 Indira Tirado-Hurtado
Indira Tirado-Hurtado Williams Fajardo
Williams Fajardo Joseph A. Pinto
Joseph A. Pinto- 1Unidad de Investigación Básica y Traslacional, Oncosalud-AUNA, Lima, Peru
- 2Escuela de Medicina Humana, Universidad Privada San Juan Bautista, Lima, Peru
DNA damage inducible transcript 4 (DDIT4) gene is expressed under stress situations turning off the metabolic activity triggered by the mammalian target of rapamycin (mTOR). Several in vitro and in vivo works have demonstrated the ability of DDIT4 to generate resistance to cancer therapy. The link between the metabolism suppression and aggressiveness features of cancer cells remains poorly understood since anti-mTOR agents who are part of the repertoire of drugs used for systemic treatment of cancer achieving variable results. Interestingly, the high DDIT4 expression is associated with worse outcomes compared to tumors with low DDIT4 expression, seen in a wide variety of solid and hematological tumors, which suggests the driver role of this gene and provide the basis to target it as part of a new therapeutic strategy. In this review, we highlight our current knowledge about the biology of DDIT4 and its role as a prognostic biomarker, encompassing the motives for the development of target drugs against DDIT4 as a better target than mTOR inhibitors.
Background
Cancer is a complex disease arising from the gradual accumulation of genetic changes resulting in the reprogramming of key cellular processes well described in “The Hallmarks of Cancer” written by Hanahan and Weinberg (1). Structural and functional alterations in driver genes and entire pathways to fulfill the nutritional requirements are responsible for this reprogramming, and although their mechanisms are not fully known, various drugs have been developed to target actionable mutations (2, 3).
One attractive therapeutic strategy is the inhibition of the mammalian target of rapamycin (mTOR), as well as various downstream and upstream signaling molecules (4). The mTOR pathway has evolved as nutrient sensing to promote cell proliferation under adequate nutritional and environmental conditions (5). The activation of mTOR depends on the formation of two complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) that are activated in distinct ways. mTORC1 controls the protein synthesis and cell survival through the phosphorylation of its substrates, 4EBP1, p70S6K and factor 4G. mTORC1 is inactivated by rapamycin but is activated by growth factors, nutrients, energy and stress signals, and essential signaling pathways (PI3K, MAPK, and AMPK). In contrast to mTORC1, mTORC2 is not inactivated by rapamycin and generally, it is not affected by nutrients and energy signals. mTORC2 regulates cytoskeleton organization and promotes cell survival through the phosphorylation of protein kinase B (Akt) and protein kinase C (6–8). The key role of mTOR in these processes explains its association in pathologies such as cancer (9, 10).
Several works and recent cancer genomic projects described high rates of mutations in genes involved in the mTOR pathway, including PI3K, PTEN, AKT, and S6K1, 4EBP1, and EIF4E. Based in this data, mTOR inhibitors (rapamycin and its analogs) have become attractive therapeutic agents tested in several clinical trials, as single agents or in combination with other types of systemic treatment (9). Unfortunately, some inhibitors of mTOR have shown lack of efficacy, including the rapamycin (11).
The DNA damage inducible transcript 4 (DDIT4) is induced by cellular stress conditions and regulates the mTOR activity (12), and also its abnormal expression has been linked to multiple diseases, including malignant tumors (13–18).
DDIT4 Gene
This gene was simultaneously discovered and cloned in 2002 by two independent research groups. Shoshani et al. using a microarray hybridization technique to investigate the hypoxia-dependent gene expression in rat glioma C6 cells reported a gene highly upregulated in response to hypoxia-inducible factor 1 (HIF-1) and regulating the generation of cellular reactive oxygen species (ROS). This gene was designated RTP801 (19). Concurrently, Ellisen et al. identified a new p53 target induced by DNA damage and regulated by p63 during embryogenesis and epidermal differentiation. In this study, this gene was alternatively designated REDD1 (regulated in development and DNA damage response 1), which was also involved in the generation of ROS (20).
Contemporarily with these works, Wang et al. analyzed which genes are potentially involved in regulating glucocorticoid-induced apoptosis in lymphoid cells. Through an oligonucleotide microarray analysis, they discovered a novel dexamethasone-induced gene designated Dig2 whose expression is significantly induced in cell lines of murine T-cell lymphoma and in normal mouse thymocytes (21). The official name given by the HUGO Gene Nomenclature Committee was DDIT4.
DDIT4 is located on chromosome 10 (10q22.1) and has a length of 2.1 kb, containing three exons and two introns (19, 20). DDIT4 has three splice variants (one it is the protein coding with 232 amino acids and the others are retained introns), 95 orthologs, one paralog (DDIT4L, DNA damage inducible transcript 4 like) and is associated with one phenotype (22). Because of this, it is presumed that it was present in the common ancestor of animals (23).
DDIT4 is ubiquitously expressed at low levels in most adult tissues (Figure 1) (19). The DDIT4 expression is induced by multiple cellular stresses, such as hypoxia (19, 24), ionizing radiation (IR) (20), methyl methane sulfonate (MMS) (25), heat shock (21), and energy depletion (12). Moreover, it is also upregulated by other chemical molecules, such as glucocorticoids (21, 26, 27), dopaminergic neurotoxins (28), endoplasmic reticulum stress inducers (21, 29), DNA damage agent etoposide (21), and arsenite (30). Conversely, DDIT4 expression decreases by testosterone, acute resistance exercise, refeeding/nutrient consumption, and suppressed mTORC1 (31).
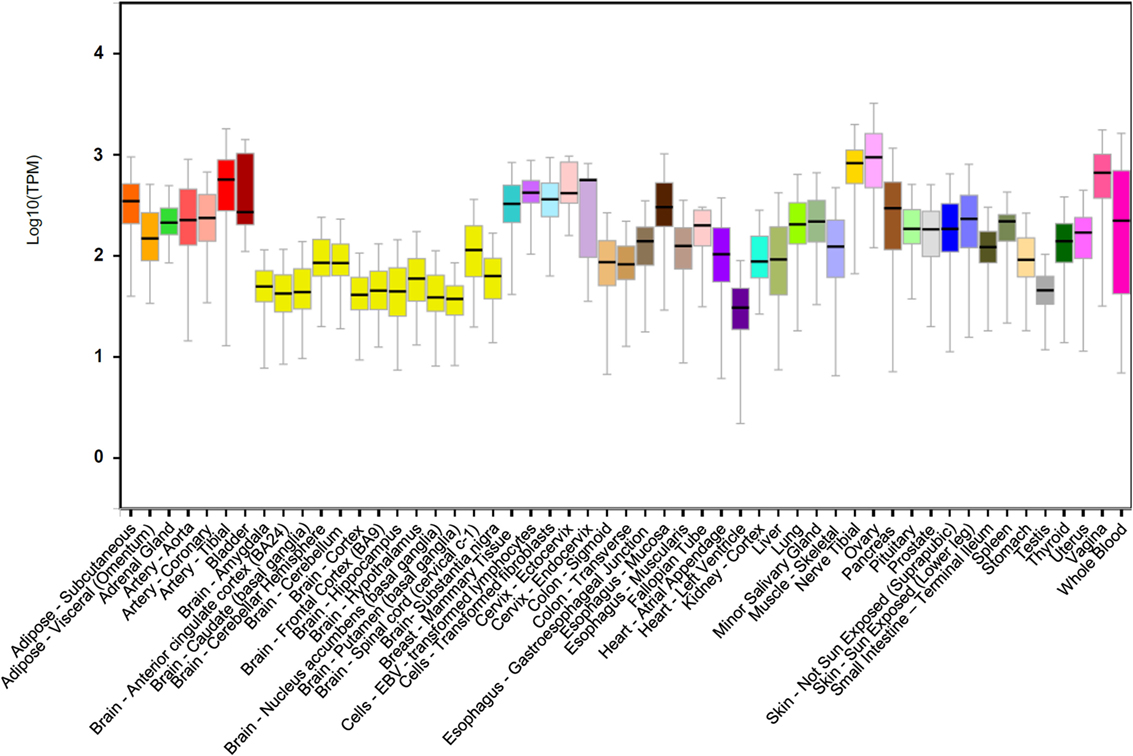
Figure 1. DNA damage inducible transcript 4 (DDIT4) gene expression. DDIT4 is ubiquitously expressed in multiple human tissues (32).
DDIT4 Protein
With an estimated half-life of approximately 5 min (33, 34), DDIT4 is a highly conserved protein composed of 232 amino acids, rich in leucine (17%) and contains conserved 9- or 10-serine stretches at its N-terminal end. Its molecular weight is 25 kDa, but it migrates around 35 kDa on Western blot because of its multiple lysine residues at the carboxyl terminus (19, 20). In addition, DDIT4 is mainly present in the cytoplasm and the nucleus (20, 30), but it has also been observed in membranes (35).
The crystal structure of DDIT4 (PDB ID# 3LQ9) shows that this protein has two chains (A and B), where each chain has antiparallel α-helices followed by four β-strands with two highly conserved residues (residues 138–141 and 218–225) that might be essential for the activity of the protein (Figure 2). Amino acids in position 85–193 and 207–225 correspond to linear segments required for its function, separated by a dispensable region. The extreme N-terminus has 84 amino acids residues poorly preserved among species and dispensable for the function of the protein. By contrast, the C-terminal region is highly conserved and essential for the correct function of DDIT4. Internal deletions and NAAIRS (the sequence Asn–Ala–Ala–Ile–Arg–Ser) substitutions are poorly tolerated because this causes the protein to change to an unphysiological conformation (36).
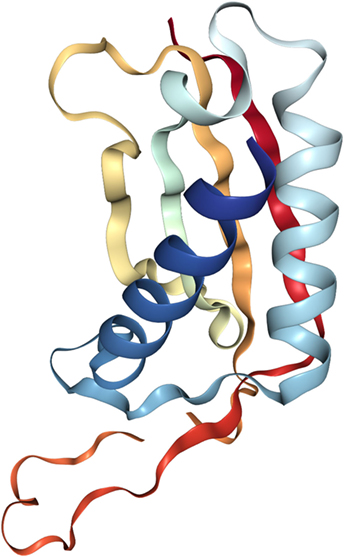
Figure 2. Crystal structure of the human protein DNA damage inducible transcript 4 (PDB ID# 3LQ9). Both of their chains have antiparallel α-helices followed by four β-strands.
Regulation of DDIT4
DDIT4 expression is upregulated by several transcription factors in response to different stressors (37). For example, under hypoxic conditions, HIF-1 increases the transcription of DDIT4 by binding to a hypoxia response element of the DDIT4 promoter causing downregulation of mTOR (19, 24). Telomere destabilization by DNA-damaging agents also increases DDIT4 expression through multiple pathways. In mouse embryo fibroblasts (MEFs), IR induces DDIT4 expression through p53 pathway in a dependent manner (20). Similarly, in HaCaT human keratinocytes, MMS induces DDIT4 expression through Elk-1 pathway and CCAAT/enhancer-binding protein (C/EBP) but in a p53-independent manner (25). Another transcription factor is the nuclear factor of activated T-cells 3, whose overexpression increases c-Myc (mTOR downstream target) and MUC2 expression (a marker of goblet cell differentiation) through the induction of DDIT4 expression; important process to the regulation of intestinal cell differentiation (38).
MicroRNAs, small and highly conserved non-coding RNA molecules, are also involved in tumorigenesis by regulating (at posttranscriptional level) specific oncogenes and tumor suppressor genes (39–41), such as DDIT4. miR-221 overexpression contributes to liver tumorigenesis through the cyclin-dependent kinase inhibitor p27 (Kip1-CDKN1B) and/or DDIT4 downregulation (42). miR-495, directly upregulated by the transcription factors E12/E47 in breast cancer stem cells promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and DDIT4 (43). miR-30c plays a key role in radiation-induced cell damage because, maybe in part, it downregulates DDIT4 expression in human hematopoietic and osteoblast cells after gamma-irradiation (44). miR-630 has a bimodal role in the regulation of apoptosis in response to DNA damage; it promotes apoptosis by downregulation of cell cycle kinase 7 kinase, and on the other hand, it reduces apoptosis by downregulating apoptotic activators, such as DDIT4, PARP3, EP300, and p53 (45).
At posttranslational level, DDIT4 is quickly degraded by the ubiquitin-proteasome system to allow cells to restore mTOR signaling once the stress conditions have been mitigated. One of the models reported by Katiyar et al. consists in the phosphorylation of DDIT4 by GSK3-β, which causes the recruitment of the Cullin 4A (CUL4A)–DNA damage-binding protein 1–regulator of cullins 1–β-transducin repeat containing protein (β-TRCP) E3 ligase complex, that results in DDIT4 ubiquitination and degradation by the proteasome (34). Amplification and overexpression of CUL4A have been observed in primary breast cancers (46) and others types of cancers such as hepatocellular carcinomas (47), so it could be considered as a potential predictive and prognostic indicator of some cancers. In addition, increased β-TRCP mRNA and protein expression have also been found in colorectal and pancreatic cancers (48, 49). By contrast, Tan and Hagen reported that there is an alternative E3 ligase (currently unknown) responsible for both basal DDIT4 ubiquitination and ubiquitination that is induced upon mTORC1 inhibition (50). These processes sustain that the dysregulation of DDIT4 degradation could be a common event that elevates mTOR signaling during tumor development.
DDIT4 Inhibits the Activity of mTOR VIA Tuberous Sclerosis Complex (TSC1/TSC2 Complex)
All the stressors mentioned above, via different transcription factors, elevate the DDIT4 expression to fulfill its main function, inhibit mTORC1 to regulate key cellular processes, such as cell growing, proliferation, and survival (12, 24). This inhibitory effect was initially identified in Drosophila, where expression of Scylla (homologous protein of DDIT4 in Drosophila) suppress the phosphorylation of S6K (a known substrate of TOR) (51); a similar process that was later confirmed in mammalian cells (52). In addition, TSC1/TSC2 complex does not interact directly with mTORC1, but it functions as a GTPase, inactivating to Rheb, converting Rheb-GTP into Rheb-GDP, unable to activate the mTORC1 complex (53).
Gordon et al. proposed two models of the effect of DDIT4 in the mTOR pathway. One model proposes that DDIT4 competes with TSC2 to bind with the 14-3-3 proteins. DDIT4 expression increases causing the dissociation of the 14-3-3 proteins with TSC2, so TSC2 is released to form a functional TSC1/TSC2 complex that inhibits mTORC1 activity (31, 35). However, functional and structural analysis has concluded that it is unlikely that DDIT4 interacts directly with 14-3-3 proteins, discarding this model (36). The other model proposes that phosphatase-2A recruit to Akt causing the reduction of Akt phosphorylation, which in turn causes the reduction of phosphorylation of TSC2 and its induction. The TSC1/TSC2 complex is formed and subsequently represses mTORC1 activity. By contrast, TSC1/TSC2 complex positively regulates mTORC2 through association with rapamycin-insensitive companion of mTOR (31) (Figures 3 and 4).
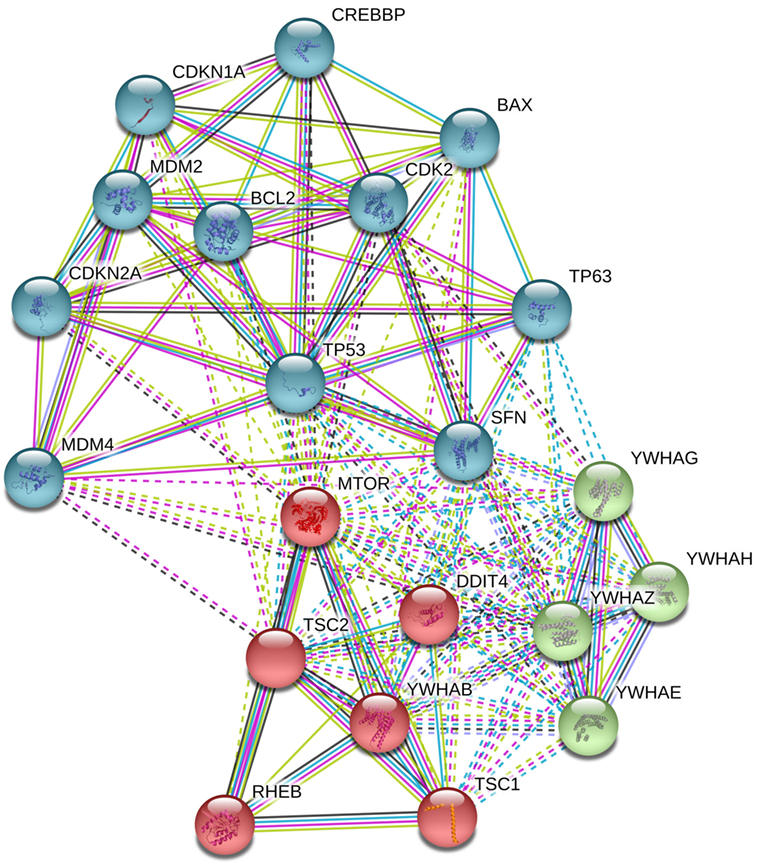
Figure 3. Analysis in STRING-DB v.10.5 describes that DNA damage inducible transcript 4 (DDIT4) is involved in the regulation of at least three clusters of proteins, (i) mammalian target of rapamycin (mTOR) pathway proteins (in red), (ii) p53 pathway (in sky blue), and (iii) 14-3-3 proteins (in green).
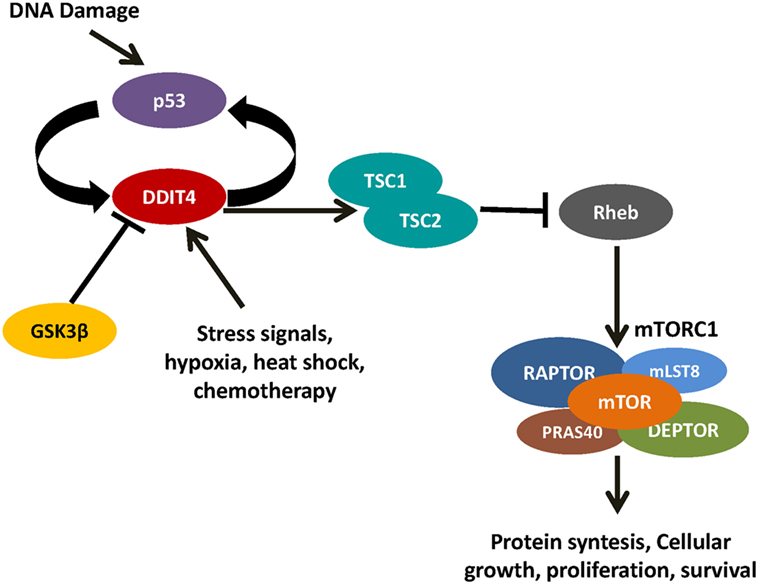
Figure 4. DNA damage inducible transcript 4 (DDIT4) controls mammalian target of rapamycin (mTOR) by the activation of the TSC1/TSC2 complex. When the TSC1/TSC2 complex inactivates Rheb, it is unable to activate mTOR complex 1.
DDIT4 and Autophagy
Autophagy is a catabolic mechanism of cellular adaptation to nutrients deprivation in which cellular components are degraded to its elementary molecules and recycled to promote the cell survival (54). In addition, autophagy participates in the removal of old or damaged cellular organelles and has been associated with the survival of cancer cells (55). Autophagy is the opposite response to cell growth and proliferation induced by mTOR activity. Under favorable nutritional conditions, the complex mTORC1 represses autophagy by phosphorylation of the ULK protein complex turning it in an open structural conformation (inactive form). DDIT4-mediated mTOR repression produces the lack of phosphorylation of the ULK complex, turning the complex in a closed structural conformation (active form) activating the mechanism of autophagy and triggering the formation of the autophagosome (56).
Role of DDIT4 in Cancer
Dysfunction of DDIT4 has been associated with multiple diseases, such as neurodegenerative disorders (28, 57), ischemic proliferative retinopathy (58), preeclampsia (59), diabetes, and other degenerative pathologies, such as cancer (10). Results of in vitro studies suggest that DDIT4 could have a context-dependent contrasting role in cancer as oncogene or tumor suppressor (19, 60).
Hyperactivation of the PI3K-Akt-mTOR signaling is a common alteration in cancer. This pathway regulates cellular processes involved in cell growth, proliferation, motility, survival, and apoptosis (61). In normal cells, PI3K-Akt-mTOR signaling is controlled by PTEN through PI3K dephosphorylation ensuring a transient and controlled activation. On the other hand, PTEN inactivation causes a chronic activation of PI3K and its downstream effectors, such as Akt, promoting the cell cycle progression, survival, and decreased apoptosis (62).
To know which genes are involved in the growth of cancer cells via chronically activated PIK-3 (such as in PTEN−/− cells), Schwarzer et al. compared PIK3 expression between cells that have active PIK-3 versus cells in which it is silenced. They found that DDIT4 mRNA was significantly downregulated in prostate cancer cells (PC-3) using LY294002 (2-morpholin-4-yl-8-phenylchromen-4-one) or rapamycin, both inhibitors of PI3K, in combination with matrigel-based 3D culture system. By contrast, DDIT4 was overexpressed under hypoxic conditions in a HIF-1α dependent manner. They also verified their results with other methods, as, for example, inhibiting the function of DDIT4 employing antisense molecules or interference RNA, which indicates that DDIT4 is a transcriptional downstream target in the PI3K pathway and essential for invasive growth of PC-3 both in vitro and in vivo (13).
On the other hand, DDIT4 has a key role in RAS signaling to transform human ovarian epithelial cells. DDIT4 is overexpressed in RAS-transformed human ovarian epithelial cells lines T29 and T80 promoting cell proliferation and colony formation. DDIT4 is overexpressed after activation of RAS oncogene, increasing levels of anti-apoptotic proteins and at the same time decreasing expression of pro-apoptotic proteins (60, 63).
Several works have described an involvement of DDIT4 in the breast cancer biology, while its expression seems to have different patterns among breast cancer subtypes. Koo and Jung characterized the expression of proteins involved in mTOR and hypoxia pathway with staining with immunohistochemistry where immunophenotypes of breast cancer were determined by the evaluation of estrogen and progesterone receptors and HER2 while breast papilloma samples were included as controls (64).
In this study, Glut-1 and HIF-1α had higher expression in triple-negative breast cancer (TNBC) and HER2 phenotypes than in the luminal A and B phenotypes. These findings could be explained by the greater hypoxia conditions present in TNBC and HER2 tumors. Likewise, Ki-67 expression in TNBC and HER2 was higher than in other phenotypes or papilloma. Interestingly, downregulation of DDIT4 does not lead to a negative feedback to HIF-1 α, so the tumorigenesis mediated by HIF-1α is constant. Moreover, this study demonstrated that HIF-1α expression was associated with reduced disease-free survival (DFS) and reduced overall survival (OS), concordant with previous studies (65–67). Concluding that TNBC and HER2 overexpression showed the highest cell proliferation and survival in a hypoxic tumor environment by activation of the mTOR pathway and HIF-1α stabilization via DDIT4 downregulation (64).
DDIT4 participate in an endogenous feedback circuit with p53. When cells and tissues of DDIT4−/− mice are exposed to IR and chemotherapy treatment, the genetic loss of DDIT4 confers an increase in DNA damage-induced apoptosis both in vitro and in vivo, which is associated with elevated levels of p53 protein following DNA damage. It was proved that deregulation of p53 is not due to increased p53 stabilization or failed DNA repair but, instead, to increased p53 translation. Consistent with this, it was demonstrated that DDIT4 loss elevates the mTORC1 activity, which explains the increased p53 translation and damage sensitivity in DDIT4−/− cells (68).
Under stress conditions mTORC1 is also inhibited by the activation of p53 both dependently and independently of Sertrin1/2, suggesting the existence of more than one pathway to inhibit mTORC1 (69–71). To test this, Cam et al. exposed MEFs and in vivo tumor models to drugs that induce DNA damage to analyze the upstream regulation of mTORC1 signaling. They found that inhibition of mTORC1 signaling to 4EBP1 requires the coordinated activity of both p53 and p63; by contrast, the inhibition of S6K1 and rpS6 phosphorylation is Akt-dependent. Concordant with this, the loss of p53 or p63 prevents the suppression of mTORC1 signaling induced by the DNA damage, supporting that both are necessary for the inhibition of mTORC1. Suggesting that there are multiples mechanisms that suppress p53/p63 responses and at the same time suppress the ability of the cancer cell to control mTORC1 (72).
In response to DNA damage, DDIT4 phosphorylates downstream Akt through the TSC1/TSC2 complex to inhibit mTORC1 signaling (73, 74). To investigate the clinical significance of this process, Wei et al. analyzed the DDIT4 and p-Akt expression in ovary cancer (primary ovarian cancer and borderline tumors) and normal fallopian tubes. Both DDIT4 and p-Akt expressions were significantly higher in patients with serous ovarian cancer and late FIGO stage; while only DDIT4 expression was significantly higher in ascites formation and only p-Akt expression was significantly histological grade and chemoresistance. Features of patients with improved outcomes in terms of DFS and OS were low DDIT4 staining or absence of p-Akt. In this work, there was no association between KRAS mutations and DDIT4 intensity staining, suggesting that KRAS is not involved in the activation of DDIT4 (15).
In contrast to the study performed by Koo and Jung, where they reported that DDIT4 is downregulated in TNBC and HER2 overexpression types (64), Pinto et al. found that DDIT4 is associated with poor prognosis in TNBC resistant to neoadjuvant chemotherapy (17).
All these data presented previously show that DDIT4 has a key role in different types of cancer and its aggressiveness. A recent in silico analysis of DDIT4 expression in several cancer types showed that the high expression of this gene was related to a bad outcome in diverse hematologic and solid tumors, such as acute myeloid leukemia, breast cancer, glioblastoma multiforme, melanoma, lung, and colon cancer. Furthermore, it was shown that response to rapamycin and others mTOR inhibitors were not influenced by DDIT4 expression (18).
Concluding Remarks and Future Perspectives
The increasing evidence about the involvement of DDIT4 in key cellular mechanisms of tumor aggressiveness suggests its driver role in cancer and consequently, its potential not only as prognostic biomarker but also as a therapeutic target. Since mTOR inhibitors have shown disappointing results in the treatment of cancer, where the main issues are the lack of a biomarker to select patients who will benefit from these drugs and the poor knowledge about mechanisms of resistance, a better therapeutic strategy would be DDIT4 inhibition. In contrast to mTOR expression, the high DDIT4 expression is related with a poor outcome. We hypothesize DDIT4 targeting could lead to cancer cells to avoid the metabolic suppression needed for cell survival under stress conditions (e.g., treatment with cytotoxic chemotherapy or radiotherapy). Combination of DDIT4 inhibitors with DNA-damaging agents, such as cisplatin, will be interesting, especially in tumors with loss of function of TP53, because wild-type p53 repress the expression of DDIT4 in a regulatory loop, potentiating the effects of DDIT4 inhibitors.
Author Contributions
IT-H gathered all the information and wrote the first draft of the manuscript. WF wrote, corrected, and also provided important contributions to the final manuscript. JP contributed to the conception and design of the project, critically reviewing the entire manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
2. Bianco R, Melisi D, Ciardiello F, Tortora G. Key cancer cell signal transduction pathways as therapeutic targets. Eur J Cancer (2006) 42(3):290–4. doi:10.1016/j.ejca.2005.07.034
3. Maiese K. Therapeutic targets for cancer: current concepts with PI 3-K, Akt, & mTOR. Indian J Med Res (2013) 137(2):243–6.
4. Mita MM, Mita A, Rowinsky EK. The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther (2003) 2(4 Suppl 1):S169–77. doi:10.4161/cbt.365
5. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
6. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124(3):471–84. doi:10.1016/j.cell.2006.01.016
7. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18(16):1926–45. doi:10.1101/gad.1212704
8. Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell (2002) 10(3):457–68. doi:10.1016/S1097-2765(02)00636-6
9. Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci (2012) 13(2):1886–918. doi:10.3390/ijms13021886
10. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12(1):21–35. doi:10.1038/nrm3025
11. Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy (2009) 5(5):725–6. doi:10.4161/auto.5.5.8504
12. Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol (2005) 25(14):5834–45. doi:10.1128/MCB.25.14.5834-5845.2005
13. Schwarzer R, Tondera D, Arnold W, Giese K, Klippel A, Kaufmann J. REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene (2005) 24(7):1138–49. doi:10.1038/sj.onc.1208236
14. An SJ, Chen JK, Liu LL, Zhao YF, Chen XM. Over-expressed genes detected by suppression subtractive hybridization in carcinoma derived from transformed 16HBE cells induced by BPDE. Biomed Environ Sci (2005) 18(5):302–6.
15. Jia W, Chang B, Sun L, Zhu H, Pang L, Tao L, et al. REDD1 and p-AKT over-expression may predict poor prognosis in ovarian cancer. Int J Clin Exp Pathol (2014) 7(9):5940–9.
16. Çelik H, Bulut G, Han J, Graham GT, Minas TZ, Conn EJ, et al. Ezrin inhibition up-regulates stress response gene expression. J Biol Chem (2016) 291(25):13257–70. doi:10.1074/jbc.M116.718189
17. Pinto JA, Araujo J, Cardenas NK, Morante Z, Doimi F, Vidaurre T, et al. A prognostic signature based on three-genes expression in triple-negative breast tumours with residual disease. NPJ Genom Med (2016) 1:15015. doi:10.1038/npjgenmed.2015.15
18. Pinto JA, Rolfo C, Raez LE, Prado A, Araujo JM, Bravo L, et al. In silico evaluation of DNA damage inducible transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci Rep (2017) 7(1):1526. doi:10.1038/s41598-017-01207-3
19. Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol (2002) 22(7):2283–93. doi:10.1128/MCB.22.7.2283-2293.2002
20. Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell (2002) 10(5):995–1005. doi:10.1016/S1097-2765(02)00706-2
21. Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem (2003) 278(29):27053–8. doi:10.1074/jbc.M303723200
22. Gene: DDIT4 (ENSG00000168209) – Summary – Homo sapiens – Ensembl Genome Browser 91 [Internet]. (2017). Available from: http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000168209;r=10:72273920-72276036 (Accessed: February 19, 2018).
23. Gene: DDIT4 (ENSG00000168209) – Gene Tree – Homo sapiens – Ensembl Genome Browser 90 [Internet]. (2017). Available from: http://www.ensembl.org/Homo_sapiens/Gene/Compara_Tree?db=core;g=ENSG00000168209;r=10:72273920-72276036 (Accessed: October 9, 2017).
24. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev (2004) 18(23):2893–904. doi:10.1101/gad.1256804
25. Lin L, Qian Y, Shi X, Chen Y. Induction of a cell stress response gene RTP801 by DNA damaging agent methyl methanesulfonate through CCAAT/enhancer binding protein. Biochemistry (Mosc) (2005) 44(10):3909–14. doi:10.1021/bi047574r
26. Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, Finkel TH, et al. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem (2011) 286(34):30181–9. doi:10.1074/jbc.M111.245423
27. Wolff NC, McKay RM, Brugarolas J. REDD1/DDIT4-independent mTORC1 inhibition and apoptosis by glucocorticoids in thymocytes. Mol Cancer Res (2014) 12(6):867–77. doi:10.1158/1541-7786.MCR-13-0625
28. Malagelada C, López-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA. RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci (2011) 31(9):3186–96. doi:10.1523/JNEUROSCI.4011-10.2011
29. Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem Biophys Res Commun (2009) 379(2):451–5. doi:10.1016/j.bbrc.2008.12.079
30. Lin L, Stringfield TM, Shi X, Chen Y. Arsenite induces a cell stress-response gene, RTP801, through reactive oxygen species and transcription factors Elk-1 and CCAAT/enhancer-binding protein. Biochem J (2005) 392(Pt 1):93–102. doi:10.1042/BJ20050553
31. Gordon BS, Steiner JL, Williamson DL, Lang CH, Kimball SR. Emerging role for regulated in development and DNA damage 1 (REDD1) in the regulation of skeletal muscle metabolism. Am J Physiol (2016) 311(1):E157–74. doi:10.1152/ajpendo.00059.2016
32. GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods Groups—Analysis Working Group, Enhancing GTEx (eGTEx) Groups, NIH Common Fund, NIH/NCI, NIH/NIMH, et al. Genetic effects on gene expression across human tissues. Nature (2017) 550(7675):204–13. doi:10.1038/nature24277
33. Kimball SR, Do AND, Kutzler L, Cavener DR, Jefferson LS. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem (2008) 283(6):3465–75. doi:10.1074/jbc.M706643200
34. Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S, et al. REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A–DDB1 ubiquitin ligase. EMBO Rep (2009) 10(8):866–72. doi:10.1038/embor.2009.93
35. DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2–mTOR signaling and tumor suppression through REDD1-mediated 14–3–3 shuttling. Genes Dev (2008) 22(2):239–51. doi:10.1101/gad.1617608
36. Vega-Rubin-de-Celis S, Abdallah Z, Kinch L, Grishin NV, Brugarolas J, Zhang X. Structural analysis and functional implications of the negative mTORC1 regulator REDD1. Biochemistry (Mosc) (2010) 49(11):2491–501. doi:10.1021/bi902135e
37. Canal M, Romaní-Aumedes J, Martín-Flores N, Pérez-Fernández V, Malagelada C. RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front Cell Neurosci (2014) 8:313. doi:10.3389/fncel.2014.00313
38. Zhou Y, Wang Q, Guo Z, Weiss HL, Evers BM. Nuclear factor of activated T-cell c3 inhibition of mammalian target of rapamycin signaling through induction of regulated in development and DNA damage response 1 in human intestinal cells. Mol Biol Cell (2012) 23(15):2963–72. doi:10.1091/mbc.E12-01-0037
39. MacFarlane L-A, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics (2010) 11(7):537–61. doi:10.2174/138920210793175895
40. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer (2006) 6(11):857–66. doi:10.1038/nrc1997
41. Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol (2009) 27(34):5848–56. doi:10.1200/JCO.2009.24.0317
42. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A (2010) 107(1):264–9. doi:10.1073/pnas.0907904107
43. Hwang-Verslues WW, Chang P-H, Wei P-C, Yang C-Y, Huang C-K, Kuo W-H, et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene (2011) 30(21):2463–74. doi:10.1038/onc.2010.618
44. Li XH, Ha CT, Fu D, Xiao M. Micro-RNA30c negatively regulates REDD1 expression in human hematopoietic and osteoblast cells after gamma-irradiation. PLoS One (2012) 7(11):e48700. doi:10.1371/journal.pone.0048700
45. Cao J-X, Lu Y, Qi J-J, An G-S, Mao Z-B, Jia H-T, et al. MiR-630 inhibits proliferation by targeting CDC7 kinase, but maintains the apoptotic balance by targeting multiple modulators in human lung cancer A549 cells. Cell Death Dis (2014) 5:e1426. doi:10.1038/cddis.2014.386
46. Chen LC, Manjeshwar S, Lu Y, Moore D, Ljung BM, Kuo WL, et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res (1998) 58(16):3677–83.
47. Yasui K, Arii S, Zhao C, Imoto I, Ueda M, Nagai H, et al. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology (2002) 35(6):1476–84. doi:10.1053/jhep.2002.33683
48. Müerköster S, Arlt A, Sipos B, Witt M, Grossmann M, Klöppel G, et al. Increased expression of the E3-ubiquitin ligase receptor subunit betaTRCP1 relates to constitutive nuclear factor-kappaB activation and chemoresistance in pancreatic carcinoma cells. Cancer Res (2005) 65(4):1316–24. doi:10.1158/0008-5472.CAN-04-1626
49. Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, et al. Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappaB in colorectal cancer. J Natl Cancer Inst (2004) 96(15):1161–70. doi:10.1093/jnci/djh219
50. Tan CY, Hagen T. mTORC1 dependent regulation of REDD1 protein stability. PLoS One (2013) 8(5):e63970. doi:10.1371/journal.pone.0063970
51. Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev (2004) 18(23):2879–92. doi:10.1101/gad.322704
52. Corradetti MN, Inoki K, Guan K-L. The stress-inducted proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem (2005) 280(11):9769–72. doi:10.1074/jbc.C400557200
53. Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol (2005) 15(8):702–13. doi:10.1016/j.cub.2005.02.053
54. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi:10.1038/nrm3735
55. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev (2011) 25(19):1999–2010. doi:10.1101/gad.17558811
56. Jung CH, Ro S-H, Cao J, Otto NM, Kim D-H. mTOR regulation of autophagy. FEBS Lett (2010) 584(7):1287–95. doi:10.1016/j.febslet.2010.01.017
57. Kim J-R, Lee S-R, Chung HJ, Kim S, Baek S-H, Kim JH, et al. Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: involvement of RTP801 in amyloid beta-peptide toxicity. Exp Mol Med (2003) 35(5):403–11. doi:10.1038/emm.2003.53
58. Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci (2004) 45(10):3796–805. doi:10.1167/iovs.04-0052
59. Hu YY, Liu JC, Xing AY, You Y, Wang XD. REDD1 expression in placenta during human gestation. Reprod Sci (2012) 19(9):995–1000. doi:10.1177/1933719112440054
60. Chang B, Liu G, Yang G, Mercado-Uribe I, Huang M, Liu J. REDD1 is required for RAS-mediated transformation of human ovarian epithelial cells. Cell Cycle (2009) 8(5):780–6. doi:10.4161/cc.8.5.7887
61. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol (2014) 4:64. doi:10.3389/fonc.2014.00064
62. Khan KH, Yap TA, Yan L, Cunningham D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J Cancer (2013) 32(5):253–65. doi:10.5732/cjc.013.10057
63. Smith ER, Xu X-X. REDD1, a new Ras oncogenic effector. Cell Cycle (2009) 8(5):675–6. doi:10.4161/cc.8.5.8184
64. Koo JS, Jung W. Alteration of REDD1-mediated mammalian target of rapamycin pathway and hypoxia-inducible factor-1α regulation in human breast cancer. Pathobiology (2010) 77(6):289–300. doi:10.1159/000320936
65. Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer (2003) 97(6):1573–81. doi:10.1002/cncr.11246
66. Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M, et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin Cancer Res (2002) 8(6):1831–7.
67. Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouysségur J, et al. HIF-1α and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer (2007) 120(7):1451–8. doi:10.1002/ijc.22436
68. Vadysirisack DD, Baenke F, Ory B, Lei K, Ellisen LW. Feedback control of p53 translation by REDD1 and mTORC1 limits the p53-dependent DNA damage response. Mol Cell Biol (2011) 31(21):4356–65. doi:10.1128/MCB.05541-11
69. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell (2008) 134(3):451–60. doi:10.1016/j.cell.2008.06.028
70. Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, et al. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle (2009) 8(24):4168–75. doi:10.4161/cc.8.24.10800
71. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res (2007) 67(7):3043–53. doi:10.1158/0008-5472.CAN-06-4149
72. Cam M, Bid HK, Xiao L, Zambetti GP, Houghton PJ, Cam H. p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J Biol Chem (2014) 289(7):4083–94. doi:10.1074/jbc.M113.530303
73. Dennis MD, McGhee NK, Jefferson LS, Kimball SR. Regulated in DNA damage and development 1 (REDD1) promotes cell survival during serum deprivation by sustaining repression of signaling through the mechanistic target of rapamycin in complex 1 (mTORC1). Cell Signal (2013) 25(12):2709–16. doi:10.1016/j.cellsig.2013.08.038
Keywords: DNA damage inducible transcript 4, mammalian target of rapamycin, malignant tumors, biomarkers, targeted therapies
Citation: Tirado-Hurtado I, Fajardo W and Pinto JA (2018) DNA Damage Inducible Transcript 4 Gene: The Switch of the Metabolism as Potential Target in Cancer. Front. Oncol. 8:106. doi: 10.3389/fonc.2018.00106
Received: 22 November 2017; Accepted: 26 March 2018;
Published: 12 April 2018
Edited by:
Karine A. Cohen-Solal, Rutgers University, United StatesReviewed by:
Chakrabhavi Dhananjaya Mohan, University of Mysore, IndiaAlessandro Rufini, University of Leicester, United Kingdom
Copyright: © 2018 Tirado-Hurtado, Fajardo and Pinto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph A. Pinto, jpinto@gecoperu.org