mTOR-Dependent Spine Dynamics in Autism
 Shabani Chaudry
Shabani Chaudry Nandini Vasudevan
Nandini Vasudevan- School of Biological Sciences, University of Reading, Reading, United Kingdom
Autism Spectrum Conditions (ASC) are a group of neurodevelopmental disorders characterized by deficits in social communication and interaction as well as repetitive behaviors and restricted range of interests. ASC are complex genetic disorders with moderate to high heritability, and associated with atypical patterns of neural connectivity. Many of the genes implicated in ASC are involved in dendritic spine pruning and spine development, both of which can be mediated by the mammalian target of rapamycin (mTOR) signaling pathway. Consistent with this idea, human postmortem studies have shown increased spine density in ASC compared to controls suggesting that the balance between autophagy and spinogenesis is altered in ASC. However, murine models of ASC have shown inconsistent results for spine morphology, which may underlie functional connectivity. This review seeks to establish the relevance of changes in dendritic spines in ASC using data gathered from rodent models. Using a literature survey, we identify 20 genes that are linked to dendritic spine pruning or development in rodents that are also strongly implicated in ASC in humans. Furthermore, we show that all 20 genes are linked to the mTOR pathway and propose that the mTOR pathway regulating spine dynamics is a potential mechanism underlying the ASC signaling pathway in ASC. We show here that the direction of change in spine density was mostly correlated to the upstream positive or negative regulation of the mTOR pathway and most rodent models of mutant mTOR regulators show increases in immature spines, based on morphological analyses. We further explore the idea that these mutations in these genes result in aberrant social behavior in rodent models that is due to these altered spine dynamics. This review should therefore pave the way for further research on the specific genes outlined, their effect on spine morphology or density with an emphasis on understanding the functional role of these changes in ASC.
Introduction
Autism Spectrum Conditions (ASC) or Autism Spectrum Disorders (ASD) are a group of neurodevelopmental disorders defined by the DSM-VI by the presence of two core symptoms: persistent deficits in social communication, social interaction, socio-emotional reciprocity, and restricted repetitive patterns of behavior, interests, or activities (Baron-Cohen, 2009; Mandy et al., 2012). Commonly diagnosed at ages 2–3, ASC affects around 1% of the population, with ASC traits recently understood to be continuous within the general population albeit those diagnosed with ASC being at the extreme end of this spectrum (Lenroot and Yeung, 2013; Ruzich et al., 2015). The elevation of these continuously distributed ASC traits in first degree relatives of those with ASC suggests a genetic basis for ASC (Piven et al., 1997; Eyuboglu et al., 2018). This genetic basis for ASC is useful to identify critical molecular and cellular drivers that may underlie the behavioral deficits in ASC. This review seeks to: (a) identify mTOR (mammalian target of rapamycin) and specifically the mTORC1 complex as a central convergent pathway for a group of select high risk ASC genes; (b) summarize how these genes can regulate a cellular phenotype, i.e., spine dynamics via mTORC1 mediated pathways; and (c) how spine dynamics may in turn result in the behavioral and synaptic physiology phenotypes that are seen in ASC. We do so by delineating supporting correlative evidence in human subjects in Section “Functional Connectivity in ASC Is Linked to Spine Dynamics” and “Many Genes Linked to ASC Are Regulators of mTORC1 or Targets of mTORC1” and use data from rodent models of ASC in further sections in order to explore genetic causality for both spine dynamics and behavior.
Functional Connectivity in ASC Is Linked to Spine Dynamics
Functional Connectivity Within the DMN Underlies the ASC Phenotype
ASC is understood to be a whole brain disorder, whose neural correlates range from alterations in synapses and neural transmission to brain volume and structure. Alterations in functional connectivity have long been implicated in the pathophysiology of ASC since differences in functional connectivity have been found in an estimated 90% of studies (Lau et al., 2013; Chen et al., 2015; Mohammad-Rezazadeh et al., 2016) although the direction of this difference, implicated brain areas, brain state, and age are disputed (Müller et al., 2011; King et al., 2019). A dominant but challenged theory (Picci et al., 2016) is the “Cortical Underconnectivity Theory” which states that there is long range underconnectivity particularly between hemispheres and frontal-posterior brain areas in ASC individuals which may explain symptomatology (Just et al., 2012). In support of this theory is the finding that many ASC individuals have decreased Corpus Callosum (CC) volume (Frazier and Hardan, 2009), and that both adults and children with congenital agenesis of the CC (AgCC) show deficits in social and emotional intelligence and score highly on the Autism Quotient (Lombardo et al., 2012; Lau et al., 2013) without intellectual disability (Lábadi and Beke, 2017). Furthermore, fMRI studies of individuals with AgCC show decreased long range functional connectivity, suggesting that decreased long range connectivity between hemispheres is what contributes to ASC symptomology (Owen et al., 2013).
Apart from underconnectivity driven by the corpus collosum, underconnectivity could also be seen by activity in the nodes of the Default Mode Network (DMN), a set of nuclei that are preferentially activated, as visualized by rsfMRI (resting state magnetic resonance imaging) scanning, while individuals are not focused on the external environment (i.e., “mind wandering” or “resting state”). While DMN activation ceases once cognitive effortful tasks commence (Anderson et al., 2011; Li et al., 2014; Padmanabhan et al., 2017), mind wandering involves internally generated thoughts about others, one’s self and remembering past or envisioning future events, as elucidated by designed self-report questionnaires and interviews (Andrews-Hanna et al., 2013; Smallwood and Schooler, 2015). The DMN identified in this manner consists of the medial prefrontal cortex (mPFC), the posterior cingulate cortex, precuneus, medial temporal lobe (which includes the hippocampus), and inferior parietal cortices (Li et al., 2014; Alves et al., 2019) which together incorporate memories (hippocampus) and emotions (mPFC) in order to create “mind wandering” (Poerio et al., 2017). Hence, adults with bilateral ventromedial prefrontal cortex (vmPFC) lesions show a reduction in self-reported mind wandering (Bertossi and Ciaramelli, 2016).
Decreased connectivity is often detected by fMRI in ASC individuals within the DMN, with underconnectivity having been found positively correlated to ASC social deficits, increased repetitive stereotyped behaviors measured by Autism Diagnostic Observational Score (ADOS; Yerys et al., 2015) as well the Autism Diagnostic Interview-Revised (ADI-R; Chen et al., 2021b). Though this data is necessarily correlative in nature, long range underconnectivity in DMN areas could be a neuroanatomical signature for ASC individuals who could then be tested for social deficits (Nair et al., 2020) using the Theory of Mind (ToM) tasks in which ASC individuals show marked deficits (Frith and Happé, 1994). However, other studies have shown short range, long range and overall brain connectivity to be functionally overconnected in adults (Belmonte, 2004; Kleinhans et al., 2016), though long range connections to and from the frontal cortex appear to be consistently under-connected (Courchesne and Pierce, 2005), with strength of connectivity between the PFC and right parietal cortex negatively correlated with autism symptom severity (Redcay et al., 2013). However, long range hyperconnectivity between the thalamus and auditory and somatosensory cortices as well as parietal regions has also been found in fMRI studies of adults with ASC (Tomasi and Volkow, 2019).
Varying long range connectivity in studies could occur due to the polygenetic nature of ASC with different genetic rodent models showing different functional connectivity patterns. Therefore, it may be possible to use varying patterns of connectivity as an endophenotype for identifying different forms of ASC (Zerbi et al., 2021). For example, homozygous knockout male mouse models of Fragile X Syndrome (FXS) show long range cortico-striatal connections to be functionally underconnected (Zerbi et al., 2018), whereas heterozygous knockout male TSC models mouse models show the same connections to be functionally hyperconnected (Pagani et al., 2021). This long range hyperconnectivity found within the TSC mouse model was also found to be associated with increased repetitive behaviors in the mice. Increased repetitive behaviors and increased long range hyperconnectivity are both reversed by the administration of the mTOR inhibitor rapamycin, which functionally reverses the effects of the loss of one of the two Tsc2 genes (Pagani et al., 2021).
Conversely, individuals with depression present increased rumination and self-referential mind wandering (Nejad et al., 2013; Nayda and Takarangi, 2021) that is correlated with hyperconnectivity between the long distance DMN nodes mPFC and the posterior cingulate cortex, suggesting that the DMN nuclei must maintain optimal connectivity for healthy cognition and perception of self (Wise et al., 2017). In humans the use of ketamine, a compound that is primarily an NMDA receptor antagonist (Sleigh et al., 2014) has been shown to alleviate symptoms of depression, as well as reduce functional connectivity within the DMN of healthy individuals (Scheidegger et al., 2012) and those with Major Depressive Disorder (MDD; Evans et al., 2018), suggesting that many behaviors are strongly correlated with functional connectivity. These studies show that functional connectivity in the DMN may be a marker of behaviors that denote increased or decreased self-reference.
Synaptic Physiology Underlies the Differences in Connectivity in ASC Individuals
What underlies differences in DMN connectivity in ASC individuals? Within discrete brain areas, this may be attributed to an Excitation Inhibition (E/I) imbalance and reduction in signal-to-noise ratio in neural circuitry that is implicated in ASC (Rubenstein and Merzenich, 2003; Testa-Silva et al., 2012). Indeed, the high incidence of epilepsy within the ASC population (Viscidi et al., 2013) as well a postmortem study showing upregulation of excitatory AMPA receptors (Purcell et al., 2001), may indicate that there is an increased excitation within the brain of ASC individuals. How increased excitation within the brain of ASC individuals causes local or long distance hyperconnectivity or hypoconnectivity is not fully understood though changing E/I ratios in local circuits to understand this question is now underway in a number of rodent models of ASC (See Section “Molecular Mechanisms That Link Dysfunctions in Neuromorphology to Behavior”). Interestingly, hyperconnectivity between various modes of the DMN, including that detected in a Tsc2+/− mouse model of ASC using rsFMRI is linked to an excess of mTOR signaling and increased spine density in the insular cortex. Using in silico modeling, these increases in synapse density were linked to hyperconnectivity and use of a mTORC1 inhibitor, rapamycin, reversed both the increases in spine density and the hyperconnectivity in this model (Pagani et al., 2021). This strongly suggests that connectivity in the DMN is positively correlated to spine density which is in turn driven by mTORC1.
Spine Dynamics Contributes to the E/I Ratio Within Neural Circuits
Clearly, DMN connectivity and the underlying E/I ratio could be dependent on neuronal architecture including dendritic spine dynamics reflected by spine density and spine morphology (Gao and Penzes, 2015). Dendritic spines are typically around 0.5–2 microns long, dendritic protrusions that receive signals from axonal boutons of other neurons to form synapses allowing neurons to transmit signals to one another (Horner and Arbuthnott, 1991). Human postmortem studies reveal that synapse formation increases throughout childhood from birth in both ASC and Typically Developing (TD) children, but is slightly elevated in ASC (Penzes et al., 2011). Synapses are then pruned from adolescence into adulthood, with pruning being more efficient in TD over ASC subjects, leaving ASC individuals with a greater number of synapses. In adulthood, this is followed by an equilibrium in synapse formation and elimination to maintain a stable number of synapses in TD individuals (Rakic et al., 1986; Masliah et al., 1993; Peters et al., 1998; Penzes et al., 2011); there is increased synapse maintenance during adulthood in ASC, often leading to a greater number of spines and synapses compared to TD individuals (Penzes et al., 2011). The typical development of cortical dendritic spines in rodents follows a similar pattern to humans, with an initial increase in dendritic spine density during the first two postnatal weeks, followed by an increase in spine pruning over spine formation during adolescence, with spine density then stabilizing in adulthood (Bhatt et al., 2009). Apart from spine number, the shape of spines is also important (Table 1). New immature spines have a small surface area and postsynaptic density (PSD), with decreased number of receptors; this could reflect an increased “neuroplasticity” of neural circuits, with a greater potential for strengthening, thus making them “learning” spines (Bourne and Harris, 2007). It should be noted that the classification of filipodia spines as being a defined and individual spine type or just the precursor to dendritic spines is disputed (Takahashi et al., 2003; Kanjhan et al., 2016); this is due to the dynamic nature of filipodia-type spines which give rise to all other spine types. However, since many studies investigating dendritic spine dynamics in rodent models of ASC report filipodia-type spines as part of the immature dendritic spine classification, and report changes in the quantity of filopodia type spines in various rodent ASC models, we have also decided to report filipodia type spines as a distinct spine classification within the larger category of immature spines (Dictenberg et al., 2008; Williams et al., 2008; Dunaevsky et al., 2014; Pyronneau et al., 2017; Sceniak et al., 2019; Skelton et al., 2020). Wider more mature “memory” spines have greater surface area and PSD for receptors and form very strong synapses with very little range for the synaptic strengthening/adaptation that is associated with Long Term Potentiation (LTP), on increased stimulation (Cooke and Bliss, 2006). Spine shape and consequently maturity are characterized by their length and width measurement ratios (Harris and Kater, 1994), with immature spines, i.e., filopodia, thin, long thin spines tending to be longer and thinner in size and mature spines, i.e., mushroom and stubby shorter and wider (Table 1). An increase in mushroom type mature spines and increase in overall spine width in the primate prefrontal cortex is associated with cognitive decline during aging in rhesus macaques (Dumitriu et al., 2010), suggesting inflexibility and loss of neural plasticity (Bourne and Harris, 2007, 2008).
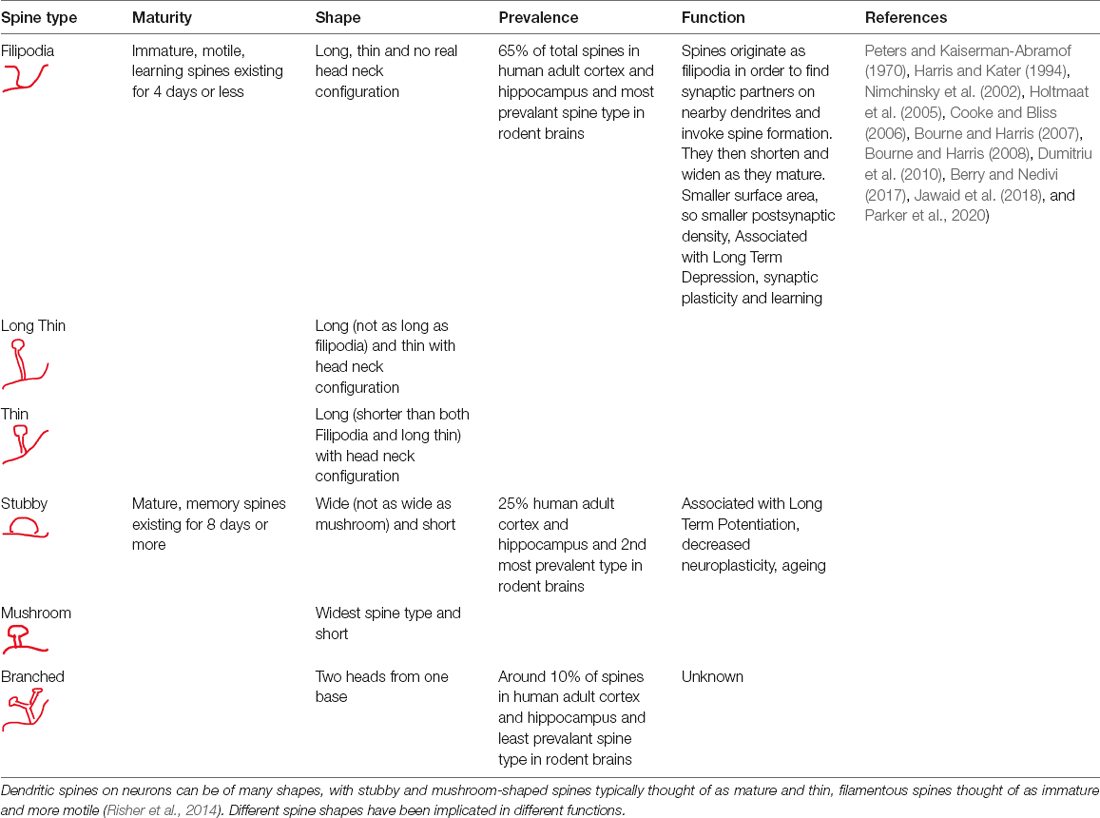
Table 1. Characteristics of dendritic spines.
A recent study using living humans showed that synaptic density (which may be indicative of spine density), as measured by PET scanning of synaptic glycoprotein 2A, is negatively correlated with symptom severity in adults with MDD, within the dorsolateral prefrontal cortex (Holmes et al., 2019). Such local hypoconnectivity is inversely related while long range hyperconnectivity between the dorsolateral prefrontal cortex and the posterior cingulate cortex is positively correlated with depression symptoms of clinically depressed individuals, suggesting that the underlying measurements at the synapse, i.e., spine density/morphology may have predictive value. In support of this idea, multiple rat models of depression also show a decrease in dendritic arbor within the hippocampal CA3 (Watanabe et al., 1992) and mPFC pyramidal cells (Goldwater et al., 2009), both key nodes involved in the DMN. Furthermore, ketamine inhibited dendritic spine loss in chronically stressed rats that displayed symptoms of depression via protection of mushroom shaped spines, and through the induction of spine formation (Ng et al., 2018).
Human postmortem studies, using Golgi-Cox staining, have shown an increase in spine density in ASC individuals compared to controls, independent of sex, within the amygdala, cortex, and temporal lobes (Hutsler and Zhang, 2010; Tang et al., 2014; Weir et al., 2018; Table 2). This increased spine density in ASC individuals (Weir et al., 2018) is correlated with increased head circumference/macrocephaly, as well as enlargement of specific brain areas including the amygdala and hippocampus (Schumann et al., 2004; Tilot et al., 2015; Weir et al., 2018). Only one human postmortem study assessed spine morphology via Golgi-cox staining in ASC vs. TD controls and found that ASC individuals had more “compact” spines with decreased spine length within the cortex, which indicates a more mature spine morphology (Hutsler and Zhang, 2010; Table 2). This along with the increased spine density found in ASC individuals may reflect an increase in local connectivity linking spine density to functional connectivity. One thing to note is that increased head size (macrocephaly) is a common finding in children and adults with ASC; toddlers with ASC show accelerated brain growth compared to TD controls to possibly result in an enlarged amygdala and hippocampus during adolescence (Groen et al., 2010) with the degree of amygdala enlargement being a predictor for ASC symptom severity (Schumann et al., 2004). A mouse ASC genetic model of Phosphatase and tensin homolog (Pten) haploinsufficiency also reflects the macrocephaly found within ASC individuals (Huang et al., 2016). Furthermore this model shows increased brain enlargement and macrocephaly that is correlated with hyperconnectivity and dendritic arborization (Huang et al., 2016), suggesting that ASC individuals with macrocephaly may also have similar hyperconnectivity with increased spine densities. A conditional knockout of Pten in a subset of auditory cortical neurons shows enhanced local and global hyperconnectivity with the strength of synaptic inputs from the thalamus and auditory neurons increased; this was decreased by the mTOR inhibitor rapamycin (Xiong et al., 2012). Together, these studies suggest that functional connectivity is correlated with both spine density and mTOR signaling and both these neural correlates may result in macrocephaly.

Table 2. Brains from human ASC individuals show increased spine density.
Many Genes Linked to ASC Are Regulators of mTORC1 Or Targets of mTORC1
Though twin and genome studies have demonstrated that ASC has a genetic component, the genetic basis could be syndromic or non-syndromic. Though syndromic forms account only for 2%–4% of ASC cases, they implicate genes that can then be used to understand underlying cellular processes (Sztainberg and Zoghbi, 2016; Ziats et al., 2021) in manipulatable rodent models. These genes tend to be very rare within the general population but they have a large effect size in that they are very penetrant (Geschwind, 2011). Therefore, syndromic forms of ASC are highly comorbid with heritable, monogenic autosomal dominant disorders caused by the dysfunction of one identified gene or chromosome section with high penetrance; examples include Fragile X syndrome (FXS), Tuberous Sclerosis Complex (TSC), Phelan-McDermid syndrome (PMS), Rett Syndrome, Smith Lemli Opitz syndrome, and Angelman/Prader-Willis Syndrome (Fernandez and Scherer, 2017). For example, 61% of TSC patients have ASC (Vignoli et al., 2015). Non-syndromic forms of ASC could be due to the mutation of many common genetic variants or Single Nucleotide Polymorphisms (SNP’s) which along with epigenetic factors summate in order for ASC symptoms to reach a threshold for diagnosis (Rylaarsdam and Guemez-Gamboa, 2019; Satterstrom et al., 2020). These genes being prevalent in the general population means that they are present in many healthy individuals and may contribute to normal variation of traits, that they have a small effect size and individually do not often result in the development of ASC in the individual. However, it is the combination of different SNP’s along with various epigenetic factors that may lead to ASC diagnosis (Geschwind, 2011). Despite several genes implicated in ASC, 75%–95% of ASC cases are idiopathic having an unidentified genetic basis (Caglayan, 2010; Richards et al., 2015; Garg and Green, 2018). The Simons Foundation Autism Research Initiative (SFARI) database contains a repository of ASC-associated genes which are categorized as either “Syndromic” (S), 1, 2 or 3 according to the strength of evidence (stronger → weaker) linking the gene to ASC (for more information see https://gene.sfari.org/about-gene-scoring/; Banerjee-Basu and Packer, 2010).
Many ASC-linked genes have been implicated in synapse development through regulating spinogenesis (Sztainberg and Zoghbi, 2016; Satterstrom et al., 2020). Using integrated transcriptomics analyses of microarray and RNA-seq data from a number of rodent ASC models, proteins expressed at glutamatergic synapses are preferentially identified as altered in ASC (Duan et al., 2019). Both spine density and dendritic arborization (Copf, 2016) have been identified as molecular processes in neurons that are important for several neurodevelopmental disorders, including ASC (Nishiyama, 2019). Together, these studies suggest that dysregulation of spine density may be causal to autistic behaviors. Since in rodents key genes and neuromorphological alterations associated with ASC are conserved (Varghese et al., 2017) with humans, and behavioral tasks such as the social discrimination and social interaction tasks and the marble burying task that mimic the ASC phenotype are well established (Crawley, 1999, 2007), focusing on genes that affect spine density or spine morphology in rodents may lead to mechanistic insight (Lazaro and Golshani, 2015) of how these can affect behavior. In order to do so, we searched the available literature on PubMed to identify 141 genes identified using the search terms “spine density” or “spine morphology” AND “autism” or “ASC” in animal models. Out of 80 genes that showed differences from the wildtype (WT) genotype in mean spine density and/or morphology, 20 have a score of 1 in the SFARI database. All these 20 genes are either regulators of the mTORC1 complex or are regulated by the mTORC1 pathway (Figure 1), a major convergent pathway for syndromic and nonsyndromic genes (Magdalon et al., 2017). Though other pathways, notably those involved in cell adhesion may also be important, we concentrate solely on the upstream regulators of mTORC1 signaling with a focus on studies where spine dynamics is altered in this review. We also discuss their effects on neuromorphology and ASC-type behaviors in genetically altered rodent models and infer mTORC1 dependent and independent molecular processes driven by them that may cause the alterations in neuromorphology and behavior.
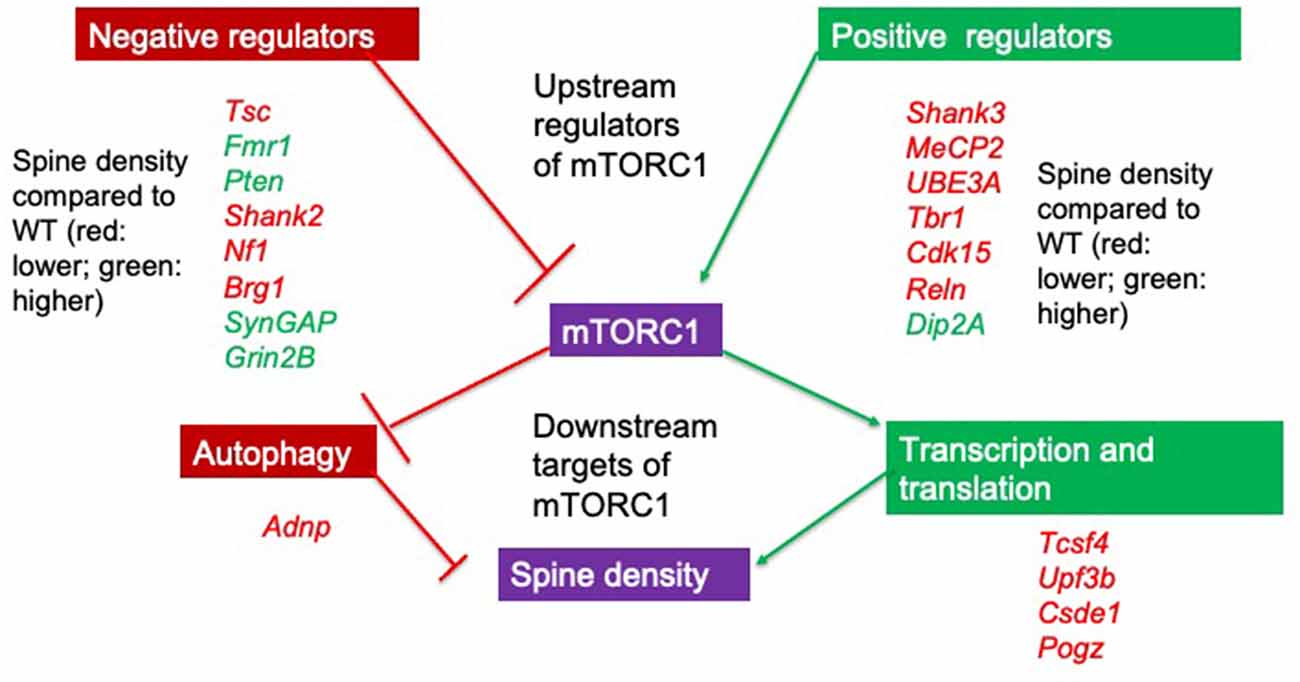
Figure 1. Upstream regulators and downstream targets of mTORC1. Positive and negative regulators of mTORC1 signaling show alterations in spine density when deleted in murine models. All identified genes with a SFARI score of 1 can regulate mTORC1 and all change spine density compared to wildtype (WT) animals. If spine density is increased compared to WT, this is denoted by the gene in green while if spine density is decreased compared to the WT, this is denoted by the gene in red. Note that positive regulators of mTORC1 (with the exception of Dip2A) all decrease spine density either when deleted or when duplicated (UBE3A and MeCP2). Though we do not focus on downstream targets of mTORC1 in this review, these targets are involved in transcription, translation, and autophagy. Adnp, Activity dependent neuroprotective protein; Tcsf4, Transcription factor 4; Upf3b, Regulator of nonsense transcripts 3B; Csde1, Cold shock domain-containing protein E1; Pogz, Pogo transposable element with ZNF domain.
The mTORC1 Pathway in ASC Is A Critical Hub for Both Neuromorphology and Behavior
mTORC1 is a serine/threonine kinase within the PI3K-related kinase (PIKK) family, made up of mTOR, Raptor, and mLST8 (mammalian lethal with Sec13 protein 8; Saxton and Sabatini, 2017) subunit and is sensitive to rapamycin inhibition. A major upstream regulator is PI3K-phosphorylated Protein Kinase B (PKB) which directly activates mTORC1 and indirectly does so by inactivating an inhibitor, i.e., the Tuberous Sclerosis 1 and 2 (TSC1/2) proteins which in turn normally inhibit Rheb GTPase from activating mTORC1 (Ersahin et al., 2015; Figure 2). In cells, the mTORC1 pathway is a critical nexus in cells linking external stimuli such as energy availability, stressors, and abundance of growth factors as well as activation of glutamate receptors (AMPA/NMDA) and associated proteins at the synapse (SYNGAP/HOMER/SHANK/PTEN) to downstream increases in protein translation and maintenance of autophagy (Winden et al., 2018). Downstream signaling targets include S6K which upregulates translation initiation and elongation via eIF4A and eEF2 respectively, as well as increases cell growth and cell survival (Ersahin et al., 2015). The mTORC1 pathway maintains autophagy by inhibiting ULK-1, an activator of BECLIN which is required for the formation of the autophagosome (Jhanwar-Uniyal et al., 2019); hence overactivation of mTORC1 results in a decrease in autophagy.
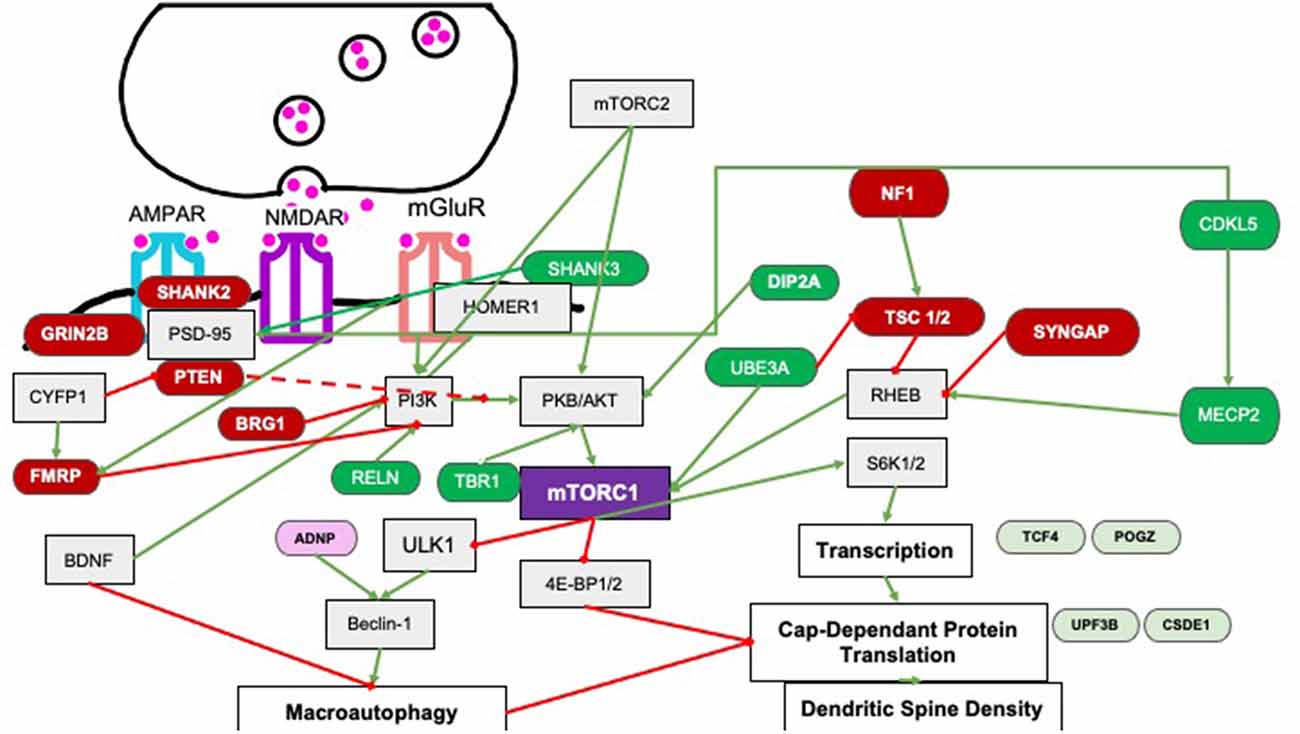
Figure 2. Pathways by which upstream regulators affect mTORC1 signaling. Several regulators affect mTORC1 signaling via PI3K signaling or via Rheb signaling. Green and red-labeled genes represent ASC related genes that are upstream regulators of mTORC1 in an either positive (dark green) or negative (dark red). Downstream target genes linked to ASC that show changes in spine density are shown as light green (positively regulated by mTORC1); or pink (genes negatively regulated by mTORC1). Downstream target genes of mTORC1 are not the focus of this review. Green arrows indicate activation and red lines represent inhibition.
In several models of autism such as deletions or mutations of PTEN, TSC, NF1 or FMRP proteins, mTORC1 activity is increased (Magdalon et al., 2017 and references therein). For example, tuberous sclerosis complex genes 1 and 2 (Tsc 1/2) code for the protein hamartin and tuberin respectively which heterodimerize to inhibit RheB, which normally directly activates mTORC1 (Franz and Capal, 2017). Therefore deletion, mutation or rearrangement of the Tsc genes leads to a loss of activity of the complex and overactivation of the mTORC1 pathway (Huang and Manning, 2008; Portocarrero et al., 2018). As expected, proteomic analyses show increases of mTOR in both the hippocampus and striatum of Tsc1+/− mice (Wesseling et al., 2017) and increases of downstream targets such as Ulk1 mRNA and phospho-S6K protein in the Tsc2+/− mouse brain (Sato et al., 2012). This shows that mTOR overactivation by the deletion of an ASC-associated gene can increase the expression of downstream targets of mTORC1 (Figure 2). Since mTORC1 overactivation in this manner causes increases in spine density that is correlated with DMN hyperconnectivity (Pagani et al., 2021), other upstream regulators of mTORC1 may also alter behavior via spine density alterations.
Autophagy, Downstream of mTORC1 Signaling, Is Required for Optimal Spine Density and Social Behavior
If mTORC1 signaling is a critical nexus for upstream regulators that are deleted or mutated in autism and if spine dynamics are an underlying mechanism for autism, then it is plausible that loss or mutation of upstream regulators may affect spinogenesis and/or spine pruning via mTORC1 signaling, just as it does social behavior (Section “Rapamycin, an Inhibitor of mTORC1, Reverses Autistic-Like Behaviors”). In ASC individuals, postmortem studies show a decrease of LC-III, an autophagosome marker, suggesting decreased pruning. Decrease of this marker is correlated with increased spine density and persistent PSD-95 marker expression in the temporal lobe throughout childhood and adolescence (Tang et al., 2014). Supporting this, a neuronal deletion of atg7, a E1 ligase required for phagosome formation in mice results in social interaction deficits and spine density on basal dendrites that stays elevated on cortical pyramidal neurons into adulthood. A second murine model that shows the significance of spine pruning is the Tsc2+/− haploinsufficient mouse where reduction of this negative regulator of mTORC1 leads to increases in spine density in cortical projection neurons (Tang et al., 2014). Though rapamycin rescues both behavioral deficits and aberrant spine density in the Tsc2+/− mouse, it does not do so in the Atg7 conditional knockout (cKO) mouse or in the double mutant Tsc2+/−, Atg7cKO mouse, demonstrating an absolute need for spine pruning in order to ameliorate social behavior deficits (Tang et al., 2014). A second upstream negative regulator of mTORC1 is the Fragile X mental retardation protein (FMRP; Sharma et al., 2010; Hoeffer et al., 2012; Darnell and Klann, 2013; Casingal et al., 2020), the Fmr1 KO shows increased spine density in the hippocampus which in turn is correlated with higher long term depression (LTD) and a lack of discrimination in the novel object recognition task. These phenotypes are ameliorated with shRNA to Raptor, an unique component of the mTORC1 complex, suggesting that overactivation of mTORC1 specifically leads to the cognitive deficit (Yan et al., 2018). This rectifying effect of decreasing Raptor levels could be abrogated by decreasing ATG7, a protein required for phagosome formation, suggesting that Raptor rescued spine and cognitive deficits via the activation of autophagy in hippocampal neurons (Yan et al., 2018). Consistent with the idea that rectifying mTORC1 activity is central to rescuing spine density and social behavior, ketamine, a mTORC1 activator because it also secondarily increases AMPA signaling, rescued the lack of fear memory and decreased spine density in the prefrontal cortex seen in a heterozygote mouse deficient in the RELN (reelin) protein. Since RELN is a positive activator of mTORC1, lack of this protein leads to lower activation of this pathway which can be rescued by activation with ketamine. Conversely, rapamycin, an inhibitor of mTORC1 can reverse the ketamine-mediated rescue of spine density and fear memory in the Reln+/− mouse, suggesting that both neuromorphology and cognitive and social behaviors are highly sensitive to mTORC1 levels (Iafrati et al., 2013; Section “Rapamycin, an Inhibitor of mTORC1, Reverses Autistic-Like Behaviors” below).
Rapamycin, an Inhibitor of mTORC1, Reverses Autistic-Like Behaviors
If mTORC1 is a critical nexus for ASC, we would also expect that alteration of this pathway results in behaviors that denote autism in rodent models. To give an example, haploinsufficiency of Tsc2 or Tsc1 in the cerebellum using a conditional knockout (KO) mouse model is sufficient for the manifestation of social preference in the 3-chamber preference test and repetitive behavior in the marble burying task and self-grooming with all behavioral phenotypes ameliorated with rapamycin treatment (Tsai et al., 2012; Reith et al., 2013). Similarly, a selective deficit in social interaction but not in locomotor or food exploration activity was noted in both Tsc1 and Tsc2 heterozygote mutant mice. This deficit is reversed in both genotypes by rapamycin (Sato et al., 2012). Tsc2+/− mice also show decreased memory in the Morris Water Maze and no discrimination between novel and training contexts in the contextual fear conditioning task, behaviors that could possibly be due to unstable late-phase LTP at the Schaeffer collateral synapse in the hippocampus; these were also reversed by rapamycin. This is a specific effect since early phase LTP, basal synaptic transmission and paired pulse facilitation are unaffected (Ehninger et al., 2008). Rapamycin reversal of specific behavioral and synaptic phenotypes demonstrates that mTORC1 signaling is specifically critical in driving autism-like behaviors in rodents.
The consequence of overactivation of the mTORC1 pathway would be an increase in translation and hence we would expect to see similar behavioral deficits in mice that overexpress or show greater activity of the downstream targets of mTORC1. Mice that are deleted for the repressor e4B-BP1, mimic mTORC1 overactivation since they have increased eIF4E activity and show increased excitation-inhibition (E/I) ratio, suggesting altered synaptic properties in the CA1 of the hippocampus. They also show decreased vocalization and socialization and no preference for conspecifics in the 3-chamber test as well as increased anxiety and repetitive behaviors such as self-grooming (Gkogkas et al., 2012). Interestingly, pharmacological inhibition of eIF4E normalized both social and repetitive behaviors and the E/I ratio. Similarly, a transgenic mouse that overexpressed eIF4E mimicking ASC individuals with a SNP in the promoter of the eIF4E gene that increases the level of this protein, also showed increased repetitive behavior and anxiety, decreased contact with conspecifics and higher mEPSCs from pyramidal neurons in the prefrontal cortex. This could be due to an increased spine density through an increase of smaller spines (Santini et al., 2012). Again, intracerebroventricular infusion of the eIF4E inhibitor, 4EGI-1, improved social behaviors and decreased anxiety (Santini et al., 2012). In Fmr1 (Fragile-X mental retardation 1) KO mice, where mTORC1 is overactive because of loss of inhibition of PI3K, S6K is also overactivated; deletion of S6K increased dendritic spine maturation with concomitant improvement in social interaction behaviors and mGluR-mediated long term depression (LTD; Bhattacharya et al., 2012). Taken together, all these studies suggest that overactivation of mTORC1 and S6K signaling is sufficient for the manifestation of autistic-type behaviors.
Many Upstream Regulators of mTORC1 Are Expressed at The Dendrite Or at The Post-Synaptic Density (PSD)
Of the upstream regulators of mTORC1 identified (Tables 3 and 4), 75% are expressed either at the membrane of neurons in the PSD or at the dendrite. These are RELN, SHANK3 (SH3 and ankyrin repeat domain protein), DIP2A, CDKL5, FMRP, SHANK2, PTEN, SYNGAP (Synaptic Ras GTPase-activating protein), GRIN2B (Glutamate ionotropic receptor NMDA type subunit 2B) proteins and include both positive and negative regulators of mTORC1 function. One percent of all ASC cases show mutations in the SHANK family of proteins which are scaffolding proteins at the PSD that link to PSD-95, Homer, Arc2/3, the WAVE regulatory complex, SHARPIN, and glutamate receptors (AMPA and NMDA) via intermediate molecules or directly via their several conserved domains (Naisbitt et al., 1999; Delling and Boeckers, 2021). The presence of most mTORC1 regulators at the synapse as well as the data in Section “The mTORC1 Pathway in ASC Is a Critical Hub for Both Neuromorphology and Behavior” using mutant mTOR regulator rodent models suggests that synaptic parameters such as spine density, spine morphology, dendritic arbor, and the functional correlate, i.e., synaptic physiology play an important and possibly causal role in autistic-like behaviors in the rodent models.

Table 3. Upstream regulators of mTORC1 implicated in ASC that act as positive regulators of mTORC1 signaling.

Table 4. Upstream regulators of mTORC1 implicated in ASC that act as negative regulators of mTORC1 signaling.
Positive and Negative Regulators of mTORC1 Have Opposing Effects on Spine Density and Synaptic Plasticity
Several reviews stress the role of mTOR signaling in autism (Magdalon et al., 2017; Winden et al., 2018) while others explore the role of spine dynamics in autism, focusing on a few genetic models (Copf, 2016; Lin et al., 2016; Martínez-Cerdeño, 2017). In these sections, we specifically focus on the effects of upstream positive and negative regulators of mTORC1 on spine density and/or spine morphology in rodent models and possible correlations with synaptic physiology [Section “Many Upstream Regulators of mTORC1 Are Expressed at the Dendrite or at the Post-Synaptic Density (PSD)”] and social behaviors (Section “Alterations of mTORC1 Activity by Alterations in the Expression of Upstream Regulators Result in Autistic Behaviors”).
Positive Regulators of mTORC1 at the PSD/Dendrite
Positive regulators of mTORC1 (Table 3) that are at the PSD/dendrite include SHANK3, DIP2A, RELN, and CDKL5. Two genes cyclin-dependent kinase-like 5 (CDKL5) encoded by the Cdlk5 gene located on the X chromosome (Montini et al., 1998) and Dip2A (Disco-interacting protein 2 homolog A), located on Chromosome 21 respectively have an important role in neuronal development and migration (Chen et al., 2010) as well as synaptogenesis and spine maturation (Della Sala and Pizzorusso, 2014). CDKL5 is localized with RAC1 in the spine of postmitotic neurons (Chen et al., 2010) whereas DIP2A is at the cell membrane and PSD (Ouchi et al., 2010). CDKL5 stimulates mTORC1 signaling through positive regulation of MeCP2, another ASC-related gene (Tsujimura et al., 2015; Section “Effects of the Loss of ‘global’ Positive Regulators of mTORC1 on Spine Density and Synaptic Physiology”) while DIP2A is a binding partner of FSTL1, which mediates Akt/PKB phosphorylation (Ouchi et al., 2010; Liang et al., 2014) and therefore increases mTORC1 activity. Unusually, RELN is an extracellular ligand that binds to the ApoER2 and VLDLR to direct cortical neuronal migration (Jossin, 2020).
Effects of the Loss of Synaptic Positive Regulators of mTORC1 in the PSD on Spine Density and Synaptic Physiology
Heterozygote CDKL5 knockout mice show lower spine density on both apical and basal dendrites of pyramidal cells in the CA1 and dentate gyrus as well as decreased arborization as shown in lower number of branches and lower dendrite lengths (Trazzi et al., 2018). Despite a small increase in immature spines, this lower spine density is driven by a larger decrease in mature spines. Juvenile haploinsufficient reeler mice also show lower spine density, most of which is attributable to lower numbers of spines with smaller head widths, accompanied by a near absence of NMDA-dependent LTP (Iafrati et al., 2013). Unlike the other positive regulators, Dip2A KO mice show increased spine density but this is confined to basal dendrites with no change in spine density on apical dendrites in cortical pyramidal neurons. However, this increased spine density is due to an increase in stubby spines albeit and a decrease in mushroom spines, flattening the PSD and possibly decreasing mEPSC amplitude due to a smaller number of NMDA and GluR subunits at the PSD (Ma et al., 2019).
Several homozygous and heterozygous rodent models use deletion or missense mutations of the scaffolding protein, SHANK, that mimic those found in human ASC individuals and that result in truncation of the protein. These are widely studied for their behavioral and neuromorphological phenotypes; for a detailed summary and a tabular synopsis of these, the reader is directed to Delling and Boeckers (2021) and Monteiro and Feng (2017) respectively. The best studied Shank isoform, SHANK 3 is uniquely expressed as part of the corticostriatal circuit in the striatum (Peça et al., 2011) and both a complete gene Shank3 KO (Shank3−/−; Wang et al., 2016) and PDZ-domain deletions (Peça et al., 2011; Demir et al., 2016) show reduced spine density in the striatum. Mice with deletions of the ankyrin repeat domain of Shank3 show reduced spine density in the hippocampus (Wang et al., 2011) and those with deletions in the Pro-rich (Zhou et al., 2016) domain also show reduced spine densities in the prefrontal cortex. In the hippocampus, LTP was also decreased in a Shank3 mutant with a deletion of the ankyrin repeat domain (Jaramillo et al., 2016). Though some studies show no difference in spine density, the weight of evidence tends towards a decrease in spine density (Figure 3) with some studies reporting a decrease in mature mushroom shaped spines or spine width and length in a fairly domain-independent fashion (Wang et al., 2014). Local removal of Shank3 in the nucleus accumbens reduces the firing rate in medium spiny neurons (MSN) of this region and whole Shank3 KO displayed reduced sEPSC and LTD that is consistent with a reduced number of glutamatergic synapses on MSN in the striatum (Verpelli et al., 2011; Wang et al., 2016). However, the intrinsic striatal excitability was higher most likely due to increased glutamatergic drive from cortical afferents since hyperactivity is seen in the cortex of Shank3−/− mice during a critical postnatal developmental period (PND14; Peixoto et al., 2016). These studies suggest that in general lower LTP or LTD along with lower spine density or less mature spines occur in mice that deleted for positive regulators of mTORC1.
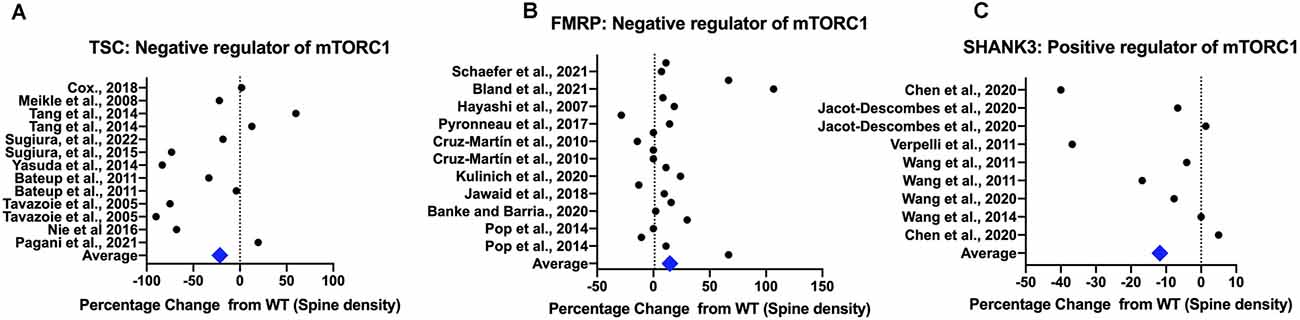
Figure 3. Rodent models show differences in spine density compared to controls. All spine density data from the TSC model are from Tsc1/2 heterozygotes and KO animals (Panel A), for the FMRP model from Fmr1 knockdown (KD) and knockout (KO) animals (Panel B) and for the SHANK3 model from Shank3 knockdown (KD) and knockout (KO) animals (Panel C). TSC1/2 and FMRP are upstream negative regulators of mTORC1 while SHANK3 is a positive regulator. Spine density values from rodent studies was extracted using WebPlot Digitiser in a semi-quantitative fashion with the magnitude of the difference from the control plotted. The average difference for all models combined across cortex and hippocampus is shown, with all studies weighted equally. References: Tavazoie et al. (2005); Hayashi et al. (2007); Meikle et al. (2008); Verpelli et al. (2011); Wang et al. (2011); Wang et al. (2014); Durand et al. (2012); Ginsberg et al. (2012); Henderson et al. (2012); Bateup et al. (2013); Dolan et al. (2013); Pop et al. (2014); Tang et al. (2014); Yasuda et al. (2014); Cochoy et al. (2015); Nie et al. (2015); Sugiura et al. (2015); Sugiura et al. (2022); Mei et al. (2016); Hodges et al. (2017); Pyronneau et al. (2017); Cox et al. (2018); Jawaid et al. (2018); Yan et al. (2018); Arroyo et al. (2019); Booker et al. (2019); Gross et al. (2019); Banke and Barria (2020); Jacot-Descombes et al. (2020); Kulinich et al. (2020); Bland et al. (2021); and Schaefer et al. (2021).
Effects of the Loss of “Global” Positive Regulators of mTORC1 on Spine Density and Synaptic Physiology
Two other positive regulators of mTORC1 are MeCP2 (Methyl CpG binding protein 2), a transcriptional repressor and UBE3A (Ubiquitin-protein ligase E3A), a transcriptional coactivator and E3 ubiquitin ligase that targets proteins for degradation (Vatsa and Jana, 2018; Figure 2). In both cases, individuals presenting with increased dosage of these genes show symptoms of autism and hence overexpression and not deletion models of mice of these genes recapitulate features of autism. For example, the 2xTg-MeCP2 mouse which is a model for both Rett syndrome and ASC shows increased spine densities with a greater number of mushroom shaped spines, increased dendritic branching at younger ages that are normalized at around 40 weeks, possibly due to increased levels of S6K (Jiang et al., 2013). A PKA-phosphorylation site in UBE3A that is mutated in an ASC proband, T485A, also shows higher spine densities in cortical neurons transfected with the mutant, suggesting that overexpression of these positive regulators of mTORC1 increases spine density (Yi et al., 2015).
However, in some mouse models, overexpression of these genes leads to lower spine densities. For example, MeCP2 overexpression in mouse cortical neuronal cultures interferes with the formation of the DiGeorge syndrome critical region 8 (DGCR8) complex with Drosha and lowers miRNA globally. This includes lowering the miRNA for CREB and LIM kinase (LIMK), whose levels rise; however, dendritic spine density decreases for reasons that are not clear (Cheng et al., 2014). Similarly, a study using inducible overexpression of UBE3A shows lower dendritic arbor, lower spine density with a lower proportion of mushroom shaped spines possibly due to an increased atypical pruning mechanism involving caspase (Khatri et al., 2018; Section “Upstream Regulators of mTORC1 Regulate Pruning by Several Pathways”).
Hence, though the loss of synaptically-localized positive regulators of mTORC1 typically leads to lower spine densities, smaller PSD and/or decreased sEPSCs/LTP, there is a lack of a clear pattern with more global positive regulators such as UBE3A and MeCP2 where overexpression rather than loss of the protein is present. This suggests that for global regulators, there are multiple targets which are critical rather than just overactivation of the mTORC1 pathway. This is exemplified by the MeCP2-mediated transcriptional repression of Reelin, where the decrease of Reelin in a MeCP2-overexpression ASC model may mimic the heterozygyous reeler mice and result in lower spine densities (Zhubi et al., 2014).
Negative Regulators of mTORC1 at the PSD/Dendrite and Their Effects on Spine Density and Physiology
Negative regulators include FMRP, SHANK2, PTEN, SYNGAP, GRIN2B, and TSC (Figure 2); many of the well-studied genes in this category when mutated or deleted in the mouse show increased spine density (Table 4) due to a combination of mTORC1-mediated lack of pruning and/or increase in spinogenesis pathways (Section “Upstream Regulators of mTORC1 Regulate Pruning by Several Pathways” and “Upstream Regulators of mTORC1 Regulate Spinogenesis by Several Pathways”).
The loss of PTEN, a negative regulator of the PI3K pathway, in hippocampal granule cells and cerebral cortex using a NSE-cre model leads to dendritic hypertrophy and macroencephaly in mice (Kwon et al., 2006). Consistent with this, a KO of Pten in granule cells at postnatal day (PND) 14 and PND 21 in a Brainbow mouse model results in longer apical dendrite lengths, greater distal arborization and increased soma size (Arafa et al., 2019). In support, E157G PTEN mutations found in ASC individuals when incorporated into iPSC-differentiated neurons also show an increase in dendritic arbor (Wong et al., 2020). This has functional implications—as expected, there is increased excitability in the postsynaptic dentate gyrus granule cell revealed by increased sEPSC, burst frequencies and reduced sIPSC in both male and females (Santos et al., 2017). This could also be due to excessive “wiring”—where competition by Pten KO cells is more efficient for the available pool of presynaptic partners resulting in increased glutamatergic excitatory afferents (Skelton et al., 2019). Similarly, truncation mutations of GRIN2B abrogated NMDA-dependent calcium influx and led to more filopodial spines in cortical neurons (Sceniak et al., 2019) even on a wildtype background, possibly due to a dominant negative effect. A third negative regulator is SynGAP which inhibits an activator of LTP, i.e., Ras-ERK signaling that in turns activates Rheb and mTOR, increasing translation of AMPA receptors (Groc et al., 2013). SynGAP+/− mice show increased glutamatergic transmission when the perforant path is stimulated during an early postnatal period of hippocampal synaptic development (PND10-PND20) possibly due to an increased proportion of mushroom spines in dentate gyrus granule cells despite similar spine densities as wildtype mice. This accelerated development of glutamatergic synapses can also be seen with persistent propagation of signal in the hippocampal tri-synaptic circuit using photolysis of caged glutamate paired with fast voltage-sensitive dye imaging, demonstrating an increased E/I ratio (Clement et al., 2012). Similar stronger excitatory synapses and stronger basal transmission were seen in neurons derived from human iPSC cells where SYNGAP expression was reduced (Llamosas et al., 2020). A fourth model of a negative mTORC1 regulator is the mouse model of Fragile X syndrome (FXS) that also shows behaviors that mirror those seen in autism. Loss of the FMRP protein, that represses translation of other dendritic spine proteins, increases spine density in the medial prefrontal cortex, basal lateral amygdala, and hippocampus compared with wild type (Qin et al., 2011) with a greater proportion of thin spines and a lower proportion of mushroom spines (Jawaid et al., 2018) as well as hyperexcitability in the somatosensory cortex and exaggerated LTD in the hippocampus (Bhattacharya et al., 2012). This could be due to the reduction in a FMRP target calsyntenin-1 in the medial prefrontal cortex which in turn represses the synaptic protein ICAM5; an increase in ICAM5 in the Fmr1 KO decreases spine maturation; overexpression of calysyntenin rescues spine maturation (Cheng et al., 2019).
Therefore overactivation of mTORC1 due to a loss of upstream negative regulators typically leads to an increase in spine density with a concomitant increase in excitability in local circuits. An outlier in this group is mice with Shank2 PDZ domain mutations which show either a decrease in spine density (Won et al., 2012) or no effect (Schmeisser et al., 2012). This could be due to compensation by the increased expression of the SHANK3 isoform (Schmeisser et al., 2012) driving the spine density effect. Another notable exception is the TSC model where studies report variable effects (Figure 3) though ASC models of TSC show increased spine density in the cortex but not in the hippocampus (Figures 4A,D). This underscores that different brain regions may show different variations in spine density.

Figure 4. Spine dynamics depends on the brain area. Semi-quantitative plots show that spine density depends on the area studied, i.e., hippocampus (A–C) or cortex (D–F) for some commonly studied ASC-related genes that are either positive (Shank3) or negative regulators (Fmr1, Tsc1/2) of the mTORC1 pathway. All spine density data from these models were extracted using WebPlot Digitiser in a semi-quantitative fashion with the magnitude of the difference from the control plotted as percentage of control. The average difference from control WT animals for each model for each different brain area is shown, with all studies weighted equally (References in Figure 3).
Alterations of mTORC1 Activity by Alterations in The Expression of Upstream Regulators Result in Autistic Behaviors
Despite the fact that spine density may increase or decrease as can be seen in Section “Many Upstream Regulators of mTORC1 are Expressed at the Dendrite or at the Post-Synaptic Density (PSD)”, there are consistent social behavior deficits reported in all rodent models of these upstream regulators of mTORC1 (Tables 3, 4). Tests range from assays of social interaction/motivation such as the 3-chamber test or ultrasonic vocalization, social recognition tests such as social novelty, anxiety measured by the open field or the elevated plus maze testing to repetitive behaviors such as self-grooming and perservatory behaviors such as longer latencies to extinguish memories. Some tasks such as the Morris Water Maze also have a large cognitive component (Miyakawa et al., 2001; Holmes et al., 2002, 2003; Tsai et al., 2012).
The Effects of Alterations of Positive Regulators of mTORC1 on Behaviors That Denote Autism
CDKL5 heterozygotes show reduced learning in the Barnes Maze and repetitive jumping behavior (Trazzi et al., 2018) while heterozygote reeler mice show decreased freezing to tone in renewal trials during fear conditioning (Iafrati et al., 2013). Similarly, the Dip2A KO shows lower social exploration time of a conspecific, increased self-grooming in both novel and home cage environments, decreased social novelty and increased marble burying indicative of higher anxiety as well as social recognition and interaction deficits (Ma et al., 2019).
Notably, rodent ASC models utilizing SHANK3 mutations have good face validity, reflecting both repetitive and altered social behavior phenotypes, including reliably increased self-grooming behavior, lower levels of social interaction and ultrasonic vocalization and increased anxiety in a domain-deletion independent manner (Wang et al., 2011; Jaramillo et al., 2016; Mei et al., 2016; Zhou et al., 2016). Most models also show decreased social recognition memory (Peça et al., 2011; Schmeisser et al., 2012; Kouser et al., 2013; Mei et al., 2016) with males scoring worse in this task than females (Wang et al., 2011). Furthermore, ankyrin-repeat and proline rich domain mutants show hypoactivity in the open field (Wang et al., 2011; Zhou et al., 2016). For the global regulators, though MeCP2 overexpression in mice recapitulates Rett syndrome, it is harder to see autistic-type behaviors. However, a recent study using MeCP2 overexpression in cynomolgus monkeys showed that these monkeys display repetitive circling in their cages and lower time with familiar or novel conspecifics, indicative of ASC (Liu et al., 2016). Similarly, UBE3A overexpression results in repetitive behaviors and social interaction deficits (Smith et al., 2011). In general, for positive regulators of mTORC1, tests of social interaction/recognition/motivation tend to report a deficit compared to tests of spatial memory where reversal of the memory is typically measured (Monteiro and Feng, 2017).
The Effects of Alterations of Negative Regulators of mTORC1 on Behaviors That Denote Autism
Pten KO mice show lower interaction with conspecific mice or novel mice and hyperactivity in the open field (Kwon et al., 2006). Unusually, in GRIN2B+/C456Y mice that mimic a human ASC mutation, NMDAR-dependent long-term depression (LTD) is decreased and though there is normal social interaction, there is an anxiolytic effect as measured by the elevated plus maze; the molecular mechanisms underlying this phenotype remain obscure (Shin et al., 2020). A similar anxiolytic effect is seen in a comprehensive panel of behavioral tests of the haploinsufficient SynGAP+/− mice which also show no deficit in social interaction but hyperactivity and cognitive deficits in working and reference memory (Ma et al., 2019) and in a dentate gyrus-specific context discrimination task (Clement et al., 2012). A similar deficit in spatial memory is seen in two Shank2 PDZ-deletion models, combined with increased self-grooming behavior and anxiety along with decreased social interaction or novelty (Schmeisser et al., 2012; Won et al., 2012). Impaired social interaction and novel object recognition (NOR) along with hyperactivity is also seen in Fmr1 KO mice (Qin et al., 2011; Bhattacharya et al., 2012). For negative regulators of mTORC1, it is notable that spatial, hippocampal-based tasks show deficit and hyperactivity is seen in several models.
Molecular Mechanisms That Link Dysfunctions in Neuromorphology to Behavior
Despite mTORC1 under and over-activation in rodent models of autism due to mutations or loss of upstream positive and negative regulators, it is notable that these models show autistic-type behaviors. Therefore, it is of interest to understand if common molecular pathways downstream of mTORC1 may underlie autistic behaviors. One of the most prevalent ideas is alteration in the excitation-inhibition (E/I) ratio (Rubenstein and Merzenich, 2003) because of functionally altered excitatory (Gibson et al., 2008) and inhibitory synapses (Wöhr et al., 2015; Vogt et al., 2018), or in some cases alteration of both. Though somewhat simplistic, a number of reviews suggest this as an useful framework to understand neurodevelopmental disorders (Howell and Smith, 2019; Sohal and Rubenstein, 2019). One caveat to keep in mind is that alteration of E/I ratio could be a mechanism for homeostatic synaptic plasticity that arises because of the primary deficit caused by mutation in a gene that is a risk for ASC (Nelson and Valakh, 2015 and references therein).
Though E/I ratio can be altered by several mechanisms, reduction of GABAergic afferents has been shown in several models, including those with over-activation and under-activation of mTORC1. For example, a meta-analyses of different mouse ASC models including MeCP2 (a positive regulator of mTORC1) and FMRP (a negative regulator of mTORC1) reduction reveals a reduction of parvalbumin-positive (PV) GABAergic interneurons such as chandelier and basket calls in the CA1 hippocampus or somatosensory cortex suggesting increased excitability (Gogolla et al., 2009). Supporting the critical role of inhibitory afferents, a knockout of PV inhibitory interneurons results in mice whose behavioral anomalies recapitulate that seen in ASC, i.e., atypical social interactions and repetitive behavior (Wöhr et al., 2015; Filice et al., 2020). In four models of ASC including Fmr1−/y and Tsc2+/–, both of which are negative regulators of mTORC1, there is greatly reduced inhibition and a small reduction in excitation measured in the feedforward circuit in L2/L3 pyramidal neurons of the somatosensory cortex, suggesting that E/I ratio is altered (Antoine et al., 2019) primarily due to decreases in inhibitory feedforward mechanisms onto the postsynaptic neuron. Also, hyperexcitability in the hippocampus of Tsc1 KO is due to decreases in inhibitory afferents onto Tsc1 KO neurons and not due to increases in intrinsic excitability, synaptic glutamatergic afferents, or regulation of Arc-dependent synaptic homeostasis (Zhao and Yoshii, 2019). However, the importance of an optimal level of inhibitory transmission is shown by a recent study whereby increasing hippocampal astrogenesis that in turn increases GABAergic transmission induces ASC-like behavior in mice such as lack of social preference in the 3-chambered test and an increase in repetitive grooming (Chen et al., 2021a). Contrary to the decreased inhibition seen in the above models, the granule cells in the dentate gyrus of the Pten KO show increased density of synapses, greater spike rates and are more sensitive to depolarizing input, demonstrating hyperexcitability (Williams et al., 2015). In addition, loss of PTEN causes increases in basal dendrites that are contacted by other mossy fiber axons leading to recurrent circuits (Pun et al., 2012). Though many models of ASC that are mutated in genes that are negative regulators of mTORC1 appear to show altered E/I ratio due to decreased inhibition, the Pten example indicates that this is not an universal mechanism.
Upstream Regulators of mTORC1 Regulate Molecules at the PSD to Effect Changes in Synaptic Physiology
One of the reasons for E/I imbalance could be altered spine density/morphology or synaptic proteins, such as glutamate receptors at the PSD that are often altered in rodent models of autism that have alterations of the upstream regulator proteins of mTORC1.
For positive upstream regulators of mTORC1: Hyperexcitability and a higher E/I ratio in the CA1 is due to higher localization of GluN2B and SAP102 in CdKl5−/y mice, as revealed by subcellular fractionation of the hippocampal PSD (Okuda et al., 2017). In Shank3 KO mouse striatum, the disrupted interaction between Homer1b/c-mGluR5 also results in increased mGluR5 at the PSD. Antagonists of the mGluR5 receptor such as MPEP could reverse the increased self-grooming in these mice, suggesting that receptor density at the PSD is critical for behavior (Wang et al., 2016). In mice carrying SHANK3 deletion of different domains, several receptors such as NR1, NR2A, GluR2, and GluA2 are decreased (Monteiro and Feng, 2017 and references therein). Conversely, the decreased mEPSC amplitude in the Dip2a KO is due to a smaller number of NMDA subunits at the PSD (Ma et al., 2019). In MeCP2 overexpressing mice, despite an increase in both mGluR and NMDA receptors at the synapse in the somatosensory cortex (Chahrour et al., 2008; Jiang et al., 2013), there is a reduction in the EPSCs because NMDA/AMPA currents correlate to reduced GluN1 expression; this is not due to reduction in GABAergic interneurons (Sceniak et al., 2016) in the prefrontal cortex. Rather, this suggests, as borne out by brain activity c-Fos mapping, that reduced functional excitation rather than reduced inhibition drives the decrease in E/I ratio in these animals since this is reversed by ketamine, showing the importance of post-synaptic glutamate receptors (Kron et al., 2012). This could demonstrate that alteration in E/I ratio is driven by different mechanisms in different brain regions since reduction in the MeCP2 overexpressing somatosensory cortex is due to a reduction in GABAergic parvalbumin neurons in that region (Gogolla et al., 2009) but this is not true in the prefrontal cortex (Sceniak et al., 2016).
For negative upstream regulators of mTORC1: In Shank2 PDZ-deletion mice, the predominant pathway driving the atypical (for a negative regulator) decrease in spine density and social interaction deficit appears to be NMDA hypofunction since cycloserine, a NMDA agonist, can restore functionality (Won et al., 2012). However, AMPA receptors are also important; for example, SynGAP dispersal from the synapse, results in loss of Ras-ERK inhibition and leads to subsequent mTOR-mediated increased AMPA receptor insertion at the PSD, a prerequisite for LTP. Therefore, in SynGAP-deficient mice, though the proportion of mature spines and basal synaptic transmission are enhanced, no further enhancement is possible since increased AMPA receptor insertion in the basal state occludes LTP (Gamache et al., 2020). A third negative regulator is Tsc1/2; a pan-neuronal KO of Tsc1 shows reduced frequency of GABAergic mIPSC, resulting in increased excitability in the cortex and an increased E/I ratio; however, a PV-specific Tsc1 KO does not show altered E/I balance, suggesting that the local hyperexcitability is not due to loss of TSC signaling in PV interneurons (Zhao and Yoshii, 2019). Similarly in the hippocampus, though homeostatic plasticity via decreased synaptic AMPA receptors is seen, in experiments where homeostatic plasticity is not initiated, the increased E/I ratio is primarily due to impaired mGluR5-LTD at hippocampal synapses that may be because of reduced Arc and other proteins in the Tsc1 KO that are required to stabilize LTD (Bateup et al., 2013). In support of this idea, the Tsc1 mutant phenotype of reduced LTD and context discrimination can be rescued by an Fmr1−/y mutant that shows the opposite, i.e., exaggerated mGluR5-LTD, suggesting that levels of PSD proteins maintained by these mTORC1 regulators underlie the deficit despite the fact that loss of Tsc1 and FRMP proteins both lead to overactivation of mTORC1 (Auerbach et al., 2011). In the amygdala of the Fmr1 KO mouse, principal cells show intrinsic hyperexcitability and increased synaptic plasticity (Svalina et al., 2021), suggesting that the enhancement of PSD-95 and Arc levels seen in the PSD density of spines of hippocampal neurons (Yan et al., 2018) may also be true of other limbic regions to result in greater excitability.
Upstream Regulators of mTORC1 Regulate Spine Morphology and Density via Pruning and/or Spinogenesis
Though the relationship between spine density and synaptic physiology appears to be complex, looking at spine morphology paints a clearer picture (Tables 3 and 4), with an increase in immature spines in many models. A number of knockout rodent models of negative regulators such as FMRP, GRIN2B, and PTEN show an increase in the proportion of thin or filopodial spines vs. mushroom spines. Similarly, in the Dip2a KO, there is an increase in spine density but a decrease in mushroom shaped spines and a flattening of the PSD with a decrease of NMDA receptors in a smaller spine volume, possibly leading to a lower E/I ratio (Ma et al., 2019).
Some of this could also be due to spine instability. This is seen in Fmr1 KO mice where there is a developmental delay in the transition from immature to mature spines in Layer2/3 pyramidal neurons of the cortex; thin spines are unusually unstable. Normally, these spines search for presynaptic partners in a lengthening process facilitated by mGluR5; however, mGluR5 agonists are unable to lengthen spines and achieve this in Fmr1 KO mice (Ginsberg et al., 2012) suggesting that stabilization via contact with pre-synaptic partners is deficient when FMRP is absent. Interestingly, spines are stabilized in an ERK-dependent manner solely in clusters whose numbers are increased in mice overexpressing MeCP2 resulting in a higher proportion of mushroom shaped spines; isolated spines are formed and stabilized at the same rate as the wildtype mouse. Such stabilization of spines at a young age is correlated with increased stability on the rotarod, a marker of perseverative behavior in these mice (Ash et al., 2021a, b) though it is unclear if this is cause of effect. For example, repetitive task learning that is cortex-dependent induced spines preferentially in clusters; these clustered spines showed greater persistence past learning than non-clustered spines (Fu et al., 2012) and hence the functional interpretation of spine clustering in ASC models is difficult to ascertain.
Spine morphology also has functional consequences; shRNA targeting Pten results in increased spine density in the basolateral amygdala (BLA) of the mouse with an increased mEPSC amplitude and frequency in the BLA. This is most likely due to a shift in spine morphology with an increased proportion of mushroom spines but decreased filopodia (Haws et al., 2014). Consistent with this finding, though granule cells of neonatal Pten KO mice show hypertrophy and therefore might be expected to be less excitable, increased density of mushroom shaped spines concomitant with increased dendrites and protrusions drives the hyperexcitability observed in this ASC model (Williams et al., 2015). This increase in spines is larger when Pten KO granule cells are sparsely surrounded by wildtype cells when compared to when they are densely surrounded by other Pten KO cells (Skelton et al., 2019) though the mechanism underlying this and its significance is unknown. It could be that secondary effects on network activity amongst surrounding cells and subsequent tertiary feedback effects by these neighboring cells on the Pten KO neuron drive the ASC phenotype (LaSarge and Danzer, 2014).
Upstream Regulators of mTORC1 Regulate Pruning by Several Pathways
Though Tang et al. elegantly showed that pruning deficits are critical for the increase in spine density in the Tsc2+/− mice (Tang et al., 2014), other studies have also shown pruning deficits. In some cases, such as MeCP2 overexpression, 2-photon imaging of L5 apical dendrites emanating from cortical pyramidal neurons reveals a slowing down of pruning and increased spinogenesis with unstable spines as development proceeds, most likely due to mTORC1 overactivation (Jiang et al., 2013). Microglia-activated pruning is lower in the CA1 hippocampal neuron in a mouse FXS model (Jawaid et al., 2018). Delivery of shRNA to Raptor into the CA1 of FXS mice decreases the PSD-95 and ARC proteins that are elevated in these mice and decreases the spine density and reduces cognitive deficits. Importantly, shRNA to Raptor in Fmr1 KO mice also decreases filopodial spines and increases the proportion of mushroom shaped spines (Yan et al., 2018). In Fmr1 KO neurons, higher levels of eIF1α sequester the E3 ubiquitin ligase, murine double minute2 (mdm2) which targets PSD-95 for degradation. Hence, in response to neuronal activity in these Fmr1 KO neurons, myocyte enhancer factor (MEF)-induced degradation of PSD-95 degradation and synapse elimination/pruning does not occur leading to higher spine densities (Pfeiffer et al., 2010). Therefore, in the Fmr1 KO mouse, overactivation of mTORC1 directly results in a decrease in pruning and higher spine densities.
The converse—an increase in pruning leading to a decrease in spines also exists. For example, in an inducible UBE3A overexpression model of autism, an increase in pruning over spine growth results in a decrease in spines. UBE3A overexpression results in increased targeting and degradation of a caspase inhibitor, XIAP, which leads to increased caspase levels with concomitant increases in tubulin cleavage and spine retraction (Khatri et al., 2018). Conversely, expression of a dominant negative caspase could decrease tubulin cleavage and allow for more mature spines and increased spine density (Khatri et al., 2018).
Upstream Regulators of mTORC1 Regulate Spinogenesis by Several Pathways
Apart from pruning, a second pathway to alter spine density is spinogenesis, driven by several Rho/ROCK pathways that activate LIMK; these include the Rac/p21-activated kinase (PAK) pathways that can activate cortactin and/or LIM kinase (LIMK; Costa et al., 2020). For example, in the Dip2A KO which shows increased spine density but a flattened PSD, there is a reduction of acetylation of cortactin, a protein activated by PAK that increases spine stability (Schnoor et al., 2018); DIP2A mutations implicated in ASC are in regions of the protein that contact cortactin. Administration of an acetylated cortactin mimic into the cerebral cortex rescues not only the flattened PSD and decreases spine density but also decreases the self-grooming and marble burying behavioral phenotype (Ma et al., 2019). Similarly, a knockdown of all Shank isoforms using miRNA in rat hippocampal neurons resulted in a decrease in spine density (specifically of mushroom shaped spines) and cortactin levels, that could not be rescued by a SHANK2 isoform deficient in cortactin binding, suggesting that SHANK isoforms recruit and stabilize cortactin in the PSD (MacGillavry et al., 2016) to grow mushroom-shaped spines. Indeed, Shank3 KO show lower levels of Rac1 and PAK proteins and an increase in actin depolymerization and behavioral ASC-like phenotypes in this mouse could be rescued by increasing Rac1, suggesting that actin dynamics and spine density were critical in driving behavior (Duffney et al., 2015). Interestingly, S6K activation activates Rac/PAK in ovarian cancer cells (Ip et al., 2011) while loss of mTORC1 leads to constitutive activation of ROCK in the epidermis (Asrani et al., 2017), suggesting that these pathways may also be present in the CNS, to regulate spinogenesis.
Amongst negative regulators of mTORC1 such as FMRP, a decrease in phosphorylated active SLINGSHOT protein, an inhibitor of LIMK combined with an increased phospho-LIMK due to increased Rac/PAK levels, leads to a decreased level of active cofilin and subsequent actin depolymerization (Pyronneau et al., 2017). This could be the mechanism that underlies the increase in filamentous spines in this model. Sequestration of proteins in the spinogenesis pathway is also a mechanism for regulation by mTORC1 regulators. For example, the cytoplasmic FMRP interacting protein (CYFIP1) sequesters and inhibits eIF4E to repress translation along with FMRP but this protein by itself is also part of the WAVE-Rac complex that increases spine protrusion. In the absence of FMRP in hippocampal neurons, the balance shifts towards spine protrusion and away from translation repression to increase spine growth, a phenomenon that can be reversed by the administration of 4EGI-1, which creates more free eIF4E that can bind CYFIP1 away from the WAVE complex (Santini et al., 2017). A caveat to these studies is that direct inhibition of spinogenesis pathways may be more important in some cases than regulation via mTORC1. NF-1, a negative regulator of mTORC1, leads to an overall decreased spine density when deleted in neurons, possibly due to inhibition of the ERK pathway which is required for spine stabilization (Ash et al., 2021a), suggesting that ERK-driven spinogenesis may supercede mTORC1-driven pruning. Indeed, as can be seen many of the upstream regulators of mTORC1 employ several mTORC1-independent means to regulate spine density.
Upstream Regulators of mTORC1 May Preferentially Regulate Corticostriatal Circuitry
Corticostriatal circuitry which underlies motivated sensorimotor by interacting with the limbic system is the focus of several neurodevelopmental disorders, including autism (Shepherd, 2013), with several alterations to this circuit. In ASC individuals, intrinsic functional connectivity studies done using fMRI imaging shows that those with highly repetitive behaviors show reduced frontoparietal and motor connectivity with the striatum but increased connectivity with the limbic system compared to those with low repetitive behaviors, underscoring the importance of the corticostriatal circuit (Abbott et al., 2018). These differences in neuroanatomy particularly for the striatum are underscored by a longitudinal structural imaging study which revealed a faster growth rate of the caudate nucleus during late childhood to early puberty in ASC individuals that was strongly correlated with repetitive behaviors seen in these individuals that developed during the preschool period (Langen et al., 2014). These alterations in mice are linked to spines; conditional dorsal pallium-specific KOs of Met, a tyrosine kinase that regulates spinogenesis, demonstrates local hyperconnectivity in this circuit with stronger afferents from corticostriatal neurons in layer 2/3 to layer 5 projection neurons in the anterior frontal cortex in heterozygotes and KOs compared to WT controls (Qiu et al., 2011).
Developmental studies of Shank3, a positive regulator of mTORC1, show that the striatal excitability is higher due to increased cortical glutamatergic efferents to the striatum (Peixoto et al., 2016) at early postnatal stages but there is reduced corticostriatal connectivity and EPSCs as adults (Mei et al., 2016; Wang et al., 2016). In addition, disruption of the dopaminergic receptor D1R-SYNGAP complex in the prefrontal cortex during development results in decreased migration of GABAergic interneurons with concomitant increased cortical excitability. This cortical excitability that develops as a result of disruption of the function of SYNGAP and D1R is instrumental in the social deficits seen in this model during adulthood (Lai et al., 2021). In Tsc2+/− mice, resting state fMRI reveals functional hyperconnectivity within the corticostriatal circuit due to increased synaptic coupling possibly as a result of increased spine density; this was strongly correlated to repetitive behaviors. All parameters, i.e., connectivity, spine density, synaptic coupling, resting state default mode network signatures, and social behaviors are rescued to normal levels with rapamycin, suggesting that mTORC1 overactivation in this circuit is crucial to spine dynamics that underlie social behaviors. This hyperconnectivity signature is valid cross-species—children with ASC showed similar fronto-insular-striatal hyperconnectivity and mTORC1 interacting genes are enriched in the ASC cortical transcriptome, suggesting that this Tsc-mTORC1 pathway is a critical driver of functional hyperconnectivity (Pagani et al., 2021). Cross-species connectivity is also shown with mutations of NF1, a negative regulator of mTOR similar to Tsc1/2, though Nf1 KO mice show decreased spine density. Children with NF1 mutations with ASC show reduced functional connectivity between the striatum and the frontoparietal network and increased striatal functional connectivity with the limbic system. Similar to the human scenario, Nf1+/− mice also show disrupted corticostriatal connectivity, as revealed by resting state imaging (Shofty et al., 2019). This shows that both positive, e.g., SHANK and negative regulators. e.g., TSC of mTORC1 typically show spine density alterations in the corticostriatal circuit that are commensurate with local connectivity signatures and may underlie the repetitive behaviors seen in both humans and rodent models.
Conclusion/Future Directions
Despite a number of studies that show both spine dynamics and social behavior alterations in ASC rodent models, a number of questions persist. Many models are homozygous deletion models in order to obtain more robust effects whereas they tend to be haploinsufficient conditions in the human (Monteiro and Feng, 2017) and data from ASC rodent models that identify molecular processes that are dysregulated may not translate to the human. For example, though reduction in GABAergic transmission has been identified in FXS (Section “Upstream Regulators of mTORC1 Regulate Molecules at the PSD to Effect Changes in Synaptic Physiology”), use of a selective GABA-B receptor agonist arbaclofen showed limited improvements in social behaviors only in young but not older children and adults in a Phase 3 clinical trial (Berry-Kravis et al., 2017). Similarly, mTOR inhibition in TSC patients has shown improvements for epileptic symptoms (Mizuguchi et al., 2019) but not for ASD-associated behaviors (Overwater et al., 2019), suggesting that treatment must be given in the critical period or that mTOR signaling is less important in ASC symptoms in humans.
Furthermore, though several of the upstream regulators such as SynGAP and SHANK have isoforms, the differential contribution of isoforms is not clear and deserves more attention. For example, SynGAPα1 is the only isoform capable of being anchored at the PSD due to the presence of its unique C-terminal PDZ binding domain and is sufficient for the enlargement of spine heads by AMPA receptor insertion upon chemically induced LTP as well as increase in PSD-95-positive puncta. SynGAPβ isoform rescues the reduction in dendritic arborization seen in cultured hippocampal neurons when SynGAP levels are reduced (Araki et al., 2020). Compensation by isoforms also confounds interpretation of the results, particularly in the case of conserved SHANK proteins.
In addition to isoform-specific functions of mTORC1 upstream regulators, the role of these genes has typically been studied in two major brain regions, i.e., the hippocampus and cortex and region-specific differences exist (Figure 4). For example, in Shank3−/− mice, mGluR5 is increased in the striatum but not in the cortex (Wang et al., 2016), possibly pointing to the importance of the corticostriatal synapse in modulation of social behaviors that have reward potential. Shank3 mice deleted for the ankyrin repeat domain show reduced excitation but increased frequency of sIPSCs at Schaffer collateral synapses in pyramidal neurons in the hippocampus. However, prelimbic layer 2/3 pyramidal neurons in the medial prefrontal cortex show decreased frequency of sIPSCs, though the basis of this difference is not known (Lee et al., 2015). In Shank2 PDZ-deletion mutant mice, NMDA hypofunction also leads to lower LTD and lower LTP at Schaffer collateral synapses but not in the prefrontal cortex (Won et al., 2012), suggesting that microcircuitry in the hippocampus and cortex may lead to different E/I ratios. There are also cell-specific differences in regulation. For example, FMRP transcripts are greatly reduced in cerebellar Purkinje cells in the Tsc1 mutant (Dalal et al., 2021) and theoretically could lead to elevated levels of FMRP target genes such as mTOR (Casingal et al., 2020), leading to an overactivation of mTORC1. However, although FMRP functions as a general translational repressor, it may also regulate ribosome binding to specific transcripts; indeed a recent study in cortical neurons saw destabilization of specific but not all transcripts (Shu et al., 2020) despite FMRP loss. However, in hippocampal slices derived from Tsc2+/− mice, there was increased expression of FMRP targets (Hien et al., 2020), most likely due to differences in Tsc-Fmr1 regulation in the cerebellum vs. the hippocampus. Apart from the hippocampus, cortex and cerebellum, many social behaviors are regulated by nuclei in the social behavior network which includes hypothalamic nuclei (Newman, 1999; Greenberg and Trainor, 2016) in a sexually dimorphic manner and spine dynamics in these regions in these ASC models is under-explored. Given the difference in incidence of ASC symptoms between the sexes with males showing greater incidence than females (Zhang et al., 2020), this would be timely to investigate.
Though the picture for changes in spine dynamics upon deletion or duplication of these genes is complex, it can be simplified when seen through the lens of regulation of mTORC1 as part of a larger grouping (Tables 3 and 4); this is especially true of positive regulators of mTORC1 (Table 3; Figures 4C,F). For some genes such as FMRP, it is clear that both mTORC1-dependent and mTORC1-independent means of regulating spine density and synaptic physiology exist with the contribution of each not clearly understood though the weight of studies suggest an overall increase in spine density (Figure 3). In addition, spine density and in particular spine morphology studies tend to be time consuming and analyses is frequently done manually. In the coming years, it would be advantageous to investigate spine dynamics using 2-photon imaging as well as artificial intelligence algorithms (Mancuso et al., 2013) for additional information on spine motility and stability, including spine clustering, in several of these rodent ASC models to probe the relationship between spine morphology, stability, neurocircuitry, and behavior.
Author Contributions
NV framed and analyzed most of the scientific content in the manuscript. SC did the analyses of individual mTORC1 regulators including the summary of the spine density and morphology in tabular form. Both SC and NV discussed and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Open access fees are paid by the University of Reading.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Bhismadev Chakrabarti for his helpful comments on the manuscript.
References
Abbott, A. E., Linke, A. C., Nair, A., Jahedi, A., Alba, L. A., Keown, C. L., et al. (2018). Repetitive behaviors in autism are linked to imbalance of corticostriatal connectivity: a functional connectivity MRI study. Soc. Cogn. Affect. Neurosci. 13, 32–42. doi: 10.1093/scan/nsx129
Alves, P. N., Foulon, C., Karolis, V., Bzdok, D., Margulies, D. S., Volle, E., et al. (2019). An improved neuroanatomical model of the default-mode network reconciles previous neuroimaging and neuropathological findings. Commun. Biol. 2, 1–14. doi: 10.1038/s42003-019-0611-3
Anderson, J. S., Druzgal, T. J., Froehlich, A., DuBray, M. B., Lange, N., Alexander, A. L., et al. (2011). Decreased interhemispheric functional connectivity in autism. Cereb. Cortex 21, 1134–1146. doi: 10.1093/cercor/bhq190
Anderson, G. R., Galfin, T., Xu, W., Aoto, J., Malenka, R. C., and Südhof, T. C. (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. U S A 109, 18120–18125. doi: 10.1073/pnas.1216398109
Andrews-Hanna, J. R., Kaiser, R. H., Turner, A. E. J., Reineberg, A. E., Godinez, D., Dimidjian, S., et al. (2013). A penny for your thoughts: dimensions of self-generated thought content and relationships with individual differences in emotional wellbeing. Front. Psychol. 4:900. doi: 10.3389/fpsyg.2013.00900
Antoine, M. W., Langberg, T., Schnepel, P., and Feldman, D. E. (2019). Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101, 648–661.e4. doi: 10.1016/j.neuron.2018.12.026
Arafa, S. R., LaSarge, C. L., Pun, R. Y. K., Khademi, S., and Danzer, S. C. (2019). Self-reinforcing effects of mTOR hyperactive neurons on dendritic growth. Exp. Neurol. 311, 125–134. doi: 10.1016/j.expneurol.2018.09.019
Araki, Y., Hong, I., Gamache, T. R., Ju, S., Collado-Torres, L., Shin, J. H., et al. (2020). SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. eLife 9, 1–28. doi: 10.7554/eLife.56273
Arroyo, E. D., Fiole, D., Mantri, S. S., Huang, C., and Portera-Cailliau, C. (2019). Dendritic spines in early postnatal fragile X mice are insensitive to novel sensory experience. J. Neurosci. 39, 412–419. doi: 10.1523/JNEUROSCI.1734-18.2018
Ash, R. T., Buffington, S. A., Park, J., Suter, B., Costa-Mattioli, M., Zoghbi, H. Y., et al. (2021a). Inhibition of elevated ras-mapk signaling normalizes enhanced motor learning and excessive clustered dendritic spine stabilization in the MECP2-duplication syndrome mouse model of autism. eNeuro 8:ENEURO.0056-21.2021. doi: 10.1523/ENEURO.0056-21.2021
Ash, R. T., Park, J., Suter, B., Zoghbi, H. Y., and Smirnakis, S. M. (2021b). Excessive formation and stabilization of dendritic spine clusters in the MECP2-duplication syndrome mouse model of autism. eNeuro 8:ENEURO.0282-20.2020. doi: 10.1523/ENEURO.0282-20.2020
Asrani, K., Sood, A., Torres, A., Georgess, D., Phatak, P., Kaur, H., et al. (2017). mTORC1 loss impairs epidermal adhesion via TGF-β/Rho kinase activation. J. Clin. Invest. 127, 4001–4017. doi: 10.1172/JCI92893
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Banerjee-Basu, S., and Packer, A. (2010). SFARI Gene: an evolving database for the autism research community. Dis. Model Mech. 3, 133–135. doi: 10.1242/dmm.005439
Banke, T. G., and Barria, A. (2020). Transient enhanced GluA2 expression in young hippocampal neurons of a fragile X mouse model. Front. Synaptic Neurosci. 12:588295. doi: 10.3389/fnsyn.2020.588295
Barnes, S. A., Wijetunge, L. S., Jackson, A. D., Katsanevaki, D., Osterweil, E. K., Komiyama, N. H., et al. (2015). Convergence of hippocampal pathophysiology in Syngap+/− and Fmr1−/y mice. J. Neurosci. 35, 15073–15081. doi: 10.1523/JNEUROSCI.1087-15.2015
Baron-Cohen, S. (2009). Autism: research into causes and intervention. Pediatr. Rehabil. 7, 73–78. doi: 10.1080/13638490310001654790
Bateup, H. S., Johnson, C. A., Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Belmonte, M. K. (2004). Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231. doi: 10.1523/JNEUROSCI.3340-04.2004
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491. doi: 10.1038/ng.589
Berry, K. P., and Nedivi, E. (2017). Spine dynamics: are they all the same. Neuron 96, 43–55. doi: 10.1016/j.neuron.2017.08.008
Berry-Kravis, E., Hagerman, R., Visootsak, J., Budimirovic, D., Kaufmann, W. E., Cherubini, M., et al. (2017). Arbaclofen in fragile X syndrome: results of phase 3 trials. J. Neurodev. Disord. 9:3. doi: 10.1186/s11689-016-9181-6
Bertossi, E., and Ciaramelli, E. (2016). Ventromedial prefrontal damage reduces mind-wandering and biases its temporal focus. Soc. Cogn. Affect. Neurosci. 11, 1783–1791. doi: 10.1093/scan/nsw099
Bhatt, D. H., Zhang, S., and Gan, W.-B. (2009). Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282. doi: 10.1146/annurev.physiol.010908.163140
Bhattacharya, A., Kaphzan, H., Alvarez-Dieppa, A. C., Murphy, J. P., Pierre, P., and Klann, E. (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. doi: 10.1016/j.neuron.2012.07.022
Bland, K. M., Aharon, A., Widener, E. L., Song, M. I., Casey, Z. O., Zuo, Y., et al. (2021). FMRP regulates the subcellular distribution of cortical dendritic spine density in a non-cell-autonomous manner. Neurobiol. Dis. 150, 1–25. doi: 10.1016/j.nbd.2021.105253
Booker, S. A., Domanski, A. P. F., Dando, O. R., Jackson, A. D., Isaac, J. T. R., Hardingham, G. E., et al. (2019). Altered dendritic spine function and integration in a mouse model of fragile X syndrome. Nat. Commun. 10, 1–14. doi: 10.1038/s41467-019-11891-6
Bourne, J., and Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember. Curr. Opin. Neurobiol. 17, 381–386. doi: 10.1016/j.conb.2007.04.009
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67. doi: 10.1146/annurev.neuro.31.060407.125646
Caglayan, A. O. (2010). Genetic causes of syndromic and non-syndromic autism. Dev. Med. Child. Neurol. 52, 130–138. doi: 10.1111/j.1469-8749.2009.03523.x
Casingal, C. R., Kikkawa, T., Inada, H., Sasaki, Y., and Osumi, N. (2020). Identification of FMRP target mRNAs in the developmental brain: FMRP might coordinate Ras/MAPK, Wnt/β-catenin and mTOR signaling during corticogenesis. Mol. Brain 13:167. doi: 10.1186/s13041-020-00706-1
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chen, L., Chen, Y., Zheng, H., Zhang, B., Wang, F., Fang, J., et al. (2021b). Changes in the topological organization of the default mode network in autism spectrum disorder. Brain Imaging. Behav. 15, 1058–1067. doi: 10.1007/s11682-020-00312-8
Cheng, K., Chen, Y. S., Yue, C. X., Zhang, S. M., Pei, Y. P., Cheng, G. R., et al. (2019). Calsyntenin-1 negatively regulates ICAM5 accumulation in postsynaptic membrane and influences dendritic spine maturation in a mouse model of fragile X syndrome. Front. Neurosci. 13:1098. doi: 10.3389/fnins.2019.01098
Chen, C. P., Keown, C. L., Jahedi, A., Nair, A., Pflieger, M. E., Bailey, B. A., et al. (2015). Diagnostic classification of intrinsic functional connectivity highlights somatosensory, default mode and visual regions in autism. Neuroimage Clin. 8, 238–245. doi: 10.1016/j.nicl.2015.04.002
Chen, J., Ma, X. L., Zhao, H., Wang, X. Y., Xu, M. X., Wang, H., et al. (2021a). Increasing astrogenesis in the developing hippocampus induces autistic-like behavior in mice via enhancing inhibitory synaptic transmission. Glia 70, 106–122. doi: 10.1002/glia.24091
Chen, D., Ren, K., Liu, H., Mao, H., Li, Z., Mo, H., et al. (2020). A whole-brain cell-type-specific sparse neuron labeling method and its application in a Shank3 autistic mouse model. Front. Cell. Neurosci. 14:145. doi: 10.3389/fncel.2020.00145
Chen, Q., Zhu, Y. C., Yu, J., Miao, S., Zheng, J., Xu, L., et al. (2010). CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 30, 12777–12786. doi: 10.1523/JNEUROSCI.1102-10.2010
Cheng, T.-L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W.-H., Xu, W., et al. (2014). MeCP2 suppresses nuclear MicroRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. doi: 10.1016/j.cell.2012.08.045
Cochoy, D. M., Kolevzon, A., Kajiwara, Y., Schoen, M., Pascual-Lucas, M., Lurie, S., et al. (2015). Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Mol. Autism 6:23. doi: 10.1186/s13229-015-0020-5
Cooke, S. F., and Bliss, T. V. (2006). Plasticity in the human central nervous system. Brain 129, 1659–1673. doi: 10.1093/brain/awl082
Copf, T. (2016). Impairments in dendrite morphogenesis as etiology for neurodevelopmental disorders and implications for therapeutic treatments. Neurosci. Biobehav. Rev. 68, 946–978. doi: 10.1016/j.neubiorev.2016.04.008
Costa, J. F., Dines, M., and Lamprecht, R. (2020). The role of Rac GTPase in dendritic spine morphogenesis and memory. Front. Synaptic Neurosci. 12:12. doi: 10.3389/fnsyn.2020.00012
Courchesne, E., and Pierce, K. (2005). Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 15, 225–230. doi: 10.1016/j.conb.2005.03.001
Cox, R. L., Calderon de Anda, F., Mangoubi, T., and Yoshii, A. (2018). Multiple critical periods for rapamycin treatment to correct structural defects in Tsc-1-suppressed brain. Front. Mol. Neurosci. 11:409. doi: 10.3389/fnmol.2018.00409
Crawley, J. N. (1999). Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities and specific behavioral tests. Brain Res. 835, 18–26. doi: 10.1016/s0006-8993(98)01258-x
Crawley, J. N. (2007). Mouse behavioral assays relevant to the symptoms of autism*. Brain Pathol. 17, 448–459. doi: 10.1111/j.1750-3639.2007.00096.x
Dalal, J. S., Winden, K. D., Salussolia, C. L., Sundberg, M., Singh, A., Pham, T. T., et al. (2021). Loss of Tsc1 in cerebellar Purkinje cells induces transcriptional and translation changes in FMRP target transcripts. eLife 10:e67399. doi: 10.7554/eLife.67399
Darbandi, S. F., Schwartz, S. E. R., Pai, E.L.-L., Everitt, A., Turner, M. L., Cheyette, B. N. R., et al. (2020). Enhancing WNT signaling restores cortical neuronal spine maturation and synaptogenesis in Tbr1 mutants. Cell Rep. 31:107495. doi: 10.1016/j.celrep.2020.03.059
Darnell, J. C., and Klann, E. (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1530–1536. doi: 10.1038/nn.3379
Della Sala, G., and Pizzorusso, T. (2014). Synaptic plasticity and signaling in Rett syndrome. Dev. Neurobiol. 74, 178–196. doi: 10.1002/dneu.22114
Delling, J. P., and Boeckers, T. M. (2021). Comparison of SHANK3 deficiency in animal models: phenotypes, treatment strategies and translational implications. J. Neurodev. Disord. 13, 1–37. doi: 10.1186/s11689-021-09397-8
Demir, K., van Gucht, A. L. M., Büyükinan, M., Çatli, G., Ayhan, Y., Nijat Baş, V., et al. (2016). Diverse genotypes and phenotypes of three novel thyroid hormone receptor-α mutations. J. Clin. Endocrinol. Metab. 101, 2945–2954. doi: 10.1210/jc.2016-1404
Dictenberg, J. B., Swanger, S. A., Antar, L. N., Singer, R. H., and Bassell, G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939. doi: 10.1016/j.devcel.2008.04.003
Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., and Beaudet, A. L. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118. doi: 10.1093/hmg/ddm288
Dolan, B. M., Duron, S. G., Campbell, D. A., Vollrath, B., Rao, B. S. S., Ko, H.-Y., et al. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. U S A 110, 5671–5676. doi: 10.1073/pnas.1219383110
Duan, W., Wang, K., Duan, Y., Chu, X., Ma, R., Hu, P., et al. (2019). Integrated transcriptome analyses revealed key target genes in mouse models of autism. Autism Res. 13, 352–368. doi: 10.1002/aur.2240
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in Shank3-Deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Dumitriu, D., Hao, J., Hara, Y., Kaufmann, J., Janssen, W. G. M., Lou, W., et al. (2010). Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J. Neurosci. 30, 7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010
Dunaevsky, A., Risher, W. C., Ustunkaya, T., Singh Alvarado, J., and Eroglu, C. (2014). Rapid golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9:e107591. doi: 10.1371/journal.pone.0107591
Durand, C. M., Perroy, J., Loll, F., Perrais, D., Fagni, L., Bourgeron, T., et al. (2012). SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 71–84. doi: 10.1038/mp.2011.57
Ehninger, D., Han, S., Shilyansky, C., Zhou, Y., Li, W., Kwiatkowski, D. J., et al. (2008). Reversal of learning deficits in a Tsc2+/– mouse model of tuberous sclerosis. Nat. Med. 14, 843–848. doi: 10.1038/nm1788
Ersahin, T., Tuncbag, N., and Cetin-Atalay, R. (2015). The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 11, 1946–1954. doi: 10.1039/c5mb00101c
Evans, J. W., Szczepanik, J., Brutsché, N., Park, L. T., Nugent, A. C., and Zarate, C. A. (2018). Default mode connectivity in major depressive disorder measured up to 10 days after ketamine administration. Biol. Psychiatry 84, 582–590. doi: 10.1016/j.biopsych.2018.01.027
Eyuboglu, M., Baykara, B., and Eyuboglu, D. (2018). Broad autism phenotype: theory of mind and empathy skills in unaffected siblings of children with autism spectrum disorder. Psychiatry Clin. Psychopharmacol. 28, 36–42. doi: 10.1080/24750573.2017.1379714
Fernandez, B. A., and Scherer, S. W. (2017). Syndromic autism spectrum disorders: moving from a clinically defined to a molecularly defined approach. Dialogues Clin. Neurosci. 19, 353–371. doi: 10.31887/DCNS.2017.19.4/sscherer
Filice, F., Janickova, L., Henzi, T., Bilella, A., and Schwaller, B. (2020). The parvalbumin hypothesis of autism spectrum disorder. Front. Cell. Neurosci. 14:577525. doi: 10.3389/fncel.2020.577525
Franz, D. N., and Capal, J. K. (2017). mTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders. Orphanet J. Rare Dis. 12:51. doi: 10.1186/s13023-017-0596-2
Frazier, T. W., and Hardan, A. Y. (2009). A meta-analysis of the corpus callosum in autism. Biol. Psychiatry 66, 935–941. doi: 10.1016/j.biopsych.2009.07.022
Frith, U., and Happé, F. (1994). Autism: beyond “theory of mind”. Cognition 50, 115–132. doi: 10.1016/0010-0277(94)90024-8
Fu, M., Yu, X., Lu, J., and Zuo, Y. (2012). Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature 483, 92–95. doi: 10.1038/nature10844
Fuchs, C., Gennaccaro, L., Trazzi, S., Bastianini, S., Bettini, S., Lo Martire, V., et al. (2018). Heterozygous CDKL5 knockout female mice are a valuable animal model for CDKL5 disorder. Neural Plast. 2018:9726950. doi: 10.1155/2018/9726950
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y.-i. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-Deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544. doi: 10.1093/jnen/64.6.537
Gamache, T. R., Araki, Y., and Huganir, R. L. (2020). Twenty years of SynGAP research: from synapses to cognition. J. Neurosci. 40, 1596–1605. doi: 10.1523/JNEUROSCI.0420-19.2020
Gao, R., and Penzes, P. (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167. doi: 10.2174/1566524015666150303003028
Garg, S., and Green, J. (2018). Studying child development in genetic models of ASD. Prog. Brain Res. 241, 159–192. doi: 10.1016/bs.pbr.2018.09.009
Geschwind, D. H. (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci. 15, 409–416. doi: 10.1016/j.tics.2011.07.003
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Ginsberg, S. D., Cruz-Martín, A., Crespo, M., and Portera-Cailliau, C. (2012). Glutamate induces the elongation of early dendritic protrusions via mGluRs in wild type mice, but not in fragile X mice. PLoS One 7:e32446. doi: 10.1371/journal.pone.0032446
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2012). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Gogolla, N., LeBlanc, J. J., Quast, K. B., Südhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 1, 172–181. doi: 10.1007/s11689-009-9023-x
Goldwater, D. S., Pavlides, C., Hunter, R. G., Bloss, E. B., Hof, P. R., McEwen, B. S., et al. (2009). Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164, 798–808. doi: 10.1016/j.neuroscience.2009.08.053
Greenberg, G. D., and Trainor, B. C. (2016). “Sex differences in the social behavior network and mesolimbic dopamine system,” in Sex Differences in the Central Nervous System, (San Diego, CA: Academic Press), 77–106.
Groc, L., Wang, C.-C., Held, R. G., and Hall, B. J. (2013). SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One 8:e83941. doi: 10.1371/journal.pone.0083941
Groen, W., Teluij, M., Buitelaar, J., and Tendolkar, I. (2010). Amygdala and hippocampus enlargement during adolescence in autism. J. Am. Acad. Child Adolesc. Psychiatry 49, 552–560. doi: 10.1016/j.jaac.2009.12.023
Gross, C., Banerjee, A., Tiwari, D., Longo, F., White, A. R., Allen, A. G., et al. (2019). Isoform-selective phosphoinositide 3-kinase inhibition ameliorates a broad range of fragile X syndrome-associated deficits in a mouse model. Neuropsychopharmacology 44, 324–333. doi: 10.1038/s41386-018-0150-5
Harris, K. M., and Kater, S. B. (1994). Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17, 341–371. doi: 10.1146/annurev.ne.17.030194.002013
Haws, M. E., Jaramillo, T. C., Espinosa, F., J. Widman, A., Stuber, G. D., Sparta, D. R., et al. (2014). PTEN knockdown alters dendritic spine/protrusion morphology, not density. J. Comp. Neurol. 522, 1171–1190. doi: 10.1002/cne.23488
Hayashi, M. L., Rao, B. S. S., Seo, J.-S., Choi, H.-S., Dolan, B. M., Choi, S.-Y., et al. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U S A 104, 11489–11494. doi: 10.1073/pnas.0705003104
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Translat. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Hien, A., Molinaro, G., Liu, B., Huber, K. M., and Richter, J. D. (2020). Ribosome profiling in mouse hippocampus: plasticity-induced regulation and bidirectional control by TSC2 and FMRP. Mol. Autism 11:78. doi: 10.1186/s13229-020-00384-9
Hodges, J. L., Yu, X., Gilmore, A., Bennett, H., Tjia, M., Perna, J. F., et al. (2017). Astrocytic contributions to synaptic and learning abnormalities in a mouse model of fragile X syndrome. Biol. Psychiatry 82, 139–149. doi: 10.1016/j.biopsych.2016.08.036
Hoeffer, C. A., Sanchez, E., Hagerman, R. J., Mu, Y., Nguyen, D. V., Wong, H., et al. (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 11, 332–341. doi: 10.1111/j.1601-183X.2012.00768.x
Holmes, A., Lit, Q., Murphy, D. L., Gold, E., and Crawley, J. N. (2003). Abnormal anxiety-related behavior in serotonin transporter null mutant mice: the influence of genetic background. Genes Brain Behav. 2, 365–380. doi: 10.1046/j.1601-1848.2003.00050.x
Holmes, S. E., Scheinost, D., Finnema, S. J., Naganawa, M., Davis, M. T., DellaGioia, N., et al. (2019). Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 10:1529. doi: 10.1038/s41467-019-09562-7
Holmes, A., Wrenn, C. C., Harris, A. P., Thayer, K. E., and Crawley, J. N. (2002). Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice. Genes Brain Behav. 1, 55–69. doi: 10.1046/j.1601-1848.2001.00005.x
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W., et al. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291. doi: 10.1016/j.neuron.2005.01.003
Horner, C. H., and Arbuthnott, E. (1991). Methods of estimation of spine density–are spines evenly distributed throughout the dendritic field. J. Anat. 177, 179–184.
Howell, B. W., and Smith, K. M. (2019). Synaptic structural protein dysfunction leads to altered excitation inhibition ratios in models of autism spectrum disorder. Pharmacol. Res. 139, 207–214. doi: 10.1016/j.phrs.2018.11.019
Huang, W.-C., Chen, Y., and Page, D. T. (2016). Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun. 7:13421. doi: 10.1038/ncomms13421
Huang, J., and Manning, B. D. (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190. doi: 10.1042/BJ20080281
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Iafrati, J., Orejarena, M. J., Lassalle, O., Bouamrane, L., and Chavis, P. (2013). Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 19, 417–426. doi: 10.1038/mp.2013.66
Ip, C. K. M., Cheung, A. N. Y., Ngan, H. Y. S., and Wong, A. S. T. (2011). p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 30, 2420–2432. doi: 10.1038/onc.2010.615
Jacot-Descombes, S., Keshav, N. U., Dickstein, D. L., Wicinski, B., Janssen, W. G. M., Hiester, L. L., et al. (2020). Altered synaptic ultrastructure in the prefrontal cortex of Shank3-deficient rats. Mol. Autism 11:89. doi: 10.1186/s13229-020-00393-8
Jaramillo, T. C., Speed, H. E., Xuan, Z., Reimers, J. M., Liu, S., and Powell, C. M. (2016). Altered striatal synaptic function and abnormal behaviour inShank3Exon4–9 deletion mouse model of autism. Autism Res. 9, 350–375. doi: 10.1002/aur.1529
Jawaid, S., Kidd, G. J., Wang, J., Swetlik, C., Dutta, R., and Trapp, B. D. (2018). Alterations in CA1 hippocampal synapses in a mouse model of fragile X syndrome. Glia 66, 789–800. doi: 10.1002/glia.23284
Jhanwar-Uniyal, M., Wainwright, J. V., Mohan, A. L., Tobias, M. E., Murali, R., Gandhi, C. D., et al. (2019). Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 72, 51–62. doi: 10.1016/j.jbior.2019.03.003
Jiang, M., Ash, R. T., Baker, S. A., Suter, B., Ferguson, A., Park, J., et al. (2013). Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci. 33, 19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013
Jossin, Y. (2020). Reelin functions, mechanisms of action and signaling pathways during brain development and maturation. Biomolecules 10:964. doi: 10.3390/biom10060964
Just, M. A., Keller, T. A., Malave, V. L., Kana, R. K., and Varma, S. (2012). Autism as a neural systems disorder: a theory of frontal-posterior underconnectivity. Neurosci. Biobehav. Rev. 36, 1292–1313. doi: 10.1016/j.neubiorev.2012.02.007
Kanjhan, R., Noakes, P. G., and Bellingham, M. C. (2016). Emerging Roles of filopodia and dendritic spines in motoneuron plasticity during development and disease. Neural Plast. 2016:3423267. doi: 10.1155/2016/3423267
Khatri, N., Gilbert, J. P., Huo, Y., Sharaflari, R., Nee, M., Qiao, H., et al. (2018). The autism protein Ube3A/E6AP remodels neuronal dendritic arborization via caspase-dependent microtubule destabilization. J. Neurosci. 38, 363–378. doi: 10.1523/JNEUROSCI.1511-17.2017
Kim, H., Kunz, P. A., Mooney, R., Philpot, B. D., and Smith, S. L. (2016). Maternal loss of Ube3a impairs experience-driven dendritic spine maintenance in the developing visual cortex. J. Neurosci. 36, 4888–4894. doi: 10.1523/JNEUROSCI.4204-15.2016
King, J. B., Prigge, M. B. D., King, C. K., Morgan, J., Weathersby, F., Fox, J. C., et al. (2019). Generalizability and reproducibility of functional connectivity in autism. Mol. Autism 10:27. doi: 10.1186/s13229-019-0273-5
Kleinhans, N. M., Reiter, M. A., Neuhaus, E., Pauley, G., Martin, N., Dager, S., et al. (2016). Subregional differences in intrinsic amygdala hyperconnectivity and hypoconnectivity in autism spectrum disorder. Autism Res. 9, 760–772. doi: 10.1002/aur.1589
Kouser, M., Speed, H. E., Dewey, C. M., Reimers, J. M., Widman, A. J., Gupta, N., et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci. 33, 18448–18468. doi: 10.1523/JNEUROSCI.3017-13.2013
Kron, M., Howell, C. J., Adams, I. T., Ransbottom, M., Christian, D., Ogier, M., et al. (2012). Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J. Neurosci. 32, 13860–13872. doi: 10.1523/JNEUROSCI.2159-12.2012
Kulinich, A. O., Reinhard, S. M., Rais, M., Lovelace, J. W., Scott, V., Binder, D. K., et al. (2020). Beneficial effects of sound exposure on auditory cortex development in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 134:104622‥ doi: 10.1016/j.nbd.2019.104622
Kwon, C.-H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Lábadi, B., and Beke, A. (2017). Mental state understanding in children with agenesis of the corpus callosum. Front. Psychol. 8:94. doi: 10.3389/fpsyg.2017.00094
Lai, T. K. Y., Abela, A. R., Su, P., Fletcher, P. J., and Liu, F. (2021). Prenatal disruption of D1R-SynGAP complex causes cognitive deficits in adulthood. Prog. Neuropsychopharmacol. Biol. Psychiatry 105:110122. doi: 10.1016/j.pnpbp.2020.110122
Langen, M., Bos, D., Noordermeer, S. D. S., Nederveen, H., van Engeland, H., and Durston, S. (2014). Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76, 405–411. doi: 10.1016/j.biopsych.2013.08.013
LaSarge, C. L., and Danzer, S. C. (2014). Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation, 1–15. Front. Mol. Neurosci. 7:18. doi: 10.3389/fnmol.2014.00018
Lau, Y. C., Hinkley, L. B. N., Bukshpun, P., Strominger, Z. A., Wakahiro, M. L. J., Baron-Cohen, S., et al. (2013). Autism traits in individuals with agenesis of the corpus callosum. J. Autism. Dev. Disord. 43, 1106–1118. doi: 10.1007/s10803-012-1653-2
Lazaro, M., and Golshani, P. (2015). The utility of rodent models of autism spectrum disorders. Curr. Opin. Neurol. 28, 103–109. doi: 10.1097/WCO.0000000000000183
Lee, J., Chung, C., Ha, S., Lee, D., Kim, D.-Y., Kim, H., et al. (2015). Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing and spatial memory deficit. Front. Cell. Neurosci. 9, 1–14. doi: 10.3389/fncel.2015.00094
Lenroot, R. K., and Yeung, P. K. (2013). Heterogeneity within autism spectrum disorders: what have we learned from neuroimaging studies. Front. Hum. Neurosci. 7:733. doi: 10.3389/fnhum.2013.00733
Li, W., Mai, X., and Liu, C. (2014). The default mode network and social understanding of others: what do brain connectivity studies tell us. Front. Hum. Neurosci. 8:74. doi: 10.3389/fnhum.2014.00074
Liang, X., Hu, Q., Li, B., McBride, D., Bian, H., Spagnoli, P., et al. (2014). FSTL1 attenuates apoptosis via DIP2A/Akt pathway after MCAO in rats. Stroke 45, 3048–3054. doi: 10.1161/STROKEAHA.114.006092
Lin, Y.-C., Frei, J. A., Kilander, M. B. C., Shen, W., and Blatt, G. J. (2016). A subset of autism-associated genes regulate the structural stability of neurons. Front. Cell. Neurosci. 10:263. doi: 10.3389/fncel.2016.00263
Liu, Z., Li, X., Zhang, J.-T., Cai, Y.-J., Cheng, T.-L., Cheng, C., et al. (2016). Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530, 98–102. doi: 10.1038/nature16533
Liu, S., Zhou, L., Yuan, H., Vieira, M., Sanz-Clemente, A., Badger, J. D., et al. (2017). A rare variant identified within the GluN2B C-Terminus in a patient with autism affects NMDA receptor surface expression and spine density. J. Neurosci. 37, 4093–4102. doi: 10.1523/JNEUROSCI.0827-16.2017
Llamosas, N., Arora, V., Vij, R., Kilinc, M., Bijoch, L., Rojas, C., et al. (2020). SYNGAP1 controls the maturation of dendrites, synaptic function and network activity in developing human neurons. J. Neurosci. 40, 7980–7994. doi: 10.1523/JNEUROSCI.1367-20.2020
Lombardo, M. V., Chakrabarti, B., Lai, M.-C., and Baron-Cohen, S. (2012). Self-referential and social cognition in a case of autism and agenesis of the corpus callosum. Mol. Autism 3:14. doi: 10.1186/2040-2392-3-14
Luikart, B. W., Schnell, E., Washburn, E. K., Bensen, A. L., Tovar, K. R., and Westbrook, G. L. (2011). Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 31, 4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011
Müller, R.-A., Shih, P., Keehn, B., Deyoe, J. R., Leyden, K. M., and Shukla, D. K. (2011). Underconnected, but how? a survey of functional connectivity MRI studies in autism spectrum disorders. Cereb. Cortex 21, 2233–2243. doi: 10.1093/cercor/bhq296
Ma, J., Zhang, L.-Q., He, Z.-X., He, X.-X., Wang, Y.-J., Jian, Y.-L., et al. (2019). Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 17:e3000461. doi: 10.1371/journal.pbio.3000461
MacGillavry, H. D., Kerr, J. M., Kassner, J., Frost, N. A., Blanpied, T. A., and Fagni, L. (2016). Shank-cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur. J. Neurosci. 43, 179–193. doi: 10.1111/ejn.13129
Magdalon, J., Sánchez-Sánchez, S., Griesi-Oliveira, K., and Sertié, A. (2017). Dysfunctional mTORC1 signaling: a convergent mechanism between syndromic and nonsyndromic forms of autism spectrum disorder. Int. J. Mol. Sci. 18:659. doi: 10.3390/ijms18030659
Mancuso, J. J., Chen, Y., Li, X., Xue, Z., and Wong, S. T. C. (2013). Methods of dendritic spine detection: from Golgi to high-resolution optical imaging. Neuroscience 251, 129–140. doi: 10.1016/j.neuroscience.2012.04.010
Mandy, W. P. L., Charman, T., and Skuse, D. H. (2012). Testing the construct validity of proposed criteria for DSM-5 autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry 51, 41–50. doi: 10.1016/j.jaac.2011.10.013
Martínez-Cerdeño, V. (2017). Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 77, 393–404. doi: 10.1002/dneu.22417
Masliah, E., Mallory, M., Hansen, L., DeTeresa, R., and Terry, R. D. (1993). Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 43, 192–197. doi: 10.1212/wnl.43.1_part_1.192
Mei, Y., Monteiro, P., Zhou, Y., Kim, J.-A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Meikle, L., Pollizzi, K., Egnor, A., Kramvis, I., Lane, H., Sahin, M., et al. (2008). Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J. Neurosci. 28, 5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008
Miyakawa, T., Yared, E., Pak, J. H., Huang, F. L., Huang, K. P., and Crawley, J. N. (2001). Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus 11, 763–775. doi: 10.1002/hipo.1092
Mizuguchi, M., Ikeda, H., Kagitani-Shimono, K., Yoshinaga, H., Suzuki, Y., Aoki, M., et al. (2019). Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain Dev. 41, 1–10. doi: 10.1016/j.braindev.2018.07.003
Mohammad-Rezazadeh, I., Frohlich, J., Loo, S. K., and Jeste, S. S. (2016). Brain connectivity in autism spectrum disorder. Curr. Opin. Neurol. 29, 137–147. doi: 10.1097/WCO.0000000000000301
Monteiro, P., and Feng, G. (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157. doi: 10.1038/nrn.2016.183
Montini, E., Andolfi, G., Caruso, A., Buchner, G., Walpole, S. M., Mariani, M., et al. (1998). Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 51, 427–433. doi: 10.1006/geno.1998.5391
Nair, A., Jolliffe, M., Lograsso, Y. S. S., and Bearden, C. E. (2020). A review of default mode network connectivity and its association with social cognition in adolescents with autism spectrum disorder and early-onset psychosis. Front. Psychiatry 11:614. doi: 10.3389/fpsyt.2020.00614
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/s0896-6273(00)80809-0
Nayda, D. M., and Takarangi, M. K. T. (2021). The cost of being absent: Is meta-awareness of mind-wandering related to depression symptom severity, rumination tendencies and trauma intrusions? J. Affect. Disord. 292, 131–138. doi: 10.1016/j.jad.2021.05.053
Nejad, A., Fossati, P., and Lemogne, C. (2013). Self-referential processing, rumination and cortical midline structures in major depression. Front. Hum. Neurosci. 7:666. doi: 10.3389/fnhum.2013.00666
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698. doi: 10.1016/j.neuron.2015.07.033
Newman, S. W. (1999). The medial extended amygdala in male reproductive behavior. a node in the mammalian social behavior network. Ann. N Y Acad. Sci. 877, 242–257. doi: 10.1111/j.1749-6632.1999.tb09271.x
Ng, L. H. L., Huang, Y., Han, L., Chang, R. C.-C., Chan, Y. S., and Lai, C. S. W. (2018). Ketamine and selective activation of parvalbumin interneurons inhibit stress-induced dendritic spine elimination. Transl. Psychiatry 8, 1–15. doi: 10.1038/s41398-018-0321-5
Nguyen, M. V. C., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Nie, D., Chen, Z., Ebrahimi-Fakhari, D., Di Nardo, A., Julich, K., Robson, V. K., et al. (2015). The stress-induced Atf3-Gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci. 35, 10762–10772. doi: 10.1523/JNEUROSCI.4796-14.2015
Nimchinsky, E. A., Sabatini, B. L., and Svoboda, K. (2002). Structure and function of dendritic spines. Annu. Rev. Physiol. 64, 313–353. doi: 10.1146/annurev.physiol.64.081501.160008
Nishiyama, J. (2019). Plasticity of dendritic spines: Molecular function and dysfunction in neurodevelopmental disorders. Psychiatry Clin. Neurosci. 73, 541–550. doi: 10.1111/pcn.12899
Okuda, K., Kobayashi, S., Fukaya, M., Watanabe, A., Murakami, T., Hagiwara, M., et al. (2017). CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 106, 158–170. doi: 10.1016/j.nbd.2017.07.002
Oliveira, A. F., and Yasuda, R. (2014). Neurofibromin is the major ras inactivator in dendritic spines. J. Neurosci. 34, 776–783. doi: 10.1523/JNEUROSCI.3096-13.2014
Ouchi, N., Asaumi, Y., Ohashi, K., Higuchi, A., Sono-Romanelli, S., Oshima, Y., et al. (2010). DIP2A functions as a FSTL1 receptor*. J. Biol. Chem. 285, 7127–7134. doi: 10.1074/jbc.M109.069468
Overwater, I. E., Rietman, A. B., Mous, S. E., Bindels-de Heus, K., Rizopoulos, D., ten Hoopen, L. W., et al. (2019). A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 93, e200–e209. doi: 10.1212/WNL.0000000000007749
Owen, J. P., Li, Y.-O., Ziv, E., Strominger, Z., Gold, J., Bukhpun, P., et al. (2013). The structural connectome of the human brain in agenesis of the corpus callosum. Neuroimage 70, 340–355. doi: 10.1016/j.neuroimage.2012.12.031
Padmanabhan, A., Lynch, C. J., Schaer, M., and Menon, V. (2017). The default mode network in autism. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 476–486. doi: 10.1016/j.bpsc.2017.04.004
Pagani, M., Barsotti, N., Bertero, A., Trakoshis, S., Ulysse, L., Locarno, A., et al. (2021). mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat. Commun. 12:6084. doi: 10.1038/s41467-021-26131-z
Parker, E. M., Kindja, N. L., Cheetham, C. E. J., and Sweet, R. A. (2020). Sex differences in dendritic spine density and morphology in auditory and visual cortices in adolescence and adulthood. Sci. Rep. 10:9442. doi: 10.1038/s41467-021-26131-z
Peça, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peixoto, R. T., Wang, W., Croney, D. M., Kozorovitskiy, Y., and Sabatini, B. L. (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B−/− mice. Nat. Neurosci. 19, 716–724. doi: 10.1038/nn.4260
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J.-E., and Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Peters, A., and Kaiserman-Abramof, I. R. (1970). The small pyramidal neuron of the rat cerebral cortex. the perikaryon, dendrites and spines. Am. J. Anat. 127, 321–355. doi: 10.1002/aja.1001270402
Peters, A., Sethares, C., and Moss, M. B. (1998). The effects of aging on layer 1 in area 46 of prefrontal cortex in the rhesus monkey. Cereb. Cortex 8, 671–684. doi: 10.1093/cercor/8.8.671
Pfeiffer, B. E., Zang, T., Wilkerson, J. R., Taniguchi, M., Maksimova, M. A., Smith, L. N., et al. (2010). Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66, 191–197. doi: 10.1016/j.neuron.2010.03.017
Picci, G., Gotts, S. J., and Scherf, K. S. (2016). A theoretical rut: revisiting and critically evaluating the generalized under/over-connectivity hypothesis of autism. Dev. Sci. 19, 524–549. doi: 10.1111/desc.12467
Piven, J., Palmer, P. D. P., Jacobi, D., Childress, D., and Arndt, S.. (1997). Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am. J. Psychiatry 154, 185–190. doi: 10.1176/ajp.154.2.185
Poerio, G. L., Sormaz, M., Wang, H.-T., Margulies, D., Jefferies, E., and Smallwood, J. (2017). The role of the default mode network in component processes underlying the wandering mind. Soc. Cogn. Affect. Neurosci. 12, 1047–1062. doi: 10.1093/scan/nsx041
Pop, A. S., Levenga, J., de Esch, C. E. F., Buijsen, R. A. M., Nieuwenhuizen, I. M., Li, T., et al. (2014). Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology (Berl) 231, 1227–1235. doi: 10.1007/s00213-012-2947-y
Portocarrero, L. K. L., Quental, K. c. N., Samorano, L. P., de Oliveira, Z. N. P., and Rivitti-Machado, M. C. l. d. M. (2018). Tuberous sclerosis complex: review based on new diagnostic criteria. An. Bras. Dermatol. 93, 323–331. doi: 10.1590/abd1806-4841.20186972
Pun, R. Y. K., Rolle, I. J., LaSarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Purcell, A. E., Jeon, O. H., Zimmerman, A. W., Blue, M. E., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628. doi: 10.1212/wnl.57.9.1618
Pyronneau, A., He, Q., Hwang, J.-Y., Porch, M., Contractor, A., and Zukin, R. S. (2017). Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function and sensory perception in fragile X syndrome. Sci. Signal. 10:eaan0852. doi: 10.1126/scisignal.aan0852
Qin, M., Entezam, A., Usdin, K., Huang, T., Liu, Z.-H., Hoffman, G. E., et al. (2011). A mouse model of the fragile X premutation: effects on behavior, dendrite morphology and regional rates of cerebral protein synthesis. Neurobiol. Dis. 42, 85–98. doi: 10.1016/j.nbd.2011.01.008
Qiu, S., Anderson, C. T., Levitt, P., and Shepherd, G. M. G. (2011). Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated met receptor tyrosine kinase. J. Neurosci. 31, 5855–5864. doi: 10.1523/JNEUROSCI.6569-10.2011
Rakic, P., Bourgeois, J. P., Eckenhoff, M. F., Zecevic, N., and Goldman-Rakic, P. S. (1986). Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232, 232–235. doi: 10.1126/science.3952506
Redcay, E., Moran, J., Mavros, P., Tager-Flusberg, H., Gabrieli, J., and Whitfield-Gabrieli, S. (2013). Intrinsic functional network organization in high-functioning adolescents with autism spectrum disorder. Front. Hum. Neurosci. 7:573. doi: 10.3389/fnhum.2013.00573
Reith, R. M., McKenna, J., Wu, H., Hashmi, S. S., Cho, S.-H., Dash, P. K., et al. (2013). Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 51, 93–103. doi: 10.1016/j.nbd.2012.10.014
Richards, C., Jones, C., Groves, L., Moss, J., and Oliver, C. (2015). Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry 2, 909–916. doi: 10.1016/S2215-0366(15)00376-4
Risher, W. C., Ustunkaya, T., Singh Alvarado, J., and Eroglu, C. (2014). Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9:e107591. doi: 10.1371/journal.pone.0107591
Rubenstein, J. L. R., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. doi: 10.1034/j.1601-183x.2003.00037.x
Ruzich, E., Allison, C., Smith, P., Watson, P., Auyeung, B., Ring, H., et al. (2015). Measuring autistic traits in the general population: a systematic review of the Autism-Spectrum Quotient (AQ) in a nonclinical population sample of 6,900 typical adult males and females. Mol. Autism 6:2. doi: 10.1186/2040-2392-6-2
Rylaarsdam, L., and Guemez-Gamboa, A. (2019). Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 13:385. doi: 10.3389/fncel.2019.00385
Santini, E., Huynh, T. N., Longo, F., Koo, S. Y., Mojica, E., D’Andrea, L., et al. (2017). Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci. Signal. 10:eaan0665. doi: 10.1126/scisignal.aan0665
Santini, E., Huynh, T. N., MacAskill, A. F., Carter, A. G., Pierre, P., Ruggero, D., et al. (2012). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. doi: 10.1038/nature11782
Santos, V. R., Pun, R. Y. K., Arafa, S. R., LaSarge, C. L., Rowley, S., Khademi, S., et al. (2017). PTEN deletion increases hippocampal granule cell excitability in male and female mice. Neurobiol. Dis. 108, 339–351. doi: 10.1016/j.nbd.2017.08.014
Sato, A., Kasai, S., Kobayashi, T., Takamatsu, Y., Hino, O., Ikeda, K., et al. (2012). Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 3:1292. doi: 10.1038/ncomms2295
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J.-Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23. doi: 10.1016/j.cell.2019.12.036
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Sceniak, M. P., Fedder, K. N., Wang, Q., Droubi, S., Babcock, K., Patwardhan, S., et al. (2019). An autism-associated mutation in GluN2B prevents NMDA receptor trafficking and interferes with dendrite growth. J. Cell Sci. 132:jcs232892. doi: 10.1242/jcs.232892
Sceniak, M. P., Lang, M., Enomoto, A. C., James Howell, C., Hermes, D. J., and Katz, D. M. (2016). Mechanisms of functional hypoconnectivity in the medial prefrontal cortex ofMecp2Null mice. Cereb. Cortex 26, 1938–1956. doi: 10.1093/cercor/bhv002
Schaefer, T. L., Ashworth, A. A., Tiwari, D., Tomasek, M. P., Parkins, E. V., White, A. R., et al. (2021). GABAA alpha 2,3 modulation improves select phenotypes in a mouse model of fragile X syndrome. Front. Psychiatry 12:678090. doi: 10.3389/fpsyt.2021.678090
Scheidegger, M., Walter, M., Lehmann, M., Metzger, C., Grimm, S., Boeker, H., et al. (2012). Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS One 7:e44799. doi: 10.1371/journal.pone.0044799
Schmeisser, M. J., Ey, E., Wegener, S., Bockmann, J., Stempel, A. V., Kuebler, A., et al. (2012). Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. doi: 10.1038/nature11015
Schnoor, M., Stradal, T. E., and Rottner, K. (2018). Cortactin: cell functions of a multifaceted actin-binding protein. Trends Cell Biol. 28, 79–98. doi: 10.1016/j.tcb.2017.10.009
Schumann, C. M., Hamstra, J., Goodlin-Jones, B. L., Lotspeich, L. J., Kwon, H., Buonocore, M. H., et al. (2004). The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci. 24, 6392–6401. doi: 10.1523/JNEUROSCI.1297-04.2004
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/JNEUROSCI.3696-09.2010
Shepherd, G. M. G. (2013). Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 14, 278–291. doi: 10.1038/nrn3469
Shih, Y.-T., Huang, T.-N., Hu, H.-T., Yen, T.-L., and Hsueh, Y.-P. (2020). Vcp overexpression and leucine supplementation increase protein synthesis and improve fear memory and social interaction of Nf1 mutant mice. Cell Rep. 31:107835. doi: 10.1016/j.celrep.2020.107835
Shin, W., Kim, K., Serraz, B., Cho, Y. S., Kim, D., Kang, M., et al. (2020). Early correction of synaptic long-term depression improves abnormal anxiety-like behavior in adult GluN2B-C456Y-mutant mice. PLoS Biol. 18:e3000717. doi: 10.1371/journal.pbio.3000717
Shofty, B., Bergmann, E., Zur, G., Asleh, J., Bosak, N., Kavushansky, A., et al. (2019). Autism-associated Nf1 deficiency disrupts corticocortical and corticostriatal functional connectivity in human and mouse. Neurobiol. Dis. 130:104479. doi: 10.1016/j.nbd.2019.104479
Shu, H., Donnard, E., Liu, B., Jung, S., Wang, R., and Richter, J. D. (2020). FMRP links optimal codons to mRNA stability in neurons. Proc. Natl. Acad. Sci. U S A 117, 30400–30411. doi: 10.1073/pnas.2009161117
Skelton, P. D., Frazel, P. W., Lee, D., Suh, H., and Luikart, B. W. (2019). Pten loss results in inappropriate excitatory connectivity. Mol. Psychiatry 24, 1627–1640. doi: 10.1038/s41380-019-0412-6
Skelton, P. D., Poquerusse, J., Salinaro, J. R., Li, M., and Luikart, B. W. (2020). Activity-dependent dendritic elaboration requires pten. Neurobiol. Dis. 134:104703. doi: 10.1016/j.nbd.2019.104703
Sleigh, J., Harvey, M., Voss, L., and Denny, B. (2014). Ketamine - more mechanisms of action than just NMDA blockade. Trends Anaesth. Crit. Care 4, 76–81. doi: 10.1016/j.tacc.2014.03.002
Smallwood, J., and Schooler, J. W. (2015). The science of mind wandering: empirically navigating the stream of consciousness. Annu. Rev. Psychol. 66, 487–518. doi: 10.1146/annurev-psych-010814-015331
Smith, S. E. P., Zhou, Y.-D., Zhang, G., Jin, Z., Stoppel, D. C., and Anderson, M. P. (2011). Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra97. doi: 10.1126/scitranslmed.3002627
Sohal, V. S., and Rubenstein, J. L. R. (2019). Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 24, 1248–1257. doi: 10.1038/s41380-019-0426-0
Spinelli, L., Black, F. M., Berg, J. N., Eickholt, B. J., and Leslie, N. R. (2015). Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J. Med. Genet. 52, 128–134. doi: 10.1136/jmedgenet-2014-102803
Sugiura, H., Shimada, T., Moriya-Ito, K., Goto, J.-I., Fujiwara, H., Ishii, R., et al. (2022). A farnesyltransferase inhibitor restores cognitive deficits in Tsc2+/− mice through inhibition of Rheb1. J. Neurosci. 42, 2598–2612. doi: 10.1523/JNEUROSCI.0449-21.2022
Sugiura, H., Yasuda, S., Katsurabayashi, S., Kawano, H., Endo, K., Takasaki, K., et al. (2015). Rheb activation disrupts spine synapse formation through accumulation of syntenin in tuberous sclerosis complex. Nat. Commun. 6:6842. doi: 10.1038/ncomms7842
Svalina, M. N., Guthman, E. M., Cea-Del Rio, C. A., Kushner, J. K., Baca, S. M., Restrepo, D., et al. (2021). Hyperexcitability and loss of feedforward inhibition contribute to aberrant plasticity in the Fmr1KO amygdala. eNeuro 8:ENEURO.0113-21.2021. doi: 10.1523/ENEURO.0113-21.2021
Sztainberg, Y., and Zoghbi, H. Y. (2016). Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 19, 1408–1417. doi: 10.1038/nn.4420
Takahashi, H., Sekino, Y., Tanaka, S., Mizui, T., Kishi, S., and Shirao, T. (2003). Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic Density-95 and dendritic spine morphogenesis. J. Neurosci. 23, 6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003
Tang, G., Gudsnuk, K., Kuo, S.-H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
Testa-Silva, G., Loebel, A., Giugliano, M., de Kock, C. P. J., Mansvelder, H. D., and Meredith, R. M. (2012). Hyperconnectivity and slow synapses during early development of medial prefrontal cortex in a mouse model for mental retardation and autism. Cereb. Cortex 22, 1333–1342. doi: 10.1093/cercor/bhr224
Tilot, A. K., Frazier, T. W., and Eng, C. (2015). Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics 12, 609–619. doi: 10.1007/s13311-015-0356-8
Tomasi, D., and Volkow, N. D. (2019). Reduced local and increased long-range functional connectivity of the thalamus in autism spectrum disorder. Cereb. Cortex 29, 573–585. doi: 10.1093/cercor/bhx340
Trazzi, S., De Franceschi, M., Fuchs, C., Bastianini, S., Viggiano, R., Lupori, L., et al. (2018). CDKL5 protein substitution therapy rescues neurological phenotypes of a mouse model of CDKL5 disorder. Hum. Mol. Genet. 27, 1572–1592. doi: 10.1093/hmg/ddy064
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tsujimura, K., Irie, K., Nakashima, H., Egashira, Y., Fukao, Y., Fujiwara, M., et al. (2015). miR-199a links MeCP2 with mTOR signaling and its dysregulation leads to rett syndrome phenotypes. Cell Rep. 12, 1887–1901. doi: 10.1016/j.celrep.2015.08.028
Valluy, J., Bicker, S., Aksoy-Aksel, A., Lackinger, M., Sumer, S., Fiore, R., et al. (2015). A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci. 18, 666–673. doi: 10.1038/nn.3996
Varea, O., Martin-de-Saavedra, M. D., Kopeikina, K. J., Schürmann, B., Fleming, H. J., Fawcett-Patel, J. M., et al. (2015). Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl. Acad. Sci. U S A 112, 6176–6181. doi: 10.1073/pnas.1423205112
Varghese, M., Keshav, N., Jacot-Descombes, S., Warda, T., Wicinski, B., Dickstein, D. L., et al. (2017). Autism spectrum disorder: neuropathology and animal models. Acta. Neuropathol. 134, 537–566. doi: 10.1007/s00401-017-1736-4
Vatsa, N., and Jana, N. R. (2018). UBE3A and its link with autism. Front. Mol. Neurosci. 11:448. doi: 10.3389/fnmol.2018.00448
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850. doi: 10.1074/jbc.M111.258384
Vignoli, A., La Briola, F., Peron, A., Turner, K., Vannicola, C., Saccani, M., et al. (2015). Autism spectrum disorder in tuberous sclerosis complex: searching for risk markers. Orphanet J. Rare Dis. 10:154. doi: 10.1186/s13023-015-0371-1
Viscidi, E. W., Triche, E. W., Pescosolido, M. F., McLean, R. L., Joseph, R. M., Spence, S. J., et al. (2013). Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS One 8:e67797. doi: 10.1371/journal.pone.0067797
Vogt, D., Cho, K. K. A., Shelton, S. M., Paul, A., Huang, Z. J., Sohal, V. S., et al. (2018). MouseCntnap2and HumanCNTNAP2ASD alleles cell autonomously regulate PV+ cortical interneurons. Cereb. Cortex 28, 3868–3879. doi: 10.1093/cercor/bhx248
Wöhr, M., Orduz, D., Gregory, P., Moreno, H., Khan, U., Vörckel, K. J., et al. (2015). Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl. Psychiatry 5:e525. doi: 10.1038/tp.2015.19
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7, 1–18. doi: 10.1038/ncomms11459
Wang, M., Li, H., Takumi, T., Qiu, Z., Xu, X., Yu, X., et al. (2017). Distinct defects in spine formation or pruning in two gene duplication mouse models of autism. Neurosci. Bull. 33, 143–152. doi: 10.1007/s12264-017-0111-8
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wang, L., Pang, K., Han, K., Adamski, C. J., Wang, W., He, L., et al. (2019). An autism-linked missense mutation in SHANK3 reveals the modularity of Shank3 function. Mol. Psychiatry 25, 2534–2555. doi: 10.1038/s41380-018-0324-x
Wang, X., Xu, Q., Bey, A. L., Lee, Y., and Jiang, Y.-h. (2014). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5:30. doi: 10.1186/2040-2392-5-30
Watanabe, Y., Gould, E., and McEwen, B. S. (1992). Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 588, 341–345. doi: 10.1016/0006-8993(92)91597-8
Weir, R. K., Bauman, M. D., Jacobs, B., and Schumann, C. M. (2018). Protracted dendritic growth in the typically developing human amygdala and increased spine density in young ASD brains. J. Comp. Neurol. 526, 262–274. doi: 10.1002/cne.24332
Wesseling, H., Elgersma, Y., and Bahn, S. (2017). A brain proteomic investigation of rapamycin effects in the Tsc1+/− mouse model. Mol. Autism 8:41. doi: 10.1186/s13229-017-0151-y
Williams, C. A., Dagli, A., and Battaglia, A. (2008). Genetic disorders associated with macrocephaly. Am. J. Med. Genet. A 146A, 2023–2037. doi: 10.1002/ajmg.a.32434
Williams, M. R., DeSpenza, T., Li, M., Gulledge, A. T., and Luikart, B. W. (2015). Hyperactivity of newborn pten knock-out neurons results from increased excitatory synaptic drive. J. Neurosci. 35, 943–959. doi: 10.1523/JNEUROSCI.3144-14.2015
Winden, K. D., Ebrahimi-Fakhari, D., and Sahin, M. (2018). Abnormal mTOR activation in autism. Annu. Rev. Neurosci. 41, 1–23. doi: 10.1146/annurev-neuro-080317-061747
Wise, T., Marwood, L., Perkins, A. M., Herane-Vives, A., Joules, R., Lythgoe, D. J., et al. (2017). Instability of default mode network connectivity in major depression: a two-sample confirmation study. Transl. Psychiatry 7:e1105. doi: 10.1038/tp.2017.40
Won, H., Lee, H.-R., Gee, H. Y., Mah, W., Kim, J.-I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Wong, C. W., Wang, Y., Liu, T., Li, L., Cheung, S. K. K., Or, P. M. Y., et al. (2020). Autism-associated PTEN missense mutation leads to enhanced nuclear localization and neurite outgrowth in an induced pluripotent stem cell line. FEBS J. 287, 4848–4861. doi: 10.1111/febs.15287
Xiong, Q., Oviedo, H. V., Trotman, L. C., and Zador, A. M. (2012). PTEN regulation of local and long-range connections in mouse auditory cortex. J. Neurosci. 32, 1643–1652. doi: 10.1523/JNEUROSCI.4480-11.2012
Yan, J., Porch, M. W., Court-Vazquez, B., Bennett, M. V. L., and Zukin, R. S. (2018). Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. U S A 115, E9707–E9716. doi: 10.1073/pnas.1808247115
Yasuda, S., Sugiura, H., Katsurabayashi, S., Shimada, T., Tanaka, H., Takasaki, K., et al. (2014). Activation of Rheb, but not of mTORC1, impairs spine synapse morphogenesis in tuberous sclerosis complex. Sci. Rep. 4:5155. doi: 10.1038/srep05155
Yerys, B. E., Gordon, E. M., Abrams, D. N., Satterthwaite, T. D., Weinblatt, R., Jankowski, K. F., et al. (2015). Default mode network segregation and social deficits in autism spectrum disorder: evidence from non-medicated children. Neuroimage Clin. 9, 223–232. doi: 10.1016/j.nicl.2015.07.018
Yi, J. J., Berrios, J., Newbern, J. M., Snider, W. D., Philpot, B. D., Hahn, K. M., et al. (2015). An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162, 795–807. doi: 10.1016/j.cell.2015.06.045
Zerbi, V., Ielacqua, G. D., Markicevic, M., Haberl, M. G., Ellisman, M. H., A-Bhaskaran, A., et al. (2018). Dysfunctional autism risk genes cause circuit-specific connectivity deficits with distinct developmental trajectories. Cereb. Cortex 28, 2495–2506. doi: 10.1093/cercor/bhy046
Zerbi, V., Pagani, M., Markicevic, M., Matteoli, M., Pozzi, D., Fagiolini, M., et al. (2021). Brain mapping across 16 autism mouse models reveals a spectrum of functional connectivity subtypes. Mol. Psychiatry 26, 7610–7620. doi: 10.1038/s41380-021-01245-4
Zhang, Y., Li, N., Li, C., Zhang, Z., Teng, H., Wang, Y., et al. (2020). Genetic evidence of gender difference in autism spectrum disorder supports the female-protective effect. Transl. Psychiatry 10:4. doi: 10.1038/s41398-020-0699-8
Zhang, Y., Xiang, Z., Jia, Y., He, X., Wang, L., and Cui, W. (2019). The Notch signaling pathway inhibitor Dapt alleviates autism-like behavior, autophagy and dendritic spine density abnormalities in a valproic acid-induced animal model of autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 94:109644. doi: 10.1016/j.pnpbp.2019.109644
Zhao, J.-P., and Yoshii, A. (2019). Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol. Brain 12, 1–13. doi: 10.1186/s13041-019-0427-6
Zhou, Y., Kaiser, T., Monteiro, P., Zhang, X., Van der Goes, M. S., Wang, D., et al. (2016). Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162. doi: 10.1016/j.neuron.2015.11.023
Zhubi, A., Chen, Y., Dong, E., Cook, E. H., Guidotti, A., and Grayson, D. R. (2014). Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 4:e349. doi: 10.1038/tp.2013.123
Keywords: autism spectrum conditions, social behaviors, rodent models, spine density, synaptic transmission, neurocircuitry, mTORC1, autophagy
Citation: Chaudry S and Vasudevan N (2022) mTOR-Dependent Spine Dynamics in Autism. Front. Mol. Neurosci. 15:877609. doi: 10.3389/fnmol.2022.877609
Received: 16 February 2022; Accepted: 25 April 2022;
Published: 15 June 2022.
Edited by:
Christos G. Gkogkas, Biomedical Research Institute (BRI), GreeceReviewed by:
Bryan Weston Luikart, Dartmouth College, United StatesWen-Jie Bian, Stanford University, United States
Copyright © 2022 Chaudry and Vasudevan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nandini Vasudevan, n.vasudevan@reading.ac.uk