 Sining Zhu1,2
Sining Zhu1,2 Jaeyong Jung
Jaeyong Jung Eton Victor
Eton Victor Johann Arceo
Johann Arceo Samantha Gokhale
Samantha Gokhale Ping Xie
Ping Xie- 1Department of Cell Biology and Neuroscience, Rutgers University, Piscataway, NJ, United States
- 2Graduate Program in Cellular and Molecular Pharmacology, Rutgers University, Piscataway, NJ, United States
- 3Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, United States
The BTK inhibitors ibrutinib and acalabrutinib are FDA-approved drugs for the treatment of B cell malignances. Both drugs have demonstrated clinical efficacy and safety profiles superior to chemoimmunotherapy regimens in patients with chronic lymphocytic leukemia. Mounting preclinical and clinical evidence indicates that both ibrutinib and acalabrutinib are versatile and have direct effects on many immune cell subsets as well as other cell types beyond B cells. The versatility and immunomodulatory effects of both drugs have been exploited to expand their therapeutic potential in a wide variety of human diseases. Over 470 clinical trials are currently registered at ClinicalTrials.gov to test the efficacy of ibrutinib or acalabrutinib not only in almost every type of B cell malignancies, but also in hematological malignancies of myeloid cells and T cells, solid tumors, chronic graft versus host disease (cGHVD), autoimmune diseases, allergy and COVID-19 (http:www.clinicaltrials.gov). In this review, we present brief discussions of the clinical trials and relevant key preclinical evidence of ibrutinib and acalabrutinib as monotherapies or as part of combination therapies for the treatment of human diseases beyond B cell malignancies. Adding to the proven efficacy of ibrutinib for cGVHD, preliminary results of clinical trials have shown promising efficacy of ibrutinib or acalabrutinib for certain T cell malignancies, allergies and severe COVID-19. However, both BTK inhibitors have no or limited efficacy for refractory or recurrent solid tumors. These clinical data together with additional pending results from ongoing trials will provide valuable information to guide the design and improvement of future trials, including optimization of combination regimens and dosing sequences as well as better patient stratification and more efficient delivery strategies. Such information will further advance the precise implementation of BTK inhibitors into the clinical toolbox for the treatment of different human diseases.
Introduction
Bruton’s tyrosine kinase (BTK), a member of the TEC kinase family, was originally identified as a non-receptor protein tyrosine kinase that is mutated and defective in patients with X−linked agammaglobulinemia (XLA) (1–3). BTK is predominantly expressed in hematopoietic cells (4, 5). In B lymphocytes, BTK is primarily required for B cell receptor (BCR) signaling (2, 3, 6). Upon BCR activation, BTK is recruited to the BCR signaling complex, where Btk activity is positively regulated by phosphorylation events. Activated BTK in turn phosphorylates PLCγ2 to induce downstream transcription factors such as NF−κB, NF-AT and ERK1/2 (1–3, 6). In addition to BCR, BTK also participates in the signaling pathways of chemokine receptors in B cells, including CXCR4 and CXCR5 (1, 7). Therefore, BTK plays essential roles in regulating B cell development, survival, proliferation, differentiation, activation and chemotaxis (2, 3, 6). Elevated expression and activity of BTK have been ubiquitously documented in many types of B cell malignancies (1, 8, 9). Aberrant BTK activities play crucial driving roles in the pathogenesis of B cell malignancies, including malignant B cell survival, proliferation and migration, and thus have been recognized as a prime therapeutic target for the treatment of B cell malignancies (1–3, 6).
Successful milestones have been achieved in the pursuit of selective BTK inhibitors. The first-in-class BTK inhibitor ibrutinib and the pioneer second-generation BTK inhibitor acalabrutinib (ACP-196) are two US Food and Drug Administration (FDA)-approved drugs for the treatment of B cell malignances (2, 6, 10). Both ibrutinib and acalabrutinib irreversibly inactivate BTK by covalently binding to Cys481 in the ATP-binding site of BTK (1, 2, 11, 12). The structures of ibrutinib and acalabrutinib as well as their interactions with BTK have been elucidated and depicted in several excellent publications (12–16). Since their approval by FDA, ibrutinib and acalabrutinib have demonstrated clinical efficacy and safety profiles superior to conventional chemoimmunotherapy (CIT) regimens in patients with chronic lymphocytic leukemia (CLL) and relapsed mantle cell lymphoma (MCL), especially in high-risk patients, bringing a major breakthrough in the field (10, 17–20). As a result, ibrutinib and acalabrutinib are currently recommended as the standard of care and preferred choice of treatment in CLL and relapsed MCL, and have also transformed the treatment options for other B cell malignancies (10, 17, 20–23).
Interestingly, mounting preclinical and clinical evidence indicates that both BTK inhibitors are much more versatile than initially envisioned and have direct effects on many cell types beyond B lymphocytes (24). In addition to B cells, many other cell types express BTK under physiological or pathological conditions, including T cells, monocytes, macrophages, granulocytes, myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), osteoclasts, mast cells, erythrocytes, platelets, epithelial cells, neurons and astrocytes (3, 24–31). Most notably, both ibrutinib and acalabrutinib have complex immunomodulatory effects on various non-B immune cell subsets by inhibiting BTK-dependent signaling pathways of specific immune receptors, including T cell receptor (TCR), Toll-like receptors (TLRs), NLRP3, TREM-1, Dectin-1, CXCR4, CXCR5, RANK, Fc receptors and CLEC-2, among others (2, 3, 6, 24–26, 32, 33). Due to its off-target inhibition of other kinases such as ITK, TEC, the SRC family kinases, EGFR, PDGF-R, VEGF-R2 and CSK, ibrutinib has additional distinct effects on T cells, natural killer (NK) cells, myeloid cells, platelets, epithelial cells, endothelial cells and cardiomyocytes (27–29, 34–40). Such mechanisms of action contribute to the exceptionally high clinical efficacy as well as the unique profiles of adverse effects observed for ibrutinib and acalabrutinib in CLL and MCL patients. Furthermore, these findings have vastly expanded the therapeutic potential of both drugs in human diseases.
Owing to their complex immunomodulatory effects together with the ease of their oral administration, their well-tolerated toxicity profiles and their potential for long-term treatment in patients, ibrutinib and acalabrutinib have become very popular in the management of B cell malignancies (6, 41). Meanwhile, the favorable and versatile features of the two drugs have attracted great interests to explore their repurposing opportunities for the treatment of a wide variety of other human diseases. Over 470 clinical trials are currently registered at ClinicalTrials.gov to test the efficacy of ibrutinib or acalabrutinib not only in almost every type of B cell malignancies, but also in hematological malignancies of myeloid cells and T cells, solid tumors, chronic graft versus host disease, autoimmune diseases, allergy and COVID-19 (http:www.clinicaltrials.gov). In this review, we present a brief discussion of the clinical trials and relevant key preclinical evidence of ibrutinib and acalabrutinib as monotherapies or as part of combination therapies for the treatment of human diseases beyond B cell malignancies. We hope such discussion will promote further explorations on the multifaceted therapeutic potential and repurposing opportunities of BTK inhibitors.
Hematological Malignancies of Myeloid Cells and T Cells
BTK is upregulated and constitutively phosphorylated in the majority of primary AML samples (42–44). In AML cells, BTK phosphorylation can be induced by SCF-CD117 signaling or activating mutations of G-CSFR (T618I or truncated Q741x) or FLT3 (with internal tandem duplication of the juxtamembrane region, FLT3-ITD) to mediate cell survival, proliferation and adhesion, and can also be elicited by SDF1-CXCR4 signaling to mediate cell migration and adhesion to stroma cells (44–47). Inhibition of BTK by ibrutinib or knockdown of BTK by siRNA in primary AML cells and AML cell lines decreases NF-κB survival pathways, SCF-CD117-BTK-MAPK/ATK signaling, G-CSFR mutants-BTK signaling, FLT3-ITD-BTK-NF-κB/MAPK/AKT/STAT5 signaling, and SDF1/CXCR4-BTK-AKT/MAPK signaling, leading to reduced cell survival and proliferation as well as migration (43–48). Together, these data reveal the pathogenic roles of BTK in AML, identifying BTK as a therapeutic target in myeloid cell malignancies.
Based on the above preclinical evidence, two clinical trials are testing the efficacy of ibrutinib for the prevention and treatment of AML and two additional trials are evaluating the effects of ibrutinib in patients with myelodysplastic syndrome (MDS) (Tables 1 and 2). Among these, one Phase II trial (NCT03267186) is studying the efficacy of ibrutinib in preventing AML relapse after reduced-intensity conditioning and allogeneic hematopoietic cell transplantation (allo-HCT) for AML. A Phase II/III trial (NCT03642236) is evaluating the potential of ibrutinib in combination with conventional chemotherapy (decitabine, aclacinomycin, cytarabine, G-CSF and sorafenib with or without FLT3 inhibitor) to overcome drug-resistance in relapsed or refractory (R/R) FLT3 mutant AML. For the treatment of MDS, a Phase Ib trial (NCT02553941) is testing the effects of ibrutinib in combination with azacitidine in higher risk MDS patients. Another Phase I trial (NCT03359460) is investigating the efficacy of ibrutinib in combination with lenalidomide in MDS patients that have failed or refused standard therapy.

Table 1 Clinical trials of BTK inhibitors in human diseases beyond B cell malignancies that are currently registered at ClinicalTrials.gov.
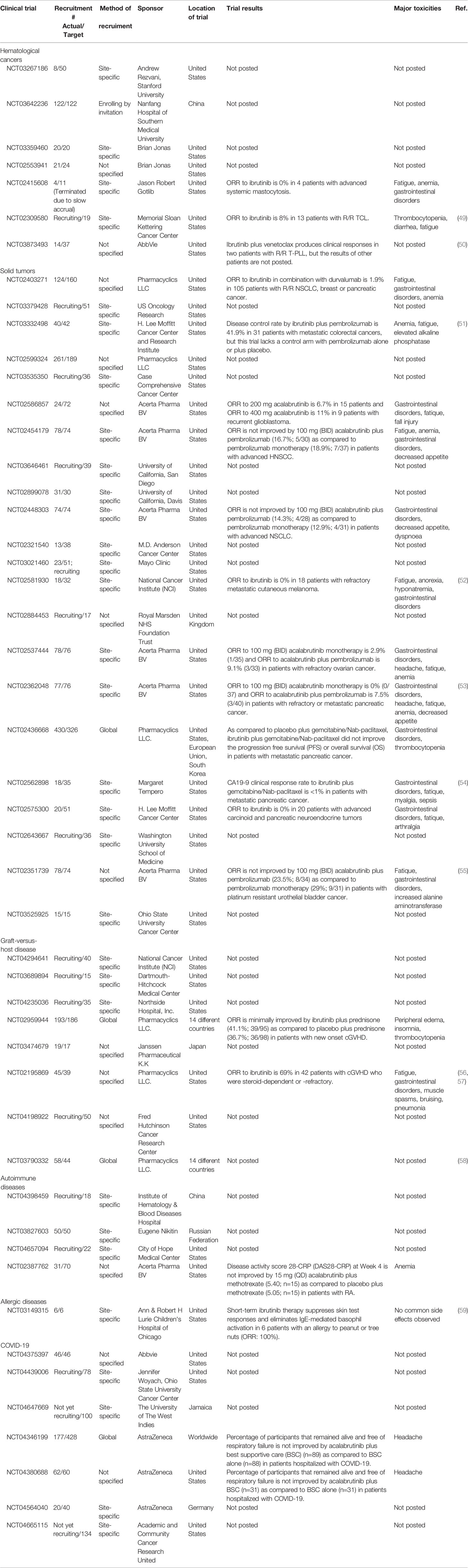
Table 2 Recruiting info and preliminary results of clinical trials with ibruitinib and acalabrutinib in human diseases beyond B cell malignancies.
However, a recently completed Phase IIa clinical trial (NCT02351037) showed limited efficacy of ibrutinib alone or in combination with cytarabine or azacitidine in patients with AML (60). Another completed Phase II clinical trial (registered in the Netherlands NL5751 [NTR6017] and EudraCT number 2015-002855-85) reported that addition of ibrutinib to decitabine does not improve the therapeutic efficacy of decitabine in unfit patients with AML and higher risk MDS (61). Although the efficacy of ibrutinib in AML and MDS awaits clarification with additional clinical data from ongoing trials, it is possible that ibrutinib may be effective only in combination with appropriate drugs or only for patients with specific oncogenic mutations or genetic alterations. In this regard, Li et al. found that ibrutinib acts synergistically with the cytotoxic alkaloid homoharringtonine to inhibit cell proliferation and induce apoptosis in AML cells with FLT3-ITD mutations (62). Another study by Eide et al. recently reported that the primary AML samples with 11q23 MLL rearrangements are highly sensitive to ibrutinib in combination with the BCL-2 inhibitor venetoclax (63). Furthermore, both ibrutinib and acalabrutinib exhibit synergistic effects with daunorubicin on inducing cytotoxicity in AKR1C3-expressing AML cells via efficiently preventing daunorubicin inactivation mediated by AKR1C3, an enzyme associated with the emergence of multidrug resistance (MDR) (64). Therefore, clinical data from ongoing trials and better understanding of the mechanisms of action for ibrutinib and acalabrutinib will guide the design and improvement of future trials, including optimization of the combination regimens and appropriate patient stratification according to relevant genetic alterations and contexts.
In addition to AML and MDS, aberrant activation of BTK has been detected in patients of chronic neutrophilic leukemia (CNL) with G-CSFR mutations and these cells show high sensitivity to ibrutinib treatment (45). Ibrutinib also suppresses BTK phosphorylation, induces apoptosis and decreases proliferation in canine neoplastic mast cells (65). Although a Phase II clinical trial (NCT02415608) of ibrutinib in systemic mastocytosis was recently terminated due to slow accrual, these preclinical findings suggest a potential of ibrutinib and other BTK inhibitors as new therapeutic agents for CNL and mast cell neoplasms.
Because of its off-target inhibition on ITK, ibrutinib is being tested in T cell malignancies (Tables 1 and 2). One Phase I clinical trial (NCT02309580) is investigating the efficacy of ibrutinib in adult patients with R/R T-cell lymphoma (TCL), including peripheral TCL (PTCL) and cutaneous TCL (CTCL). Another Phase II clinical trial (NCT03873493) is evaluating the efficacy of ibrutinib in combination with venetoclax in adult patients with R/R T-cell prolymphocytic leukemia (T-PLL). Mechanistically, ITK is required for TCR signaling and CXCR4 signaling, and thus critical for malignant T cell proliferation, differentiation and migration, while preventing anti-tumor immune responses by inhibiting TH1 CD4 T cell differentiation (49, 50, 66). Treatment with ibrutinib or knockdown of ITK by siRNA results in reduced ITK phosphorylation and decreased activation of downstream MEK1/2 and AKT, leading to compromised survival and cytokine production as well as migration of PTCL cells (66). A large proportion of PTCL cases are derived from follicular helper T cells (Tfh), which express high levels of ITK proteins (66). In a mouse model of Tfh-derived lymphoma, ibrutinib effectively induces lymphoma regression (67). However, the preliminary results of the Phase I trial NCT02309580 suggest that ibrutinib has limited clinical benefits in 13 patients with R/R TCL (49) (Table 2). Interestingly, although ibrutinib alone has very modest effects on T-PLL cells, the combination of ibrutinib and the BCL-2 inhibitor venetoclax exhibits strong synergism at inducing apoptosis in primary T-PLL cells (50). This synergism is because venetoclax monotherapy leads to ITK activation and ibrutinib-mediated ITK inhibition enhances the dependence of T-PLL cells on BCL-2 for survival (50). An anecdotal case of combinatorial treatment of one T-PLL patient was recently published by Oberbeck et al., which reported disease stabilization in the patient after short-term treatment with ibrutinib plus venetoclax but progression after cessation of treatment (68). In line with this, preliminary clinical data of the Phase II trial NCT03873493 demonstrated that ibrutinib in combination with venetoclax produces clinical responses in two patients with R/R T-PLL (50). Thus, it is promising that more clinical data from ongoing trials will advance the development and addition of ibrutinib into the treatment regimens for T cell malignancies.
Solid Tumors
Currently, 24 clinical trials are registered at ClinicalTrials.gov to test the efficacy of ibrutinib (18 trials) and acalabrutinib (6 trials), alone or in combination therapy, in patients with a variety of solid tumors, including breast, prostate, lung, kidney, head and neck, pancreatic, colorectal, oesophagogastric, urothelial, ovarian, gastrointestinal and genitourinary cancers, glioblastoma, melanoma and metastatic solid tumors (Tables 1 and 2). These clinical trials are elicited by strong preclinical evidence indicating that ibrutinib and acalabrutinib will potentially have broad applications in the treatment of various solid tumors because of their multi-layered mechanisms of action (24, 28, 29, 53, 69–79). These include both direct tumoricidal activities on cancer cells and indirect immunomodulatory effects on different immune cell subsets as well as other relevant cell types in the tumor microenvironment (TME) via on-target inhibition of BTK (for both drugs) or off-target inhibition of ITK or EGFR (for ibrutinib) (2, 24, 28, 29).
Ibrutinib and acalabrutinib exhibit direct tumoricidal activities on certain types of solid tumor cells, including neuroblastoma, glioblastoma, breast cancer, prostate cancer, bladder cancer and advanced oral squamous cell carcinoma (OSCC) (30, 31, 78, 80–82). Accumulating studies report that BTK is highly expressed in certain solid tumor cells and increased BTK levels are associated with a poor prognosis in patients (30, 31, 78, 80–82). Gene silencing of BTK or inhibition of BTK with ibrutinib or acalabrutinib attenuates the proliferation, migration, invasion and stemness of these cancer cells both in vitro and in vivo (30, 31, 78, 80–82). Mechanistically, ibrutinib or acalabrutinib potently inhibits BTK phosphorylation and its downstream oncogenic pathways such as the PI3K-mTOR and MEK/MAPK pathways, resulting in impaired tumorigenicity of these tumor cells (30, 31, 78, 80–82). However, it should be noted that there are controversial reports regarding the expression of BTK in tumor cells derived from non-hematopoietic lineages. For example, Li et al. reported that the expression of BTK is detectable in human neuroblastoma cell lines IMR32, LAN2, NBL-S and SHSY5Y (83), and Pikatan et al. also detected the expression of BTK in human neuroblastoma cell lines SK-N-BE2 and SH-5YSY by Western blot analysis (30). In contrast, Ishfaq et al. reported that the expression of BTK is not detectable in human neuroblastoma cell lines SK-N-BE2, IMR32, SH-SY-5Y and SKNSH or a murine neuroblastoma cell line NB9464 by Western blot analysis (84). In addition to the contradictory observations in BTK expression, these three groups also reported contradictory effects of ibrutinib on neuroblastoma cell lines. Ishfaq et al. did not observe any effect of ibrutinib on the proliferation of murine and human neuroblastoma cell lines (84). In sharp contrast, Li et al. and Pikatan et al. reported that silencing of BTK by siRNAs or inhibition of BTK by ibrutinib or acalabrutinib significantly reduces the proliferation and viability of human neuroblastoma cell lines (30, 83). It remains unclear what factors may cause such contradictory results in the same cell lines. In this context, the expression of BTK in solid tumors derived from non-hematopoietic lineage cells requires more stringent scrutiny in future studies, especially given the frequent presence of BTK-expressing infiltrating immune cells in the TME of primary tumor specimens.
Indeed, BTK is critical for the function of multiple cell types representing important components of the TME, including macrophages, MDSCs, DCs, neutrophils, B cells and endothelial cells (2, 24, 28, 29). In particular, BTK is overexpressed in TAMs and MDSCs, which regulate tumor progression, immunosuppression and angiogenesis (71, 84, 85). Ibrutinib treatment, Btk deficiency or siRNA-mediated knockdown of BTK shifts macrophage polarization from tumor-promoting M2-like macrophages toward inflammatory M1-like and diminishes PD-1 and SIRPα expression on macrophages (24, 77, 86, 87). Ibrutinib treatment also inhibits MDSC generation, induces MDSC differentiation to mature DCs, and impairs IDO expression and NO production in MDSCs, resulting in reduced MDSC-mediated immunosuppression and increased CD8 T cell proliferation, infiltration and effector function in the TME (71, 80, 84, 88). These observations are consistent with the previous evidence that Btk-/- DCs exhibit a more mature phenotype, expressing higher levels of MHC class II and co-stimulatory molecules, than wild type DCs (89, 90). Interestingly, emerging evidence reveals complex roles of different B cell subsets in the progression of solid tumors (77, 91, 92). BTK regulates the crosstalk between B cells and FcRγ+ TAMs in the TME and thus TAM-mediated immunosuppression of T cells in pancreatic adenocarcinomas, which can be reverted by treatment with ibrutinib or siRNA-mediated knockdown of BTK (77). Ibrutinib also inhibits the production of immunosuppressive adenosine by regulatory B cells (Breg) and increases the infiltration of effector B cells into the TME (74). Taken together, ibrutinib-mediated inhibition of BTK in TAMs, MDSCs, DCs and B cells indirectly promote anti-tumor immunity by augmenting TH1 CD4 T cell response and increasing the infiltration of CD8 cytotoxic T cells and effector B cells into the TME (24, 69, 74, 77, 80, 84, 93).
An additional indirect anti-tumor mechanism via on-target inhibition of BTK by ibrutinib or acalabrutinib is realized through the regulation of cytokines, chemokines and growth factors. Btk deficiency or inhibition of BTK with ibrutinib or acalabrutinib impairs TLR signaling and inflammasome activation in TAMs, MDSCs, DCs and B cells, thereby efficiently suppressing the production of specific cytokines, chemokines and growth factors such as IL-1β, IL-6, CXCL12, CXCL13, CCL19 and VEGF (2, 24, 28, 29, 69, 94, 95). Reduced levels of these molecules significantly compromise the adhesion, migration and invasion of tumor cells, and also impair the ability of endothelial cells to undergo angiogenic tube formation (2, 28, 29, 69, 95). Thus, ibrutinib and acalabrutinib can be of clinical use in abrogating inflammation-associated cancer progression and angiogenesis in the TME (2, 28, 29, 69, 95).
It has been well-documented that off-target inhibition of ITK allows ibrutinib to modulate CD4 T cell differentiation and CD8 T cell effector function (24, 28, 29, 34, 96). ITK is required for CD4 TH2 differentiation and negatively regulates the expression of the transcription factor Eomes in CD8 T cells (34, 96). Eomes inhibits the expression of checkpoint receptors (PD-1, TIM-3, and LAG-3) and induces the expression of effector cytokines (IFNγ and TNFα) in CD8 T cells (96). Therefore, ibrutinib-mediated inhibition of ITK can robustly enhance anti-tumor immune responses by favoring TH1 differentiation and promoting the effector functions of CD8 cytotoxic T cells (96). On the other hand, off-target inhibition of EGFR allows ibrutinib to directly suppress the proliferation, growth and stemness of certain types of solid tumors that are dependent on EGFR oncogenic pathways, including pancreatic cancer, hepatocellular carcinoma (HCC) and esophageal squamous cell carcinoma (ESCC) (97–99). Ibrutinib represses the phosphorylation of EGFR and thus inhibits the downstream activation of AKT and ERK signaling in these cancer cells, leading to reduced tumorigenicity both in vitro and in vivo (97–99). In addition, off-target inhibition of EGFR by ibrutinib also abrogates the pro-tumoral function of glioma-derived pericytes with EGFR mutations (100). Thus, through its off-target inhibition of EGFR in cancer cells or pericytes and ITK in T cells, ibrutinib can directly suppress the tumorigenicity and growth of EGFR-dependent solid tumors and dampen the pro-tumoral function of pericytes with EGFR mutations, while indirectly promoting anti-tumor TH1 and CD8 T cell responses to attenuate tumor progression.
Both their direct cytotoxic effects on tumor cells and their indirect immunomodulatory effects on various immune cell subsets in the TME provide strong rationales for a potentially broad use of ibrutinib and acalabrutinib in solid tumors (2, 28, 29, 69). However, preliminary clinical data of several trials show no or limited clinical benefit of ibrutinib and acalabrutinib at improving the objective response rates (ORRs), progression-free survival (PFS) or overall survival (OS) in patients with several solid tumors (Table 2). These include ibrutinib plus durvalumab in R/R non-small cell lung cancer (NSCLC), breast cancer and pancreatic cancer (NCT02403271); acalabrutinib in recurrent glioblastoma (NCT02586857); ibrutinib in refractory metastatic cutaneous melanoma (NCT02581930) (52); acalabrutinib in combination with pembrolizumab in advanced head and neck squamous cell carcinoma (HNSCC) (NCT02454179), non-small cell lung cancer (NCT02448303) and platinum-refractory metastatic urothelial carcinoma (NCT02351739) (55); acalabrutinib alone or in combination with pembrolizumab in refractory ovarian cancer (NCT02537444) and metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma (NCT02362048) (53); ibrutinib in combination with Nab-paclitaxel and gemcitabine in metastatic pancreatic cancer (NCT02436668) and (NCT02562898) (54); and ibrutinib alone in advanced carcinoid and pancreatic neuroendocrine tumors (Table 2) (www.clinicaltrials.com). Nonetheless, peripheral reductions in MDSCs and increases in proliferating CD8 T cell subsets are observed in some of these cases (53, 55). It has been noticed that lack of efficacy is at least partially due to the rapid clearance in vivo and low accumulation of ibrutinib and acalabrutinib in solid tumors (12, 85). To circumvent this hurdle, Qiu et al. has recently developed novel nanocomplexes coated with a sialic acid (SA)-stearic acid conjugate-decorated surface to encapsulate ibrutinib, which demonstrates prolonged blood circulation and efficient delivery of ibrutinib to the tumor, consequently enhancing anti-tumor immunity, reducing angiogenesis and suppressing tumor growth (85). Therefore, the challenging tasks in future clinical trials are to develop more efficient delivery strategies, implement better patient stratification according to their genetic contexts and tumor stage, and optimize the regimens as well as the sequence of combination therapies to improve the efficacy of ibrutinib and acalabrutinib in the treatment of solid tumors.
Chronic Graft Versus Host Disease (cGVHD)
Based on the favorable results of a completed clinical trial (NCT02195869), ibrutinib has been approved by the FDA in 2017 for the treatment of chronic graft versus host disease (cGVHD) in adult patients after failure of one or more first-line therapy (56, 57, 101–103). cGVHD is one of the major complications in patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT) and represents a significant cause of morbidity and mortality after allo-HSCT (104, 105). cGVHD often requires enduring immunosuppressive treatment and corticosteroids remain the cornerstone therapy (103, 104). For steroid-refractory cGVHD, ibrutinib has become one of the preferred options (103, 105, 106). Mechanistically, the key kinases BTK and ITK are critical to cGVHD development, as animals lacking BTK or ITK do not develop alloantibody-driven cGVHD and T cell-mediated sclerodermatous cGVHD (107). Ibrutinib inhibits BTK in B cells and ITK in T cells that mediate the pathogenesis of cGVHD, leading to reduced B cell and T cell proliferation and co-stimulatory molecule activation, decreased germinal center (GC) reactions and tissue immunoglobulin (Ig) deposition, TH2 cell depletion, reduced cytokine production and lymphocyte infiltration, ameliorated skin lesions and multiorgan inflammation and fibrosis, which all contribute to improved patient survival (103–105, 107–109).
Currently, there are 7 additional clinical trials of ibrutinib (6 trials) and acalabrutinib (1 trial) in cGVHD registered at ClinicalTrials.gov (Tables 1 and 2). One Phase II clinical trial (NCT04198922) is testing the efficacy of acalabrutinib for the treatment of cGVHD caused by stem cell transplants in steroid-refractory adult patients. A phase III clinical trial (NCT03474679) is comparing the effects of ibrutinib between steroid-dependent and steroid-refractory cGVHD patients. Four ongoing clinical trials of ibrutinib are testing the efficacy of ibrutinib as front-line therapy for cGVHD in patients that receive a stem cell or bone marrow transplant, either alone in monotherapy (NCT04294641) or in combination therapy with corticosteroids (NCT02959944) or rituximab (NCT04235036: in newly diagnosed cGVHD; NCT03689894: in steroid-dependent and steroid-refractory cGVHD). Moreover, initial clinical data of ibrutinib in pediatric patients with cGVHD are promising and one Phase I/II clinical trial (NCT03790332) is determining the optimal dosing and further evaluating the efficacy and safety of ibrutinib in pediatric cGVHD patients after failure of 1 or more lines of systemic therapy (58).
Compared to the standard immunosuppressive therapy of cGVHD, corticosteroids (103, 104), ibrutinib has different mechanisms of action and toxicity profiles. Corticosteroids are synthetic analogues of the natural steroid hormones produced by the adrenal cortex. Commonly used glucocorticoids (such as prednisone, prednisolone and dexamethasone) are predominantly involved in carbohydrate, lipid and protein metabolism, and have immunosuppressive, anti-inflammatory, anti-proliferative, and vasoconstrictive effects (110). Most of the immunosuppressive and anti-inflammatory actions of glucocorticoids are mediated by their interactions with the glucocorticoid receptors, which subsequently alter gene transcription in both inflammatory leukocytes and structural cells (such as epithelial and endothelial cells) (110). The well-known adverse effects of corticosteroids include osteoporosis, fractures, adrenal suppression, hyperglycemia, diabetes, cushingoid appearance, weight gain, cataracts, glaucoma, dyslipidemia, psychiatric and cognitive disturbances (110), which are not commonly observed in ibrutinib-treated patients (38) (Table 2). Interestingly, the results of NCT02195869 demonstrated that ibrutinib is effective for both corticosteroid-dependent and corticosteroid-refractory cGVHD patients (56, 57). Because of their complementary mechanisms of action, ibrutinib and corticosteroids were hypothesized to have combinatory effects on cGVHD, which would improve clinical benefits in cGVHD patients. However, the trial results of NCT02959944 showed that the ORR is minimally improved by ibrutinib plus prednisone (41.1%) as compared to placebo plus prednisone (36.7%) in patients with new onset cGVHD (Table 2). These data together with the results of other ongoing trials will provide new insights to better design future trials to improve the efficacy and safety of ibrutinib as monotherapy or in combination therapy with corticosteroids for the treatment of cGVHD.
In addition to cGVHD, preclinical evidence demonstrates the efficacy of ibrutinib at alleviating the disease manifestations in mouse models of acute GVHD (aGVHD) and allo-skin transplantation, suggesting potential expansion of its use to the treatment of aGVHD and allo-skin graft rejection (108, 111).
Autoimmune Diseases
Increased BTK protein expression in patients with systemic autoimmune diseases appears to be correlated with autoantibody production (3, 112, 113). Mechanistically, BTK is required for the proliferation and activation of autoreactive B cells (3, 112, 113). Although BTK expression was not initially detected in T cells (114) and XLA patients with BTK mutations do not exhibit apparent T cell defects (115–117), Xia et al. recently reported that BTK is indeed expressed in T cells and further upregulated in effector and memory T cells (25). Btk-/- T cells exhibit defective proliferation and reduced expression of the activation marker CD69 as well as production of cytokines following CD3 and CD28 stimulation (25). Inhibition of BTK by acalabrutinib suppresses CD3 plus CD28-induced proliferation of WT but not Btk-/- T cells in vitro (25). Notably, Btk-/- donor T cells fail to mount graft-versus-host responses and cannot cause bone marrow destruction or blood pancytopenia in a mouse model of autoimmune aplastic anemia (25). This recent finding, which needs to be further confirmed, suggests that BTK may also be implicated in the proliferation and activation of autoreactive T cells. Furthermore, BTK may promote autoimmunity as an important driver of myeloid cell inflammation, osteoclast differentiation and altered B cell-T cell interactions (3, 112, 113). Together, these findings reveal multiple pathogenic roles of BTK in autoimmune diseases.
Based on the above evidence of the pathogenic roles for BTK in autoimmunity, 4 clinical trials are testing the efficacy of ibrutinib (2 trials) and acalabrutinib (2 trials) in autoimmune diseases, including autoimmune hemolytic anemia (AIHA) and rheumatoid arthritis (RA) (Tables 1 and 2). One Phase II clinical trial (NCT04398459) is evaluating the safety and efficacy of ibrutinib in R/R AIHA patients that are previously treated with glucocorticoids and rituximab. Two Phase II trials are assessing the efficacy of ibrutinib (NCT03827603) and acalabrutinib (NCT04657094) in R/R AIHA patients with underlying CLL. A recently completed clinical trial (NCT02387762) compared the effects of acalabrutinib versus placebo in combination with methotrexate in RA patients, but the initial results of primary outcome do not show beneficial effects of acalabrutinib (Table 2). Interestingly, the preliminary results of a Phase IIb clinical trial (NCT03233230) on another BTK inhibitor evobrutinib (also known as M2951 or MSC2364447C) demonstrate that it is effective in RA patients with an inadequate response to methotrexate (www.clinicaltrials.gov). Additional clinical data from these trials will inform the design of future trials to improve the efficacy of BTK inhibitors, alone or in combination with other drugs, for the treatment of AIHA and RA.
Several clinical trials are studying the effects of other BTK inhibitors such as evobrutinib, tirabrutinib (also called ONO/GS-4059) and AC0058TA for the treatment of systemic lupus erythematosus (SLE) and relapsing multiple sclerosis (MS) (www.clinicaltrials.gov) (3, 112, 118–120). In addition, preclinical evidence has shown that ibrutinib effectively ameliorates disease symptoms in patient samples or animal models of SLE, MS, systemic sclerosis (SSc), neuropathy with anti-myelin-associated glycoprotein (MAG), type 2 diabetes (T2D) and obesity (118, 119, 121–125). Such ongoing clinical studies and preclinical evidence demonstrate the immunosuppressive effects of BTK inhibitors in autoimmune diseases and warrant further considerations of new clinical trials on BTK inhibitors as therapeutic agents for these autoimmune diseases.
Commonly used and recently developed drugs for autoimmune diseases include glucocorticoids, non-steroidal anti-inflammatory drugs (NSAIDs; such as aspirin, acetaminophen and selective COX-2 inhibitors), conventional disease-modifying anti-rheumatic drugs (cDMARDs; such as methotrexate and leflunomide), mTOR inhibitors (such as sirolimus and everolimus), JAK inhibitors (such as tofacitinib and baricitinib), biologics (such as etanercept, anakinra and abatacept) and monoclonal antibodies (such as rituximab, infliximab and belimumab) (126, 127). All these drugs have different mechanisms of action and toxicity profiles (126, 127). Traditional NSAIDs mainly inhibits the activity of COX-1 and COX-2, two enzymes responsible for the production of prostaglandins, which mediate inflammation and pain (126). The representative cDMARD methotrexate is an antagonist of folic acid and able to competitively inhibit the activity of folate-dependent enzymes required for DNA and RNA synthesis (126). mTOR inhibitors target essential metabolic pathways in autoimmune lymphocytes such as CD4 T cells (127). JAK inhibitors suppresses the JAK-STAT-dependent signaling pathways of many inflammatory cytokines (127). Biologics typically block specific inflammatory cytokines or immune receptors, including TNFα (such as etanercept), IL-1 (such as anakinra) and CD80/CD86 (such as abatacept), etc. (126, 127). Monoclonal antibodies act to deplete autoimmune B cells (such as rituximab) or block specific inflammatory cytokines (such as infliximab for TNFα, secukinumab for IL-17A and belimumab for BAFF) or immune receptors (such as natalizumab and vedolizumab for integrins) (126, 127). Compared to the above drugs, BTK inhibitors have distinct mechanisms of action. In addition, oral administration of BTK inhibitors is more convenient than subcutaneous injection or i.v. infusion required for biologics or monoclonal antibodies. Therefore, if their efficacy in autoimmune diseases is proven by ongoing trials, BTK inhibitors will significantly enrich the treatment options for patients with autoimmune diseases, especially given the possibility of various combination therapies.
Allergy
One recently completed Phase II clinical trial (NCT03149315) has tested the efficacy of ibrutinib on inhibiting food-induced anaphylaxis in adult patients. The initial results of this trial showed that short-term ibrutinib therapy (as few as 2 doses) suppresses skin test responses and eliminates IgE-mediated basophil activation in adults with peanut or tree nut allergy (59, 128). Mechanistically, BTK is essential for FcϵRI signaling and allergic responses in human mast cells and basophils (59, 128–131). Bone marrow-derived mast cells of Btk-/- mice exhibit impaired FcϵRI-mediated production of eicosanoid, leukotriene C4 (LTC4) and reactive oxygen species (ROS) in vitro (132). Both ibrutinib and acalabrutinib can prevent IgE-mediated degranulation, histamine release, cytokine production and upregulation of activation markers (such as CD63, CD164, CD203c or LAMP1) in primary human mast cells and basophils, and thus block allergen-induced contraction of isolated human bronchi in vitro (59, 129–131). Preclinical evidence demonstrates that two oral doses of acalabrutinib potently inhibits anaphylaxis in a humanized mouse model of systemic anaphylaxis by suppressing IgE-evoked activation of mast cells and basophils in vivo, and also significantly protects mice against death during severe anaphylaxis (129). In addition, ibrutinib attenuates both TH2/TH17 and neutrophilic/eosinophilic airway inflammation in a mouse model of CAE-induced mixed granulocytic asthma by inhibiting BTK phosphorylation in neutrophils and ITK activation in CD4 T cells (133). Thus, these clinical data and preclinical evidence support additional clinical trials of ibrutinib and acalabrutinib in allergic diseases.
Compared to other drugs commonly used for allergy, ibrutinib and acalabrutinib offer several unique benefits, including the ability to effectively inhibit IgE-FcϵRI-mediated activation of both mast cells and basophils, rapid onset of action and transient efficacy (59, 128). Other commonly used allergy drugs do not have the capability to effectively reduce both mast cell and basophil activation (59, 128). For example, antihistamines target only one mediator, histamine, of allergy and cannot prevent anaphylaxis, which could be triggered by other mediators (such as prostaglandins, leukotrienes and cytokines) released by activated mast cells and basophils (59, 128). Anti-IgE immunotherapy such as omalizumab has shown efficacy in reducing circulating IgE to improve urticaria and asthma, but they cannot completely suppress IgE-dependent mast cell and basophil activation (128). Although omalizumab has been shown to increase the threshold dose of allergen consumption in food-allergic subjects, it takes weeks to months for omalizumab to obtain efficacy. In contrast, two doses of ibrutinib effectively reduce or eliminate skin prick test responses to foods and aeroallergens in allergic subjects (59, 128). Within 1 week after cessation of ibrutinib, these responses are returned to baseline. This transient efficacy suggests that short courses or episodic treatment of ibrutinib could be used to prevent anaphylaxis to foods or drugs (59, 128). However, BTK inhibitors would not likely have efficacy on IgE-independent allergic diseases, as BTK is not known to be involved in IgE-independent pathways of mast cell or basophil activation (128). Because of their distinct mechanisms of action, both ibrutinib and acalabrutinib have the potential to improve the efficacy and reduce adverse events of other commonly used allergy drugs in combination therapies for patients with IgE-dependent allergic diseases.
In addition to ibrutinib and acalabrutinib, several other BTK inhibitors are currently in clinical trials as potential drugs for chronic spontaneous urticaria (CSU) (128). Phase IIa data from a clinical trial (NCT03137069) recently showed impressive efficacy for fenebrutinib (GDC-0853), a highly selective non-covalent BTK inhibitor, at improving clinical scores in patients with antihistamine-refractory CSU, especially in a subgroup of patients with auto-antibodies to FcϵRI, who are refractory to current CSU therapies including omalizumab (128). Another covalent BTK inhibitor, remibrutinib (LOU064), is currently being tested in a Phase IIb clinical trial in patients with antihistamine-resistant CSU (NCT03926611) (128). Collectively, the above preclinical evidence and clinical studies have paved the way for additional clinical trials of ibrutinib and acalabrutinib as well as other BTK inhibitors for the treatment of IgE-dependent anaphylaxis, food allergy, drug allergy, asthma, CSU and other difficult-to-treat allergic diseases.
COVID-19, Sepsis and Other Infectious and Inflammatory Diseases
Notably, 7 clinical trials are registered to examine the efficacy of acalabrutinib (4 trials) and ibrutinib (3 trials) on COVID-19 (Tables 1 and 2), the current pandemic caused by the SARS-CoV-2 virus that has posed a global health threat concerning high mortality rate, economic meltdown and daily life distress (134, 135). Respiratory failure due to acute respiratory distress syndrome (ARDS) is the leading cause of mortality (136, 137). The main mechanism of ARDS is uncontrolled systemic inflammatory response, termed cytokine storm (136, 137). Given the suppressive effects of BTK inhibitors or Btk deficiency on cytokine production and inflammatory responses (24, 94), the clinical trials on COVID-19 are evaluating if acalabrutinib or ibrutinib can lessen the inflammatory responses in the lungs and reduce respiratory failure in patients, while preserving overall immune function (138–143). Emerging clinical evidence shows that both acalabrutinib and ibrutinib have protective effects against pulmonary injury, decreasing the duration of mechanical ventilation and mortality rate for hospitalized patients with severe COVID-19 (138–141). The protective effects of BTK inhibitors have also been observed in CLL patients with COVID-19, as the hospitalization rate and duration for severe COVID-19 is lower and shorter for CLL patients on ibrutinib versus those on other regimens or off treatment (142, 144–146).
Mechanistically, elevated levels of BTK activity have been reported in blood monocytes from patients with severe COVID-19 compared with those from healthy volunteers (138, 139, 141, 147, 148). Btk-/- monocytes, macrophages and neutrophils show defects in TLR- and NLRP3- induced NF-κB activation as well as impaired production of inflammatory cytokines and chemokines (24). Acalabrutinib and ibrutinib are able to inhibit monocyte, macrophage and neutrophil activation, and thus decrease the levels of inflammatory cytokines and chemokines such as IL-6, TNFα, IL-1, IFNγ and MCP-1 in patients with severe COVID-19 (Figure 1) (138–140, 142). In addition, ITK-dependent effects of ibrutinib on T cell differentiation, effector function and survival may also contribute to its modulatory effects on immunopathology and lymphopenia in COVID-19 therapy, skewing T cells from a TH2-dominant to a TH1 and CD8 cytotoxic populations and thus promoting a TH1 antiviral immunity (Figure 1) (24, 142, 143). However, two recently completed phase II trials of acalabrutinib (NCT04346199 and NCT04380688) in COVID-19 patients hospitalized with respiratory symptoms did not meet the primary efficacy endpoint of increasing the proportion of patients who remained alive and free of respiratory failure (142) (Table 2). Therefore, the exact efficacy of BTK inhibitors on COVID-19 remains to be clarified with additional clinical data from the recently completed and ongoing trials, and more mechanistic studies are needed to better understand how the drugs inhibit cytokine storm and improve lymphopenia in patients with severe COVID-19.
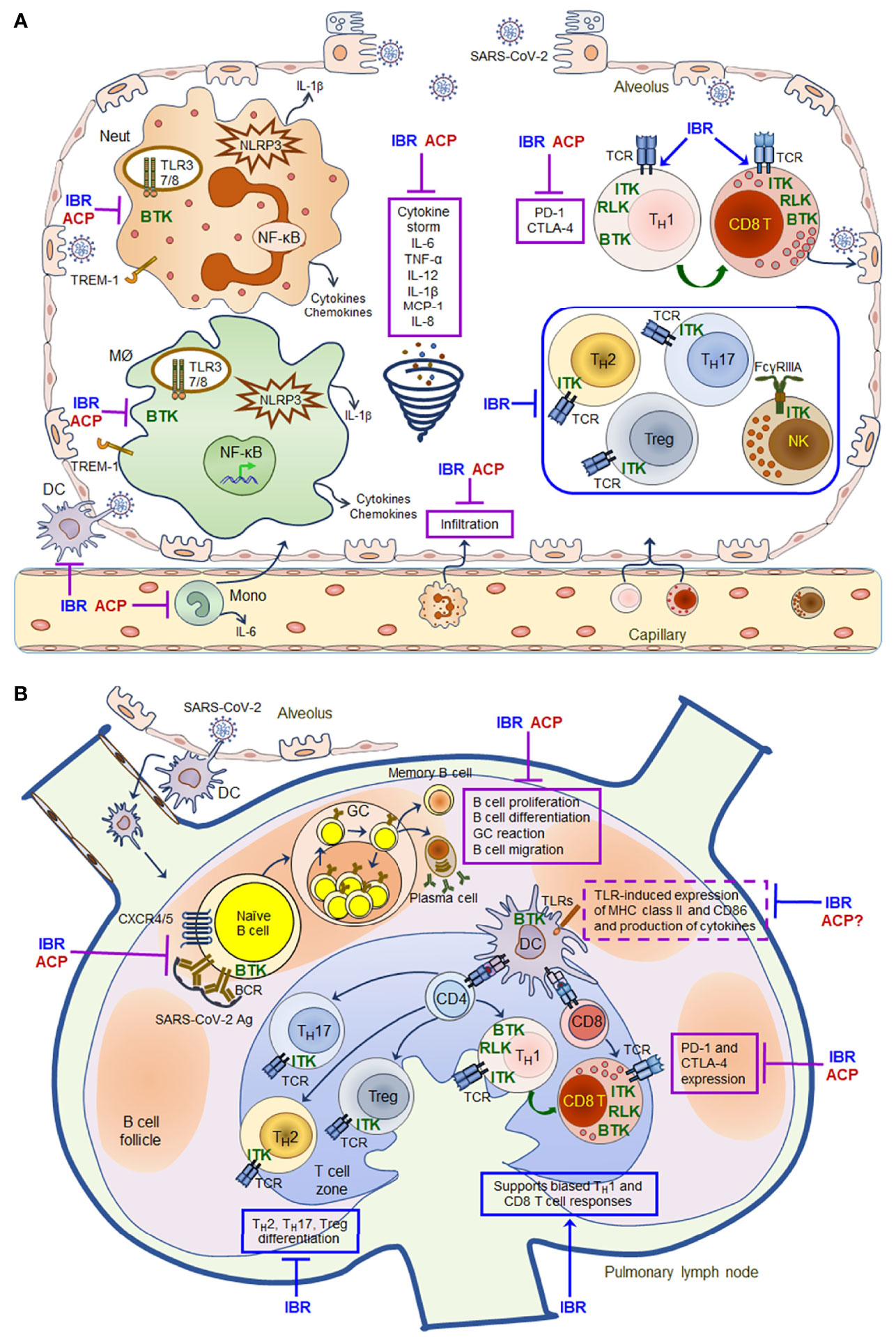
Figure 1 Schematic mechanisms of action for ibrutinib and acalabrutinib in the lung of patients with severe COVID-19. A model of mechanisms of action for ibrutinib (IBR) and acalabrutinib (ACP) is proposed based on the evidence that both drugs have protective effects on ARDS in patients with severe COVID-19 and that both drugs have multifaceted immunomodulatory effects on different immune cell subsets. (A) Inhibitory effects of ibrutinib and acalabrutinib on cytokine storm in the alveolus. (B) Immunomodulatory effects of ibrutinib and acalabrutinib on B cell and T cell responses in pulmonary lymph nodes. Both drugs can inhibit monocyte, macrophage and neutrophil activation induced by TLRs, NLRP3, TREM-1 and Dectin-1 via BTK-dependent mechanisms, thereby decreasing the production of inflammatory cytokines and chemokines such as IL-6, TNFα, IL-12, IL-1β, MCP-1 and IL-8 to ameliorate cytokine storm. Both drugs also inhibit the expression of PD-1 and CTLA-4 on activated T cells via BTK-dependent mechanisms, thus enhancing T cell anti-viral immunity. In addition, ITK-dependent effects of ibrutinib on T cell differentiation, effector function and survival may also contribute to its modulatory effects on immunopathology and lymphopenia in COVID-19 therapy, skewing T cells to a TH1- and CD8 cytotoxic T cell-dominant populations by inhibiting TH2/TH17/Treg differentiation, and thus promoting TH1 immunity and CD8 cytotoxic effector functions. However, both drugs can suppress the proliferation, differentiation, GC reaction and migration of SARS-CoV-2-specific B cells by inhibiting BCR and CXCR4/CXCR5 signaling via BTK-dependent mechanisms, thereby inhibiting the production of antibodies and memory B cell responses. All the shared effects of both drugs are depicted in solid purple boxes, while the distinct ITK-dependent effects of ibrutinib are depicted in solid blue boxes in the figure. Ibrutinib (and likely acalabrutinib) can also affect TLR-induced DC activation and expression of MHC class II and CD86 via BTK-dependent mechanisms (depicted in dashed purple boxes in the figure), and therefore may alter DC-mediated antigen presentation. Given the complex protective and undesired immunomodulatory effects of both drugs, the timing and duration of their administration need to be appropriately adjusted and tailored to improve patient outcome according to clinical data obtained from recently completed and ongoing clinical trials.
The promising clinical data of ibrutinib and acalabrutinib at inhibiting cytokine storm and improving lymphopenia in COVID-19 patients will likely promote further development of additional trials to expand the use of BTK inhibitors to sepsis and other infectious and inflammatory diseases. In particular, abundant preclinical evidence has demonstrated the efficacy of both ibrutinib and acalabrutinib at preventing and inhibiting cytokine storm as well as protecting against organ damage in several animal models of sepsis, including polymicrobial sepsis, cecal ligation and puncture (CLP)-induced sepsis, and LPS or LPS/galactosamine-induced sepsis (149–152). Evidence obtained from Xid mice with an inactivating mutation of Btk verified that the protective effects of BTK inhibitors in polymicrobial sepsis are mediated solely by inhibition of BTK (149). Mechanistically, BTK inhibitors suppresses the activation of TLR-NF-κB and NLRP3 inflammasomes in myeloid cells, leading to decreased release of cytokines and chemokines, reduced innate immune cell recruitment and a switch of macrophages from M1 to M2 phenotype (24, 149–151). In line with the animal evidence, increased BTK expression in blood cells is associated with poor survival in patients with sepsis (149). Together, all the above preclinical evidence supports the development of clinical trials to test ibrutinib and acalabrutinib for the treatment of sepsis, a major life-threatening health burden worldwide.
Interestingly, preclinical evidence also suggests potential application of ibrutinib and acalabrutinib in a number of other infectious and inflammatory diseases. This has been shown with animal models of Mycobacterium tuberculosis (Mtb) infection, visceral leishmaniasis caused by Leishmania donovani, Influenza A infection-induced acute lung injury, Streptococcus pneumoniae-induced acute pulmonary inflammation, high-fat-diet (HFD)-induced metabolic inflammation, endometriosis, post-ischemic brain inflammation after stroke and neuroinflammation-induced depression (26, 124, 153–159). On the other hand, CLL and MCL patients treated with BTK inhibitors exhibit increased risk of invasive fungal infection, bacterial infection and hepatitis B reactivation (32, 160–168). Relevant to these clinical observations, Btk-/- or ibrutinib/acalabrutinib-treated monocytes and macrophages exhibit defective TLR9-, TREM-1 and Dectin-1-mediated phagocytosis in response to fungal infections (24, 163, 169–172). In this scenario, clinical use of ibrutinib and acalabrutinib in infectious and inflammatory diseases requires prudent considerations of both the benefits and risks depending on the specific disease settings and patients’ immune cell and genetic contexts.
Major Toxicities and Potential Limitations of Ibrutinib and Acalabrutinib in Therapeutic Use
Clinical data obtained from patients with CLL and MCL have shown that compared to conventional chemoimmunotherapy regimens, ibrutinib has a favorable safety profile and is generally well-tolerated (10, 17, 19, 20, 38). However, a unique set of toxicities has been reported for ibrutinib. Common adverse effects of ibrutinib observed in CLL and MCL patients include bleeding, atrial fibrillation, hypertension, neutropenia, arthralgias, myalgias, headache, diarrhea, nausea, fatigue, rash and infection (38, 162, 166, 173–180). Based on the preliminary results posted at ClinicalTrials.gov, commonly observed adverse effects of ibrutinib in patients with other human diseases include fatigue, anemia, gastrointestinal disorders, thrombocytopenia, myalgia, arthralgia, bleeding and infection (Table 2). These toxicities are mediated by both on-target inhibition of BTK and variable off-target inhibition of other kinases such as ITK, TEC, CSK, ERBB2/HER2, ERBB4/HER4, SRC, BMX, JAK3, EGFR, PTK6, c-Kit and PDGFR, etc. (38, 173, 178, 181, 182). Severe adverse events lead to discontinuation of ibrutinib treatment in 9 – 23% of CLL and MCL patients (38) and in occasional cases of other human diseases (ClinicalTrials.gov).
Because of its higher biochemical and cellular selectivity, acalabrutinib has an improved safety profile and exhibits a high efficacy in ibrutinib-intolerant CLL patients (6, 11, 12, 183–187). Despite its improved specificity and toxicity profile, common adverse effects of acalabrutinib have also been reported, including headache, diarrhea, fatigue, myalgias, cough, neutropenia, nausea, skin rash and infection (181, 186, 188–191) (Table 2). Severe adverse events of acalabrutinib are relatively rarer than those of ibrutinib in patients with B cell malignancies and other diseases (181, 186, 188–191) (ClinicalTrials.gov). These data suggest that acalabrutinib may be an option for ibrutinib-intolerant patients with B cell malignancies as well as other diseases.
Due to its off-target inhibition of ITK, TEC and other kinases, ibrutinib has stronger and broader immunomodulatory effects than acalabrutinib (24). Of clinical importance, one major difference between the immunomodulatory effects of these two BTK inhibitors is that ibrutinib, but not acalabrutinib, impairs FcγR-mediated antibody-dependent cellular cytotoxicity (ADCC) in human NK cells and antibody-dependent cellular phagocytosis (ADCP) in human macrophages and neutrophils (24, 35, 36, 192–198). In line with this notion, a recent phase Ib/II study (NCT02296918) showed that acalabrutinib in combination with obinutuzumab (an anti-CD20 with enhanced ADCC activity) produce high and durable responses that deepen over time in CLL patients, while ibrutinib plus rituximab or obinutuzumab do not show benefits over the respective monotherapies (199, 200). Therefore, ibrutinib might have relatively limited potential in combination therapies with a variety of antibodies used for the treatment of different human diseases whose mechanisms of action depend on ADCC or ADCP. When designing combination therapies involving ibrutinib and antibodies, appropriate sequential or alternate dosing schedules of ibrutinib versus antibody treatment episodes should be considered and would be more effective than concurrent administration of the drugs.
For both ibrutinib and acalabrutinib, one limitation is that after long-term treatment many patients acquire resistance caused by mutations of Cys481 in the kinase domain of BTK, the covalent binding site for both drugs (38). This limitation could be potentially overcome by the development of non-covalent binding BTK inhibitors such as fenebrutinib (GDC-0853), ARQ 531 (ArQule 531) or LOXO-305 (RXC005, REDDX08608) (38). Another potential common limitation for all BTK inhibitors is that long-term treatment with BTK inhibitors may affect bone homoeostasis and bone structure due to inhibition of osteoclast differentiation and function, as BTK is essential for RANKL-induced signaling pathways in osteoclasts (24, 113, 201, 202). These limitations suggest that combination therapies exploiting BTK inhibitors and other effective drugs with distinct mechanisms of activation and toxicity profiles, provided with appropriate dosing and treatment schedules, will help to enhance the efficacy while reducing the severity of adverse effects induced by each individual drug, thereby improving patient outcome.
Summary
The BTK inhibitors ibrutinib and acalabrutinib are FDA-approved drugs for the treatment of B cell malignances and have demonstrated unprecedented success in CLL patients. Mounting preclinical and clinical evidence indicates that both ibrutinib and acalabrutinib are versatile and have direct effects on many immune cell subsets as well as other cell types beyond B cells. The versatility and immunomodulatory effects of both drugs have been exploited to expand their therapeutic potential in a wide variety of human diseases beyond B cell malignancies. Ibrutinib and acalabrutinib, as monotherapies or as part of combination therapies, are being tested in clinical trials in patients with hematological malignancies of myeloid cells and T cells, various solid tumors, cGVHD, autoimmune diseases, anaphylaxis and COVID-19. Clinical results obtained from these ongoing trials will provide valuable information to guide the design and improvement of future trials, including optimization of combination regimens and dosing sequences as well as better patient stratification and more efficient delivery strategies. Such information will further advance the precise implementation of ibrutinib and acalabrutinib into the clinical toolbox for the treatment of different human diseases.
Author Contributions
PX, SZ, and SG have taken the leading roles in designing and writing this manuscript. JJ, EV, and JA have also made significant contributions to writing this manuscript, especially the section of COVID-19 and sepsis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a research grant from Acerta Pharma, the National Institutes of Health grants R01 CA158402 and R21 AI128264 (PX), the Department of Defense grant W81XWH-13-1-0242 (PX), a New Jersey Commission on Cancer Research (NJCCR) grant DCHS19CRF005 (PX), a Busch Biomedical Grant (PX) and a Pilot Award from the Cancer Institute of New Jersey through Grant Number P30CA072720 from the National Cancer Institute (PX). Acerta Pharma, LLC. was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s Tyrosine Kinase in B Cell Malignancies. Nat Rev Cancer (2014) 14:219–32. doi: 10.1038/nrc3702
2. Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol Cancer (2018) 17:57. doi: 10.1186/s12943-018-0779-z
3. Rip J, van der Ploeg EK, Hendriks RW, Corneth OBJ. The Role of Bruton’s Tyrosine Kinase in Immune Cell Signaling and Systemic Autoimmunity. Crit Rev Immunol (2018) 38:17–62. doi: 10.1615/CritRevImmunol.2018025184
4. Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, et al. Bruton’s Tyrosine Kinase (Btk): Function, Regulation, and Transformation With Special Emphasis on the PH Domain. Immunol Rev (2009) 228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x
5. Wang X, Kokabee L, Kokabee M, Conklin DS. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front Cell Dev Biol (2021) 9:668996. doi: 10.3389/fcell.2021.668996
6. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia (2021) 35:312–32. doi: 10.1038/s41375-020-01072-6
7. de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton’s Tyrosine Kinase and Phospholipase Cgamma2 Mediate Chemokine-Controlled B Cell Migration and Homing. Immunity (2007) 26:93–104. doi: 10.1016/j.immuni.2006.11.012
8. Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton Tyrosine Kinase Represents a Promising Therapeutic Target for Treatment of Chronic Lymphocytic Leukemia and is Effectively Targeted by PCI-32765. Blood (2011) 117:6287–96. doi: 10.1182/blood-2011-01-328484
9. Merolle MI, Ahmed M, Nomie K, Wang ML. The B Cell Receptor Signaling Pathway in Mantle Cell Lymphoma. Oncotarget (2018) 9:25332–41. doi: 10.18632/oncotarget.25011
10. Lucas F, Woyach JA. Inhibiting Bruton’s Tyrosine Kinase in CLL and Other B-Cell Malignancies. Target Oncol (2019) 14:125–38. doi: 10.1007/s11523-019-00635-7
11. Wu JJ, Zhang MZ, Liu DL. Acalabrutinib (ACP-196): A Selective Second-Generation BTK Inhibitor. J Hematol Oncol (2016) 9:21. doi: 10.1186/s13045-016-0250-9
12. Barf T, Covey T, Izumi R, van de Kar B, Gulrajani M, van Lith B, et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor With a Differentiated Selectivity and In Vivo Potency Profile. J Pharmacol Exp Ther (2017) 363:240–52. doi: 10.1124/jpet.117.242909
13. Roskoski R Jr. Ibrutinib Inhibition of Bruton Protein-Tyrosine Kinase (BTK) in the Treatment of B Cell Neoplasms. Pharmacol Res (2016) 113:395–408. doi: 10.1016/j.phrs.2016.09.011
14. Joseph RE, Amatya N, Fulton DB, Engen JR, Wales TE, Andreotti A. Differential Impact of BTK Active Site Inhibitors on the Conformational State of Full-Length BTK. Elife (2020) 9:e60470. doi: 10.7554/eLife.60470
15. Gu D, Tang H, Wu J, Li J, Miao Y. Targeting Bruton Tyrosine Kinase Using non-Covalent Inhibitors in B Cell Malignancies. J Hematol Oncol (2021) 14:40. doi: 10.1186/s13045-021-01049-7
16. Zain R, Vihinen M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front Immunol (2021) 12:694853. doi: 10.3389/fimmu.2021.694853
17. Liu D, Zhao J. Frontline Therapies for Untreated Chronic Lymphoid Leukemia. Exp Hematol Oncol (2019) 8:15. doi: 10.1186/s40164-019-0139-8
18. Telford C, Kabadi SM, Abhyankar S, Song J, Signorovitch J, Zhao J, et al. Matching-Adjusted Indirect Comparisons of the Efficacy and Safety of Acalabrutinib Versus Other Targeted Therapies in Relapsed/Refractory Mantle Cell Lymphoma. Clin Ther (2019) 41:2357–79.e1. doi: 10.1016/j.clinthera.2019.09.012
19. Bond DA, Alinari L, Maddocks K. Bruton Tyrosine Kinase Inhibitors for the Treatment of Mantle Cell Lymphoma: Review of Current Evidence and Future Directions. Clin Adv Hematol Oncol (2019) 17:223–33 https://www.hematologyandoncology.net/archives/april-2019/bruton-tyrosine-kinase-inhibitors-for-the-treatment-of-mantle-cell-lymphoma-review-of-current-evidence-and-future-directions/.
20. Rhodes JM, Barrientos JC. Chemotherapy-Free Frontline Therapy for CLL: Is it Worth it? Hematol Am Soc Hematol Educ Program (2020) 2020:24–32. doi: 10.1182/hematology.2020000085
21. Iovino L, Shadman M. Novel Therapies in Chronic Lymphocytic Leukemia: A Rapidly Changing Landscape. Curr Treat Options Oncol (2020) 21:24. doi: 10.1007/s11864-020-0715-5
22. Bond DA, Maddocks KJ. Current Role and Emerging Evidence for Bruton Tyrosine Kinase Inhibitors in the Treatment of Mantle Cell Lymphoma. Hematol Oncol Clin North Am (2020) 34:903–21. doi: 10.1016/j.hoc.2020.06.007
23. Hanna KS, Campbell M, Husak A, Sturm S. The Role of Bruton’s Tyrosine Kinase Inhibitors in the Management of Mantle Cell Lymphoma. J Oncol Pharm Pract (2020) 26:1190–9. doi: 10.1177/1078155220915956
24. Zhu S, Gokhale S, Jung J, Spirollari E, Tsai J, Arceo J, et al. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets - Beyond B Lymphocytes. Front Cell Dev Biol (2021) 9:727531. doi: 10.3389/fcell.2021.727531
25. Xia S, Liu X, Cao X, Xu S. T-Cell Expression of Bruton’s Tyrosine Kinase Promotes Autoreactive T-Cell Activation and Exacerbates Aplastic Anemia. Cell Mol Immunol (2019) 17:1042–52. doi: 10.1038/s41423-019-0270-9
26. Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front Immunol (2017) 8:1454. doi: 10.3389/fimmu.2017.01454
27. Berglof A, Hamasy A, Meinke S, Palma M, Krstic A, Mansson R, et al. Targets for Ibrutinib Beyond B Cell Malignancies. Scand J Immunol (2015) 82:208–17. doi: 10.1111/sji.12333
28. Molina-Cerrillo J, Alonso-Gordoa T, Gajate P, Grande E. Bruton’s Tyrosine Kinase (BTK) as a Promising Target in Solid Tumors. Cancer Treat Rev (2017) 58:41–50. doi: 10.1016/j.ctrv.2017.06.001
29. Metzler JM, Burla L, Fink D, Imesch P. Ibrutinib in Gynecological Malignancies and Breast Cancer: A Systematic Review. Int J Mol Sci (2020) 21:4154 doi: 10.3390/ijms21114154
30. Pikatan NW, Liu YL, Bamodu OA, Hsiao M, Hsu WM, Haryana SM, et al. Aberrantly Expressed Bruton’s Tyrosine Kinase Preferentially Drives Metastatic and Stem Cell-Like Phenotypes in Neuroblastoma Cells. Cell Oncol (Dordr) (2020) 43:1067–84. doi: 10.1007/s13402-020-00541-5
31. Wang J, Liu X, Hong Y, Wang S, Chen P, Gu A, et al. Ibrutinib, a Bruton’s Tyrosine Kinase Inhibitor, Exhibits Antitumoral Activity and Induces Autophagy in Glioblastoma. J Exp Clin Cancer Res (2017) 36:96. doi: 10.1186/s13046-017-0549-6
32. Maffei R, Maccaferri M, Arletti L, Fiorcari S, Benatti S, Potenza L, et al. Immunomodulatory Effect of Ibrutinib: Reducing the Barrier Against Fungal Infections. Blood Rev (2020) 40:100635. doi: 10.1016/j.blre.2019.100635
33. Nicolson PLR, Nock SH, Hinds J, Garcia-Quintanilla L, Smith CW, Campos J, et al. Low-Dose Btk Inhibitors Selectively Block Platelet Activation by CLEC-2. Haematologica (2021) 106:208–19. doi: 10.3324/haematol.2019.218545
34. Mhibik M, Wiestner A, Sun C. Harnessing the Effects of BTKi on T Cells for Effective Immunotherapy Against CLL. Int J Mol Sci (2019) 21:68. doi: 10.3390/ijms21010068
35. Flinsenberg TWH, Tromedjo CC, Hu N, Liu Y, Guo Y, Thia KYT, et al. Differential Effects of BTK Inhibitors Ibrutinib and Zanubrutinib on NK Cell Effector Function in Patients With Mantle Cell Lymphoma J Haematologica (2019) 105:e76–9. doi: 10.3324/haematol.2019.220590%
36. Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib Interferes With the Cell-Mediated Anti-Tumor Activities of Therapeutic CD20 Antibodies: Implications for Combination Therapy. Haematologica (2015) 100:77–86. doi: 10.3324/haematol.2014.107011
37. Bye AP, Unsworth AJ, Desborough MJ, Hildyard CAT, Appleby N, Bruce D, et al. Severe Platelet Dysfunction in NHL Patients Receiving Ibrutinib is Absent in Patients Receiving Acalabrutinib. Blood Adv (2017) 1:2610–23. doi: 10.1182/bloodadvances.2017011999
38. Estupinan HY, Berglof A, Zain R, Smith CIE. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front Cell Dev Biol (2021) 9:630942. doi: 10.3389/fcell.2021.630942
39. Adasme MF, Parisi D, Van Belle K, Salentin S, Haupt VJ, Jennings GS, et al. Structure-Based Drug Repositioning Explains Ibrutinib as VEGFR2 Inhibitor. PloS One (2020) 15:e0233089. doi: 10.1371/journal.pone.0233089
40. Xiao L, Salem JE, Clauss S, Hanley A, Bapat A, Hulsmans M, et al. Ibrutinib-Mediated Atrial Fibrillation Attributable to Inhibition of C-Terminal Src Kinase. Circulation (2020) 142:2443–55. doi: 10.1161/CIRCULATIONAHA.120.049210
41. da Cunha-Bang C, Niemann CU. Targeting Bruton’s Tyrosine Kinase Across B-Cell Malignancies. Drugs (2018) 78:1653–63. doi: 10.1007/s40265-018-1003-6
42. Tomasson MH, Xiang Z, Walgren R, Zhao Y, Kasai Y, Miner T, et al. Somatic Mutations and Germline Sequence Variants in the Expressed Tyrosine Kinase Genes of Patients With. Novo Acute Myeloid Leukemia Blood (2008) 111:4797–808. doi: 10.1182/blood-2007-09-113027
43. Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton’s Tyrosine Kinase as a Therapeutic Target in Acute Myeloid Leukemia. Blood (2014) 123:1229–38. doi: 10.1182/blood-2013-06-511154
44. Rushworth SA, Pillinger G, Abdul-Aziz A, Piddock R, Shafat MS, Murray MY, et al. Activity of Bruton’s Tyrosine-Kinase Inhibitor Ibrutinib in Patients With CD117-Positive Acute Myeloid Leukaemia: A Mechanistic Study Using Patient-Derived Blast Cells. Lancet Haematol (2015) 2:e204–11. doi: 10.1016/S2352-3026(15)00046-0
45. Dwivedi P, Muench DE, Wagner M, Azam M, Grimes HL, Greis KD. Time Resolved Quantitative Phospho-Tyrosine Analysis Reveals Bruton’s Tyrosine Kinase Mediated Signaling Downstream of the Mutated Granulocyte-Colony Stimulating Factor Receptors. Leukemia (2019) 33:75–87. doi: 10.1038/s41375-018-0188-8
46. Pillinger G, Abdul-Aziz A, Zaitseva L, Lawes M, MacEwan DJ, Bowles KM, et al. Targeting BTK for the Treatment of FLT3-ITD Mutated Acute Myeloid Leukemia. Sci Rep (2015) 5:12949. doi: 10.1038/srep12949
47. Zaitseva L, Murray MY, Shafat MS, Lawes MJ, MacEwan DJ, Bowles KM, et al. Ibrutinib Inhibits SDF1/CXCR4 Mediated Migration in AML. Oncotarget (2014) 5:9930–8. doi: 10.18632/oncotarget.2479
48. Dwivedi P, Chutipongtanate S, Muench DE, Azam M, Grimes HL, Greis KD. SWATH-Proteomics of Ibrutinib’s Action in Myeloid Leukemia Initiating Mutated G-CSFR Signaling. Proteomics Clin Appl (2020) 14:e1900144. doi: 10.1002/prca.201900144
49. Kumar A, Vardhana S, Moskowitz AJ, Porcu P, Dogan A, Dubovsky JA, et al. Pilot Trial of Ibrutinib in Patients With Relapsed or Refractory T-Cell Lymphoma. Blood Adv (2018) 2:871–6. doi: 10.1182/bloodadvances.2017011916
50. Kornauth C, Herbaux C, Boidol B, Guillemette C, Caron P, Mayerhofer ME, et al. Rationale for the Combination of Venetoclax and Ibrutinib in T-prolymphocytic Leukemia. Haematologica (2021) 106:2251–6. doi: 10.3324/haematol.2020.271304
51. Kim DW, Tan E, Zhou JM, Schell MJ, Martinez M, Yu J, et al. A Phase 1/2 Trial of Ibrutinib in Combination With Pembrolizumab in Patients With Mismatch Repair Proficient Metastatic Colorectal Cancer. Br J Cancer (2021) 124:1803–8. doi: 10.1038/s41416-021-01368-z
52. Moschos SJ, Eroglu Z, Khushalani NI, Kendra KL, Ansstas G, In GK, et al. Targeting the IL-2 Inducible Kinase in Melanoma; a Phase 2 Study of Ibrutinib in Systemic Treatment-Refractory Distant Metastatic Cutaneous Melanoma: Preclinical Rationale, Biology, and Clinical Activity (NCI9922). Melanoma Res (2021) 31:162–72. doi: 10.1097/CMR.0000000000000726
53. Overman M, Javle M, Davis RE, Vats P, Kumar-Sinha C, Xiao L, et al. Randomized Phase II Study of the Bruton Tyrosine Kinase Inhibitor Acalabrutinib, Alone or With Pembrolizumab in Patients With Advanced Pancreatic Cancer. J Immunother Cancer (2020) 8:e000587. doi: 10.1136/jitc-2020-000587
54. Tempero M, Oh DY, Tabernero J, Reni M, Van Cutsem E, Hendifar A, et al. Ibrutinib in Combination With Nab-Paclitaxel and Gemcitabine for First-Line Treatment of Patients With Metastatic Pancreatic Adenocarcinoma: Phase III RESOLVE Study. Ann Oncol (2021) 32:600–8. doi: 10.1016/j.annonc.2021.01.070
55. Zhang T, Harrison MR, O’Donnell PH, Alva AS, Hahn NM, Appleman LJ, et al. A Randomized Phase 2 Trial of Pembrolizumab Versus Pembrolizumab and Acalabrutinib in Patients With Platinum-Resistant Metastatic Urothelial Cancer. Cancer (2020) 126:4485–97. doi: 10.1002/cncr.33067
56. Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for Chronic Graft-Versus-Host Disease After Failure of Prior Therapy. Blood (2017) 130:2243–50. doi: 10.1182/blood-2017-07-793786
57. Waller EK, Miklos D, Cutler C, Arora M, Jagasia MH, Pusic I, et al. Ibrutinib for Chronic Graft-Versus-Host Disease After Failure of Prior Therapy: 1-Year Update of a Phase 1b/2 Study. Biol Blood Marrow Transplant (2019) 25:2002–7. doi: 10.1016/j.bbmt.2019.06.023
58. Teusink-Cross A, Davies SM, Grimley MS, Chandra S, Flannery A, Dandoy CE, et al. Ibrutinib for the Treatment of Chronic Graft-vs-Host Disease in Pediatric Hematopoietic Stem Cell Transplant Patients: A Single-Center Experience. Pediatr Transplant (2020) 24:e13692. doi: 10.1111/petr.13692
59. Dispenza MC, Pongracic JA, Singh AM, Bochner BS. Short-Term Ibrutinib Therapy Suppresses Skin Test Responses and Eliminates IgE-Mediated Basophil Activation in Adults With Peanut or Tree Nut Allergy. J Allergy Clin Immunol (2018) 141:1914–6 e7. doi: 10.1016/j.jaci.2017.12.987
60. Cortes JE, Jonas BA, Graef T, Luan Y, Stein AS. Clinical Experience With Ibrutinib Alone or in Combination With Either Cytarabine or Azacitidine in Patients With Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk (2019) 19:509–15.e1. doi: 10.1016/j.clml.2019.05.008
61. Huls G, Chitu DA, Pabst T, Klein SK, Stussi G, Griskevicius L, et al. Ibrutinib Added to 10-Day Decitabine for Older Patients With AML and Higher Risk MDS. Blood Adv (2020) 4:4267–77. doi: 10.1182/bloodadvances.2020002846
62. Li X, Yin X, Wang H, Huang J, Yu M, Ma Z, et al. The Combination Effect of Homoharringtonine and Ibrutinib on FLT3-ITD Mutant Acute Myeloid Leukemia. Oncotarget (2017) 8:12764–74. doi: 10.18632/oncotarget.14463
63. Eide CA, Kurtz SE, Kaempf A, Long N, Agarwal A, Tognon CE, et al. Simultaneous Kinase Inhibition With Ibrutinib and BCL2 Inhibition With Venetoclax Offers a Therapeutic Strategy for Acute Myeloid Leukemia. Leukemia (2020) 34:2342–53. doi: 10.1038/s41375-020-0764-6
64. Morell A, Cermakova L, Novotna E, Lastovickova L, Haddad M, Haddad A, et al. Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing Akr1c3. Cancers (Basel) (2020) 12:3731. doi: 10.3390/cancers12123731
65. Gamperl S, Stefanzl G, Peter B, Smiljkovic D, Bauer K, Willmann M, et al. Effects of Ibrutinib on Proliferation and Histamine Release in Canine Neoplastic Mast Cells. Vet Comp Oncol (2019) 17:553–61. doi: 10.1111/vco.12520
66. Mamand S, Allchin RL, Ahearne MJ, Wagner SD. Comparison of Interleukin-2-Inducible Kinase (ITK) Inhibitors and Potential for Combination Therapies for T-Cell Lymphoma. Sci Rep (2018) 8:14216. doi: 10.1038/s41598-018-32634-5
67. Allchin RL, Kelly ME, Mamand S, Doran AG, Keane T, Ahearne MJ, et al. Structural and Diffusion Weighted MRI Demonstrates Responses to Ibrutinib in a Mouse Model of Follicular Helper (Tfh) T-Cell Lymphoma. PloS One (2019) 14:e0215765. doi: 10.1371/journal.pone.0215765
68. Oberbeck S, Schrader A, Warner K, Jungherz D, Crispatzu G, von Jan J, et al. Noncanonical Effector Functions of the T-Memory-Like T-PLL Cell are Shaped by Cooperative TCL1A and TCR Signaling. Blood (2020) 136:2786–802. doi: 10.1182/blood.2019003348
69. Ping L, Ding N, Shi Y, Feng L, Li J, Liu Y, et al. The Bruton’s Tyrosine Kinase Inhibitor Ibrutinib Exerts Immunomodulatory Effects Through Regulation of Tumor-Infiltrating Macrophages. Oncotarget (2017) 8:39218–29. doi: 10.18632/oncotarget.16836
70. Wang X, Wong J, Sevinsky CJ, Kokabee L, Khan F, Sun Y, et al. Bruton’s Tyrosine Kinase Inhibitors Prevent Therapeutic Escape in Breast Cancer Cells. Mol Cancer Ther (2016) 15:2198–208. doi: 10.1158/1535-7163.MCT-15-0813
71. Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res (2016) 76:2125–36. doi: 10.1158/0008-5472.CAN-15-1490
72. Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic Antitumor Immunity by Checkpoint Blockade is Enhanced by Ibrutinib, an Inhibitor of Both BTK and ITK. Proc Natl Acad Sci U States America (2015) 112:E966–72. doi: 10.1073/pnas.1500712112
73. Guerra DAP, Paiva AE, Sena IFG, Azevedo PO, Silva WN, Mintz A, et al. Targeting Glioblastoma-Derived Pericytes Improves Chemotherapeutic Outcome. Angiogenesis (2018) 21:667–75. doi: 10.1007/s10456-018-9621-x
74. Jeske SS, Brand M, Ziebart A, Laban S, Doescher J, Greve J, et al. Adenosine-Producing Regulatory B Cells in Head and Neck Cancer. Cancer Immunol Immunother (2020) 69:1205–16. doi: 10.1007/s00262-020-02535-6
75. Sun FD, Wang PC, Shang J, Zou SH, Du X. Ibrutinib Presents Antitumor Activity in Skin Cancer and Induces Autophagy. Eur Rev Med Pharmacol Sci (2018) 22:561–6. doi: 10.26355/eurrev_201801_14210
76. Zucha MA, Wu AT, Lee WH, Wang LS, Lin WW, Yuan CC, et al. Bruton’s Tyrosine Kinase (Btk) Inhibitor Ibrutinib Suppresses Stem-Like Traits in Ovarian Cancer. Oncotarget (2015) 6:13255–68. doi: 10.18632/oncotarget.3658
77. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discovery (2016) 6:270–85. doi: 10.1158/2159-8290.CD-15-0827
78. Zhu Z, Ling L, Qi L, Chong Y, Xue L. Bruton’s Tyrosine Kinase (BTK) Inhibitor (Ibrutinib)-Suppressed Migration and Invasion of Prostate Cancer. Onco Targets Ther (2020) 13:4113–22. doi: 10.2147/OTT.S245848
79. Chong IY, Aronson L, Bryant H, Gulati A, Campbell J, Elliott R, et al. Mapping Genetic Vulnerabilities Reveals BTK as a Novel Therapeutic Target in Oesophageal Cancer. Gut (2018) 67:1780–92. doi: 10.1136/gutjnl-2017-314408
80. Varikuti S, Singh B, Volpedo G, Ahirwar DK, Jha BK, Saljoughian N, et al. Ibrutinib Treatment Inhibits Breast Cancer Progression and Metastasis by Inducing Conversion of Myeloid-Derived Suppressor Cells to Dendritic Cells. Br J Cancer (2020) 122:1005–13. doi: 10.1038/s41416-020-0743-8
81. Pan Y, Chiu YH, Chiu SC, Cho DY, Lee LM, Wen YC, et al. Inhibition of Bruton’s Tyrosine Kinase Suppresses Cancer Stemness and Promotes Carboplatin-Induced Cytotoxicity Against Bladder Cancer Cells. Anticancer Res (2020) 40:6093–9. doi: 10.21873/anticanres.14630
82. Liu SC, Wu YC, Huang CM, Hsieh MS, Huang TY, Huang CS, et al. Inhibition of Bruton’s Tyrosine Kinase as a Therapeutic Strategy for Chemoresistant Oral Squamous Cell Carcinoma and Potential Suppression of Cancer Stemness. Oncogenesis (2021) 10:20. doi: 10.1038/s41389-021-00308-z
83. Li T, Deng Y, Shi Y, Tian R, Chen Y, Zou L, et al. Bruton’s Tyrosine Kinase Potentiates ALK Signaling and Serves as a Potential Therapeutic Target of Neuroblastoma. Oncogene (2018) 37:6180–94. doi: 10.1038/s41388-018-0397-7
84. Ishfaq M, Pham T, Beaman C, Tamayo P, Yu AL, Joshi S. BTK Inhibition Reverses MDSC-Mediated Immunosuppression and Enhances Response to Anti-PDL1 Therapy in Neuroblastoma. Cancers (Basel) (2021) 13:817. doi: 10.3390/cancers13040817
85. Qiu Q, Li C, Song Y, Shi T, Luo X, Zhang H, et al. Targeted Delivery of Ibrutinib to Tumor-Associated Macrophages by Sialic Acid-Stearic Acid Conjugate Modified Nanocomplexes for Cancer Immunotherapy. Acta Biomater (2019) 92:184–95. doi: 10.1016/j.actbio.2019.05.030
86. Ren L, Campbell A, Fang H, Gautam S, Elavazhagan S, Fatehchand K, et al. Analysis of the Effects of the Bruton’s Tyrosine Kinase (Btk) Inhibitor Ibrutinib on Monocyte Fcgamma Receptor (FcgammaR) Function. J Biol Chem (2016) 291:3043–52. doi: 10.1074/jbc.M115.687251
87. Jimenez I, Carabia J, Bobillo S, Palacio C, Abrisqueta P, Pages C, et al. Repolarization of Tumor Infiltrating Macrophages and Increased Survival in Mouse Primary CNS Lymphomas After XPO1 and BTK Inhibition. J Neurooncol (2020) 149:13–25. doi: 10.1007/s11060-020-03580-y
88. Conniot J, Scomparin A, Peres C, Yeini E, Pozzi S, Matos AI, et al. Immunization With Mannosylated Nanovaccines and Inhibition of the Immune-Suppressing Microenvironment Sensitizes Melanoma to Immune Checkpoint Modulators. Nat Nanotechnol (2019) 14:891–901. doi: 10.1038/s41565-019-0512-0
89. Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of Dendritic Cell Maturation and Function by Bruton’s Tyrosine Kinase via IL-10 and Stat3. J Proc Nat Acad Sci U S A (2006) 103:153–8. doi: 10.1073/pnas.0509784103%
90. Singhal E, Kumar P, Sen P. A Novel Role for Bruton’s Tyrosine Kinase in Hepatocyte Growth Factor-Mediated Immunoregulation of Dendritic Cells. J Biol Chem (2011) 286:32054–63. doi: 10.1074/jbc.M111.271247
91. Flynn NJ, Somasundaram R, Arnold KM, Sims-Mourtada J. The Multifaceted Roles of B Cells in Solid Tumors: Emerging Treatment Opportunities. Target Oncol (2017) 12:139–52. doi: 10.1007/s11523-017-0481-x
92. Campbell R, Chong G, Hawkes EA. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, Beyond Hematological Malignancies. J Clin Med (2018) 7:62. doi: 10.3390/jcm7040062
93. Pleyer C, Wiestner A, Sun C. Immunological Changes With Kinase Inhibitor Therapy for Chronic Lymphocytic Leukemia. Leuk Lymphoma (2018) 59:2792–800. doi: 10.1080/10428194.2018.1457147
94. Mulder TA, Pena-Perez L, Berglof A, Meinke S, Estupinan HY, Heimersson K, et al. Ibrutinib Has Time-Dependent On- and Off-Target Effects on Plasma Biomarkers and Immune Cells in Chronic Lymphocytic Leukemia. Hemasphere (2021) 5:e564. doi: 10.1097/HS9.0000000000000564
95. Benner B, Scarberry L, Stiff A, Duggan MC, Good L, Lapurga G, et al. Evidence for Interaction of the NLRP3 Inflammasome and Bruton’s Tyrosine Kinase in Tumor-Associated Macrophages: Implications for Myeloid Cell Production of Interleukin-1beta. Oncoimmunology (2019) 8:1659704. doi: 10.1080/2162402X.2019.1659704
96. Emerson DA, Rolig AS, Redmond WL. Enhancing the Generation of Eomeshi CD8+ T Cells Augment the Efficacy of OX40- and CTLA-4-Targeted Immunotherapy. Cancer Immunol Res (2021) 9:430–40. doi: 10.1158/2326-6066.CIR-20-0338
97. Tan B, Huang Y, Zhang B, Lin N. The Effect of Ibrutinib on Radiosensitivity in Pancreatic Cancer Cells by Targeting EGFR/AKT/mTOR Signaling Pathway. BioMed Pharmacother (2020) 128:110133. doi: 10.1016/j.biopha.2020.110133
98. Lin CH, Elkholy KH, Wani NA, Li D, Hu P, Barajas JM, et al. Ibrutinib Potentiates Antihepatocarcinogenic Efficacy of Sorafenib by Targeting EGFR in Tumor Cells and BTK in Immune Cells in the Stroma. Mol Cancer Ther (2020) 19:384–96. doi: 10.1158/1535-7163.MCT-19-0135
99. Luo QY, Zhou SN, Pan WT, Sun J, Yang LQ, Zhang L, et al. A Multi-Kinase Inhibitor APG-2449 Enhances the Antitumor Effect of Ibrutinib in Esophageal Squamous Cell Carcinoma via EGFR/FAK Pathway Inhibition. Biochem Pharmacol (2021) 183:114318. doi: 10.1016/j.bcp.2020.114318
100. Segura-Collar B, Garranzo-Asensio M, Herranz B, Hernandez-SanMiguel E, Cejalvo T, Casas BS, et al. Tumor-Derived Pericytes Driven by EGFR Mutations Govern the Vascular and Immune Microenvironment of Gliomas. Cancer Res (2021) 81:2142–56. doi: 10.1158/0008-5472.CAN-20-3558
101. Jaglowski SM, Blazar BR. How Ibrutinib, a B-Cell Malignancy Drug, Became an FDA-Approved Second-Line Therapy for Steroid-Resistant Chronic GVHD. Blood Adv (2018) 2:2012–9. doi: 10.1182/bloodadvances.2018013060
102. King-Kallimanis BL, Wroblewski T, Kwitkowski V, De Claro RA, Gwise T, Bhatnagar V, et al. FDA Review Summary of Patient-Reported Outcome Results for Ibrutinib in the Treatment of Chronic Graft Versus Host Disease. Qual Life Res (2020) 29:1903–11. doi: 10.1007/s11136-020-02448-y
103. Gonzalez RM, Pidala J. Evolving Therapeutic Options for Chronic Graft-Versus-Host Disease. Pharmacotherapy (2020) 40:756–72. doi: 10.1002/phar.2427
104. Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, et al. New Approaches for the Treatment of Chronic Graft-Versus-Host Disease: Current Status and Future Directions. Front Immunol (2020) 11:578314. doi: 10.3389/fimmu.2020.578314
105. Vaidya T, Menzer C, Ponce DM, Markova A. Inpatient Management of Mucocutaneous GVHD. Curr Dermatol Rep (2019) 8:258–78. doi: 10.1007/s13671-019-00280-3
106. Shapiro RM, Antin JH. Therapeutic Options for Steroid-Refractory Acute and Chronic GVHD: An Evolving Landscape. Expert Rev Hematol (2020) 13:519–32. doi: 10.1080/17474086.2020.1752175
107. Dubovsky JA, Flynn R, Du J, Harrington BK, Zhong Y, Kaffenberger B, et al. Ibrutinib Treatment Ameliorates Murine Chronic Graft-Versus-Host Disease. J Clin Invest (2014) 124:4867–76. doi: 10.1172/JCI75328
108. Schutt SD, Fu J, Nguyen H, Bastian D, Heinrichs J, Wu Y, et al. Inhibition of BTK and ITK With Ibrutinib Is Effective in the Prevention of Chronic Graft-Versus-Host Disease in Mice. PloS One (2015) 10:e0137641. doi: 10.1371/journal.pone.0137641
109. Ryan CE, Sahaf B, Logan AC, O’Brien S, Byrd JC, Hillmen P, et al. Ibrutinib Efficacy and Tolerability in Patients With Relapsed Chronic Lymphocytic Leukemia Following Allogeneic HCT. Blood (2016) 128:2899–908. doi: 10.1182/blood-2016-06-715284
110. Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, et al. A Practical Guide to the Monitoring and Management of the Complications of Systemic Corticosteroid Therapy. Allergy Asthma Clin Immunol (2013) 9:30. doi: 10.1186/1710-1492-9-30
111. Zhang Q, Chen J, Gao H, Zhang S, Zhao C, Zhou C, et al. Drug Repurposing: Ibrutinib Exhibits Immunosuppressive Potential in Organ Transplantation. Int J Med Sci (2018) 15:1118–28. doi: 10.7150/ijms.24460
112. Lorenzo-Vizcaya A, Fasano S, Isenberg DA. Bruton’s Tyrosine Kinase Inhibitors: A New Therapeutic Target for the Treatment of SLE? Immunotargets Ther (2020) 9:105–10. doi: 10.2147/ITT.S240874
113. Liu YT, Ding HH, Lin ZM, Wang Q, Chen L, Liu SS, et al. A Novel Tricyclic BTK Inhibitor Suppresses B Cell Responses and Osteoclastic Bone Erosion in Rheumatoid Arthritis. Acta Pharmacol Sin (2021) 42:1653–64. doi: 10.1038/s41401-020-00578-0
114. Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The Gene Involved in X-Linked Agammaglobulinaemia is a Member of the Src Family of Protein-Tyrosine Kinases. Nature (1993) 361:226–33. doi: 10.1038/361226a0
115. Plebani A, Fischer MB, Meini A, Duse M, Thon V, Eibl MM. T Cell Activity and Cytokine Production in X-Linked Agammaglobulinemia: Implications for Vaccination Strategies. Int Arch Allergy Immunol (1997) 114:90–3. doi: 10.1159/000237649
116. Paroli M, Accapezzato D, Francavilla V, Insalaco A, Plebani A, Balsano F, et al. Long-Lasting Memory-Resting and Memory-Effector CD4+ T Cells in Human X-Linked Agammaglobulinemia. Blood (2002) 99:2131–7. doi: 10.1182/blood.v99.6.2131
117. Liu Y, Wu Y, Lam KT, Lee PP, Tu W, Lau YL. Dendritic and T Cell Response to Influenza is Normal in the Patients With X-Linked Agammaglobulinemia. J Clin Immunol (2012) 32:421–9. doi: 10.1007/s10875-011-9639-y
118. Bag-Ozbek A, Hui-Yuen JS. Emerging B-Cell Therapies in Systemic Lupus Erythematosus. Ther Clin Risk Manag (2021) 17:39–54. doi: 10.2147/TCRM.S252592
119. Torke S, Weber MS. Inhibition of Bruton s Tyrosine Kinase as a Novel Therapeutic Approach in Multiple Sclerosis. Expert Opin Investig Drugs (2020) 29:1143–50. doi: 10.1080/13543784.2020.1807934
120. Carnero Contentti E, Correale J. Bruton’s Tyrosine Kinase Inhibitors: A Promising Emerging Treatment Option for Multiple Sclerosis. Expert Opin Emerg Drugs (2020) 25:377–81. doi: 10.1080/14728214.2020.1822817
121. Einhaus J, Pecher AC, Asteriti E, Schmid H, Secker KA, Duerr-Stoerzer S, et al. Inhibition of Effector B Cells by Ibrutinib in Systemic Sclerosis. Arthritis Res Ther (2020) 22:66. doi: 10.1186/s13075-020-02153-8
122. Castellani F, Visentin A, Campagnolo M, Salvalaggio A, Cacciavillani M, Candiotto C, et al. The Bruton Tyrosine Kinase Inhibitor Ibrutinib Improves Anti-MAG Antibody Polyneuropathy. Neurol Neuroimmunol Neuroinflamm (2020) 7:e720. doi: 10.1212/NXI.0000000000000720
123. Yang X, Cao Z, Wu P, Li Z. Effect and Mechanism of the Bruton Tyrosine Kinase (Btk) Inhibitor Ibrutinib on Rat Model of Diabetic Foot Ulcers. Med Sci Monit (2019) 25:7951–7. doi: 10.12659/MSM.916950
124. Purvis GSD, Collino M, Aranda-Tavio H, Chiazza F, O’Riordan CE, Zeboudj L, et al. Inhibition of Bruton’s TK Regulates Macrophage NF-kappaB and NLRP3 Inflammasome Activation in Metabolic Inflammation. Br J Pharmacol (2020) 177:4416–32. doi: 10.1111/bph.15182
125. Althubiti M, Almaimani R, Eid SY, Elzubaier M, Refaat B, Idris S, et al. BTK Targeting Suppresses Inflammatory Genes and Ameliorates Insulin Resistance. Eur Cytokine Netw (2020) 31:168–79. doi: 10.1684/ecn.2020.0454
126. Li P, Zheng Y, Chen X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front Pharmacol (2017) 8:460. doi: 10.3389/fphar.2017.00460
127. Fugger L, Jensen LT, Rossjohn J. Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases. Cell (2020) 181:63–80. doi: 10.1016/j.cell.2020.03.007
128. Dispenza MC. The Use of Bruton’s Tyrosine Kinase Inhibitors to Treat Allergic Disorders. Curr Treat Options Allergy (2021) 1–13. doi: 10.1007/s40521-021-00286-y
129. Dispenza MC, Krier-Burris RA, Chhiba KD, Undem BJ, Robida PA, Bochner BS. Bruton’s Tyrosine Kinase Inhibition Effectively Protects Against Human IgE-Mediated Anaphylaxis. J Clin Invest (2020) 130:4759–70. doi: 10.1172/JCI138448
130. MacGlashan D Jr., Honigberg LA, Smith A, Buggy J, Schroeder JT. Inhibition of IgE-Mediated Secretion From Human Basophils With a Highly Selective Bruton’s Tyrosine Kinase, Btk, Inhibitor. Int Immunopharmacol (2011) 11:475–9. doi: 10.1016/j.intimp.2010.12.018
131. Smiljkovic D, Blatt K, Stefanzl G, Dorofeeva Y, Skrabs C, Focke-Tejkl M, et al. BTK Inhibition is a Potent Approach to Block IgE-Mediated Histamine Release in Human Basophils. Allergy (2017) 72:1666–76. doi: 10.1111/all.13166
132. Kuehn HS, Swindle EJ, Kim MS, Beaven MA, Metcalfe DD, Gilfillan AM. The Phosphoinositide 3-Kinase-Dependent Activation of Btk is Required for Optimal Eicosanoid Production and Generation of Reactive Oxygen Species in Antigen-Stimulated Mast Cells. J Immunol (2008) 181:7706–12. doi: 10.4049/jimmunol.181.11.7706
133. Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim KE, Siddiqui N, Al-Harbi MM, et al. Inhibition of Bruton’s Tyrosine Kinase and IL-2 Inducible T-Cell Kinase Suppresses Both Neutrophilic and Eosinophilic Airway Inflammation in a Cockroach Allergen Extract-Induced Mixed Granulocytic Mouse Model of Asthma Using Preventative and Therapeutic Strategy. Pharmacol Res (2019) 148:104441. doi: 10.1016/j.phrs.2019.104441
134. Ranney ML, Griffeth V, Jha AK. Critical Supply Shortages - The Need for Ventilators and Personal Protective Equipment During the Covid-19 Pandemic. N Engl J Med (2020) 382:e41. doi: 10.1056/NEJMp2006141
135. Wolfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Muller MA, et al. Virological Assessment of Hospitalized Patients With COVID-2019. Nature (2020) 581:465–9. doi: 10.1038/s41586-020-2196-x
136. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
137. Rabaan AA, Al-Ahmed SH, Muhammad J, Khan A, Sule AA, Tirupathi R, et al. Role of Inflammatory Cytokines in COVID-19 Patients: A Review on Molecular Mechanisms, Immune Functions, Immunopathology and Immunomodulatory Drugs to Counter Cytokine Storm. Vaccines (Basel) (2021) 9:436. doi: 10.3390/vaccines9050436
138. Roschewski M, Lionakis MS, Sharman JP, Roswarski J, Goy A, Monticelli MA, et al. Inhibition of Bruton Tyrosine Kinase in Patients With Severe COVID-19. Sci Immunol (2020) 5:eabd0110. doi: 10.1126/sciimmunol.abd0110
139. Rada M, Qusairy Z, Massip-Salcedo M, Macip S. Relevance of the Bruton Tyrosine Kinase as a Target for COVID-19 Therapy. Mol Cancer Res (2020) 19:549–54. doi: 10.1158/1541-7786.MCR-20-0814
140. Benner B, Carson WE. Observations on the Use of Bruton’s Tyrosine Kinase Inhibitors in SAR-CoV-2 and Cancer. J Hematol Oncol (2021) 14:15. doi: 10.1186/s13045-020-00999-8
141. Treon SP, Castillo JJ, Skarbnik AP, Soumerai JD, Ghobrial IM, Guerrera ML, et al. The BTK Inhibitor Ibrutinib may Protect Against Pulmonary Injury in COVID-19-Infected Patients. Blood (2020) 135:1912–5. doi: 10.1182/blood.2020006288
142. Fiorcari S, Atene CG, Maffei R, Debbia G, Potenza L, Luppi M, et al. Ibrutinib Interferes With Innate Immunity in Chronic Lymphocytic Leukemia Patients During COVID-19 Infection. Haematologica (2021) 106:2265–8. doi: 10.3324/haematol.2020.277392
143. McGee MC, August A, Huang W. BTK/ITK Dual Inhibitors: Modulating Immunopathology and Lymphopenia for COVID-19 Therapy. J Leukoc Biol (2021) 109:49–53. doi: 10.1002/JLB.5COVR0620-306R
144. Scarfo L, Chatzikonstantinou T, Rigolin GM, Quaresmini G, Motta M, Vitale C, et al. COVID-19 Severity and Mortality in Patients With Chronic Lymphocytic Leukemia: A Joint Study by ERIC, the European Research Initiative on CLL, and CLL Campus. Leukemia (2020) 34:2354–63. doi: 10.1038/s41375-020-0959-x
145. Molina-Cerrillo J, Marquet-Palomanes J, Alonso-Gordoa T, Lopez-Jimenez J, Grande E. May Ibrutinib Have Activity in Respiratory Complications by SARS-CoV-2? Clinical Experience in a Patient With Chronic Lymphocytic Leukemia. Healthcare (Basel) (2021) 9:78. doi: 10.3390/healthcare9010078
146. Lin AY, Cuttica MJ, Ison MG, Gordon LI. Ibrutinib for Chronic Lymphocytic Leukemia in the Setting of Respiratory Failure From Severe COVID-19 Infection: Case Report and Literature Review. EJHaem (2020). doi: 10.1002/jha2.98
147. Thibaud S, Tremblay D, Bhalla S, Zimmerman B, Sigel K, Gabrilove J. Protective Role of Bruton Tyrosine Kinase Inhibitors in Patients With Chronic Lymphocytic Leukaemia and COVID-19. Br J Haematol (2020) 190:e73–e6. doi: 10.1111/bjh.16863
148. Chong EA, Roeker LE, Shadman M, Davids MS, Schuster SJ, Mato AR. BTK Inhibitors in Cancer Patients With COVID-19: “The Winner Will be the One Who Controls That Chaos” (Napoleon Bonaparte). Clin Cancer Res (2020) 26:3514–6. doi: 10.1158/1078-0432.CCR-20-1427
149. O’Riordan CE, Purvis GSD, Collotta D, Krieg N, Wissuwa B, Sheikh MH, et al. X-Linked Immunodeficient Mice With No Functional Bruton’s Tyrosine Kinase Are Protected From Sepsis-Induced Multiple Organ Failure. Front Immunol (2020) 11:581758. doi: 10.3389/fimmu.2020.581758
150. O’Riordan CE, Purvis GSD, Collotta D, Chiazza F, Wissuwa B, Al Zoubi S, et al. Bruton’s Tyrosine Kinase Inhibition Attenuates the Cardiac Dysfunction Caused by Cecal Ligation and Puncture in Mice. Front Immunol (2019) 10:2129. doi: 10.3389/fimmu.2019.02129
151. Shaker ME, Gomaa HAM, Alharbi KS, Al-Sanea MM, El-Mesery ME, Hazem SH. Inhibition of Bruton Tyrosine Kinase by Acalabrutinib Dampens Lipopolysaccharide/Galactosamine-Induced Hepatic Damage. BioMed Pharmacother (2020) 131:110736. doi: 10.1016/j.biopha.2020.110736
152. Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim KE, Alqahtani F, Alanazi WA, et al. Bruton’s Tyrosine Kinase Inhibition Attenuates Oxidative Stress in Systemic Immune Cells and Renal Compartment During Sepsis-Induced Acute Kidney Injury in Mice. Int Immunopharmacol (2021) 90:107123. doi: 10.1016/j.intimp.2020.107123
153. Hu Y, Wen Z, Liu S, Cai Y, Guo J, Xu Y, et al. Ibrutinib Suppresses Intracellular Mycobacterium Tuberculosis Growth by Inducing Macrophage Autophagy. J Infect (2020) 80:e19–26. doi: 10.1016/j.jinf.2020.03.003
154. Florence JM, Krupa A, Booshehri LM, Davis SA, Matthay MA, Kurdowska AK. Inhibiting Bruton’s Tyrosine Kinase Rescues Mice From Lethal Influenza-Induced Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol (2018) 315:L52–L8. doi: 10.1152/ajplung.00047.2018
155. de Porto AP, Liu Z, de Beer R, Florquin S, de Boer OJ, Hendriks RW, et al. Btk Inhibitor Ibrutinib Reduces Inflammatory Myeloid Cell Responses in the Lung During Murine Pneumococcal Pneumonia. Mol Med (2019) 25:3. doi: 10.1186/s10020-018-0069-7
156. Varikuti S, Volpedo G, Saljoughian N, Hamza OM, Halsey G, Ryan NM, et al. The Potent ITK/BTK Inhibitor Ibrutinib Is Effective for the Treatment of Experimental Visceral Leishmaniasis Caused by Leishmania Donovani. J Infect Dis (2019) 219:599–608. doi: 10.1093/infdis/jiy552
157. Riccio LGC, Jeljeli M, Santulli P, Chouzenoux S, Doridot L, Nicco C, et al. B Lymphocytes Inactivation by Ibrutinib Limits Endometriosis Progression in Mice. Hum Reprod (2019) 34:1225–34. doi: 10.1093/humrep/dez071
158. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s Tyrosine Kinase is Essential for NLRP3 Inflammasome Activation and Contributes to Ischaemic Brain Injury. Nat Commun (2015) 6:7360. doi: 10.1038/ncomms8360
159. Li W, Ali T, He K, Liu Z, Shah FA, Ren Q, et al. Ibrutinib Alleviates LPS-Induced Neuroinflammation and Synaptic Defects in a Mouse Model of Depression. Brain Behav Immun (2021) 92:10–24. doi: 10.1016/j.bbi.2020.11.008
160. Blez D, Blaize M, Soussain C, Boissonnas A, Meghraoui-Kheddar A, Menezes N, et al. Ibrutinib Induces Multiple Functional Defects in the Neutrophil Response Against Aspergillus Fumigatus. Haematologica (2020) 105:478–89. doi: 10.3324/haematol.2019.219220
161. Brochard J, Morio F, Mahe J, Le Pape P, Guimard T, Mahe B, et al. Ibrutinib, a Bruton’s Tyrosine Kinase Inhibitor, a New Risk Factor for Cryptococcosis. Med Mal Infect (2020) 50:742–5. doi: 10.1016/j.medmal.2020.07.005
162. Ball S, Das A, Vutthikraivit W, Edwards PJ, Hardwicke F, Short NJ, et al. Risk of Infection Associated With Ibrutinib in Patients With B-Cell Malignancies: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Clin Lymphoma Myeloma Leuk (2020) 20:87–97.e5. doi: 10.1016/j.clml.2019.10.004
163. Fiorcari S, Maffei R, Vallerini D, Scarfo L, Barozzi P, Maccaferri M, et al. BTK Inhibition Impairs the Innate Response Against Fungal Infection in Patients With Chronic Lymphocytic Leukemia. Front Immunol (2020) 11:2158. doi: 10.3389/fimmu.2020.02158
164. Stadler N, Hasibeder A, Lopez PA, Teschner D, Desuki A, Kriege O, et al. The Bruton Tyrosine Kinase Inhibitor Ibrutinib Abrogates Triggering Receptor on Myeloid Cells 1-Mediated Neutrophil Activation. Haematologica (2017) 102:e191–4. doi: 10.3324/haematol.2016.152017
165. Tillman BF, Pauff JM, Satyanarayana G, Talbott M, Warner JL. Systematic Review of Infectious Events With the Bruton Tyrosine Kinase Inhibitor Ibrutinib in the Treatment of Hematologic Malignancies. Eur J Haematol (2018) 100:325–34. doi: 10.1111/ejh.13020
166. Los-Arcos I, Aguilar-Company J, Ruiz-Camps I. Risk of Infection Associated With New Therapies for Lymphoproliferative Syndromes. Med Clin (Barc) (2020) 154:101–7. doi: 10.1016/j.medcli.2019.07.026
167. Marchesini G, Nadali G, Facchinelli D, Candoni A, Cattaneo C, Laurenti L, et al. Infections in Patients With Lymphoproliferative Diseases Treated With Targeted Agents: SEIFEM Multicentric Retrospective Study. Br J Haematol (2020) 193:316–24. doi: 10.1111/bjh.17145
168. Risnik D, Elias EE, Keitelman I, Colado A, Podaza E, Cordini G, et al. The Effect of Ibrutinib on Neutrophil and Gammadelta T Cell Functions. Leuk Lymphoma (2020) 61:2409–18. doi: 10.1080/10428194.2020.1753043
169. Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, et al. Macrophages Eat Cancer Cells Using Their Own Calreticulin as a Guide: Roles of TLR and Btk. Proc Natl Acad Sci USA (2015) 112:2145–50. doi: 10.1073/pnas.1424907112
170. Bercusson A, Colley T, Shah A, Warris A, Armstrong-James D. Ibrutinib Blocks Btk-Dependent NF-kB and NFAT Responses in Human Macrophages During Aspergillus Fumigatus Phagocytosis. Blood (2018) 132:1985–8. doi: 10.1182/blood-2017-12-823393
171. Ormsby T, Schlecker E, Ferdin J, Tessarz AS, Angelisova P, Koprulu AD, et al. Btk is a Positive Regulator in the TREM-1/DAP12 Signaling Pathway. Blood (2011) 118:936–45. doi: 10.1182/blood-2010-11-317016
172. Strijbis K, Tafesse FG, Fairn GD, Witte MD, Dougan SK, Watson N, et al. Bruton’s Tyrosine Kinase (BTK) and Vav1 Contribute to Dectin1-Dependent Phagocytosis of Candida Albicans in Macrophages. PloS Pathog (2013) 9:e1003446. doi: 10.1371/journal.ppat.1003446
173. Lipsky A, Lamanna N. Managing Toxicities of Bruton Tyrosine Kinase Inhibitors. Hematol Am Soc Hematol Educ Program (2020) 2020:336–45. doi: 10.1182/hematology.2020000118
174. Lasica M, Tam CS. Management of Ibrutinib Toxicities: A Practical Guide. Curr Hematol Malig Rep (2020) 15:177–86. doi: 10.1007/s11899-020-00576-3
175. Pellegrini L, Novak U, Andres M, Suter T, Nagler M. Risk of Bleeding Complications and Atrial Fibrillation Associated With Ibrutinib Treatment: A Systematic Review and Meta-Analysis. Crit Rev Oncol Hematol (2021) 159:103238. doi: 10.1016/j.critrevonc.2021.103238
176. Rhodes JM, LoRe VA 3rd, Mato AR, Chong EA, Barrientos JC, Gerson JN, et al. Ibrutinib-Associated Arthralgias/Myalgias in Patients With Chronic Lymphocytic Leukemia: Incidence and Impact on Clinical Outcomes. Clin Lymphoma Myeloma Leuk (2020) 20:438–44.e1. doi: 10.1016/j.clml.2020.02.001
177. Pileri A, Guglielmo A, Agostinelli C, Evangelista V, Bertuzzi C, Alessandrini A, et al. Cutaneous Adverse-Events in Patients Treated With Ibrutinib. Dermatol Ther (2020) 33:e14190. doi: 10.1111/dth.14190
178. Bitar C, Sadeghian A, Sullivan L, Murina A. Ibrutinib-Associated Pityriasis Rosea-Like Rash. JAAD Case Rep (2018) 4:55–7. doi: 10.1016/j.jdcr.2017.06.035
179. Steingrimsson V, Gislason GK, Thornorsteinsdottir S, Rognvaldsson S, Gottfreethsson M, Aspelund T, et al. A Nationwide Study on Inpatient Opportunistic Infections in Patients With Chronic Lymphocytic Leukemia in the Pre-Ibrutinib Era. Eur J Haematol (2021) 106:346–53. doi: 10.1111/ejh.13553
180. Kin A, Schiffer CA. Infectious Complications of Tyrosine Kinase Inhibitors in Hematological Malignancies. Infect Dis Clin North Am (2020) 34:245–56. doi: 10.1016/j.idc.2020.02.008
181. Sibaud V, Beylot-Barry M, Protin C, Vigarios E, Recher C, Ysebaert L. Dermatological Toxicities of Bruton’s Tyrosine Kinase Inhibitors. Am J Clin Dermatol (2020) 21:799–812. doi: 10.1007/s40257-020-00535-x
182. Liclican A, Serafini L, Xing W, Czerwieniec G, Steiner B, Wang T, et al. Biochemical Characterization of Tirabrutinib and Other Irreversible Inhibitors of Bruton’s Tyrosine Kinase Reveals Differences in on - and Off - Target Inhibition. Biochim Biophys Acta Gen Subj (2020) 1864:129531. doi: 10.1016/j.bbagen.2020.129531
183. Covey T, Barf T, Gulrajani M, Krantz F, van Lith B, Bibikova E, et al. ACP-196: A Novel Covalent Bruton’s Tyrosine Kinase (Btk) Inhibitor With Improved Selectivity and In Vivo Target Coverage in Chronic Lymphocytic Leukemia (CLL) Patients. Cancer Res (2015) 75:2596. doi: 10.1158/1538-7445.AM2015-2596
184. Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. New Engl J Med (2016) 374:323–32. doi: 10.1056/Nejmoa1509981
185. Davids MS, Waweru C, Le Nouveau P, Padhiar A, Singh G, Abhyankar S, et al. Comparative Efficacy of Acalabrutinib in Frontline Treatment of Chronic Lymphocytic Leukemia: A Systematic Review and Network Meta-Analysis. Clin Ther (2020) 42:1955–74.e15. doi: 10.1016/j.clinthera.2020.08.017
186. Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, et al. Acalabrutinib Monotherapy in Patients With Chronic Lymphocytic Leukemia Who are Intolerant to Ibrutinib. Blood Adv (2019) 3:1553–62. doi: 10.1182/bloodadvances.2018030007
187. Isaac K, Mato AR. Acalabrutinib and Its Therapeutic Potential in the Treatment of Chronic Lymphocytic Leukemia: A Short Review on Emerging Data. Cancer Manag Res (2020) 12:2079–85. doi: 10.2147/CMAR.S219570
188. Khan Y, O’Brien S. Acalabrutinib and its Use in Treatment of Chronic Lymphocytic Leukemia. Future Oncol (2019) 15:579–89. doi: 10.2217/fon-2018-0637
189. Byrd JC, Wierda WG, Schuh A, Devereux S, Chaves JM, Brown JR, et al. Acalabrutinib Monotherapy in Patients With Relapsed/Refractory Chronic Lymphocytic Leukemia: Updated Phase 2 Results. Blood (2020) 135:1204–13. doi: 10.1182/blood.2018884940
190. Owen RG, McCarthy H, Rule S, D’Sa S, Thomas SK, Tournilhac O, et al. Acalabrutinib Monotherapy in Patients With Waldenstrom Macroglobulinemia: A Single-Arm, Multicentre, Phase 2 Study. Lancet Haematol (2020) 7:e112–21. doi: 10.1016/S2352-3026(19)30210-8
191. Delgado J, Josephson F, Camarero J, Garcia-Ochoa B, Lopez-Anglada L, Prieto-Fernandez C, et al. EMA Review of Acalabrutinib for the Treatment of Adult Patients With Chronic Lymphocytic Leukemia. Oncologist (2021) 26:242–9. doi: 10.1002/onco.13685
192. Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SEM, Butchar JP, Cheney C, et al. Ibrutinib Antagonizes Rituximab-Dependent NK Cell–Mediated Cytotoxicity. Blood (2014) 123:1957–60. doi: 10.1182/blood-2014-01-547869%
193. Hassenrück F, Knödgen E, Göckeritz E, Midda SH, Vondey V, Neumann L, et al. Sensitive Detection of the Natural Killer Cell-Mediated Cytotoxicity of Anti-CD20 Antibodies and Its Impairment by B-Cell Receptor Pathway Inhibitors. BioMed Res Int (2018) 2018:9. doi: 10.1155/2018/1023490
194. Borge M, Belen Almejun M, Podaza E, Colado A, Fernandez Grecco H, Cabrejo M, et al. Ibrutinib Impairs the Phagocytosis of Rituximab-Coated Leukemic Cells From Chronic Lymphocytic Leukemia Patients by Human Macrophages. Haematologica (2015) 100:e140–2. doi: 10.3324/haematol.2014.119669
195. Golay J, Ubiali G, Introna M. The Specific Bruton Tyrosine Kinase Inhibitor Acalabrutinib (ACP-196) Shows Favorable In Vitro Activity Against Chronic Lymphocytic Leukemia B Cells With CD20 Antibodies. Haematologica (2017) 102:e400–3. doi: 10.3324/haematol.2017.169334
196. VanDerMeid KR, Elliott MR, Baran AM, Barr PM, Chu CC, Zent CS. Cellular Cytotoxicity of Next-Generation CD20 Monoclonal Antibodies. Cancer Immunol Res (2018) 6:1150–60. doi: 10.1158/2326-6066.CIR-18-0319
197. Duong MN, Matera EL, Mathe D, Evesque A, Valsesia-Wittmann S, Clemenceau B, et al. Effect of Kinase Inhibitors on the Therapeutic Properties of Monoclonal Antibodies. MAbs (2015) 7:192–8. doi: 10.4161/19420862.2015.989020
198. Prezzo A, Cavaliere FM, Bilotta C, Pentimalli TM, Iacobini M, Cesini L, et al. Ibrutinib-Based Therapy Impaired Neutrophils Microbicidal Activity in Patients With Chronic Lymphocytic Leukemia During the Early Phases of Treatment. Leuk Res (2019) 87:106233. doi: 10.1016/j.leukres.2019.106233
199. Woyach JA, Blachly JS, Rogers KA, Bhat SA, Jianfar M, Lozanski G, et al. Acalabrutinib Plus Obinutuzumab in Treatment-Naive and Relapsed/Refractory Chronic Lymphocytic Leukemia. Cancer Discov (2020) 10:394–405. doi: 10.1158/2159-8290.CD-19-1130
200. Rogers A, Woyach JA. BTK Inhibitors and Anti-CD20 Monoclonal Antibodies for Treatment-Naive Elderly Patients With CLL. Ther Adv Hematol (2020) 11:2040620720912990. doi: 10.1177/2040620720912990
201. Shinohara M, Chang BY, Buggy JJ, Nagai Y, Kodama T, Asahara H, et al. The Orally Available Btk Inhibitor Ibrutinib (PCI-32765) Protects Against Osteoclast-Mediated Bone Loss. Bone (2014) 60:8–15. doi: 10.1016/j.bone.2013.11.025
202. Pokhrel NK, Kim YG, Kim HJ, Kim HJ, Lee JH, Choi SY, et al. A Novel Bruton’s Tyrosine Kinase Inhibitor, Acalabrutinib, Suppresses Osteoclast Differentiation and Porphyromonas Gingivalis Lipopolysaccharide-Induced Alveolar Bone Resorption. J Periodontol (2019) 90:546–54. doi: 10.1002/JPER.18-0334
Keywords: BTK, ibrutinib, acalabrutinib, immunomodulation, immune responses, inflammation, cancers, COVID-19
Citation: Zhu S, Jung J, Victor E, Arceo J, Gokhale S and Xie P (2021) Clinical Trials of the BTK Inhibitors Ibrutinib and Acalabrutinib in Human Diseases Beyond B Cell Malignancies. Front. Oncol. 11:737943. doi: 10.3389/fonc.2021.737943
Received: 16 July 2021; Accepted: 11 October 2021;
Published: 28 October 2021.
Edited by:
Rodabe N. Amaria, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
C.I. Edvard Smith, Karolinska Institutet (KI), SwedenMahmoud Kandeel, King Faisal University, Saudi Arabia
Copyright © 2021 Zhu, Jung, Victor, Arceo, Gokhale and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Xie, xie@dls.rutgers.edu