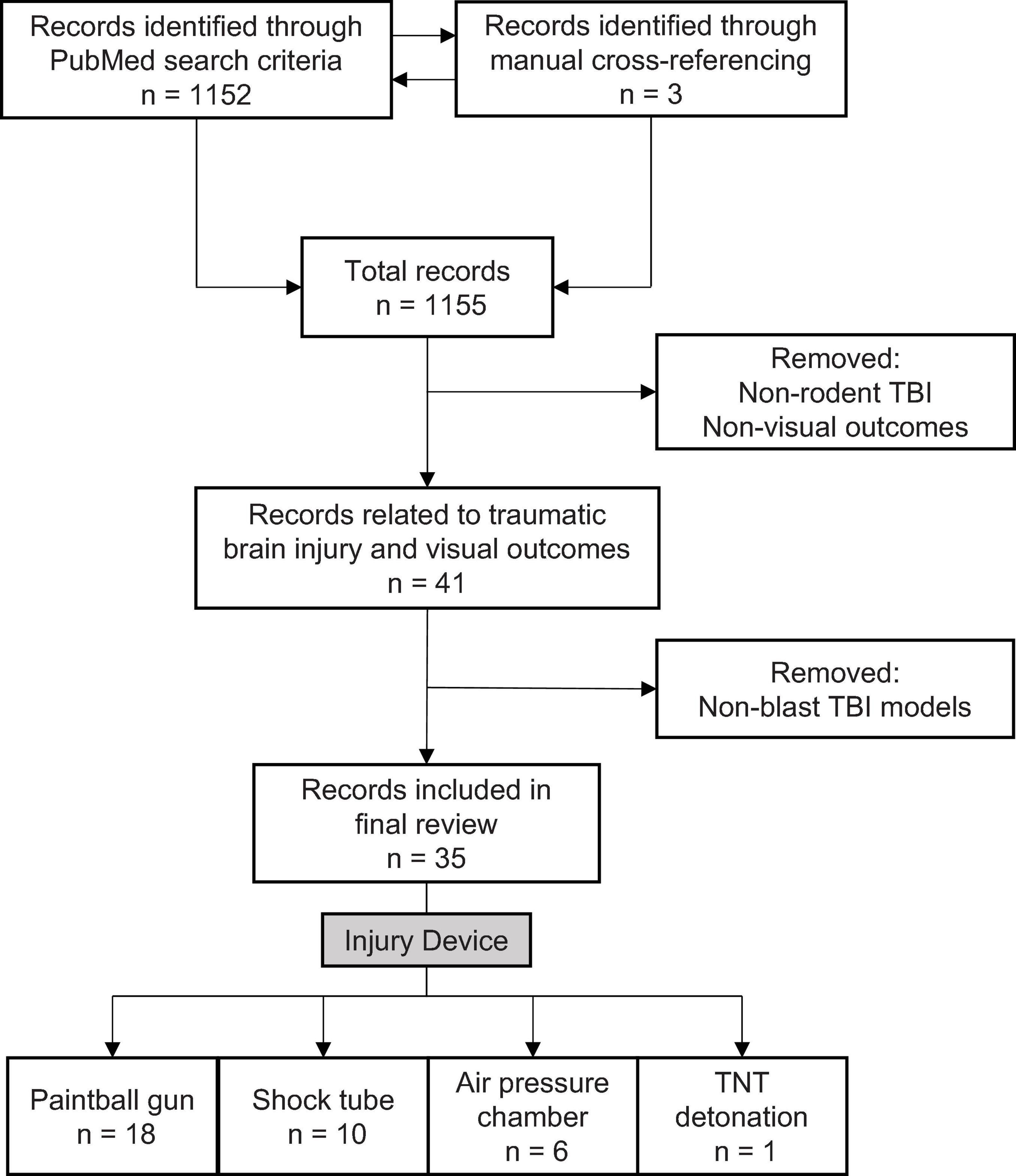
Figure 1. Literature search and selection criteria for rodent models examining blast-mediated TBI and visual outcomes.
Results
Experimental bTBI Induction
TBI Subjects
Although all studies used adult rodent models for bTBI, sex and strain varied. A striking 88.57% (31) of the studies used male subjects only; 8.57% (3) used male and female; and 2.86% (1) used females only. C57BL/6 mice were used in 57.14% (20) of the studies, Long-Evans rats in 14.29% (5), Sprague-Dawley rats in 11.43% (4), Balb/c mice in 5.71% (2), BXD recombinant inbred mice in 2.86% (2), and transgenic mice in 17.14% (6; Table 1).
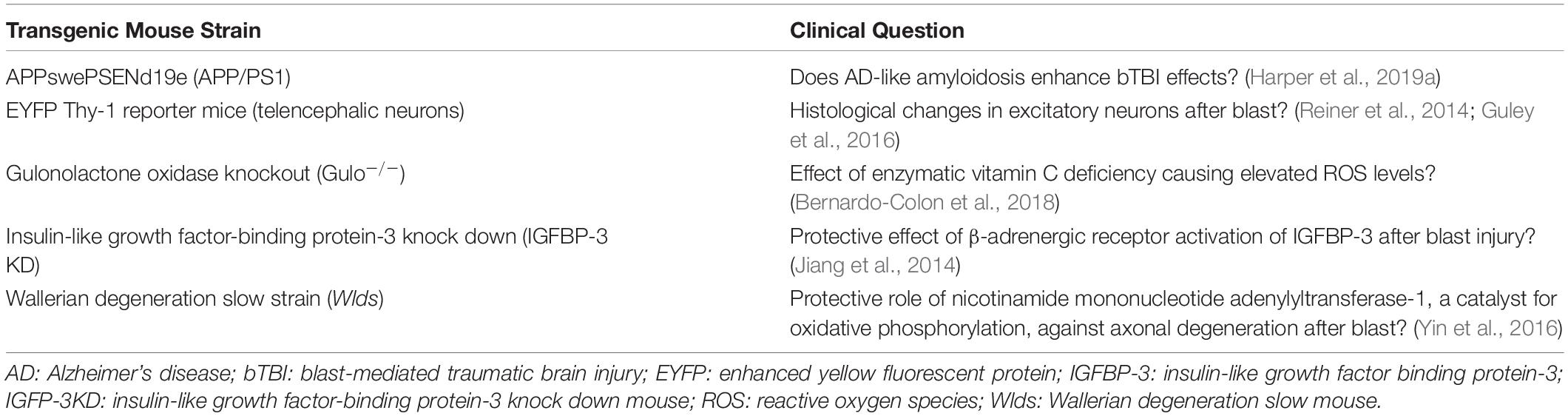
Table 1. Transgenic mouse models.
Blast Injury Devices
Devices used to administer bTBI fall into four classes (Figure 2): 51.43% (18) of studies used a modified paintball gun, 28.57% (10) used a compressed air shock tube, 17.14% (6) used an air pressure chamber, and 2.86% (1) used a TNT detonation model (injury parameters found in Table 2). Importantly, there is no commercially available standardized blast equipment; many groups have built or made adjustments to their own injury-induction device, leading to slight variations even within each sub-type of bTBI. However, the majority of groups studying bTBI in this review generally accept that the blast wave created in their model should mimic the Friedlander waveform, modeling the primary blast exposure dynamics that victims are exposed to in the field (Friedlander, 1946; Kuriakose et al., 2016). The Friedlander waveform consists of the shock front, which is an immediate sharp rise in pressure, followed by the blast wind, which is an exponential decay in pressure (Cullis, 2001). All devices induced bTBI with a pressure wave between 3.916 and 80 psi, but target injury locations varied. Control mice underwent an identical process but were not exposed to a blast wave.
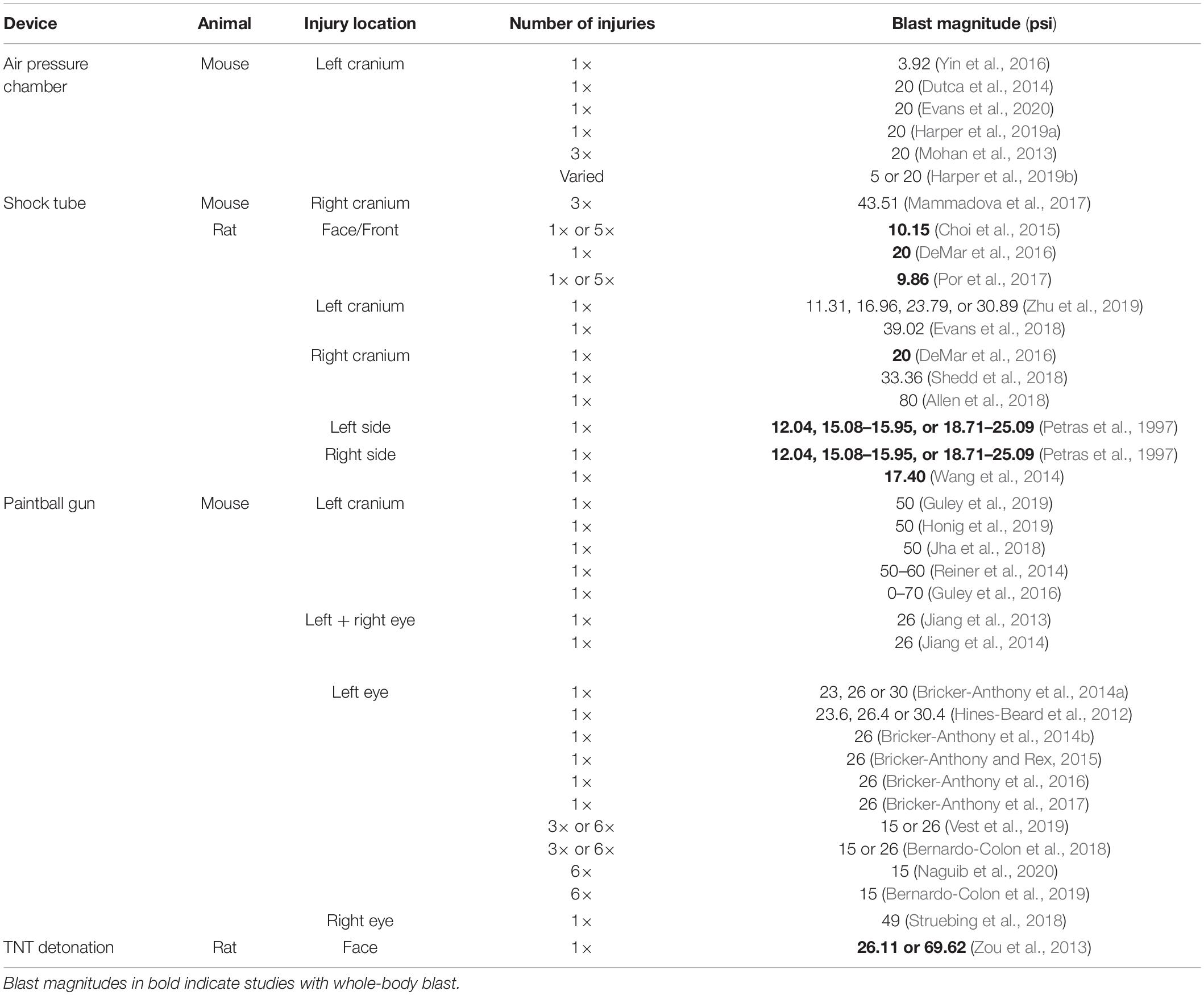
Table 2. Summary of the parameters used in rodent models of bTBI.
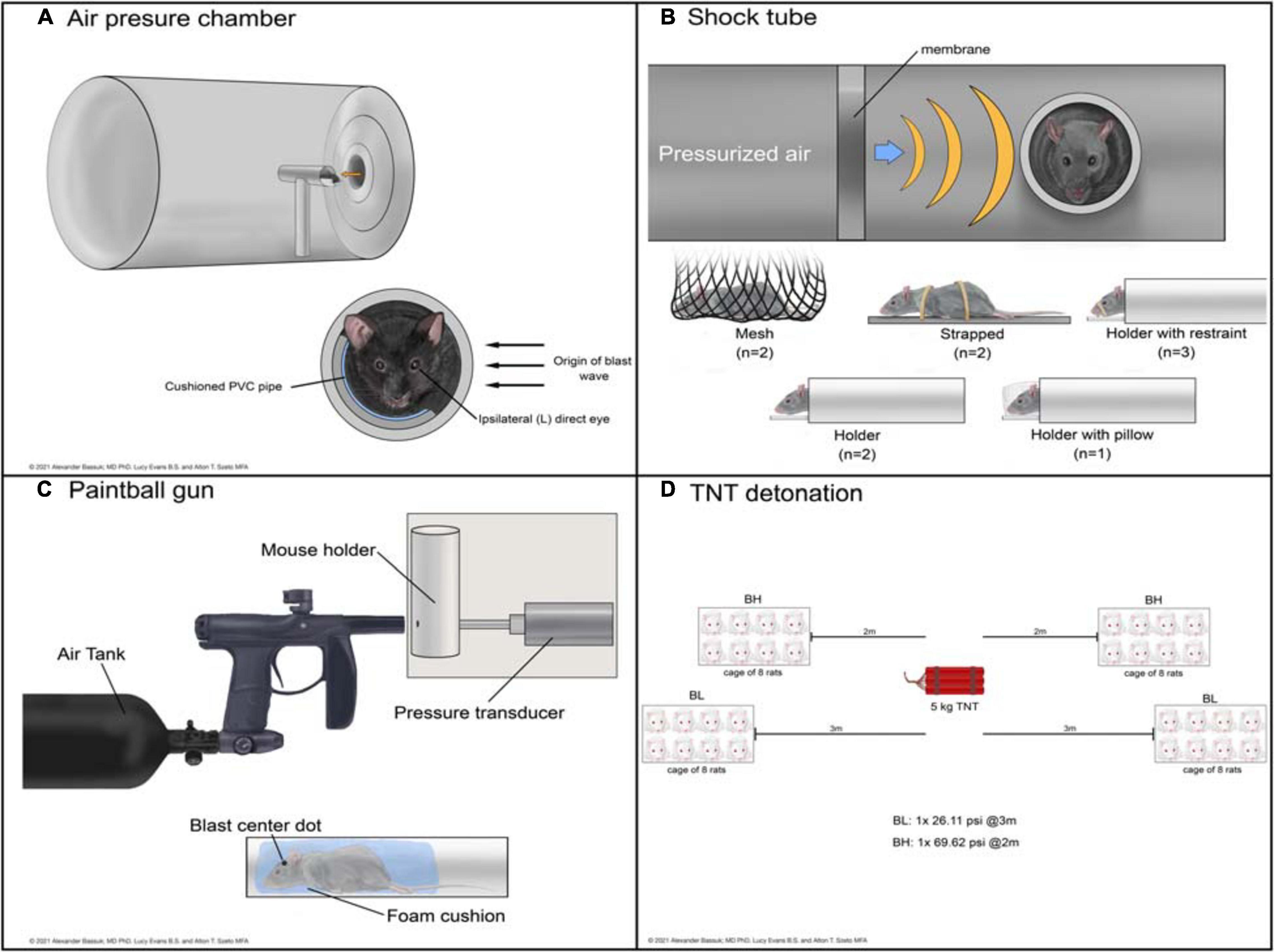
Figure 2. Schematic representations of devices used to deliver bTBI in rodent models. Blast waves were generated by an air pressure chamber (A), through a shock tube (B), from a paintball gun (C), or by TNT detonation (D). (BH): blast high; (BL): blast low.
The air pressure chamber houses the subject lateral to the blast-wave origin and exposes the left cranium to a long-duration (10–15 ms) blast wave between 3.92 and 20 psi (Figure 2A; Goldstein et al., 2012; Mohan et al., 2013). Anesthetized subjects are in a cushioned PVC pipe with their heads unrestrained to allow for full injury to the left side of the head while preventing injury to the right side. The shock tube is a long chamber that generates a short-duration (2–3 ms) blast overpressure wave, ranging from 9.14 to 80 psi, that travels the length of the tube (Figure 2B; Panzer et al., 2012; Swietek et al., 2019). Impact was delivered to the right or left cranium, or left, right, or front side of the body in anesthetized subjects with various levels of protection.
Shock tube devices administer short (2 to 3 ms) blast overpressures (Panzer et al., 2012; Swietek et al., 2019) while air pressure chambers administer longer (10 to 15 ms) biphasic blast overpressures (Goldstein et al., 2012; Mohan et al., 2013). Whether from expanding gaseous chemicals in shock tubes or pressurized air in pressure chambers, both deliver an initial sharp rise in pressure. This difference in achieving air compression causes the peak in overpressure to be slightly delayed in the air pressure chamber compared to the shock tube. As the wave propagates away from its source, the pressure drops in both devices; in a shock tube this drop occurs exponentially, while in an air pressure chamber the drop is biphasic. The shock wave ends with subsonic particle velocities creating a vacuum with slightly negative pressure, until the wave dissipates (Panzer et al., 2012; Mohan et al., 2013; Swietek et al., 2019).
The modified paintball gun air-tank device emits a brief high pressure air blast that can be calibrated to a specific pressure by adjusting the output from a pressurized air tank, striking the target location on the mouse (Figure 2C; Hines-Beard et al., 2012). It administers bTBI with magnitudes between 15 and 70 psi, with blasts directed at the left eye, right eye, or left cranium. Anesthetized bTBI subjects were placed in a small chamber with a foam cushion to prevent secondary somatic injuries. One study from our literature search utilized 5 kg of 2,4,6-trinitrotoluene (TNT) to generate a blast wave and induce bTBI (Figure 2D; Zou et al., 2013). Exposure conditions were manipulated by varying the distance from detonation. At two meters from the source, mice experienced a blast overpressure of 69.62 psi, whereas at three meters blast overpressure was 26.11 psi (blast high [BH] and blast low [BL], respectively). Anesthetized mice were positioned in metal cages facing the detonation site and secured to the cage loosely with Velcro to prevent movement.
Of note, mice were exposed to whole-body blast in only a handful of studies (bolded studies in Table 2), which can also serve as another variable within bTBI models. Damage to other organ systems can cause alterations in the body’s response to injury and subsequently the systemic environment that the brain and eyes are exposed to after blood-brain-barrier and blood-retina-barrier damage due to blast.
Overall summary
Mouse models of bTBI include modified paintball guns, air shock tubes, air pressure chambers, or TNT explosives; while these devices vary, many aim to recreate the Friedlander waveform victims of blast injury experience.
Pharmacologic Interventions
Pharmacologic intervention was the focus of 34.29% (12) of the studies (see Table 3). Of these, 75% (9) reduced inflammation, 33.33% (4) reduced apoptotic pathways, 25% (3) reduced oxidative stress levels, 8.33% (1) decreased excitotoxicity, and 8.33% (1) reduced tryptophan oxidative degradation. Galantamine, an acetylcholinesterase inhibitor, blocked inflammation, oxidative stress, and excitotoxicity (Naguib et al., 2020). Inflammation was blocked with cannabinoid type-2 receptor inverse agonists, raloxifene (Honig et al., 2019) and SMM-189 (Reiner et al., 2014; Guley et al., 2019) the paracrine factors secreted by adipose stem cell concentrated conditioned media (ASC-CCM) pre-stimulated with inflammatory cytokines (Honig et al., 2019) and interleukin-1 receptor (IL-1RI) antagonist anakinra (Evans et al., 2020). Two approaches were used to deliver erythropoietin (EPO) to treat retinal oxidative stress and neuroinflammation. One method injected exogenous EPO into DBA/2J mice, a short-term exposure; and the second delivered EPO via adeno-associated virus (rAAV), a longer-term EPO treatment. Importantly, the rAAV form of EPO had attenuated erythropoietic activity, so it would not stimulate RBC production and trigger retinal oxidative stress (Bricker-Anthony et al., 2017). Apoptosis was decreased by P7C3-S243, an activator of nicotinamide phosphoribosyltransferase (NAMPT) (Dutca et al., 2014). Both apoptosis and inflammation were reduced by the β-adrenergic receptor agonist, Compound 49b (Jiang et al., 2013, 2014). Inflammation and reactive oxygen species (ROS) were reduced by Vitamin E supplements and a ketogenic diet, while a deficiency in vitamin C that elevates inflammation and ROS was used to study how antioxidants might be therapeutic for bTBI (Bernardo-Colon et al., 2018). Finally, a downregulation of a neurotoxic gene encoding kynurenine 3-monooxygenase (KMO) protected blast mice that were preconditioned (exposed to smaller, five psi blasts prior to a higher-intensity 20 psi blast). A KMO inhibitor (Ro-61-8048) blocked oxidative degradation of tryptophan and formation of neurotoxic intermediates (Harper et al., 2019b).
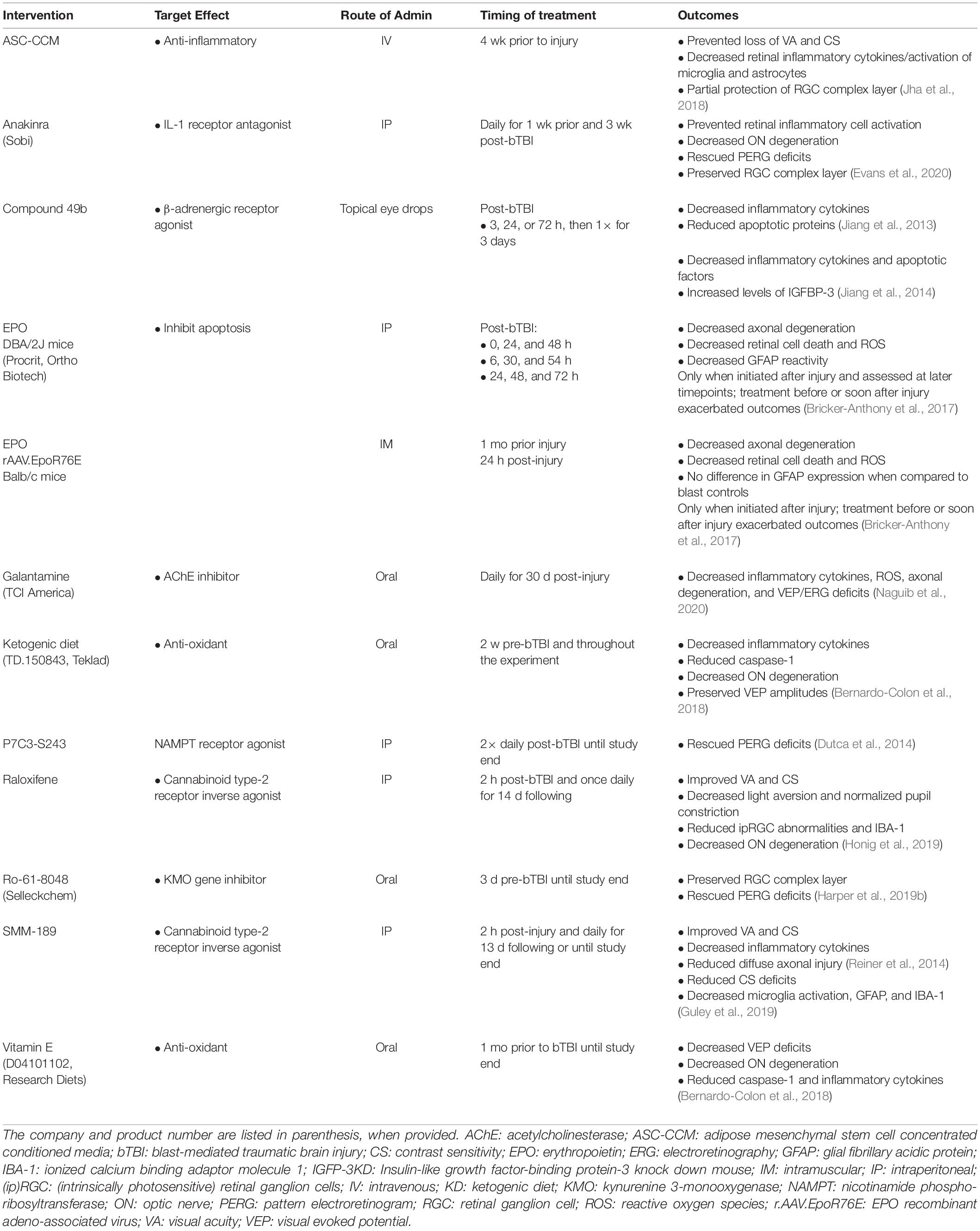
Table 3. Summary of the pharmacological interventions and outcomes observed following bTBI.
Overall summary
Most pharmaceutical interventions aimed to reduce retinal inflammation or apoptosis after blast injury.
Structural Outcomes (Supplementary Table 1)
The Eye and the Retina
Blast injury resulted in a wide range of pathologic findings, including corneal edema (Hines-Beard et al., 2012; Bricker-Anthony et al., 2014a, 2016; Bricker-Anthony and Rex, 2015), cataracts (Bricker-Anthony et al., 2014a, 2016), corneal epithelial thinning (Bricker-Anthony et al., 2016), retinal pigment epithelial (RPE) vacuoles (Bricker-Anthony and Rex, 2015; Bricker-Anthony et al., 2016), neovascularization (Bricker-Anthony and Rex, 2015; Shedd et al., 2018), abrasions (Hines-Beard et al., 2012; Zhu et al., 2019), vitreous detachments (Bricker-Anthony et al., 2014a; Bricker-Anthony and Rex, 2015; Evans et al., 2018), and hemorrhage (Evans et al., 2018). Additionally, RGC dendritic rearrangement (Dutca et al., 2014), severity-dependent retinal lesioning and disorganization (Zou et al., 2013), and increased retinal pyknotic nuclei were reported (Bricker-Anthony et al., 2014a, b, 2016; Bricker-Anthony and Rex, 2015). One commonality across several exposure paradigms was progressive RGC complex layer thinning (Dutca et al., 2014; Evans et al., 2018, 2020; Jha et al., 2018; Struebing et al., 2018; Harper et al., 2019a, b; Honig et al., 2019). Retinal thickening was observed in three studies, as bTBI was proposed to stimulate astrocytes and increase expression of vascular endothelial growth factor (VEGF) (Zou et al., 2013; Reiner et al., 2014; Allen et al., 2018; Shedd et al., 2018). Additionally, retinal detachment was reported (Bricker-Anthony et al., 2014a, 2016; Bricker-Anthony and Rex, 2015). Treatment with anti-inflammatory anakinra (Evans et al., 2020), anti-inflammatory ASC-CCM (Jha et al., 2018), and blast preconditioning (Harper et al., 2019b) preserved RGC complex layer thickness; the AD model exacerbated RGC complex layer loss (Harper et al., 2019a).
Intraocular pressure (IOP) was measured in several bTBI rodent models, but the direct effects of blast injury on IOP remain unclear as studies reported increased (Dutca et al., 2014; Bricker-Anthony et al., 2016; Shedd et al., 2018; Bernardo-Colon et al., 2019; Zhu et al., 2019), decreased (Hines-Beard et al., 2012), or unchanged (Hines-Beard et al., 2012; Guley et al., 2016; Zhu et al., 2019) IOP measurements. Some studies detected a change and found IOP returned to baseline between 7 and 60 days post-injury (Bricker-Anthony et al., 2016; Bernardo-Colon et al., 2019). There was no clear association with injury location or type, as IOP measurements varied within each sub-group: increased with paintball gun (Bricker-Anthony et al., 2016; Bernardo-Colon et al., 2019), shock tube (Shedd et al., 2018; Zhu et al., 2019), and air chamber (Dutca et al., 2014) decreased with paintball gun (Hines-Beard et al., 2012) no change with shock tube (Zhu et al., 2019) and paintball gun (Hines-Beard et al., 2012; Guley et al., 2016). Additionally, there was no linear association with increased IOP and increased blast intensity.
Optic Nerve
Optic nerve degeneration in blast-injured mice was consistently identified (Petras et al., 1997; Mohan et al., 2013; Bricker-Anthony et al., 2014a, b, 2016, 2017; Bernardo-Colon et al., 2018, 2019; Guley et al., 2019; Honig et al., 2019; Vest et al., 2019; Evans et al., 2020; Naguib et al., 2020). The optic nerve was protected pharmacologically with anakinra (Evans et al., 2020), EPO (Bricker-Anthony et al., 2017), raloxifene (Honig et al., 2019), SMM-189 (Reiner et al., 2014; Guley et al., 2019), and galantamine (Naguib et al., 2020) along with dietary changes such as vitamin E supplements and a ketogenic diet (Bernardo-Colon et al., 2018). Again, the AD model of amyloidosis increased optic nerve degeneration after blast injury (Harper et al., 2019a). SMM-189 (Guley et al., 2019) and raloxifene (Honig et al., 2019) may prevent secondary neuroinflammation in the optic nerve. Similarly, galantamine treatment (Naguib et al., 2020), sufficient levels of vitamin C, vitamin E, and a ketogenic diet (Bernardo-Colon et al., 2018) may protect against optic nerve degeneration by decreasing the presence of ROS and the subsequent upregulation of inflammatory cytokines.
Higher Visual Loci and Pathways
Blasted rodents demonstrated edematous forebrain enlargement (Guley et al., 2016), cerebral cortical neuronal loss (Petras et al., 1997; Guley et al., 2016; Yin et al., 2016), microglial activation (Reiner et al., 2014), and axonal transport defects (Bernardo-Colon et al., 2019). One study of repetitive 43.51 psi blasts to the right cranium reported significantly decreased glial fibrillary acidic protein (GFAP) protein expression in the prefrontal cortex after 30 days, while they found no difference in protein levels of ionized calcium binding adapter molecule 1 (IBA-1) or phosphorylated tau. This study found striatal neurotransmitter levels unchanged following repetitive injury, suggesting that neurons in this location were not damaged in their model (Mammadova et al., 2017). Likewise, another group reported no histological changes including tissue destruction or inflammatory cell accumulation 24 days after a single 39.02 psi blast to the left cranium (Evans et al., 2018). Of note, these groups did observe retinal changes described in other portions of this review.
Overall summary
The majority of structural changes after blast injury were seen in the eye, retina, and optic nerve, with RGC and optic nerve damage frequently reported.
Objective Functional Outcomes (Supplementary Table 2)
The functional integrity of photoreceptor and retinal bipolar cells is measured by electroretinogram (ERG), recorded as a- and b- sine waves, respectively (Perlman, 1995). These metrics of retinal health and visual ability offer an in vivo diagnostic for both murine and human subjects, however, the results in these studies were inconsistent. When compared to baseline and/or sham controls, the a-wave and b-wave amplitudes in bTBI subjects either increased (Bricker-Anthony et al., 2014b; Allen et al., 2018), decreased (Honig et al., 2019; Naguib et al., 2020), remained unchanged (Mohan et al., 2013), or exhibited both decreased or unchanged values (Bricker-Anthony and Rex, 2015; DeMar et al., 2016; Bricker-Anthony et al., 2017; Zhu et al., 2019). These disparities might be caused by variations in light intensity (Bricker-Anthony et al., 2014a), mouse strain (Bricker-Anthony and Rex, 2015), orientation to the blast wave (DeMar et al., 2016), and/or blast magnitude (Zhu et al., 2019). In terms of pharmacological interventions, raloxifene effectively restored ERG amplitudes (Honig et al., 2019), while galantamine partially prevented waveform reductions (Naguib et al., 2020).
While an ERG reflects the ability of the retina to respond to incoming light, a pattern ERG (PERG) is a functional readout of RGC signaling, providing information about visual transduction from the retina to the brain (Bach et al., 2013). Consistently decreased PERG amplitudes were seen in blast-injured rodents, for up to 16 weeks post-injury (Mohan et al., 2013; Dutca et al., 2014; Yin et al., 2016; Harper et al., 2019a, b; Evans et al., 2020). Interestingly, two studies reported temporary recovery in PERG amplitudes: one at 24 h post-injury (Mohan et al., 2013) and another at 4 weeks with a reoccurrence of impaired signaling again at 16 weeks (Dutca et al., 2014). The Wlds genotype (Yin et al., 2016), blast preconditioning (Harper et al., 2019b), and P7C3-S243 treatment (Dutca et al., 2014) preserved PERG amplitudes, hypothetically by promoting survival of the visual circuitry. The Wlds genotype protects against axonal degeneration and inflammatory proliferation at the site of injury (Yin et al., 2016), while blast preconditioning is thought to upregulate endogenous survival factors or downregulate harmful ones. In retinas of mice preconditioned with a small blast, RNA sequencing showed KMO was downregulated. Mice receiving daily oral treatments of Ro-61-8048, a KMO inhibitor, had improved PERG signaling (Harper et al., 2019b). Similarly, P7C3-S243 might preserve visual system integrity by activating metabolic cofactors (Dutca et al., 2014). The anti-inflammatory drug, anakinra, is also partially protective of impaired PERG signaling after blast injury via IL-1RI antagonism, preventing the propagation of inflammatory signaling through this pathway (Evans et al., 2020). An AD model developed worse PERG deficits after blast exposure, suggesting AD amplifies the pathologic retinal effects of bTBI (Harper et al., 2019a). Notably, all of the studies that measured PERG administered blast injury to the left cranium using an air pressure chamber with relatively low pressures (psi between 3.92 and 20).
Visually evoked potentials (VEP) via flash stimulation were also assessed. The VEP N1 amplitude, an early response to visual stimuli (Creel, 1995), was normalized by galantamine (Naguib et al., 2020) and vitamin E treatment (Bernardo-Colon et al., 2018), suggesting these pharmacologic agents protect against VEP response deficits. One study examining RGC physiology reported temporary, spontaneous hyperactivity at 1 and 16 weeks after one 20 psi blast to the left cranium (Dutca et al., 2014). RGC hyperactivity is linked to photoreceptor dystrophic disorders and can significantly decrease the quality of vision (Barrett et al., 2015).
Overall summary
PERG was a functional outcome commonly investigated after bTBI, with many studies describing impaired PERG and RGC signaling due to blast injury. ERG changes were inconsistent when compared between studies.
Subjective and Behavioral Visual Outcomes (Supplementary Table 3)
Bilateral contrast sensitivity and visual acuity were consistently damaged by blast exposure, despite differences in blast magnitude and injury location (Bricker-Anthony et al., 2014a, b; Reiner et al., 2014; Bricker-Anthony and Rex, 2015; Guley et al., 2016, 2019; Allen et al., 2018; Jha et al., 2018; Shedd et al., 2018; Struebing et al., 2018; Honig et al., 2019). Deficits were reported as early as 1 day (Shedd et al., 2018) and up to 8 months (Allen et al., 2018) following injury. Some groups found contrast sensitivity and visual acuity improved over time (Hines-Beard et al., 2012; Bricker-Anthony et al., 2014b) while others conversely found it declined, particularly with age (Bricker-Anthony et al., 2014a; Bricker-Anthony and Rex, 2015; Allen et al., 2018). Treatment with raloxifene (Honig et al., 2019) and SMM-189 (Reiner et al., 2014; Guley et al., 2019) promoted full recovery of both contrast sensitivity and visual acuity, while ASC-CCM treatment provided partial recovery of both parameters (Jha et al., 2018).
Pupillary light constriction produced contrasting findings. One day post-20 psi blast to the left cranium, diminished pupillary constriction was seen that resolved after 10 months (Mohan et al., 2013). At 7 months post-injury, another group reported elevated pupillary constriction that was normalized by treatment with raloxifene (Honig et al., 2019). AD mice had an impaired pupillary light response, possibly due to amyloid deposits in the retina (Harper et al., 2019a).
Overall summary
Contrast sensitivity and visual acuity were frequently impaired after bTBI, while studies produced conflicting data on changes in pupillary light constriction.
Subjective and Behavioral General Outcomes (Supplementary Table 4)
Spatial-learning and memory was unaffected by bTBI, as assessed via Morris water maze, at 30 days (Mammadova et al., 2017) and Y maze at 3, 6, and 8 months (Allen et al., 2018). At 7 days post-injury, however, significant behavioral deficits were detectable in Barnes maze performance and could be rescued through a Wlds genotype, which prevents axonal degeneration and inflammatory infiltration to the injuries (Yin et al., 2016). That this genotype protects learning and memory implies potential for post-bTBI therapies that preserve axonal integrity and prevent inflammatory infiltration. Depressive behavior and contextual fear (at 6 to 8 weeks post-injury) were identified in blast rodents and were alleviated by SMM-189, which prevents blast-induced loss of Thy-1 fear-suppressing neurons (Reiner et al., 2014). Of note, blast-related vision loss can affect the assessment of cognitive function, as visual and spatial cues guide subjects throughout the task for many readouts.
Blast-injured rodents showed marked decreases in motor coordination and activity at acute time points (≤ 14 days) (Reiner et al., 2014; Guley et al., 2016) but other studies showed no deficits 30 days after injury (Yin et al., 2016; Mammadova et al., 2017; Harper et al., 2019a) suggesting that findings resolved over time. SMM-189 treatment (Reiner et al., 2014) and the Wlds genotype (Yin et al., 2016) were protective against motor deficits, while an AD model exacerbated deficits (Harper et al., 2019a). In particular, SMM-189 treatment protected corticospinal tract integrity and cerebellar and motivational circuitry (Reiner et al., 2014), while the Wlds genotype protected axonal integrity in the brain and spinal cord (Yin et al., 2016). In the AD model, β-amyloid peptide (Aβ), amyloid precursor protein (APP), and tau protein exacerbated bTBI pathologies (Harper et al., 2019a).
Overall summary
Studies have found that blast causes deficits in a wide range of behavioral outcomes, but consistent trends and methodology have not been established.
Inflammatory Over-Activation Following bTBI
Post-bTBI inflammation was consistently detected in the included studies by identifying activated cellular inflammatory modulators or directly measuring inflammatory molecules (Supplementary Tables 1, 5). In the inflamed retina, activated resident immune cells attract peripheral immune cells to the site of injury, amplifying the inflammatory response. Inflammatory cytokine levels rise and immunomodulatory cells are activated, triggering changes in cellular morphology or protein expression (Simon et al., 2017). Various post-bTBI-retina immunohistochemistry data detected inflammation-associated upregulation of IBA-1 (Bricker-Anthony et al., 2016, 2017; Guley et al., 2016), GFAP (Zou et al., 2013; Choi et al., 2015; Allen et al., 2018; Honig et al., 2019), or both (Bricker-Anthony et al., 2014a; Bricker-Anthony and Rex, 2015; Mammadova et al., 2017; Jha et al., 2018; Guley et al., 2019; Evans et al., 2020), markers of microglial and macroglial activation, respectively, suggesting these inflammatory modulators are responding to stress and propagating inflammatory signals post-injury.
bTBI can activate resident immune cells to release pro-inflammatory cytokines, signaling for prolonged retinal inflammation and exacerbating visual damage (Li et al., 2015; Fehily and Fitzgerald, 2017). Normally, IBA-1 is expressed in quiescent microglia, but following a TBI, activated microglia proliferate, migrate to injured tissue, and exhibit morphological changes (Shapiro et al., 2009). Many studies detected upregulated IBA-1 in the bTBI retina, both as an acute and chronic indicator of ocular trauma and stress (Bricker-Anthony et al., 2014a, b; Bricker-Anthony and Rex, 2015; Guley et al., 2016, 2019; Mammadova et al., 2017; Jha et al., 2018; Honig et al., 2019; Evans et al., 2020). The transition of microglia from the pro-inflammatory M1 state to the reparative M2 state is expressed as the M1/M2 ratio. A prolonged M1 state can damage retinal tissues due to its swelling injury response and downstream release of pro-inflammatory cytokines and free radicals (Loane and Kumar, 2016; Fehily and Fitzgerald, 2017). The M1/M2 ratio decreased after treatment with both raloxifene (Honig et al., 2019) and SMM-189 (Guley et al., 2019), suggesting microglia in blast retinas transitioned toward a reparative phenotype after pharmacologic treatment.
Macroglia (astrocytes and Müller glia) are supportive glial cells within the retina. Astrocytes are located throughout the CNS including in the retina, while Müller glia are uniquely retinal (Sofroniew and Vinters, 2010). Together, their activation in the retina promotes reactive gliosis, a beneficial repair mechanism following injury; over-activation, however, can cause glial scarring, (Pekny et al., 2014) disruption of neural plasticity, and damage to visual circuitry (Sardar Pasha et al., 2017). Although healthy astrocytes strongly express GFAP, gliotic changes and astrocyte hypertrophy can induce pathological levels of GFAP expression. Müller glia significantly increase GFAP expression due to retinal stress; high GFAP expression in Müller glia and their processes is indicative of injury (Eisenfeld et al., 1984). bTBI retinas consistently demonstrated increased GFAP immunoreactivity, suggestive of macroglial activation in this stressed tissue (Zou et al., 2013; Bricker-Anthony et al., 2014a, 2016, 2017; Bricker-Anthony and Rex, 2015; Choi et al., 2015; Mammadova et al., 2017; Jha et al., 2018; Guley et al., 2019; Evans et al., 2020).
Several post-injury drug interventions suppressed activation of microglia and macroglia in the retina. EPO given at least 1 day after injury in DBA/2J mice decreased GFAP expression (Bricker-Anthony et al., 2017). Notably, Balb/c mice treated with rAAV EPO did not show a difference in retinal GFAP between sham and blast, suggesting the timing of EPO therapy is important. ASC-CCM (Jha et al., 2018), anakinra (Evans et al., 2020), and SMM-189 (Guley et al., 2019) downregulated GFAP and IBA-1 expression after blast.
Activated resident microglia and macroglia recruit infiltrating systemic inflammatory cells and upregulate pro-inflammatory cytokines (IL-1α, IL-1β, IL-6, IL-18, IL-33, IFN-γ, TNFα), contributing to retinal pathogenesis, as all of these cell types are activated by and can propagate retinal inflammation (Allan et al., 2005; Holan et al., 2019). Though multiple studies found acute increases in pro-inflammatory cytokines post-blast (Jiang et al., 2013, 2014; Zou et al., 2013; Bernardo-Colon et al., 2018, 2019; Shedd et al., 2018; Struebing et al., 2018; Harper et al., 2019b; Evans et al., 2020) vitamin E was shown to decrease IL-1β (Bernardo-Colon et al., 2018), while ASC-CCM treatment (Jha et al., 2018) and Compound 49b (Jiang et al., 2013, 2014) decreased both IL-1β and TNFα expression. One study also reported galantamine suppressed pathologic elevation in IL-1α and IL-1β (Naguib et al., 2020). Interestingly, retinal expression of IL-1α and IL-1β increased in vitamin C-deficient mice, which had worse post-bTBI outcomes (Bernardo-Colon et al., 2018).
Neutrophil infiltrates were also found in corneal stromal layers following blast injury with concurrent increases in pain and inflammatory signaling mediators such as the transient receptor potential vanilloid 1 (TRPV1) channel, calcitonin gene-related peptide (CGRP), substance P (SP), and endothelin-1 (ET-1). Increases in hematic myeloperoxidase (MPO), a peroxidase enzyme released during the degranulation and activation of neutrophils, were also reported (Por et al., 2017).
Optic nerves of blast-injured mice showed signs of aberrant inflammatory signaling—axonal degeneration (Petras et al., 1997; Mohan et al., 2013; Bricker-Anthony et al., 2014a, b, 2016, 2017; Bernardo-Colon et al., 2018, 2019; Guley et al., 2019; Honig et al., 2019; Vest et al., 2019; Evans et al., 2020; Naguib et al., 2020), microglial activation (Reiner et al., 2014), astrocytic activation/glial scarring (Mohan et al., 2013; Choi et al., 2015; Bernardo-Colon et al., 2019; Vest et al., 2019), and infiltration of CD68-positive cells (indicative of an inflammatory response) (Choi et al., 2015). Infiltration and morphological changes in microglia and astrocytes in the optic nerve appeared to increase acutely, but decrease as early as 1 week post-injury (Reiner et al., 2014; Bernardo-Colon et al., 2019). However, in one study, the astrocyte percent area significantly increased again 30 days after injury, while percent astrocyte parallelism (a measure of astrocyte process orientation and orderliness) remained abnormally low (Bernardo-Colon et al., 2019).
Overall summary
Significant evidence in the literature suggests post-blast inflammation contributes to visual dysfunction after bTBI.
bTBI Upregulates Apoptotic, Necroptotic, and Pyroptotic Mediators (Supplementary Table 6)
bTBI activates programmed cell death pathways, such as apoptosis, necroptosis, or pyroptosis. Caspase activation initiates apoptosis causing DNA cleavage and genome fragmentation (Reed, 2000). Modified DNA is packed into apoptotic bodies that await engulfment by phagocytes resulting in cellular death (Elmore, 2007). The terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay detects apoptotic DNA fragments. Caspase-3 functions as an effector caspase, killing the cell by cleaving specific intracellular targets (Pasinelli et al., 2000; Wang et al., 2014). The necroptotic pathway is characterized by plasma membrane rupture that renders the extracellular environment for nearby cells toxic with inflammatory cytokines (Dhuriya and Sharma, 2018; Chen et al., 2019). Necroptosis is associated with the family of receptor interacting protein kinases (RIPs); RIP1 and RIP3 are two critical signaling molecules and markers of necroptosis (Liu et al., 2019). Pyroptosis also features plasma membrane rupture and cytokine release affecting neighboring tissues, but caspase-1 initiates pyroptosis by cleaving the substrate gasdermin D, which creates pores and then ruptures the plasma membrane (Bergsbaken et al., 2009; Man et al., 2017).
Following injury, DNA damage detected via TUNEL assays was suggestive of apoptotic cell death (Jiang et al., 2013; Zou et al., 2013; Bricker-Anthony et al., 2014a, 2016, 2017; Wang et al., 2014; Bricker-Anthony and Rex, 2015). The presence of caspase-3 in the retina (Zou et al., 2013; Bricker-Anthony et al., 2014a; Wang et al., 2014; Choi et al., 2015) and optic nerve (Wang et al., 2014; Choi et al., 2015) after blast was similarly indicative of apoptosis. Additionally, post-bTBI necroptosis was detected via increased retinal RIP1 and RIP3 expression (Bricker-Anthony et al., 2014a, b; Bricker-Anthony and Rex, 2015) while the upregulation of caspase-1 was suggestive of increased pyroptotic activity (Bricker-Anthony et al., 2014a; Bernardo-Colon et al., 2018).
Three studies investigated therapeutic reduction in apoptosis to increase retinal cell survival after blast injury. In the injured retina, Compound 49b stimulated β-adrenergic receptor activation and IGFBP-3 production, which in turn decreased the level of cleaved caspase-3 and decreased TUNEL labeling, reducing retinal apoptosis (Jiang et al., 2013). A ketogenic diet decreased inflammation and ROS levels, leading to a reduction in the level of cleaved retinal caspase-1 following blast (Bernardo-Colon et al., 2018). Finally, an acute increase in EPO in DBA/2J mice exacerbated retinal cell death after bTBI, possibly due to increased oxidative stress from amplified RBC formation and retinal iron levels. However, when analyzed 1 week after injury, this treatment was protective for cell death when compared to controls. EPO treatment with an rAAV with attenuated erythropoietic activity promoted retinal cell survival better if treatment was delayed (Bricker-Anthony et al., 2017).
Overall summary
Cell death pathways are frequently implicated in post-blast visual pathophysiology.
Discussion
We reviewed the devices and exposure paradigms employed in bTBI research and found notable interstudy variations in techniques and assessment outcomes. The variability of bTBI experimental models’ blast magnitude, location of injury, and device biomechanics makes comparing data on visual outcomes difficult; and the picture is further complicated by inconsistencies in outcome measures. Nevertheless, bTBI consistently resulted in increased inflammation, activation of resident inflammatory mediators, impaired PERG signaling, decreased visual acuity and contrast sensitivity, decreased RGC complex thickness, and ON degeneration. This suggests that these characteristics of visual dysfunction after bTBI are reproducible regardless of the technique employed.
Blast injury in humans, as in murine models, is an extremely heterogenous and multifactorial condition that can result in a wide range of consequences. Many of the outcomes measured in animal studies are not practically measured when assessing human injury, i.e., histology or measuring retinal inflammatory modulators at multiple time points. On the other hand, several parameters used to assess human ocular injury cannot be completely recapitulated in murine studies. For example, it is not feasible to measure specific reading issues or subtle changes in color vision in mice. However, the findings in this review of decreased visual acuity, impaired contrast sensitivity, and optic nerve dysfunction have been consistently seen in human blast injury (Cockerham et al., 2009; Scott, 2011; Saunders and Echt, 2012).
While retinal and optic nerve damage was frequently identified after blast injury, many groups did not find overt damage to brain tissue. This lack of consistent changes in the brain after blast could suggest that the retina and optic nerve are more sensitive indicators of mild injury in this model. The increased vulnerability of the eye and optic nerve to bTBI specifically is a unique aspect of this injury model, as other types of TBI can experience greater damage to brain tissue. Additionally, as impaired vision can be a confounding factor for cognitive testing, we recommend that future cognitive testing should be done in concert with tests assessing basic visual performance.
The data implicate inflammatory and apoptotic pathways as playing a causal role in long-term visual dysfunction after bTBI and several targeted pharmacological interventions show promise for manipulating those pathways. Generally, inflammatory blockade protected against deficits in contrast sensitivity, visual acuity, and RGC signaling. Additionally, anti-inflammatory agents preserved RGC complex layer thickness and optic nerve integrity. Interestingly, these pharmacologic interventions targeted different portions of the inflammatory response and, in the studies that reported levels of individual inflammatory cytokines, varied in terms of the actual reduction of inflammatory molecules. This could suggest that multiple inflammatory pathways play a role after bTBI and that combination therapy using multiple agents would confer the most retinal protection after injury.
It is also clear that programmed cell death contributes to the retinal pathogenesis and subsequent visual disturbances following bTBI. Preventing cellular death is vital for vision preservation and would greatly improve outcomes post-injury. Together, these observations suggest that the overactivation of both inflammatory and apoptotic pathways contribute to visual dysfunction following blast injury (Figure 3). While the field still does not have a gold standard for a rodent blast model, making direct comparisons difficult at times, these common pathways could serve to bridge the gaps caused by variations in experimental techniques and outcome assessments.
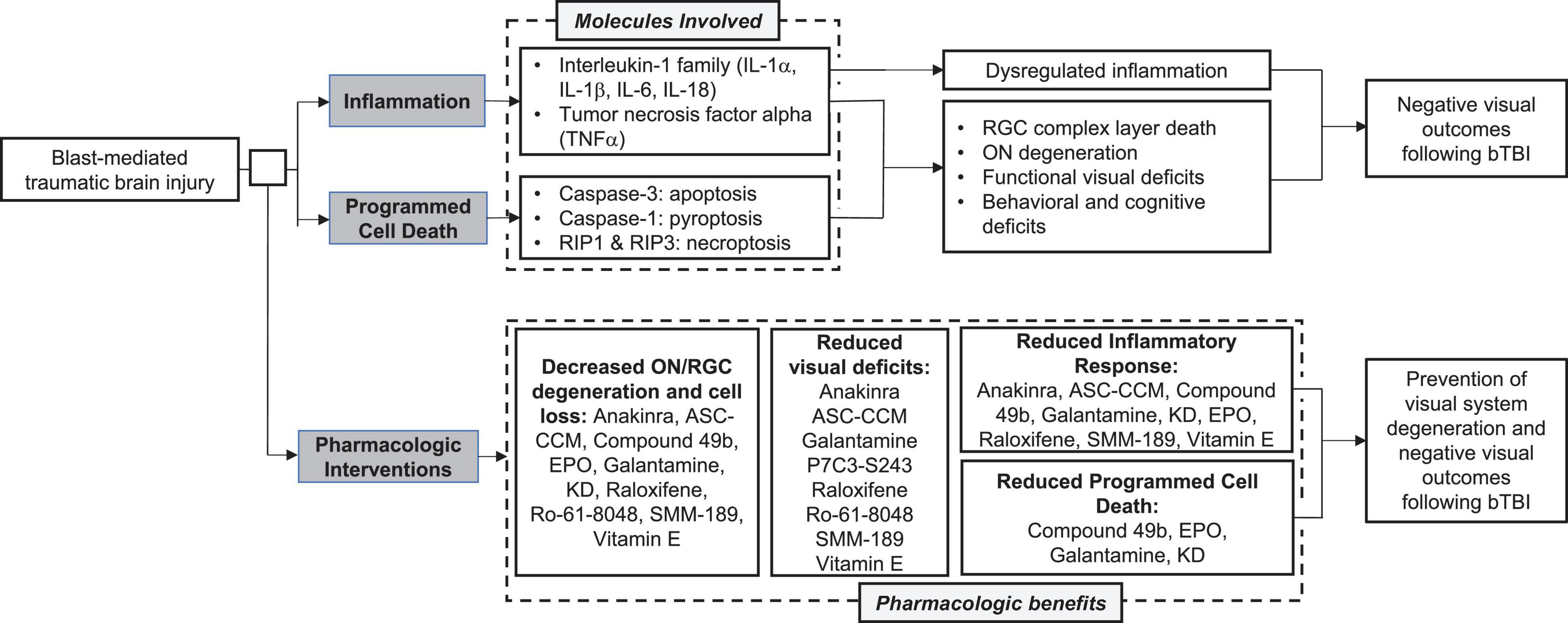
Figure 3. Summary of common molecular pathways, visual outcomes, and pharmacologic interventions following blast mediated traumatic brain injury (bTBI).
Due to the lack of commercially available equipment for blast-induction, standardization of the equipment can be difficult and could contribute to issues with reproducibility across groups using the same type of injury device. However, studies should focus on recapitulating the Friedlander waveform, mimicking the primary blast experienced in the field, enhancing their direct translational potential, and improving cross-model standardization. We recognize the vital need for a study comparing the models directly across a range of blast magnitudes and injury locations to fully understand the commonalities and differences in visual outcomes following varied blast exposure. While this would be a massive undertaking for one group to conduct, the field would benefit greatly from the creation of a large-scale data repository. Individual labs could contribute data generated from their specific parameters, outcomes, and injury type, allowing for comparisons across, as well as within, blast devices, time points, and readouts. This would provide information concerning reproducibility in addition to identifying clear commonalities that could guide research in the search for an effective intervention. We urge that future studies focus on these pathways and their downstream targets to identify specific molecules that could mediate visual protection in patients suffering from bTBI. Experiments pinpointing the anti-inflammatory mediators or survival factors that confer the greatest retinal protection would generate great strides toward translating these treatments to human use.
Author Contributions
LE, NG, AR, and AB contributed to the conception and design of the study. NG conducted the literature search. LE, NG, and AR wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
LE was supported by the NIH grant F30 EY031245. AB was supported by the NIH grants R01NS098590 and R01AR059703.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2021.659576/full#supplementary-material
Abbreviations
AChE, acetylcholinesterase; AD, Alzheimer’s disease; ASC-CCM, adipose mesenchymal stem cell concentrated conditioned media; bTBI, blast-mediated traumatic brain injury; CE, corneal edema; CGRP, calcitonin gene-related peptide; CNV, corneal neovascularization; CS, contrast sensitivity; dLGN, dorsal lateral geniculate nucleus; ELISA, enzyme-linked immunosorbent assay; EPO, erythropoietin; ERG, electroretinography; ET-1, endothelin-1; EYFP, enhanced yellow fluorescent protein; GFAP, glial fibrillary acidic protein; HPLC, high-performance liquid chromatography; IBA-1, ionized calcium binding adaptor molecule 1; IFN- γ, interferon-gamma; IGFBP-3, insulin-like growth factor binding protein-3; IGFBP-3 KD, insulin-like growth factor-binding protein-3 knock down mouse; IHC, immunohistochemistry; IL-1, Interleukin-1; IM, intramuscular; INL, inner nuclear layer; IOP, intraocular pressure; IP, intraperitoneal; (ip)RGC, (intrinsically photosensitive) retinal ganglion cells; IV, intravenous; KD, ketogenic diet; KMO, kynurenine 3-monooxygenase; LGN, lateral geniculate nucleus; MPO, myeloperoxidase; NAMPT, nicotinamide phosphoribosyltransferase; NLRP, nucleotide-binding oligomerization domain; NO, nitric oxide; NOS, nitrous oxide synthase; OCT, optical coherence tomography; ON, optic nerve; ONL, outer nuclear layer; OT, optic tract; PERG, pattern electroretinogram; qPCR, quantitative polymerase chain reaction; RGC, retinal ganglion cell; RIP, receptor interacting protein kinase; RNFL, retinal nerve fiber layer; ROS, reactive oxygen species; RPE, retinal pigment epithelium; r.AAV.EpoR76E, EPO recombinant adeno-associated virus; SC, superior colliculus; SOD2, superoxide dismutase 2; SP, substance P; TBI, traumatic brain injury; TNF α, tumor necrosis factor alpha; TRPV1, transient receptor potential vanilloid 1; TUNEL, terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling; VA, visual acuity; VEP, visual evoked potential; Wlds, Wallerian degeneration slow mouse; WT, wild-type; YFP, yellow fluorescent protein.
References
Allan, S. M., Tyrrell, P. J., and Rothwell, N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640. doi: 10.1038/nri1664
Allen, R. S., Motz, C. T., Feola, A., Chesler, K. C., Haider, R., Ramachandra Rao, S., et al. (2018). Long-term functional and structural consequences of primary blast overpressure to the eye. J. Neurotrauma 35, 2104–2116. doi: 10.1089/neu.2017.5394
Armstrong, R. A. (2018). Visual problems associated with traumatic brain injury. Clin. Exp. Optom. 101, 716–726. doi: 10.1111/cxo.12670
Bach, M., Brigell, M. G., Hawlina, M., Holder, G. E., Johnson, M. A., McCulloch, D. L., et al. (2013). ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc. Ophthalmol. 126, 1–7. doi: 10.1007/s10633-012-9353-y
Barrett, J. M., Degenaar, P., and Sernagor, E. (2015). Blockade of pathological retinal ganglion cell hyperactivity improves optogenetically evoked light responses in rd1 mice. Front. Cell. Neurosci. 9:330. doi: 10.3389/fncel.2015.00330
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Bernardo-Colon, A., Vest, V., Clark, A., Cooper, M. L., Calkins, D. J., Harrison, F. E., et al. (2018). Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis. 9:1097.
Bernardo-Colon, A., Vest, V., Cooper, M. L., Naguib, S. A., Calkins, D. J., and Rex, T. S. (2019). Progression and pathology of traumatic optic neuropathy from repeated primary blast exposure. Front. Neurosci. 13:719. doi: 10.3389/fnins.2019.00719
Bricker-Anthony, C., D’Surney, L., Lunn, B., Hines-Beard, J., Jo, M., Bernardo-Colon, A., et al. (2017). Erythropoietin either prevents or exacerbates retinal damage from eye trauma depending on treatment timing. Optom. Vis. Sci. 94, 20–32. doi: 10.1097/opx.0000000000000898
Bricker-Anthony, C., Hines-Beard, J., D’Surney, L., and Rex, T. S. (2014a). Exacerbation of blast-induced ocular trauma by an immune response. J. Neuroinflammation 11:192.
Bricker-Anthony, C., Hines-Beard, J., and Rex, T. S. (2014b). Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Invest. Ophthalmol. Vis. Sci. 55, 4853–4862. doi: 10.1167/iovs.14-14353
Bricker-Anthony, C., Hines-Beard, J., and Rex, T. S. (2016). Eye-directed overpressure airwave-induced trauma causes lasting damage to the anterior and posterior globe: a model for testing cell-based therapies. J. Ocul. Pharmacol. Ther. 32, 286–295. doi: 10.1089/jop.2015.0104
Bricker-Anthony, C., and Rex, T. S. (2015). Neurodegeneration and vision loss after mild blunt trauma in the C57Bl/6 and DBA/2J mouse. PLoS One 10:e0131921. doi: 10.1371/journal.pone.0131921
Bryden, D. W., Tilghman, J. I., and Hinds, S. R. (2019). 2nd, blast-related traumatic brain injury: current concepts and research considerations. J. Exp. Neurosci. 13:1179069519872213.
Cernak, I. (2017). Understanding blast-induced neurotrauma: how far have we come? Concussion 2:CNC42.
Chen, J., Kos, R., Garssen, J., and Redegeld, F. (2019). Molecular insights into the mechanism of necroptosis: the necrosome as a potential therapeutic target. Cells 8:1486. doi: 10.3390/cells8121486
Choi, J. H., Greene, W. A., Johnson, A. J., Chavko, M., Cleland, J. M., McCarron, R. M., et al. (2015). Pathophysiology of blast-induced ocular trauma in rats after repeated exposure to low-level blast overpressure. Clin. Exp. Ophthalmol. 43, 239–246. doi: 10.1111/ceo.12407
Cockerham, G. C., Goodrich, G. L., Weichel, E. D., Orcutt, J. C., Rizzo, J. F., Bower, K. S., et al. (2009). Eye and visual function in traumatic brain injury. J. Rehabil. Res. Dev. 46, 811–818.
Cockerham, G. C., Rice, T. A., Hewes, E. H., Cockerham, K. P., Lemke, S., Wang, G., et al. (2011). Closed-eye ocular injuries in the Iraq and Afghanistan wars. N. Engl. J. Med. 364, 2172–2173. doi: 10.1056/nejmc1010683
Creel, D. (1995). “Visually evoked potentials,” in Webvision: The Organization of the Retina and Visual System, eds H. Kolb, E. Fernandez, and R. Nelson (Salt Lake City, UT: University of Utah Health Sciences Center).
Cullis, I. G. (2001). Blast waves and how they interact with structures. J. R. Army. Med. Corps. 147, 16–26. doi: 10.1136/jramc-147-01-02
Defense and Veterans Brain Injury Center (2020). DoD Worldwide Numbers for TBI. Defense and Veterans Brain Injury Center, 17. https://dvbic.dcoe.mil/dod-worldwide-numbers-tbi (accessed June 28, 2020).
DeMar, J., Sharrow, K., Hill, M., Berman, J., Oliver, T., and Long, J. (2016). Effects of primary blast overpressure on retina and optic tract in rats. Front. Neurol. 7:59. doi: 10.3389/fneur.2016.00059
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflammation 15:199.
Dutca, L. M., Stasheff, S. F., Hedberg-Buenz, A., Rudd, D. S., Batra, N., Blodi, F. R., et al. (2014). Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Invest. Ophthalmol. Vis. Sci. 55, 8330–8341. doi: 10.1167/iovs.14-15468
Eisenfeld, A. J., Bunt-Milam, A. H., and Sarthy, P. V. (1984). Muller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Invest. Ophthalmol. Vis. Sci. 25, 1321–1328.
Evans, L. P., Newell, E. A., Mahajan, M., Tsang, S. H., Ferguson, P. J., Mahoney, J., et al. (2018). Acute vitreoretinal trauma and inflammation after traumatic brain injury in mice. Ann. Clin. Transl. Neurol. 5, 240–251. doi: 10.1002/acn3.523
Evans, L. P., Woll, A. W., Wu, S., Todd, B. P., Hehr, N., Hedberg-Buenz, A., et al. (2020). Modulation of post-traumatic immune response using the IL-1 receptor antagonist anakinra for improved visual outcomes. J. Neurotrauma 37, 1463–1480. doi: 10.1089/neu.2019.6725
Fehily, B., and Fitzgerald, M. (2017). Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant. 26, 1131–1155. doi: 10.1177/0963689717714092
Frick, K. D., and Singman, E. L. (2019). Cost of military eye injury and vision impairment related to traumatic brain injury: 2001-2017. Mil. Med. 184, e338–e343.
Friedlander, F. G. (1946). The diffraction of sound pulses; diffraction by a semi-infinite plane. Proc. R Soc. Lond. A Math. Phys. Sci. 186, 322–344. doi: 10.1098/rspa.1946.0046
Goldstein, L. E., Fisher, A. M., Tagge, C. A., Zhang, X. L., Velisek, L., Sullivan, J. A., et al. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4:134ra60.
Guley, N. H., Rogers, J. T., Del Mar, N. A., Deng, Y., Islam, R. M., D’Surney, L., et al. (2016). A novel closed-head model of mild traumatic brain injury using focal primary overpressure blast to the cranium in mice. J. Neurotrauma 33, 403–422. doi: 10.1089/neu.2015.3886
Guley, N. M., Del Mar, N. A., Ragsdale, T., Li, C., Perry, A. M., Moore, B. M., et al. (2019). Amelioration of visual deficits and visual system pathology after mild TBI with the cannabinoid type-2 receptor inverse agonist SMM-189. Exp. Eye Res. 182, 109–124. doi: 10.1016/j.exer.2019.03.013
Gupta, R., Saha, P., Sen, T., and Sen, N. (2019). An augmentation in histone dimethylation at lysine nine residues elicits vision impairment following traumatic brain injury. Free Radic. Biol. Med. 134, 630–643. doi: 10.1016/j.freeradbiomed.2019.02.015
Harper, M. M., Hedberg-Buenz, A., Herlein, J., Abrahamson, E. E., Anderson, M. G., Kuehn, M. H., et al. (2019a). Blast-mediated traumatic brain injury exacerbates retinal damage and amyloidosis in the APPswePSENd19e mouse model of alzheimer’s disease. Invest. Ophthalmol. Vis. Sci. 60, 2716–2725. doi: 10.1167/iovs.18-26353
Harper, M. M., Woll, A. W., Evans, L. P., Delcau, M., Akurathi, A., Hedberg-Buenz, A., et al. (2019b). Blast preconditioning protects retinal ganglion cells and reveals targets for prevention of neurodegeneration following blast-mediated traumatic brian injury. Invest. Ophthalmol. Vis. Sci. 60, 4159–4170. doi: 10.1167/iovs.19-27565
Hernandez, A., Tan, C., Plattner, F., Logsdon, A. F., Pozo, K., Yousuf, M. A., et al. (2018). Exposure to mild blast forces induces neuropathological effects, neurophysiological deficits and biochemical changes. Mol. Brain 11:64.
Hines-Beard, J., Marchetta, J., Gordon, S., Chaum, E., Geisert, E. E., and Rex, T. S. (2012). A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp. Eye Res. 99, 63–70. doi: 10.1016/j.exer.2012.03.013
Holan, V., Hermankova, B., Krulova, M., and Zajicova, A. (2019). Cytokine interplay among the diseased retina, inflammatory cells and mesenchymal stem cells–a clue to stem cell-based therapy. World J. Stem Cells 11, 957–967. doi: 10.4252/wjsc.v11.i11.957
Honig, M. G., Del Mar, N. A., Henderson, D. L., Ragsdale, T. D., Doty, J. B., Driver, J. H., et al. (2019). Amelioration of visual deficits and visual system pathology after mild TBI via the cannabinoid Type-2 receptor inverse agonism of raloxifene. Exp. Neurol. 322:113063. doi: 10.1016/j.expneurol.2019.113063
Jha, K. A., Pentecost, M., Lenin, R., Klaic, L., Elshaer, S. L., Gentry, J., et al. (2018). Concentrated conditioned media from adipose tissue derived mesenchymal stem cells mitigates visual deficits and retinal inflammation following mild traumatic brain injury. Int. J. Mol. Sci. 19:2016. doi: 10.3390/ijms19072016
Jiang, Y., Liu, L., Pagadala, J., Miller, D. D., and Steinle, J. J. (2013). Compound 49b protects against blast-induced retinal injury. J Neuroinflammation 10, 96.
Jiang, Y., Pagadala, J., Miller, D. D., and Steinle, J. J. (2014). Insulin-like growth factor-1 binding protein 3 (IGFBP-3) promotes recovery from trauma-induced expression of inflammatory and apoptotic factors in retina. Cytokine 70, 115–119. doi: 10.1016/j.cyto.2014.07.004
Kuriakose, M., Skotak, M., Misistia, A., Kahali, S., Sundaramurthy, A., and Chandra, N. (2016). Tailoring the blast exposure conditions in the shock tube for generating pure, primary shock waves: the end plate facilitates elimination of secondary loading of the specimen. PLoS One 11:e0161597. doi: 10.1371/journal.pone.0161597
Li, L., Eter, N., and Heiduschka, P. (2015). The microglia in healthy and diseased retina. Exp. Eye Res. 136, 116–130. doi: 10.1016/j.exer.2015.04.020
Liu, Y., Liu, T., Lei, T., Zhang, D., Du, S., Girani, L., et al. (2019). RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int. J. Mol. Med. 44, 771–786.
Loane, D. J., and Kumar, A. (2016). Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp. Neurol. 275(Pt 3), 316–327. doi: 10.1016/j.expneurol.2015.08.018
Mammadova, N., Ghaisas, S., Zenitsky, G., Sakaguchi, D. S., Kanthasamy, A. G., Greenlee, J. J., et al. (2017). Lasting retinal injury in a mouse model of blast-induced trauma. Am. J. Pathol. 187, 1459–1472. doi: 10.1016/j.ajpath.2017.03.005
Man, S. M., Karki, R., and Kanneganti, T. D. (2017). Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75. doi: 10.1111/imr.12534
McKee, A. C., and Robinson, M. E. (2014). Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 10, S242–S253.
Mohan, K., Kecova, H., Hernandez-Merino, E., Kardon, R. H., and Harper, M. M. (2013). Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest. Ophthalmol. Vis. Sci. 54, 3440–3450. doi: 10.1167/iovs.12-11522
Naguib, S., Bernardo-Colon, A., Cencer, C., Gandra, N., and Rex, T. S. (2020). Galantamine protects against synaptic, axonal, and vision deficits in experimental neurotrauma. Neurobiol. Dis. 134:104695. doi: 10.1016/j.nbd.2019.104695
Panzer, M. B., Matthews, K. A., Yu, A. W., Morrison, B. III, Meaney, D. F., and Bass, C. R. (2012). A multiscale approach to blast neurotrauma modeling: part I–development of novel test devices for in vivo and in vitro blast injury models. Front Neurol 3:46. doi: 10.3389/fneur.2012.00046
Pasinelli, P., Houseweart, M. K., Brown, R. H. Jr., and Cleveland, D. W. (2000). Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 97, 13901–13906. doi: 10.1073/pnas.240305897
Pekny, M., Wilhelmsson, U., and Pekna, M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30–38. doi: 10.1016/j.neulet.2013.12.071
Perlman, I. (1995). “The electroretinogram: ERG,” in Webvision: The Organization of the Retina and Visual System, eds H. Kolb, E. Fernandez, and R. Nelson (Salt Lake City, UT: University of Utah Health Sciences Center).
Petras, J. M., Bauman, R. A., and Elsayed, N. M. (1997). Visual system degeneration induced by blast overpressure. Toxicology 121, 41–49. doi: 10.1016/s0300-483x(97)03654-8
Por, E. D., Sandoval, M. L., Thomas-Benson, C., Burke, T. A., Doyle Brackley, A., Jeske, N. A., et al. (2017). Repeat low-level blast exposure increases transient receptor potential vanilloid 1 (TRPV1) and endothelin-1 (ET-1) expression in the trigeminal ganglion. PLoS One 12:e0182102. doi: 10.1371/journal.pone.0182102
Regasa, L. E., Agimi, Y., and Stout, K. C. (2019). Traumatic brain injury following military deployment: evaluation of diagnosis and cause of injury. J. Head Trauma Rehabil. 34, 21–29. doi: 10.1097/htr.0000000000000417
Reiner, A., Heldt, S. A., Presley, C. S., Guley, N. H., Elberger, A. J., Deng, Y., et al. (2014). 2nd, Motor, visual and emotional deficits in mice after closed-head mild traumatic brain injury are alleviated by the novel CB2 inverse agonist SMM-189. Int. J. Mol. Sci. 16, 758–787. doi: 10.3390/ijms16010758
Sardar Pasha, S. P. B., Munch, R., Schafer, P., Oertel, P., Sykes, A. M., Zhu, Y., et al. (2017). Retinal cell death dependent reactive proliferative gliosis in the mouse retina. Sci. Rep. 7:9517.
Saunders, G. H., and Echt, K. V. (2012). Blast exposure and dual sensory impairment: an evidence review and integrated rehabilitation approach. J. Rehabil. Res. Dev. 49, 1043–1058. doi: 10.1682/jrrd.2010.08.0157
Sen, N. (2017). An insight into the vision impairment following traumatic brain injury. Neurochem. Int. 111, 103–107. doi: 10.1016/j.neuint.2017.01.019
Shapiro, L. A., Perez, Z. D., Foresti, M. L., Arisi, G. M., and Ribak, C. E. (2009). Morphological and ultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res. 1266, 29–36. doi: 10.1016/j.brainres.2009.02.031
Shedd, D. F., Benko, N. A., Jones, J., Zaugg, B. E., Peiffer, R. L., and Coats, B. (2018). Long term temporal changes in structure and function of rat visual system after blast exposure. Invest. Ophthalmol. Vis. Sci. 59, 349–361. doi: 10.1167/iovs.17-21530
Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191. doi: 10.1038/nrneurol.2017.13
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Struebing, F. L., King, R., Li, Y., Chrenek, M. A., Lyuboslavsky, P. N., Sidhu, C. S., et al. (2018). Transcriptional changes in the mouse retina after ocular blast injury: a role for the immune system. J. Neurotrauma 35, 118–129. doi: 10.1089/neu.2017.5104
Swietek, B., Skotak, M., Chandra, N., and Pfister, B. J. (2019). Characterization of a controlled shock wave delivered by a pneumatic table-top gas driven shock tube. Rev. Sci. Instrum. 90:075116. doi: 10.1063/1.5099633
Vest, V., Bernardo-Colon, A., Watkins, D., Kim, B., and Rex, T. S. (2019). Rapid repeat exposure to subthreshold trauma causes synergistic axonal damage and functional deficits in the visual pathway in a mouse model. J. Neurotrauma 36, 1646–1654. doi: 10.1089/neu.2018.6046
Wang, H. C., Choi, J. H., Greene, W. A., Plamper, M. L., Cortez, H. E., Chavko, M., et al. (2014). Pathophysiology of blast-induced ocular trauma with apoptosis in the retina and optic nerve. Mil. Med. 179, 34–40. doi: 10.7205/milmed-d-13-00504
Wang, J., Fox, M. A., and Povlishock, J. T. (2013). Diffuse traumatic axonal injury in the optic nerve does not elicit retinal ganglion cell loss. J. Neuropathol. Exp. Neurol. 72, 768–781. doi: 10.1097/nen.0b013e31829d8d9d
Yin, T. C., Voorhees, J. R., Genova, R. M., Davis, K. C., Madison, A. M., Britt, J. K., et al. (2016). Acute axonal degeneration drives development of cognitive, motor, and visual deficits after blast-mediated traumatic brain injury in mice. eNeuro 3, 1–11.
Zhu, Y., Howard, J. T., Edsall, P. R., Morris, R. B., Lund, B. J., and Cleland, J. M. (2019). Blast exposure induces ocular functional changes with increasing blast over-pressures in a rat model. Curr. Eye Res. 44, 770–780.
Keywords: blast, retina, traumatic brain injury, visual function, bTBI, rodent
Citation: Evans LP, Roghair AM, Gilkes NJ and Bassuk AG (2021) Visual Outcomes in Experimental Rodent Models of Blast-Mediated Traumatic Brain Injury. Front. Mol. Neurosci. 14:659576. doi: 10.3389/fnmol.2021.659576
Received: 27 January 2021; Accepted: 18 March 2021;
Published: 15 April 2021.
Edited by:
Ashok K. Shetty, Texas A&M University College of Medicine, United StatesReviewed by:
Karen Krukowski, University of Denver, United StatesAric Flint Logsdon, University of Washington, United States
Copyright © 2021 Evans, Roghair, Gilkes and Bassuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander G. Bassuk, alexander-bassuk@uiowa.edu