Autism Spectrum Disorder Genes: Disease-Related Networks and Compensatory Strategies
 Hye Kyung Lim
Hye Kyung Lim Jong Hyuk Yoon
Jong Hyuk Yoon Minseok Song
Minseok Song- 1Department of Life Sciences, Yeungnam University, Gyeongsan, South Korea
- 2Neurodegenerative Diseases Research Group, Korea Brain Research Institute, Daegu, South Korea
The mammalian brain comprises structurally and functionally distinct regions. Each of these regions has characteristic molecular mechanisms that mediate higher-order tasks, such as memory, learning, emotion, impulse, and motor control. Many genes are involved in neuronal signaling and contribute to normal brain development. Dysfunction of essential components of neural signals leads to various types of brain disorders. Autism spectrum disorder is a neurodevelopmental disorder characterized by social deficits, communication challenges, and compulsive repetitive behaviors. Long-term genetic studies have uncovered key genes associated with autism spectrum disorder, such as SH3 and multiple ankyrin repeat domains 3, methyl-CpG binding protein 2, neurexin 1, and chromodomain helicase DNA binding protein 8. In addition, disease-associated networks have been identified using animal models, and the understanding of the impact of these genes on disease susceptibility and compensation is deepening. In this review, we examine rescue strategies using key models of autism spectrum disorder.
Introduction
Autism spectrum disorder (ASD) is a clinically heterogeneous and inherited neurodevelopmental disorder. To diagnose a broad range of clinical phenotypes of the disease, the American Psychiatric Association published the Diagnostic and Statistical Manual of Mental Disorders (Bienvenu et al., 2013). Following the criteria indicated therein, ASD can be diagnosed on the basis of core manifestations of impaired social communication and interaction and restricted, repetitive behaviors, in addition to the different degrees of concurrent conditions, including intellectual deficit, language difficulty, epilepsy, sleep disturbance, and attention disability. According to the Centers for Disease Control and Prevention, the current overall prevalence of ASD is estimated to be 1 in 44 children, and there are 4.2 times more boys than girls with ASD (Maenner et al., 2021). Despite multi-faceted research being conducted to understand the molecular mechanism of ASD, its precise etiology remains unknown; however, both genetic and environmental factors are considered to play a role. Despite decades of research on novel pharmacological treatments for core ASD symptoms, the number of medications that exhibit therapeutic effects that meet certain criteria is still limited, and there is insufficient evidence for their clinical use. Several large-scale drug development efforts for ASD based on basic science and molecular research have yielded mostly negative or ambiguous clinical trial outcomes. As a result, the existing authorized ASD treatment methods only address secondary symptoms, including restlessness and impatience.
Non-invasive imaging studies have revealed anatomical and functional alterations in the brain, associated with the disorder (Kemper and Bauman, 1998; Amaral et al., 2008; Courchesne et al., 2011). Brain hypergrowth in patients with ASD is associated with an unusually excess number of neurons in the prefrontal cortex (PFC), according to a small-scale preliminary study analyzing post-mortem prefrontal tissue in children with ASD. Although post-mortem and structural magnetic resonance imaging studies have made it difficult to establish a clear, generalized pathology for overall ASD, researchers have been able to uncover pathological abnormalities in the frontal lobe, amygdala, and cerebellum in many cases of ASD. The brains of children and adults with ASD have been shown to exhibit a variety of morphological and functional abnormalities (Sparks et al., 2002; Amaral et al., 2008). However, these defects have not been consistently identified in distinct patients with ASD and are rarely correlated with symptom severity. Genetic predisposition to ASD has been well documented through research in families and twins. Exome sequencing, which is being performed on a continuously increasing number of patient samples, has identified novel rare mutations relevant to ASD (Voineagu et al., 2011; O’Roak et al., 2012; De Rubeis et al., 2014). Researchers identifying the genetic and pathological etiologies of patients with major ASD have faced enormous difficulty owing to the high heterogeneity in the expression and severity of the core and associated symptoms. Based on genetic investigations to date, it has been predicted that approximately 1000 genes are implicated in ASD (O’Roak et al., 2012; De Rubeis et al., 2014); however, no single variants have been found to be strongly linked to ASD.
Because ASD has multigenic traits, understanding its etiology may be difficult. However, the related genes can be classified into several groups, allowing researchers to deduce the specific molecular pathways relevant to ASD (De Rubeis et al., 2014). ASD has been linked to genes involved in synaptic transmission and scaffolding, chromatin remodeling, protein synthesis and degradation, and actin cytoskeletal dynamics (Figure 1) (Bourgeron, 2015). Researchers must use experimental animal models to further understand how these genes are related to the etiology and pathophysiology of ASD. Mice are the most commonly used animal models in neuroscience. A number of characteristics explain why they are frequently used to describe human neuropsychiatric disorders. In reality, mice are social animals that live in hierarchical groups and engage in complex social activities, such as parenting and communal nesting of the young, juvenile play, and adult sexual and aggressive behaviors (Ellenbroek and Youn, 2016). They are usually selected because they can be used for neurobiological, behavioral, and pharmacological studies to examine autistic characteristics in humans and because of their rapid generation time, small size, and comprehensive genome sequencing (Wohr and Scattoni, 2013). Mice with a mutation in one of several ASD risk genes have been the most widely utilized animal models to date (Rubenstein and Merzenich, 2003; Lewis et al., 2007; Wohr and Scattoni, 2013; Ellenbroek and Youn, 2016; Chung et al., 2021). Behavioral tests used to evaluate social deficits, communication impairments, and repetitive activities have also been employed to validate the mouse models of ASD (Table 1).
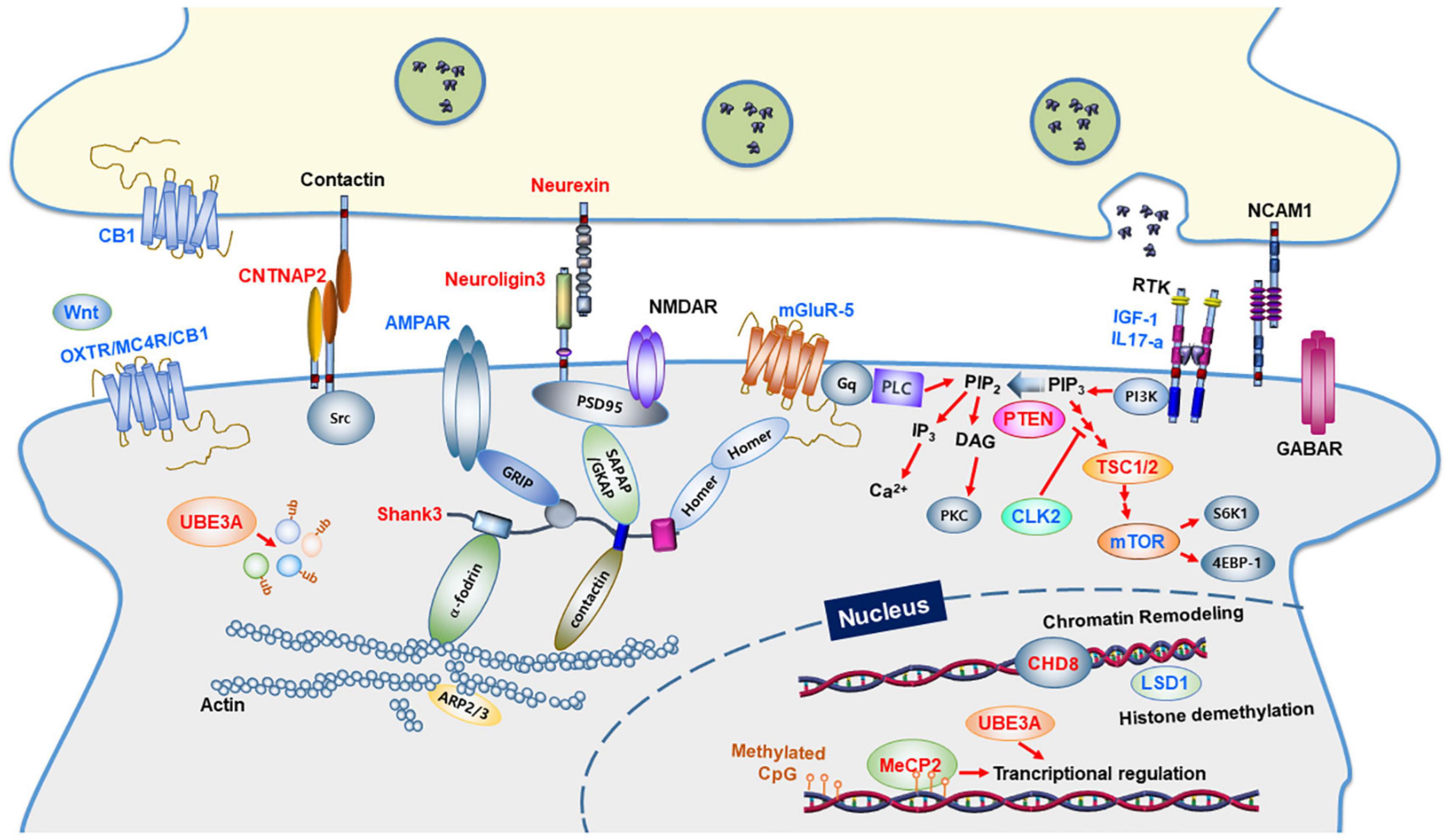
Figure 1. Synaptic proteins and signaling pathways linked to autism spectrum disorders. Synaptic proteins associated with ASD are involved in synapse formation and synaptic function. The ASD-linked proteins covered in this review article are highlighted in red. Proteins in blue indicate molecular targets that were used to ameliorate the ASD phenotype. CB1, Cannabinoid receptor type 1; OXTR, Oxytocin Receptor; MC4R, Melanocortin 4 Receptor; PLC, Phospholipase C; GABAR, GABAA receptor; AMPAR, AMPA receptor; NMDAR, NMDA receptor; mGluR, metabolic glutamate receptor; IGF-1, Insulin-like growth factor 1; IL17A, Interleukin 17A; NCAM1, Neural Cell Adhesion Molecule 1; GRIP1, Glutamate Receptor Interacting Protein 1; SHANK3, SH3 and multiple ankyrin repeat domains 3; SAPAP, Synapse-Associated Protein 90/Postsynaptic Density-95-Associated Protein; GKAP, guanylate-kinase-associated-protein; PSD-95, post-synaptic scaffolding protein 95 kDa; CNTNAP2, a contactin-associated protein-like 2 gene; PI3K, phosphoinositide-3 kinase; CLK2, CDC Like Kinase 2; PTEN, phosphatase and tensin homologs; TSC, tuberous sclerosis complex; mTOR, a mammalian target of rapamycin; CHD8, chromodomain helicase DNA binding protein 8; MeCP2, methyl CpG binding protein 2; LSD1, lysine-specific demethylase 1; UBE3A, Ubiquitin Protein Ligase E3A.
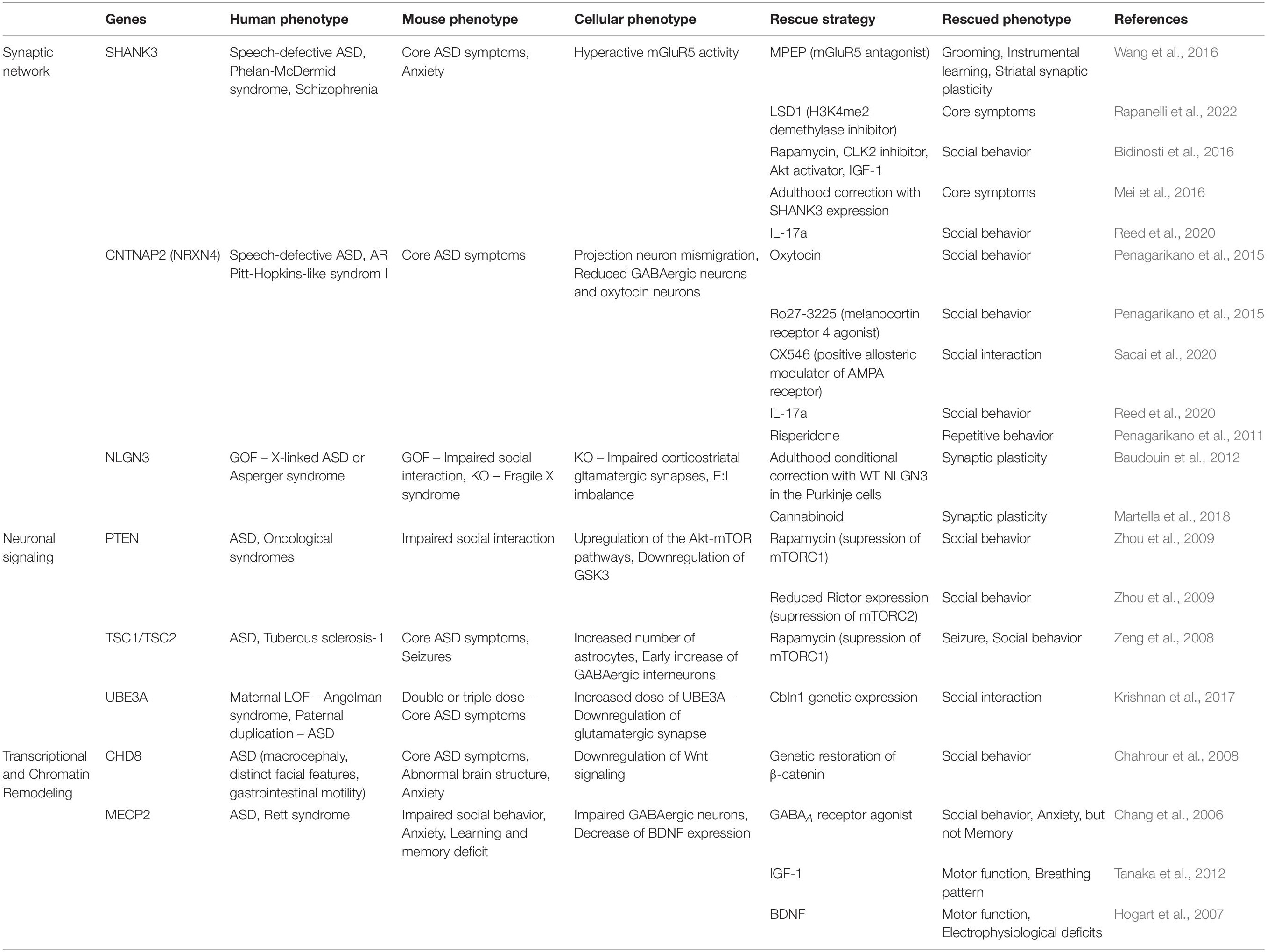
Table 1. Patients’ phenotypes, as well as phenotypes and rescue strategies shown in ASD mouse models.
The hallmark characteristic of ASD is abnormal social interaction, and several behavioral tests can be used to assess social approach, reciprocal social interactions, nesting, sexual interactions, parenting behaviors, and hostile encounters (Wohr and Scattoni, 2013; Pensado-Lopez et al., 2020). An automated three-chambered social approach task with a stranger and non-social object on the opposite end can be utilized to reveal the deficiency of sociability based on the time spent during the task (Yang et al., 2011). The social approach task can also be used to measure social recognition by replacing stimuli with familiar and novel mice. An open field apparatus can be used for social and sexual interactions by placing two unfamiliar mice together to evaluate more sophisticated social behaviors. The mice interact primarily by sniffing, following, crawling, and pushing each other. Another ASD criterion is a deficit in communication, which is the most challenging assay because mice do not use language. Instead, mice produce ultrasonic vocalizations under various conditions. Such ultrasonic vocalizations have been found in newborns at the time of birth, pups removed from their mother, juveniles during social play, and adults during mating, aggression, or exploration (Scattoni et al., 2009). Restricted, repetitive, and stereotyped patterns of behavior are fundamental characteristics of ASD. Vertical jumping, backflipping, circling, digging, marble burying, rearing, and excessive self-grooming are common behaviors in mice (Lewis et al., 2007). The open field test measures the presence and duration of locomotor activity. Reversal learning tasks assess the capacity to adapt to a new routine in the T-maze or Morris water maze (Zaccaria et al., 2010). The tendency of mice to investigate novel objects and poke their nose into holes in the wall or floor can be used to reflect restricted interests (Moy et al., 2008).
Following the establishment of genetically engineered mouse models, behavioral research, biochemical studies, and neurophysiological investigations have validated the link between candidate genes and ASD phenotypes. Various studies using animal models of ASD are currently providing fundamental knowledge regarding the biology and treatment strategies of the disease. Based on the findings of analyses of signal circuits for ASD models, it is conceivable to provide molecular targets relevant for therapeutic intervention, yielding optimism that ASD caused by defects in several genes could be treated even after brain development has been completed to some extent. In this review, we explain the mouse models that can help in understanding ASD, the pathological characteristics noted from each model, and pharmacological restoration and potential rescue tactics derived for future clinical trials.
Mouse Models
ASD-related mouse models are implicated in three generally defined genes: those engaged in the synaptic network, neuronal signaling, and transcriptional and chromatin remodeling, based on the discovered risk factors and biological etiology (De Rubeis et al., 2014; Yoo, 2015; Rylaarsdam and Guemez-Gamboa, 2019; Chung et al., 2021). Various mouse models with targeted mutations in these candidate genes have been developed and used to explore the pathophysiological mechanisms of ASD biology to better understand the genetics of ASD. Each mouse model provided novel insights into the pathophysiology of ASD, and pharmacological and genetic restoration experiments using these mouse models revealed important clues for future clinical trials.
Synaptic Network
Mutations in synapse-related genes, such as SH3 and multiple ankyrin repeat domains (SHANK), neurexin (NRXN), contactin-associated protein-like 2 (CNTNAP2), and neuroligin (NLGN), are the most commonly described genetic etiologies in ASD. Abnormalities in these synaptic genes are linked to ASD and other complex neuropsychiatric illnesses, such as locomotor difficulties, intellectual impairments, seizures, and speech issues. Mouse models that target these synaptic genes have primarily been used to precisely understand the molecular mechanisms of ASD and have made significant contributions to the discovery of relevant neuronal pathways and plausible therapeutic approaches.
Neuroligin 3
Neuroligins (NLGNs) are post-synaptic adhesion proteins that serve as ligands for NRXNs and pre-synaptically localized adhesion molecules that support Ca2+-dependent trans-synaptic binding to initiate synaptogenesis at central synapses (Reissner et al., 2008). Among the five isoforms in humans, NLGN3 is highly associated with non-syndromic ASD. A gain-of-function mutation in NLGN3 (R451C) predisposes patients to X-linked ASD or Asperger syndrome (Jamain et al., 2003). Synaptic transmission and heterosynaptic competition have been reported to be disturbed in NLGN3 knockout mice owing to disruption in metabotropic glutamate receptor-dependent synaptic plasticity. Autism is a developmental disorder that is usually diagnosed after 3 years of age. Researchers have attempted to determine whether the behavioral traits of ASD can be reversed in adults with abnormally developed brain structures over long periods of time. A resolution to this topic was provided by rearing genetically modified mice with a tetracycline transactivator to temporally control the re-expression of wild-type NLGN3 in the Purkinje cells of NLGN3 knockout mice. The synaptic abnormalities observed in NLGN3 knockout mice could be reversed when the NLGN3 gene was restored after brain development was complete, indicating that the condition could be rescued even after a period of time had elapsed since its onset (Baudouin et al., 2012). The development of long-term synaptic depression at corticostriatal glutamatergic synapses was observed in R451C-NLGN3 mutant mice, corroborating the evidence for an imbalance between excitatory and inhibitory (E:I) neurotransmissions in models of ASD. Stimulation of the cannabinoid CB1 receptor partially relieved long-term synaptic depression in this model, suggesting that altered cannabinoid signaling may underpin the synaptic plasticity impairments observed in ASD (Martella et al., 2018). A recent optogenetic study in R451C knock-in mice demonstrated that social deficiencies in autism are caused by gamma oscillation disruption in the mPFC and that modulating mPFC PV interneurons even in adulthood may help correct the deficits (Cao et al., 2018).
Contactin-Associated Protein-Like 2
Contactin-associated protein-like 2 (CNTNAP2), also known as NRXN4, is an NRXN superfamily transmembrane protein found in the CNS. During nervous system development, it influences the subcellular localization of potassium channels within developing axons and mediates synapse formation between neurons and glia (Poliak et al., 1999). In the developing human brain, CNTNAP2 is abundant in circuits that are critical for language development. Genetic abnormalities in CNTNAP2 underlie speech-defective ASD and autosomal recessive Pitt–Hopkins-like syndrome-1, characterized by severe intellectual disability, regression of speech development, and behavioral abnormalities (Alarcon et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Smogavec et al., 2016). CNTNAP2 knockout mice display all the core behavioral characteristics of ASD, with cortical projection neuron migratory impairments, a decrease in GABAergic intermediate neurons, and concurrent altered brain synchrony (Penagarikano et al., 2011). In CNTNAP2 mutant mice, the number of oxytocin-positive neurons in the paraventricular nucleus is reduced, similar to brain oxytocin levels. Consistent with these findings, postnatal treatment with the neuropeptide oxytocin improves aberrant social behaviors in CNTNAP2 mutant mice (Penagarikano et al., 2015). Social deficiency has been successfully corrected in CNTNAP2 knockout mice upon treatment with a selective melanocortin receptor 4 agonist known to promote endogenous oxytocin release, whereas the oxytocin antagonist had the opposite effect. Based on these findings, it is plausible to deduce that in the CNTNAP2 knockout model, oxytocin signaling failure causes ASD-like behavior. CNTNAP2 has been found to be important in excitatory synaptic transmission and maintenance of E:I balance in pyramidal neurons of the developing mouse PFC. The impaired social interaction shown in CNTNAP2 mutant mice was significantly restored when AMPA receptor activity was enhanced through pharmacological treatment, suggesting that disrupted E:I balance in layer 2/3 pyramidal neurons of the PFC led to impaired social interaction in CNTNAP2 knockout mice (Sacai et al., 2020).
SH3 and Multiple Ankyrin Repeat Domains 3
SH3 and multiple ankyrin repeat domains (SHANK) is a large protein with several protein interaction domains, including ankyrin repeats, an SH3 domain, a PDZ domain, a proline-rich region, and an SAM domain (Sheng and Hoogenraad, 2007). The multidomain protein isoforms are generated by three SHANK genes that are differentially expressed in developmental stages, cell types, and brain regions (Boeckers et al., 1999; Bonaglia et al., 2001). SHANK family proteins play a critical role in the organization of post-synaptic molecular complexes in excitatory synapses through interactions with cytoskeletal regulators, endocytic machinery, diverse receptors, other adapters, and synaptic scaffold proteins (Naisbitt et al., 1999; Tu et al., 1999; Yao et al., 1999; Zitzer et al., 1999a,b; Kreienkamp et al., 2000; Tobaben et al., 2000; Lim et al., 2001; Bockers et al., 2004). Studies have shown that SHANK proteins play a vital role in dendritic spine formation and neurotransmission (Hering and Sheng, 2001; Sala et al., 2001; Ehlers, 2002; Rubenstein and Merzenich, 2003; Shcheglovitov et al., 2013; Gogolla et al., 2014; Filice et al., 2016; Lu et al., 2016; Yi et al., 2016).
SH3 and multiple ankyrin repeat domains 3 (SHANK3) deficiency in humans is associated with language and social communication problems of ASD, Phelan–McDermid syndrome, and schizophrenia (Durand et al., 2007; Gauthier et al., 2010). To further understand the molecular mechanisms underlying the role of SHANK3 in ASD, several research groups have developed SHANK3 mouse models that closely resemble human ASD characteristics, making a significant contribution to the understanding of the pathophysiology of ASD. SHANK3-deficient mice, both homozygous and heterozygous, displayed repetitive grooming, anxiety, and social interaction deficits (Bangash et al., 2011; Peca et al., 2011; Wang et al., 2011). SHANK3 gene deletion also affects the spine number and size, as well as AMPA transmission, indicating that SHANK proteins play a crucial role in excitatory neurotransmission. Indeed, follow-up studies have revealed that in the absence of SHANK3, excitatory input from the cortex to the striatum develops abnormally, resulting in an altered E:I ratio in the cortex and striatum (Peixoto et al., 2016).
A mouse model was employed to study the signaling circuit related to SHANK3 and played a key role in presenting the molecular network considered to be involved in ASD, in addition to presenting the anatomical and physiological characteristics that can arise in ASD. The findings have led to the development of a novel therapeutic strategy that was immediately verified using an animal model. SHANK3 deficiency causes the mGluR5-dependent corticostriatal-thalamic circuit to become hypoactive during social behaviors (Wang et al., 2016). In the mouse model, modulation of mGluR5 activity restored excessive grooming, instrumental learning, and striatal synaptic plasticity, suggesting that mGluR5 can be used to treat ASD. In the PFC of patients with ASD and SHANK3 mutant mice, histone lysine 4 demethylation (H3K4me2) was drastically reduced. Treatment of autism models with highly selective inhibitors of the H3K4me2 demethylase LSD1 results in a robust rescue of the core symptoms of ASD, revealing an important role of H3K4me2 abnormality in ASD pathophysiology and the therapeutic potential of targeting H3K4me2 demethylase LSD1 (Rapanelli et al., 2022). SHANK3-deficient neurons showed decreased ubiquitin-dependent degradation of CLK2 and impaired activation of Akt and mammalian target of rapamycin complex 1 (mTORC1) pathway proteins (Bidinosti et al., 2016). In Phelan–McDermid syndrome-derived neurons, pharmacological activation of Akt or inhibition of CLK2, and insulin-like growth factor-1 treatment restored excitatory synaptic activity in an Akt-dependent manner. In a SHANK3-deficient mouse model, CLK2 inhibition also restored normal sociability, providing novel mechanistic and potentially therapeutic insights into SHANK3-deficient signaling.
As previously stated, expressing wild-type NLGN3 after maturity can ameliorate the ASD phenotype in NLGN3-null mice. We tested whether the reversible features of ASD pathophysiology apply to an ASD model with other genetic abnormalities, using SHANK3 knockout mice. The answer was revealed by rearing transgenic mice with SHANK3 exons flipped upside down and then utilizing genetic manipulation to restore the normal orientation of the reverse exons once the mice had reached adulthood (Mei et al., 2016). Mice with inverted exons had biochemical, anatomical, and electrophysiological defects, and behavioral characteristics of ASD, which were confirmed to be substantially reversed when the exon was corrected. These findings suggest that aberrant development of the autistic brain caused by a congenital SHANK3 gene defect has considerable plasticity and can be substantially restored with later treatment.
Immune system dysregulation is seen in certain patients with ASD, and it is hypothesized that such a dysregulation could contribute to the development of ASD (Hsiao, 2013; Gottfried et al., 2015). Surprisingly, during systemic inflammation, some children with ASD appear to have improved autistic behavioral symptoms (Curran et al., 2007). When a mouse model deficient in CNTNAP2, FMR1, and SHANK3 was compared with an environmental model of a neurodevelopmental disorder in which mice were exposed to maternal immune activation during embryonic development, it was demonstrated that interleukin (IL)-17a production during inflammation directly affects the nervous system to improve the expression of social behavioral deficits (Reed et al., 2020). This research points to a unique neuroimmune mechanism underlying neurodevelopmental disorders, and IL-17a as a potential therapeutic target for autistic behavioral improvement.
Neuronal Signaling
Downstream and upstream signaling pathways of synaptic receptors and channels influence synapse formation, development, and elimination. The signaling molecules control the size and characteristics of neurotransmission and therefore regulate gene expression programs in neurons. This gene group includes kinases, phosphatases, and adaptors, and other components of neuronal signaling, with phosphatase and tensin homolog (PTEN) and mammalian target of rapamycin (mTOR) signal complexes serving as examples.
Phosphatase and Tensin Homolog
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that encodes dual-specificity phosphatase and suppresses the PI3K-mTOR signaling pathway (Luo et al., 2003). It regulates synaptogenesis, connections, and synaptic plasticity in the brain and is a critical modulator of synaptic function. PTEN deficiency is linked to oncological syndromes in humans, and social interaction and communication disorders, repetitive behaviors, and epilepsy in some circumstances (Rademacher and Eickholt, 2019). PTEN deficiency in the mouse CNS resulted in forebrain macrocephaly and neuronal hypertrophy, aberrant dendritic and axonal development, and synapse density (Kwon et al., 2006; Zhou and Parada, 2009; Takeuchi et al., 2013). These morphological changes are linked to upregulation of the Akt-mTOR pathways and downregulation of GSK3 caused by PTEN dysfunction, resulting in aberrant social interactions and excessive responses to sensory stimuli (Kwon et al., 2006).
Rapamycin effectively inhibits the overactivation of mTORC1 in the mTOR pathway, thereby improving neuronal hypertrophy and behavioral abnormalities in PTEN knockout mice (Zhou et al., 2009). In PTEN-null mice, mTORC2, which is less sensitive to rapamycin, is also overactivated. The behavioral and neurophysiological abnormalities of PTEN-null mice were improved when Rictor expression was suppressed using an antisense oligonucleotide to selectively disrupt the function of mTORC2; this finding suggests that mTORC1 and mTORC2 hyperactivities are the primary driver of the neuropathophysiology associated with PTEN deficiency (Chen et al., 2019).
Tuberous Sclerosis 1
The tumor suppressor gene Tuberous Sclerosis 1 (TSC1) forms a molecular complex with tuberin (TSC2) to inactivate the mTOR pathway (van Slegtenhorst et al., 1997). TSC1/2 is intimately associated with ASD, and ASD-like symptoms can be seen in approximately 30–60% of patients with TSC (Numis et al., 2011; Bahl et al., 2013). To determine the role of TSC1/2 in ASD, researchers developed a mouse model and found that heterozygous TSC1/2 mice showed an increase in the number of astrocytes, implying that the TSC1/2 complex is an essential regulator of astrocyte growth (Uhlmann et al., 2002). Conditional knockout of TSC1 resulted in brain enlargement, progressive epilepsy, and premature death (Zeng et al., 2008). TSC1-haploinsufficient and -null mice displayed impaired social behaviors, as well as an early increase in terminal axonal branching and synaptic density of PV-positive GABAergic interneurons (Amegandjin et al., 2021). These findings indicate that alterations in the mTOR signaling pathway caused by TSC1/2 mutations may be linked to the development of ASD, with the aim of determining whether rapamycin can improve ASD symptoms. In heterozygous or homozygous TSC1 mutant mice, rapamycin administration suppressed mTOR, resulting in reduced S6 phosphorylation, and alleviated aberrant social behaviors and seizures (Zeng et al., 2008; Tsai et al., 2012; Abs et al., 2013).
Ubiquitin-Protein Ligase E3A
Ubiquitin-Protein Ligase E3A (UBE3A) acts as an E3 ligase in the ubiquitin-proteasome pathway and is engaged in synaptic plasticity as well as transcriptional coactivation (Scheffner et al., 1993; Nawaz et al., 1999; Margolis et al., 2010; Sun et al., 2015). Angelman syndrome is caused by a maternal genetic loss of function of chromosome 15q containing the UBE3A gene, which is characterized by severe neurological and motor impairments (Albrecht et al., 1997; Cook et al., 1997). Transgenic mice with increased UBE3A levels, which show deficiencies in social interactions and stereotyped repetitive behaviors, were reared to investigate the function of this gene in brain developmental disorders (Smith et al., 2011). In UBE3A transgenic mice, there was a decrease in excitatory synaptic transmission owing to a reduction in the probability of presynaptic glutamate release and suppression of the post-synaptic action potential (Smith et al., 2011). Social behavioral defects were induced when UBE3A was overexpressed in the midbrain ventral tegmental area owing to downregulation of the glutamatergic synaptic organizer Cbln1, which is required for sociability; however, this was resolved by genetically restoring UBE3A in glutamatergic neurons in the region (Krishnan et al., 2017). The E:I balance in the mPFC of the Angelman syndrome model was considerably disrupted, and the interneuron excitability decreased. Increased Arc expression and decreased AMPA subtypes of glutamate receptors at excitatory synapses result from UBE3A deletion (Greer et al., 2010). Even when the brain is fully mature, physiological alterations in the mPFC caused by a defect in the UBE3A gene have been shown to be fully reversible upon gene reactivation; this implies that there is no substantial developmental window for reversing the physiological deficiencies detected in UBE3A conditional knockout mice (Rotaru et al., 2018).
Transcriptional and Chromatin Remodeling
Transcriptional and chromatin remodeling are other categories of genes linked to ASD. ATP-dependent chromatin remodelers and modifiers are crucial for chromatin control (Ronan et al., 2013). Chromatin remodelers adjust the status of nucleosomes to coordinate gene transcription, whereas chromatin modifiers add or remove acetylation, methylation, ubiquitylation, and phosphorylation of histone proteins. These mechanisms, which allow neuronal growth, differentiation, and connectivity, are regulated by chromatin remodeling (Ronan et al., 2013).
Chromodomain Helicase DNA Binding Protein 8
Chromodomain Helicase DNA Binding Protein 8 (CHD8) is an ATP-dependent chromatin remodeling protein that directly recruits and binds β-catenin from the promoter region of the β-catenin target gene, thereby repressing β-catenin-mediated transcription (Thompson et al., 2008). Disruptive CHD8 mutations are strongly linked to ASD and are characterized by macrocephaly, distinct facial features, and gastrointestinal motility (Bernier et al., 2014). ASD-like symptoms, including social behavioral problems, repetitive activities, and increased anxiety, are observed in heterozygous individuals with CHD8 mutations (Katayama et al., 2016). CHD8 knockdown resulted in the downregulation of several Wnt signaling pathway transducers and effectors, including FZD1, FZD2, DVL2, DVL3, and CTNNB1, leading to abnormal neural progenitor cell proliferation and differentiation during cortical development. Furthermore, CHD8 knockdown resulted in abnormalities in brain structure, increased anxiety levels, and aberrant social interactions in adult mice (Durak et al., 2016). Finally, whether augmenting Wnt signaling could alleviate the ASD phenotype caused by CHD8 knockdown was examined. In utero electroporation was used to co-express a β-catenin construct with CHD8 shRNA at E13. Genetic restoration of β-catenin rescued the anatomical, developmental, and behavioral defects caused by CHD8 knockdown, suggesting that the ASD-related phenotype induced by CHD8 dysfunction can be improved by enhancing the Wnt signaling pathway (Thompson et al., 2008).
Methyl-CpG Binding Protein 2
Methyl-CpG Binding Protein 2 (MECP2) is a chromatin-associated protein that binds to methylated CpG and has a role in transcriptional activation or repression of possibly hundreds of genes (Nan et al., 1997; Chahrour et al., 2008). It is an X-linked gene necessary for neural maturation in both mice and humans during embryonic development (Tate et al., 1996). MECP2 inhibits the expression of BDNF by specifically regulating BDNF promoter III (Chen et al., 2003). After depolarization, neurons release more BDNF owing to the dissociation of the MECP2-histone deacetylase-mSin3A inhibitory complex from the promoter due to reduced CpG methylation, indicating that MECP2 plays a key role in BDNF expression (Martinowich et al., 2003). It is clear from these findings that DNA methylation and related chromatin remodeling in response to neural activity play a significant role in gene transcription regulation (Chen et al., 2003; Martinowich et al., 2003). Rett syndrome and ASD, which are progressive neurodevelopmental diseases marked by an increase in repetitive stereotyped behaviors and loss of social, cognitive, and language abilities, have a high prevalence of MECP2 gene mutations (Zappella et al., 2001; Heindel et al., 2006; Swanberg et al., 2009).
Methyl-CpG Binding Protein 2 (MECP2)-null mice showed impaired nest-building activity, learning and memory deficits, and defective social interaction (Moretti et al., 2005). Deletion of MECP2 in forebrain GABAergic neurons recapitulated Rett syndrome and ASD symptoms, such as repetitive behaviors, impaired motor coordination, and altered sensorimotor arousal (Chao et al., 2010). In MECP2 mutant mice, tactile over-reactivity was reduced by restricted treatment with a GABAA receptor agonist in peripheral mechanosensory neurons, and chronic treatment improved anxiety-like behaviors and social impairments, but not memory deficits (Orefice et al., 2019). Insulin-like growth factor 1 is a pleiotropic growth factor that acts similarly to BDNF in the nervous system, stimulating the PI3K/pAkt/PSD-95 and MAPK pathways and increasing excitatory post-synaptic currents (Zheng and Quirion, 2004; Ramsey et al., 2005). Insulin-like growth factor-1 treatment normalized spine density and synaptic amplitude, increased PSD-96, and stabilized cortical plasticity in MECP2 mutant mice, which led to partial improvements in motor function and breathing patterns (Tropea et al., 2009).
Furthermore, MECP2 mutant mice had lower BDNF expression in the brain, and conditional deletion of BDNF in MECP2 mutants accelerated the development of RTT-like syndrome (Chang et al., 2006). Motor impairments were restored, and electrophysiological deficits were alleviated when BDNF expression increased in MECP2 mutant mice (Chang et al., 2006). Interestingly, MECP2-deficient mice had a higher rate of neuronal BDNF secretion but a lower overall BDNF content, implying that MECP2 deficiency disrupts the balance between BDNF protein expression and secretion, impairing synaptic BDNF signaling (Wang et al., 2006).
According to several studies, MECP2 loss suppresses the expression of UBE3A and another autism-associated gene (GABRB3) in the postnatal mammalian brain (Samaco et al., 2005; Hogart et al., 2007; Tanaka et al., 2012). MECP2 loss causes epigenetic abnormalities, which result in increased antisense RNA levels and decreased UBE3A production, implying a mechanistic link between MECP2 and UBE3A-induced autism (Makedonski et al., 2005).
MECP2 transgenic mice overexpressing the human MECP2 gene displayed MECP2 replication syndrome symptoms, and by 10 weeks of age, they had increased contextual learning and motor function, and improved synaptic plasticity in the hippocampus (Collins et al., 2004). Consistent with the results of the SHANK3 knockout model, when MECP2 expression was genetically restored in adult MECP2-null mice, all neurological abnormalities were reversed, and the phenotype significantly improved; this finding implies that aberrant brain development caused by MECP2 loss can be addressed even after a certain amount of time (Guy et al., 2007).
Discussion
Several investigations have been conducted on ASD, using genome sequencing to identify gene mutations in patients with ASD, non-invasive brain imaging studies, anatomical and neurophysiological studies, and most importantly, development and examination of animal models with candidate genes. Given the genetic manipulation processes used to create transgenic animals, generating the syndrome ASD model is rather straightforward and efficient. Since each gene associated with syndromic ASD is located in one of the principal signaling pathways in neurons, dysregulation of genes upstream or downstream of the pathway might cause similar abnormalities. As a result, the syndromic ASD model meets the requirements for representativeness and convenience in ASD research. However, it must be emphasized that research using the syndrome ASD model have limitations in presenting knowledge for non-syndromic ASD, which accounts for a high percentage of cases. So far, the etiology and pathophysiology of ASD remain poorly understood. The fact that the genetic causes of ASD are diverse is the primary reason for these difficulties. As ASD is caused by a variety of genetic abnormalities, even if effective medications are developed for one subtype of ASD, they may not be successful against another subtype of ASD. Furthermore, because the genetic causes in patients are diverse, it is possible to conclude that the effectiveness of a particular drug is low; this is because it is beneficial only in a portion of patients during the development phase. Consequently, this disease needs to be characterized more precisely to create medications for treatment and gaining better knowledge of the disease itself.
To date, E:I imbalance in cortical and subcortical neuronal circuits is one of the most prominent theories underlying ASD (Rubenstein and Merzenich, 2003). This theory is notable because it could explain the frequent observation of reduced GABAergic signaling in the developmental ASD brain (Cellot and Cherubini, 2014). Any defect in GABAergic inhibition increases noise in the cortex, basal ganglia, and hippocampus. Abnormal glutamatergic neurotransmission has also been reported in adult ASD brain regions, such as the primary sensorimotor cortex (He et al., 2021). Defects in the several genes mentioned above may cause alterations in the glutamatergic and GABAergic systems, resulting in an overall increase in the E:I ratio in ASD (Canitano and Palumbi, 2021). Glutamatergic hyperactivity is associated with NLGN, NRXN, and SHANK. Genetic defects in NLGN and NRXN reduce excitatory activity and the NMDA/AMPA ratio (Etherton et al., 2009). SHANK knockout results in impaired mGluR5-dependent modulation of neural network activity and reduced NMDA receptors, specifically at synapses (Verpelli et al., 2011). GABAnergic inhibitory dysfunction has been observed in MECP2 and CNTNAP2 deficiencies. IL-17a is expected to affect the firing pattern of certain neurons, which has been shown to improve behavioral abnormalities in animal models of ASD; however, further investigation is required to fully understand its therapeutic benefits. Based on these findings, it may be determined that a group of ASD-related genes causes the disease by disturbing the E:I balance. However, further research is necessary to determine if the deletion of these genes leads to a disturbance in the overall E:I balance owing to the impairment of the same type of neurons in the same brain region.
Furthermore, certain genes implicated in the development of ASD may cause the disease through a mechanism other than E:I imbalance. For example, the molecular mechanisms through which PTEN or mTOR complex components cause ASD are not fully understood. They may have a pathogenic mechanism distinct from E:I imbalance because they are engaged in the regulation of overall translation. Additionally, oxytocin is considered a promising treatment for ASD, although the precise mechanism of action is unclear, apart from the fact that it affects the limbic system to regulate sociability and anxiety. Therefore, distinct groups of ASD-associated genes may be involved in diverse neurophysiological features that are yet to be fully delineated, and it is difficult to predict the diversity of these genes.
Although the contribution of each gene to the development of ASD is small (less than 1%), it is determined that each gene is part of a distinct biochemical pathway and is engaged in the production of a common output (Voineagu et al., 2011). This finding suggests that therapeutics that involve biological pathways in which they converge, rather than individual genes, could be effective. Therapeutic medications discovered using this approach could be widely employed in the treatment of ASD caused by defects in a specific pathway, thereby benefiting a larger patient population.
In summary, ASD should be categorized according to the molecular pathways in which each disease gene converges, and common features and potential therapeutic targets for these groups should be determined through multi-faceted studies, including animal model studies. The number of ASD-associated genes in larger patient samples will continue to increase in the future. If researchers can demonstrate the molecular connections between ASD genes and identify their function in specific brain areas, ASD can be classified more effectively. This work can serve as the foundation for developing biomarkers for each subgroup and identifying therapeutic strategies.
Author Contributions
HL, JY, and MS developed the writing plan and drafted the manuscript. HL and MS developed the figure. All authors approved the final manuscript.
Funding
This work was supported by the National Research Foundation (NRF) of Korea (NRF-2020R1A6A3A01100511 and NRF-2021R1I1A3055750), the 2020 Yeungnam University Research Grant (220A580034) and KBRI basic research program through Korea Brain Research Institute funded by Ministry of Science and ICT (22-BR-02-03).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the members of Song’s lab for their valuable comments.
Abbreviations
ASD, autism-spectrum disorder; DSM, the Diagnostic and Statistical Manual of Mental Disorders; CDC, Centers for Disease Control and Prevention; USVs, ultrasonic vocalizations; SHANK, SH3 and multiple ankyrin repeat domains; NLGN, neuroligins; LTD, long-term synaptic depression; PVN, paraventricular nucleus; PFC, prefrontal cortex; mTORC1, mTOR complex 1; CB1, Cannabinoid receptor type 1; OXTR, Oxytocin Receptor; MC4R, Melanocortin 4 Receptor; PLC, Phospholipase C; GABAR, GABAA receptor; AMPAR, AMPA receptor; NMDAR, NMDA receptor; mGluR, metabolic glutamate receptor; IGF-1, Insulin-like growth factor 1; IL17A, Interleukin 17A; NCAM1, Neural Cell Adhesion Molecule 1; GRIP1, Glutamate Receptor Interacting Protein 1; SAPAP, Synapse-Associated Protein 90/Postsynaptic Density-95-Associated Protein; GKAP, guanylate-kinase-associated-protein; PSD-95, post-synaptic scaffolding protein 95 kDa; CNTNAP2, a contactin-associated protein-like 2 gene; PI3K, phosphoinositide-3 kinase; CLK2, CDC Like Kinase 2; PTEN, phosphatase and tensin homologs; TSC, tuberous sclerosis complex; mTOR, a mammalian target of rapamycin; CHD8, chromodomain helicase DNA binding protein 8; MeCP2, methyl CpG binding protein 2; LSD1, lysine-specific demethylase 1; UBE3A, Ubiquitin Protein Ligase E3A.
References
Abs, E., Goorden, S. M., Schreiber, J., Overwater, I. E., Hoogeveen-Westerveld, M., Bruinsma, C. F., et al. (2013). TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann. Neurol. 74, 569–579. doi: 10.1002/ana.23943
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 82, 150–159. doi: 10.1016/j.ajhg.2007.09.005
Albrecht, U., Sutcliffe, J. S., Cattanach, B. M., Beechey, C. V., Armstrong, D., Eichele, G., et al. (1997). Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 17, 75–78. doi: 10.1038/ng0997-75
Amaral, D. G., Schumann, C. M., and Nordahl, C. W. (2008). Neuroanatomy of autism. Trends Neurosci. 31, 137–145.
Amegandjin, C. A., Choudhury, M., Jadhav, V., Carrico, J. N., Quintal, A., Berryer, M., et al. (2021). Sensitive period for rescuing parvalbumin interneurons connectivity and social behavior deficits caused by TSC1 loss. Nat. Commun. 12:3653. doi: 10.1038/s41467-021-23939-7
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., et al. (2008). A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 82, 160–164. doi: 10.1016/j.ajhg.2007.09.015
Bahl, S., Chiang, C., Beauchamp, R. L., Neale, B. M., Daly, M. J., Gusella, J. F., et al. (2013). Lack of association of rare functional variants in TSC1/TSC2 genes with autism spectrum disorder. Mol. Autism 4:5. doi: 10.1186/2040-2392-4-5
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., et al. (2008). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173. doi: 10.1016/j.ajhg.2007.09.017
Bangash, M. A., Park, J. M., Melnikova, T., Wang, D., Jeon, S. K., Lee, D., et al. (2011). Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell 145, 758–772.
Baudouin, S. J., Gaudias, J., Gerharz, S., Hatstatt, L., Zhou, K., Punnakkal, P., et al. (2012). Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132. doi: 10.1126/science.1224159
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Bidinosti, M., Botta, P., Kruttner, S., Proenca, C. C., Stoehr, N., Bernhard, M., et al. (2016). CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science 351, 1199–1203. doi: 10.1126/science.aad5487
Bienvenu, O. J., Needham, D. M., and Hopkins, R. O. (2013). Diagnostic and statistical manual of mental disorders, Fifth Edition, and the impact of events scale-revised response. Chest 144, 1974–1975. doi: 10.1378/chest.13-1691
Bockers, T. M., Segger-Junius, M., Iglauer, P., Bockmann, J., Gundelfinger, E. D., Kreutz, M. R., et al. (2004). Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3’ untranslated region of Shank1 mRNA. Mol. Cell. Neurosci. 26, 182–190. doi: 10.1016/j.mcn.2004.01.009
Boeckers, T. M., Kreutz, M. R., Winter, C., Zuschratter, W., Smalla, K. H., Sanmarti-Vila, L., et al. (1999). Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J. Neurosci. 19, 6506–6518.
Bonaglia, M. C., Giorda, R., Borgatti, R., Felisari, G., Gagliardi, C., Selicorni, A., et al. (2001). Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 69, 261–268. doi: 10.1086/321293
Bourgeron, T. (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 16, 551–563. doi: 10.1038/nrn3992
Canitano, R., and Palumbi, R. (2021). Excitation/inhibition modulators in autism spectrum disorder: current clinical research. Front. Neurosci. 15:753274. doi: 10.3389/fnins.2021.753274
Cao, W., Lin, S., Xia, Q. Q., Du, Y. L., Yang, Q., Zhang, M. Y., et al. (2018). Gamma oscillation dysfunction in mPFC leads to social deficits in neuroligin 3 R451C knockin mice. Neuron 97, 1253.e7–1260.e7.
Cellot, G., and Cherubini, E. (2014). GABAergic signaling as therapeutic target for autism spectrum disorders. Front. Pediatr. 2:70. doi: 10.3389/fped.2014.00070
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chen, C. J., Sgritta, M., Mays, J., Zhou, H., Lucero, R., Park, J., et al. (2019). Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 25, 1684–1690. doi: 10.1038/s41591-019-0608-y
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Chung, C., Shin, W., and Kim, E. (2021). Early and late corrections in mouse models of autism spectrum disorder. Biol. Psychiatry 91, 934–944. doi: 10.1016/j.biopsych.2021.07.021
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., et al. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689. doi: 10.1093/hmg/ddh282
Cook, E. H. Jr., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 60, 928–934.
Courchesne, E., Mouton, P. R., Calhoun, M. E., Semendeferi, K., Ahrens-Barbeau, C., Hallet, M. J., et al. (2011). Neuron number and size in prefrontal cortex of children with autism. J. Am. Med. Assoc. 306, 2001–2010. doi: 10.1001/jama.2011.1638
Curran, L. K., Newschaffer, C. J., Lee, L. C., Crawford, S. O., Johnston, M. V., and Zimmerman, A. W. (2007). Behaviors associated with fever in children with autism spectrum disorders. Pediatrics 120, e1386–e1392. doi: 10.1542/peds.2007-0360
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Durak, O., Gao, F., Kaeser-Woo, Y. J., Rueda, R., Martorell, A. J., Nott, A., et al. (2016). Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 19, 1477–1488. doi: 10.1038/nn.4400
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Ehlers, M. D. (2002). Molecular morphogens for dendritic spines. Trends Neurosci. 25, 64–67. doi: 10.1016/s0166-2236(02)02061-1
Ellenbroek, B., and Youn, J. (2016). Rodent models in neuroscience research: is it a rat race? Dis. Models Mech. 9, 1079–1087. doi: 10.1242/dmm.026120
Etherton, M. R., Blaiss, C. A., Powell, C. M., and Sudhof, T. C. (2009). Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc. Natl. Acad. Sci. U.S.A. 106, 17998–18003. doi: 10.1073/pnas.0910297106
Filice, F., Vorckel, K. J., Sungur, A. O., Wohr, M., and Schwaller, B. (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 9:10. doi: 10.1186/s13041-016-0192-8
Gauthier, J., Champagne, N., Lafreniere, R. G., Xiong, L., Spiegelman, D., Brustein, E., et al. (2010). De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 107, 7863–7868. doi: 10.1073/pnas.0906232107
Gogolla, N., Takesian, A. E., Feng, G., Fagiolini, M., and Hensch, T. K. (2014). Sensory integration in mouse insular cortex reflects GABA circuit maturation. Neuron 83, 894–905. doi: 10.1016/j.neuron.2014.06.033
Gottfried, C., Bambini-Junior, V., Francis, F., Riesgo, R., and Savino, W. (2015). The impact of neuroimmune alterations in autism spectrum disorder. Front. Psychiatry 6:121. doi: 10.3389/fpsyt.2015.00121
Greer, P. L., Hanayama, R., Bloodgood, B. L., Mardinly, A. R., Lipton, D. M., Flavell, S. W., et al. (2010). The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716. doi: 10.1016/j.cell.2010.01.026
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
He, J. L., Oeltzschner, G., Mikkelsen, M., Deronda, A., Harris, A. D., Crocetti, D., et al. (2021). Region-specific elevations of glutamate + glutamine correlate with the sensory symptoms of autism spectrum disorders. Transl. Psychiatry 11:411. doi: 10.1038/s41398-021-01525-1
Heindel, J. J., McAllister, K. A., Worth, L. Jr., and Tyson, F. L. (2006). Environmental epigenomics, imprinting and disease susceptibility. Epigenetics 1, 1–6. doi: 10.4161/epi.1.1.2642
Hering, H., and Sheng, M. (2001). Dendritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci. 2, 880–888. doi: 10.1038/35104061
Hogart, A., Nagarajan, R. P., Patzel, K. A., Yasui, D. H., and Lasalle, J. M. (2007). 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 16, 691–703. doi: 10.1093/hmg/ddm014
Hsiao, E. Y. (2013). Immune dysregulation in autism spectrum disorder. Int. Rev. Neurobiol. 113, 269–302.
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Katayama, Y., Nishiyama, M., Shoji, H., Ohkawa, Y., Kawamura, A., Sato, T., et al. (2016). CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537, 675–679. doi: 10.1038/nature19357
Kemper, T. L., and Bauman, M. (1998). Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol. 57, 645–652.
Kreienkamp, H. J., Zitzer, H., Gundelfinger, E. D., Richter, D., and Bockers, T. M. (2000). The calcium-independent receptor for alpha-latrotoxin from human and rodent brains interacts with members of the ProSAP/SSTRIP/Shank family of multidomain proteins. J. Biol. Chem. 275, 32387–32390. doi: 10.1074/jbc.C000490200
Krishnan, V., Stoppel, D. C., Nong, Y., Johnson, M. A., Nadler, M. J., Ozkaynak, E., et al. (2017). Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature 543, 507–512. doi: 10.1038/nature21678
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., et al. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. doi: 10.1016/j.neuron.2006.03.023
Lewis, M. H., Tanimura, Y., Lee, L. W., and Bodfish, J. W. (2007). Animal models of restricted repetitive behavior in autism. Behav. Brain Res. 176, 66–74. doi: 10.1016/j.bbr.2006.08.023
Lim, S., Sala, C., Yoon, J., Park, S., Kuroda, S., Sheng, M., et al. (2001). Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Mol. Cell. Neurosci. 17, 385–397. doi: 10.1006/mcne.2000.0940
Lu, C., Chen, Q., Zhou, T., Bozic, D., Fu, Z., Pan, J. Q., et al. (2016). Micro-electrode array recordings reveal reductions in both excitation and inhibition in cultured cortical neuron networks lacking Shank3. Mol. Psychiatry 21, 159–168. doi: 10.1038/mp.2015.173
Luo, J., Manning, B. D., and Cantley, L. C. (2003). Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4, 257–262. doi: 10.1016/s1535-6108(03)00248-4
Maenner, M. J., Shaw, K. A., Bakian, A. V., Bilder, D. A., Durkin, M. S., Esler, A., et al. (2021). Prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveil. Summar. 70, 1–15. doi: 10.15585/mmwr.ss7011a1
Makedonski, K., Abuhatzira, L., Kaufman, Y., Razin, A., and Shemer, R. (2005). MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 14, 1049–1058. doi: 10.1093/hmg/ddi097
Margolis, S. S., Salogiannis, J., Lipton, D. M., Mandel-Brehm, C., Wills, Z. P., Mardinly, A. R., et al. (2010). EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 143, 442–455. doi: 10.1016/j.cell.2010.09.038
Martella, G., Meringolo, M., Trobiani, L., De Jaco, A., Pisani, A., and Bonsi, P. (2018). The neurobiological bases of autism spectrum disorders: the R451C-neuroligin 3 mutation hampers the expression of long-term synaptic depression in the dorsal striatum. Eur. J. Neurosci. 47, 701–708. doi: 10.1111/ejn.13705
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Mei, Y., Monteiro, P., Zhou, Y., Kim, J. A., Gao, X., Fu, Z., et al. (2016). Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484. doi: 10.1038/nature16971
Moretti, P., Bouwknecht, J. A., Teague, R., Paylor, R., and Zoghbi, H. Y. (2005). Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 205–220. doi: 10.1093/hmg/ddi016
Moy, S. S., Nadler, J. J., Poe, M. D., Nonneman, R. J., Young, N. B., Koller, B. H., et al. (2008). Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav. Brain Res. 188, 178–194. doi: 10.1016/j.bbr.2007.10.029
Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., et al. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. doi: 10.1016/s0896-6273(00)80809-0
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/s0092-8674(00)81887-5
Nawaz, Z., Lonard, D. M., Smith, C. L., Lev-Lehman, E., Tsai, S. Y., Tsai, M. J., et al. (1999). The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19, 1182–1189. doi: 10.1128/MCB.19.2.1182
Numis, A. L., Major, P., Montenegro, M. A., Muzykewicz, D. A., Pulsifer, M. B., and Thiele, E. A. (2011). Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology 76, 981–987. doi: 10.1212/WNL.0b013e3182104347
Orefice, L. L., Mosko, J. R., Morency, D. T., Wells, M. F., Tasnim, A., Mozeika, S. M., et al. (2019). Targeting peripheral somatosensory neurons to improve tactile-related phenotypes in ASD models. Cell 178, 867.e24–886.e24. doi: 10.1016/j.cell.2019.07.024
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–U136. doi: 10.1038/nature10989
Peca, J., Feliciano, C., Ting, J. T., Wang, W., Wells, M. F., Venkatraman, T. N., et al. (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. doi: 10.1038/nature09965
Peixoto, R. T., Wang, W., Croney, D. M., Kozorovitskiy, Y., and Sabatini, B. L. (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(-/-) mice. Nat. Neurosci. 19, 716–724. doi: 10.1038/nn.4260
Penagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. doi: 10.1016/j.cell.2011.08.040
Penagarikano, O., Lazaro, M. T., Lu, X. H., Gordon, A., Dong, H., Lam, H. A., et al. (2015). Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 7:271ra8. doi: 10.1126/scitranslmed.3010257
Pensado-Lopez, A., Veiga-Rua, S., Carracedo, A., Allegue, C., and Sanchez, L. (2020). Experimental models to study autism spectrum disorders: hiPSCs, rodents and Zebrafish. Genes 11:1376. doi: 10.3390/genes11111376
Poliak, S., Gollan, L., Martinez, R., Custer, A., Einheber, S., Salzer, J. L., et al. (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. doi: 10.1016/s0896-6273(00)81049-1
Rademacher, S., and Eickholt, B. J. (2019). PTEN in Autism and neurodevelopmental disorders. Cold Spring Harb. Perspect. Med. 9:a036780. doi: 10.1101/cshperspect.a036780
Ramsey, M. M., Adams, M. M., Ariwodola, O. J., Sonntag, W. E., and Weiner, J. L. (2005). Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J. Neurophysiol. 94, 247–254. doi: 10.1152/jn.00768.2004
Rapanelli, M., Williams, J. B., Ma, K., Yang, F., Zhong, P., Patel, R., et al. (2022). Targeting histone demethylase LSD1 for treatment of deficits in autism mouse models. Mol. Psychiatry [Epub ahead of print]. doi: 10.1038/s41380-022-01508-8
Reed, M. D., Yim, Y. S., Wimmer, R. D., Kim, H., Ryu, C., Welch, G. M., et al. (2020). IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature 577, 249–253. doi: 10.1038/s41586-019-1843-6
Reissner, C., Klose, M., Fairless, R., and Missler, M. (2008). Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc. Natl. Acad. Sci. U.S.A. 105, 15124–15129. doi: 10.1073/pnas.0801639105
Ronan, J. L., Wu, W., and Crabtree, G. R. (2013). From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359. doi: 10.1038/nrg3413
Rotaru, D. C., van Woerden, G. M., Wallaard, I., and Elgersma, Y. (2018). Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J. Neurosci. 38, 8011–8030. doi: 10.1523/JNEUROSCI.0083-18.2018
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Rylaarsdam, L., and Guemez-Gamboa, A. (2019). Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 13:385. doi: 10.3389/fncel.2019.00385
Sacai, H., Sakoori, K., Konno, K., Nagahama, K., Suzuki, H., Watanabe, T., et al. (2020). Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun. 11:5140. doi: 10.1038/s41467-020-18861-3
Sala, C., Piech, V., Wilson, N. R., Passafaro, M., Liu, G., and Sheng, M. (2001). Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. doi: 10.1016/s0896-6273(01)00339-7
Samaco, R. C., Hogart, A., and LaSalle, J. M. (2005). Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492. doi: 10.1093/hmg/ddi045
Scattoni, M. L., Crawley, J., and Ricceri, L. (2009). Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci. Biobehav. Rev. 33, 508–515. doi: 10.1016/j.neubiorev.2008.08.003
Scheffner, M., Huibregtse, J. M., Vierstra, R. D., and Howley, P. M. (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. doi: 10.1016/0092-8674(93)90384-3
Shcheglovitov, A., Shcheglovitova, O., Yazawa, M., Portmann, T., Shu, R., Sebastiano, V., et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. doi: 10.1038/nature12618
Sheng, M., and Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847. doi: 10.1146/annurev.biochem.76.060805.160029
Smith, S. E., Zhou, Y. D., Zhang, G., Jin, Z., Stoppel, D. C., and Anderson, M. P. (2011). Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra97. doi: 10.1126/scitranslmed.3002627
Smogavec, M., Cleall, A., Hoyer, J., Lederer, D., Nassogne, M. C., Palmer, E. E., et al. (2016). Eight further individuals with intellectual disability and epilepsy carrying bi-allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J. Med. Genet. 53, 820–827. doi: 10.1136/jmedgenet-2016-103880
Sparks, B. F., Friedman, S. D., Shaw, D. W., Aylward, E. H., Echelard, D., Artru, A. A., et al. (2002). Brain structural abnormalities in young children with autism spectrum disorder. Neurology 59, 184–192. doi: 10.1212/wnl.59.2.184
Sun, J., Zhu, G., Liu, Y., Standley, S., Ji, A., Tunuguntla, R., et al. (2015). UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep. 12, 449–461. doi: 10.1016/j.celrep.2015.06.023
Swanberg, S. E., Nagarajan, R. P., Peddada, S., Yasui, D. H., and LaSalle, J. M. (2009). Reciprocal co-regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum. Mol. Genet. 18, 525–534. doi: 10.1093/hmg/ddn380
Takeuchi, K., Gertner, M. J., Zhou, J., Parada, L. F., Bennett, M. V., and Zukin, R. S. (2013). Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc. Natl. Acad. Sci. U.S.A. 110, 4738–4743. doi: 10.1073/pnas.1222803110
Tanaka, M., DeLorey, T. M., Delgado-Escueta, A., and Olsen, R. W. (2012). “GABRB3, epilepsy, and neurodevelopment,” in Jasper’s Basic Mechanisms of the Epilepsies, eds J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (Bethesda, MD: Oxford University Press).
Tate, P., Skarnes, W., and Bird, A. (1996). The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat. Genet. 12, 205–208. doi: 10.1038/ng0296-205
Thompson, B. A., Tremblay, V., Lin, G., and Bochar, D. A. (2008). CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell. Biol. 28, 3894–3904. doi: 10.1128/MCB.00322-08
Tobaben, S., Sudhof, T. C., and Stahl, B. (2000). The G protein-coupled receptor CL1 interacts directly with proteins of the Shank family. J. Biol. Chem. 275, 36204–36210. doi: 10.1074/jbc.M006448200
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034. doi: 10.1073/pnas.0812394106
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Tu, J. C., Xiao, B., Naisbitt, S., Yuan, J. P., Petralia, R. S., Brakeman, P., et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592. doi: 10.1016/s0896-6273(00)80810-7
Uhlmann, E. J., Apicelli, A. J., Baldwin, R. L., Burke, S. P., Bajenaru, M. L., Onda, H., et al. (2002). Heterozygosity for the tuberous sclerosis complex (TSC) gene products results in increased astrocyte numbers and decreased p27-Kip1 expression in TSC2+/- cells. Oncogene 21, 4050–4059. doi: 10.1038/sj.onc.1205435
van Slegtenhorst, M., de Hoogt, R., Hermans, C., Nellist, M., Janssen, B., Verhoef, S., et al. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. doi: 10.1126/science.277.5327.805
Verpelli, C., Dvoretskova, E., Vicidomini, C., Rossi, F., Chiappalone, M., Schoen, M., et al. (2011). Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 286, 34839–34850. doi: 10.1074/jbc.M111.258384
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wang, H., Chan, S. A., Ogier, M., Hellard, D., Wang, Q., Smith, C., et al. (2006). Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J. Neurosci. 26, 10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006
Wang, X., Bey, A. L., Katz, B. M., Badea, A., Kim, N., David, L. K., et al. (2016). Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat. Commun. 7:11459. doi: 10.1038/ncomms11459
Wang, X., McCoy, P. A., Rodriguiz, R. M., Pan, Y., Je, H. S., Roberts, A. C., et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 20, 3093–3108. doi: 10.1093/hmg/ddr212
Wohr, M., and Scattoni, M. L. (2013). Behavioural methods used in rodent models of autism spectrum disorders: current standards and new developments. Behav. Brain Res. 251, 5–17. doi: 10.1016/j.bbr.2013.05.047
Yang, M., Silverman, J. L., and Crawley, J. N. (2011). Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci. 56, 8.26.1–8.26.16. doi: 10.1002/0471142301.ns0826s56
Yao, I., Hata, Y., Hirao, K., Deguchi, M., Ide, N., Takeuchi, M., et al. (1999). Synamon, a novel neuronal protein interacting with synapse-associated protein 90/postsynaptic density-95-associated protein. J. Biol. Chem. 274, 27463–27466. doi: 10.1074/jbc.274.39.27463
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Yoo, H. (2015). Genetics of autism spectrum disorder: current status and possible clinical applications. Exp. Neurobiol. 24, 257–272. doi: 10.5607/en.2015.24.4.257
Zaccaria, K. J., Lagace, D. C., Eisch, A. J., and McCasland, J. S. (2010). Resistance to change and vulnerability to stress: autistic-like features of GAP43-deficient mice. Genes Brain Behav. 9, 985–996. doi: 10.1111/j.1601-183X.2010.00638.x
Zappella, M., Meloni, I., Longo, I., Hayek, G., and Renieri, A. (2001). Preserved speech variants of the Rett syndrome: molecular and clinical analysis. Am. J. Med. Genet. 104, 14–22. doi: 10.1002/ajmg.10005
Zeng, L. H., Xu, L., Gutmann, D. H., and Wong, M. (2008). Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 63, 444–453. doi: 10.1002/ana.21331
Zheng, W. H., and Quirion, R. (2004). Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 89, 844–852. doi: 10.1111/j.1471-4159.2004.02350.x
Zhou, J., Blundell, J., Ogawa, S., Kwon, C. H., Zhang, W., Sinton, C., et al. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 29, 1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009
Zhou, J., and Parada, L. F. (2009). A motor driving PTEN. Nat. Cell Biol. 11, 1177–1179. doi: 10.1038/ncb1009-1177
Zitzer, H., Honck, H. H., Bachner, D., Richter, D., and Kreienkamp, H. J. (1999a). Somatostatin receptor interacting protein defines a novel family of multidomain proteins present in human and rodent brain. J. Biol. Chem. 274, 32997–33001. doi: 10.1074/jbc.274.46.32997
Keywords: autism spectrum disorder, genetic mice model, pathophysiology, pharmacological restoration, genetic restoration
Citation: Lim HK, Yoon JH and Song M (2022) Autism Spectrum Disorder Genes: Disease-Related Networks and Compensatory Strategies. Front. Mol. Neurosci. 15:922840. doi: 10.3389/fnmol.2022.922840
Received: 18 April 2022; Accepted: 17 May 2022;
Published: 03 June 2022.
Edited by:
Byung Chang Suh, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South KoreaCopyright © 2022 Lim, Yoon and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong Hyuk Yoon, jhyoon@kbri.re.kr; Minseok Song, minseok@yu.ac.kr