Atypical Presentations of IPEX: Expect the Unexpected
 Filippo Consonni
Filippo Consonni Sara Ciullini Mannurita
Sara Ciullini Mannurita Eleonora Gambineri
Eleonora Gambineri- 1Anna Meyer Children's Hospital, University of Florence, Florence, Italy
- 2Division of Pediatric Oncology/Hematology, Meyer University Children's Hospital, Florence, Italy
- 3Department of Neurosciences, Psychology, Drug Research, and Child Health (NEUROFARBA), University of Florence, Florence, Italy
Immune dysregulation, polyendocrinopathy, and enteropathy, X-linked (IPEX) syndrome is a rare disorder that has become a model of monogenic autoimmunity. IPEX is caused by mutations in FOXP3 gene, a master regulator of regulatory T cells (Treg). Cases reported in the last 20 years demonstrate that IPEX clinical spectrum encompasses more than the classical triad of early-onset intractable diarrhea, type 1 diabetes (T1D) and eczema. Atypical cases of IPEX include patients with late-onset of symptoms, single-organ involvement, mild disease phenotypes or rare clinical features (e.g., atrophic gastritis, interstitial lung disease, nephropathy etc.). Several atypical presentations have recently been reported, suggesting that IPEX incidence might be underestimated. Immunosuppression (IS) treatment strategies can control the disease, however at the moment allogeneic hematopoietic stem cell transplantation (HSCT) is the only available definitive cure, therefore it is important to achieve a prompt diagnosis. This review aims to describe unusual clinical phenotypes, beyond classical IPEX. Overall, our analysis contributes to increase awareness and finally improve diagnosis and treatment intervention in IPEX in order to ensure a good quality of life.
Introduction
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare congenital disorder caused by mutations in the Forkhead Box Protein 3 (FOXP3) gene, a master regulator of regulatory T cells (Treg). Its first clinical description dates back to 1982 (1), while its genetic characterization took place in 2000 (2, 3). Since then, research has shed light on its clinical spectrum and molecular features, and IPEX has become a model of monogenic autoimmunity and immune dysregulation (4). Moreover, recent advances proposed gene editing as a feasible therapeutic perspective for this syndrome (5), in addition to current treatments such as allogeneic hematopoietic stem cell transplantation (HSCT) and immunosuppression (IS) (6).
Although IPEX is a rare disease, more than 300 affected patients have been published so far (7), indicating an increasing awareness of the disorder (8). Cases reported in the last two decades demonstrate that IPEX clinical spectrum is much more heterogeneous, suggesting that its incidence might be underestimated (8). Apart from the classical triad (i.e., intractable diarrhea, type 1 diabetes mellitus—T1DM—and eczema), other autoimmune symptoms could also develop, such as thyroiditis, cytopenias, hepatitis, nephropathy or other (9). Interestedly, some reported patients only present with single organ involvement (10) or display unusual clinical features (11). Moreover, mild cases with late disease onset (12) and less severe disease course (13) have been described.
This review aims to focus on atypical clinical presentations of IPEX, giving examples of how the disease spectrum is wider than the usual classical manifestations. Mutations underlying atypical cases will be reported, with the aim of investigating possible genotype-phenotype correlations. Given the wide range of clinical manifestations, some “red flags” will be proposed. These could be helpful tools for clinicians in order to increase awareness of this disorder and to reduce diagnostic delay.
Atypical IPEX: Clinical Phenotype
Classically, IPEX presents early in life and typical cases are characterized by the previously mentioned clinical triad often associated with failure to thrive (14). Diarrhea is due to autoimmune enteropathy, a hallmark of the disease, and it may precede or follow the onset of T1DM (8). Laboratory tests often show hypereosinophilia, elevated IgE levels and a wide variety of autoantibodies, not necessarily related to an ongoing autoimmune pathology. FOXP3 expression is variable in IPEX patients, depending on the type of mutations (4).
In the last two decades many reports have contributed to better define IPEX clinical spectrum and in particular to increase awareness of atypical presentations. Therefore, we performed an extensive literature review to summarize the main features of the atypical presentations based on cases reported. Accordingly, we propose to further classify atypical IPEX in cases characterized by late-onset (i.e., more than 1 year of age), mild disease course (i.e., long-term survival without IS or with first line IS regimens), no enteropathy and/or unusual clinical features (i.e., infrequent manifestations that go beyond the classical triad and/or involve different organs, therefore considered atypical for their unique presenting features).
Late-Onset Disease
Recent studies show that the median age at disease onset is 2 months, even though its range is broad, going from prenatal manifestations to onset in the second decade of life (6). Rare cases of prenatal IPEX have been described, presenting toward the end of the second trimester of gestation with a lethal non-immune fetal hydrops (15, 16).
On the other hand, literature shows that IPEX can make its clinical debut later in life, the oldest reported patient being 12 years old at disease onset (12, 13). Enteropathy is the first manifestation in the majority of late-onset IPEX. However, a patient reported by Duclaux-Loras et al. displayed nephropathy as a first clinical sign and eventually died at 7 years (17), demonstrating that late-onset IPEX doesn't necessarily implicate a mild clinical phenotype. Classical IPEX features (i.e., diarrhea, dermatitis, and T1DM) have been largely described in patients with a delayed onset (6, 10, 11, 18–21). However, some patients with a mild clinical behavior together with a late clinical debut have been reported (12, 17). Interestingly, some late-onset IPEX patients had been initially treated as IBD (Inflammatory Bowel Disease) (10, 21, 22) and some received anti-TNF therapy with low clinical response. This confirms that IPEX should be suspected as a monogenic cause in front of a pediatric IBD phenotype (23), especially if treatment is not effective.
Mild Disease
Recent reports revealed an increasing number of IPEX cases characterized by a less severe phenotype (e.g., mild eczema, well-tolerated diarrhea) (12, 17, 18, 24, 25). In these cases, symptoms are usually well-controlled by a single drug IS regimen (6). Moreover, in some patients no specific therapy is needed, except for hormone-replacement therapy if endocrine glands are involved (25). Mild IPEX cases are sporadic, if compared to patients with classical clinical features (17). However, such phenotypes have been increasingly described in recent years, possibly meaning that widespread use of genetic testing revealed several mild cases that had previously been underdiagnosed, since FOXP3 sequencing was only reserved for severe clinical presentations.
As their severe counterpart, reported mild IPEX cases might also have an early clinical presentation. Enteropathy is usually the first presenting sign, even though Hwang et al. reported five FOXP3-mutated patients with early-onset T1DM, no gastrointestinal involvement and no need of IS (25). Laboratory tests show that immunoglobulin levels may vary from normal to reduced (12, 18), while IgE levels could be in range or increased (24). Autoantibodies are often detected, just like in severe forms. However, rare reports of mild cases with negative autoantibodies (17) suggest that this could possibly be considered a feature of mild IPEX. Anyhow, such finding needs to be confirmed in a larger cohort. Curiously, in Duclaux-Loras et al.'s cohort, Treg cells were measured in 10 patients and found normal in 3, two of whom were affected by mild disease (17). Such findings could reveal that mild IPEX patients have normal levels of Treg cells, potentially explaining the low intensity of the autoimmune phenomena in these cases. In the same study, authors speculated a relationship between mild phenotypes and mutations within the first splice donor site (17). Such genotype-phenotype correlation will be furtherly discussed.
No-Enteropathy
Interestingly, some cases of IPEX without enteropathy are reported (24–29). In these patients, first disease manifestation is usually early-onset T1DM (25). Even in the presence of diabetes, however, diagnostic suspicion could be jeopardized by the absence of enteropathy since intractable diarrhea is universally known as a hallmark of IPEX syndrome (27). For the same reason, IPEX cases without enteropathy could be underdiagnosed.
Unusual Clinical Features
Reported cases of IPEX show an increasing number of additional clinical manifestations beyond the classical triad (4) (Figure 1A). Apart from pancreatic islets in T1DM, other endocrine organs can be involved. While the thyroid gland is the second most frequently affected one (9), rare cases of adrenal insufficiency are also reported (26). Together with eczema, infrequent cutaneous manifestations include psoriasiform and ichthyosiform dermatitis (30), alopecia (31) and bullous pemphigoid (32). Similarly, enteropathy can have different histopathologic phenotypes (i.e., GvHD-like, depletion of goblet cells and Coeliac Disease-like) (19). Notably, Coeliac Disease (CD) antibodies can also be detected. For this reason, patients with early-onset enteropathy and positive CD serology who do not respond to gluten-free diet should raise suspicion of IPEX, even in the presence of CD-like histologic findings (13). Another unusual gastrointestinal finding is gastritis, which is usually atrophic (33) and might display hemorrhagic features (34) or metaplastic epithelial changes (11).
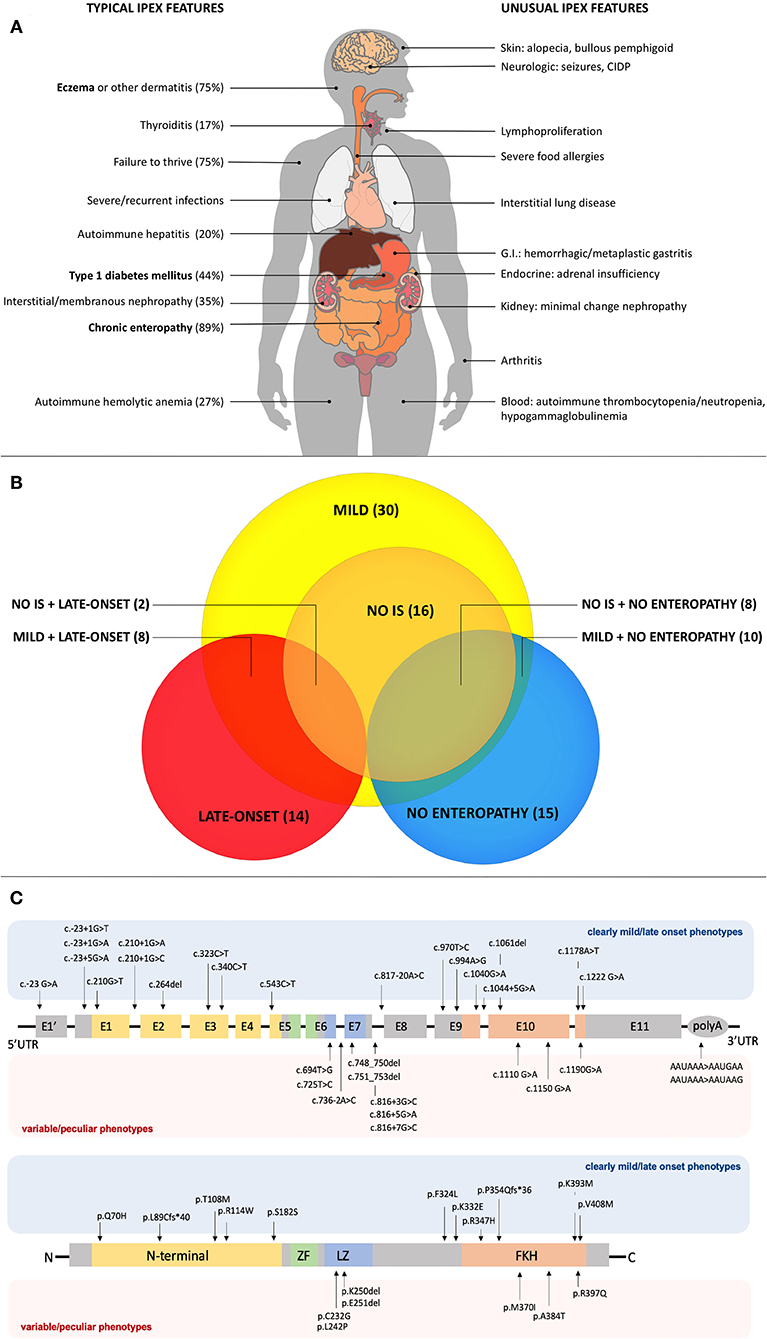
Figure 1. (A) Typical and unusual clinical features in IPEX. Percentages for typical features are based on the most recently published IPEX cohort (6). Classical triad features are in bold. CIDP, Chronic Inflammatory Demyelinating Polyneuropathy; GI, Gastrointestinal. (B) Relationships among atypical IPEX subgroups revealed by our analysis. Subgroup (number of patients); IS, Immunosuppression. (C) FOXP3 gene and protein structure showing mutations associated with mild/late-onset and variable/peculiar IPEX phenotypes.
Several reported cases of IPEX developed kidney involvement, which can infrequently be the first clinical manifestation (29). Membranous glomerulonephritis and interstitial nephritis are the most commonly reported (4, 29, 35, 36), but also Minimal-Change Nephropathy (MCN) has been described (37, 38). Anyhow, renal involvement is not only caused by underlying autoimmune processes, since IS drugs used in IPEX can induce nephrotoxicity (4). Similarly, lung involvement has been described, even though it is sometimes difficult to ascertain whether it is due to infections or to autoimmunity (4, 39). Anyhow, an autoimmune pneumopathy can be inferred if clinical signs improve with IS treatment. Such cases have been occasionally reported and are associated with fatal outcomes (39).
AutoImmune Hemolytic Anemia (AIHA) is frequent [27% of cases in a recent cohort (6)], while thrombocytopenia and neutropenia are quite unusual (4). Cytopenias can rarely coexist (29), therefore IPEX should be considered as a potential underlying cause of Evans syndrome (40) together with other Tregopathies such as CTLA-4 haploinsufficiency, LRBA, STAT3-GOF etc. (41–43). Similarly, autoimmune hepatitis is not rare, being reported in ~20% of cases (6), and it can present both with both positive and negative autoantibodies (29, 44). Signs of lymphoproliferation are occasionally described, such as splenomegaly, lymphadenopathy and lymphocytic infiltrates in multiple organs (14, 45). Finally, infrequent—yet reported—clinical findings are arthritis [whose severity and extension are quite variable (13, 45)] and severe food allergies, which can further complicate gastrointestinal symptoms (46).
Atypical IPEX: Genotype-Phenotype Correlations
Overall, our analysis of atypical forms revealed 30 mild, 14 late-onset and 15 cases with no enteropathy (Figure 1B). Moreover, several unusual clinical manifestations have been identified (Tables 1–3). Even though IPEX so far revealed an inconsistent genotype-phenotype correlation (9), we have clustered in this section reported cases that meet the criteria above, indicating—when available—information about FOXP3 protein expression and Treg percentage.
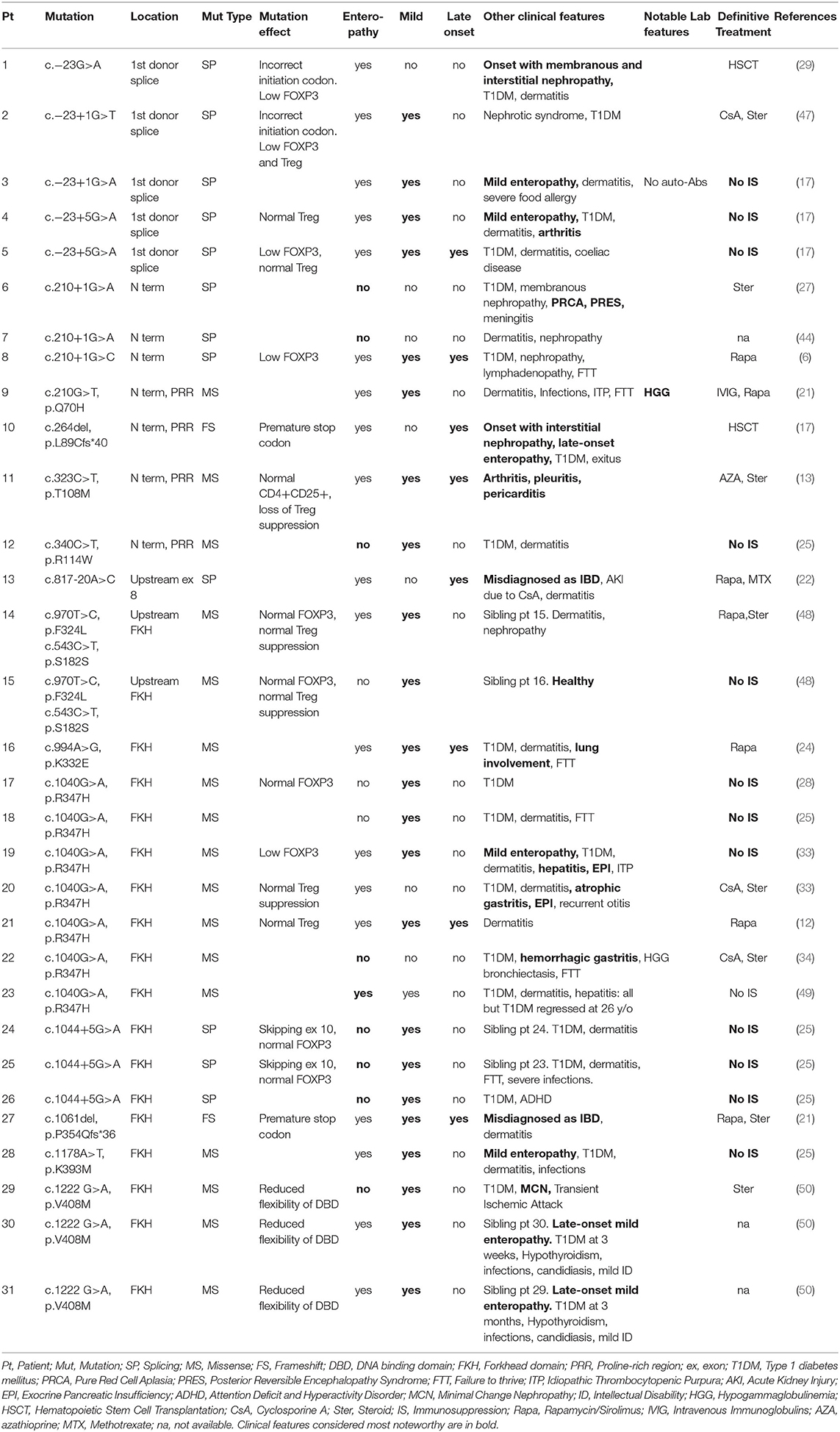
Table 1. Mutations clearly associated with a mild/late-onset IPEX phenotype.
As shown in Table 1, some FOXP3 mutations seem to be clearly associated with atypical phenotypes. Conversely, other mutations may bear from mild to severe presentations with a still debated genotype-phenotype association. Therefore, we have defined these mutations as associated with a variable disease course (Table 2). Finally, some peculiar features have shown to be recurrent in patients carrying certain mutations: these cases have been depicted in Table 3. We reported FOXP3 gene and corresponding protein mutations associated with the described phenotypes in Figure 1C.
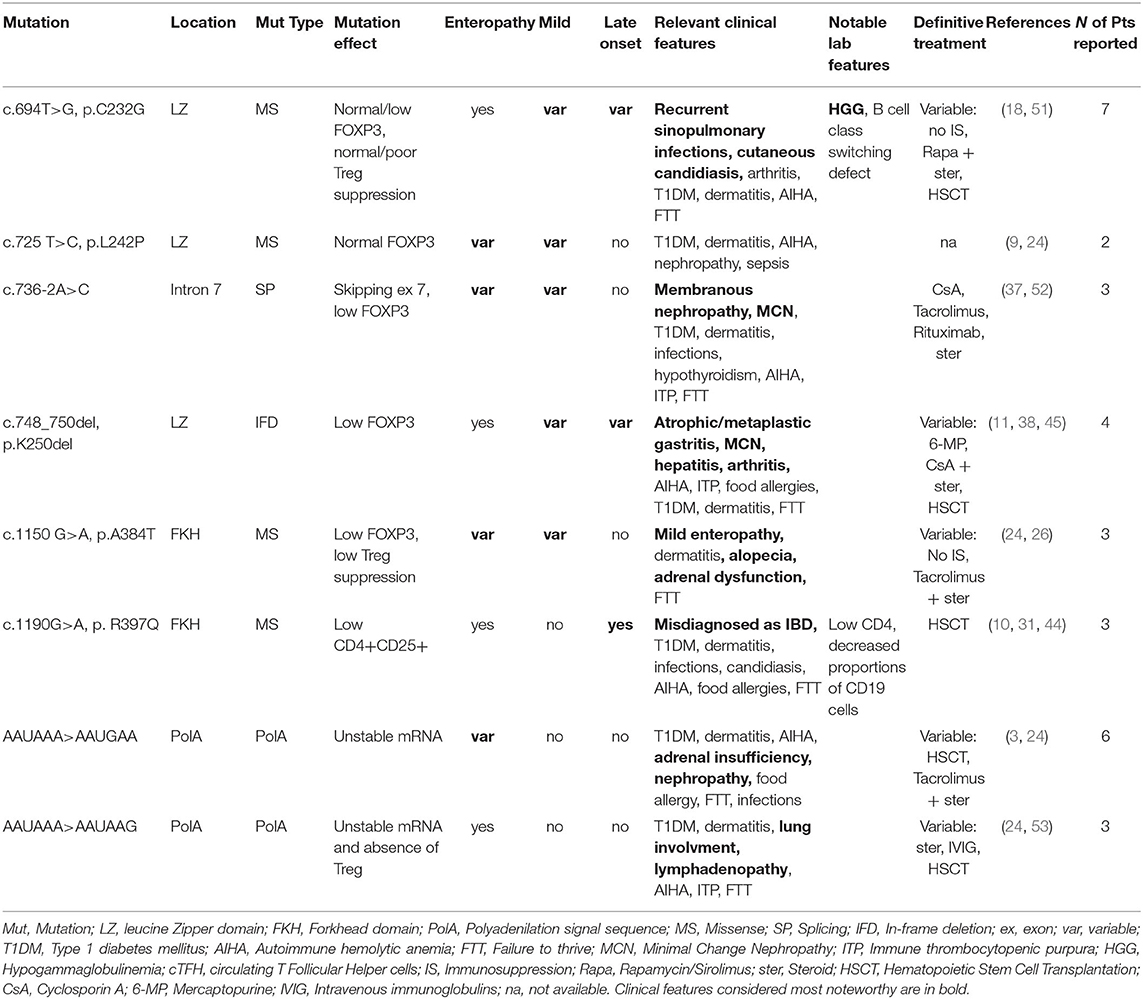
Table 2. Mutations associated with a variable IPEX phenotype.
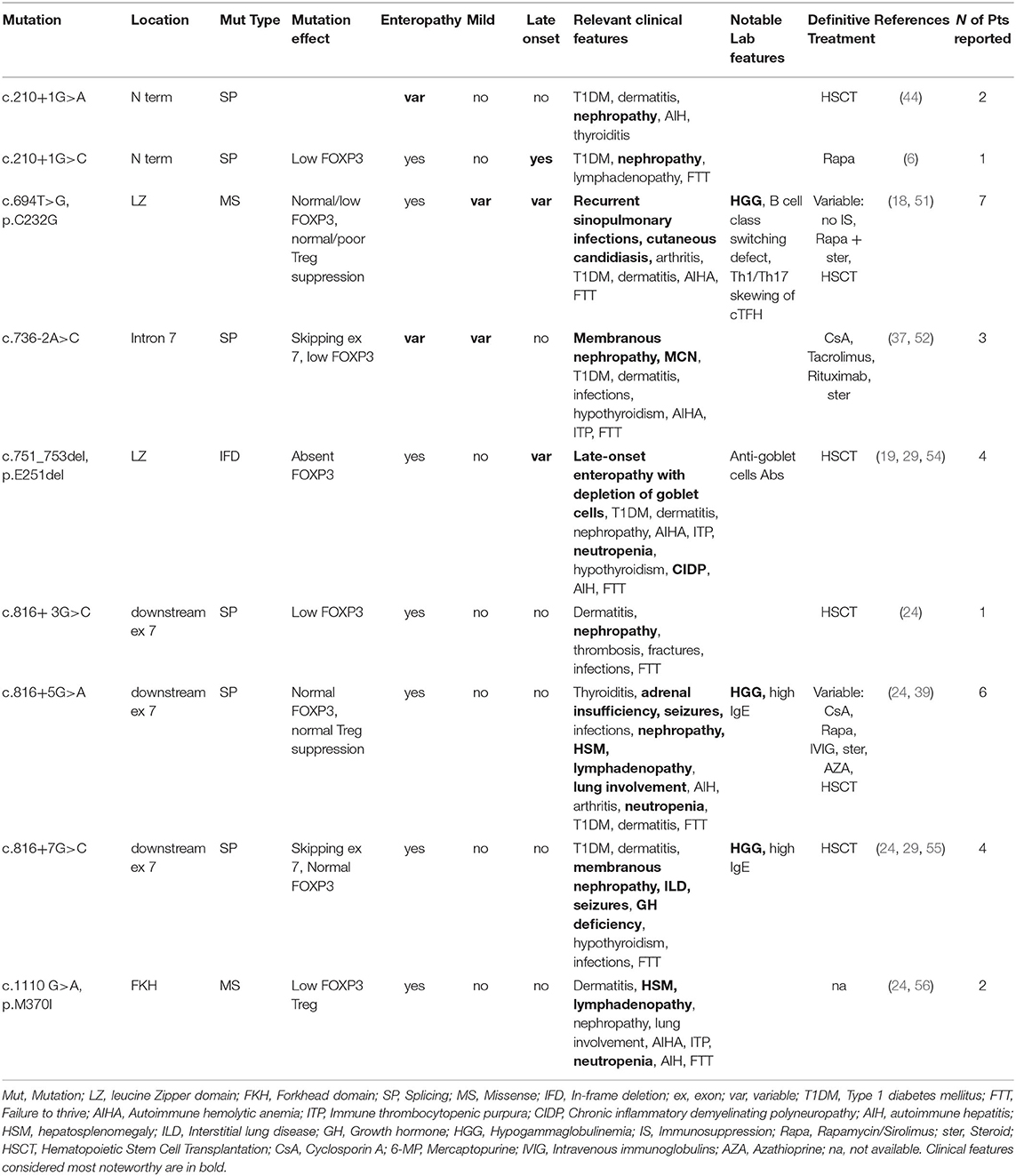
Table 3. Mutations associated with peculiar clinical features of IPEX.
Mutations Clearly Associated With Atypical Phenotypes
Mutations in Non-coding Regions
Mutations associated with mild IPEX phenotypes tend to occur outside FOXP3 coding regions (Table 1 and Figure 1C). Genetic defects located within an intron/exon splice junction or in the first polyadenylation signal might hamper gene expression and protein production (17). Besides, the first splice donor site is highly methylated due to the presence of multiple conserved non-coding enhancer sequences responsible for epigenetic regulation (57). Mutations located in this site might affect the overall methylation status leading to a decreased FOXP3 expression, thus resulting in an atypical phenotype (17).
In detail, 5 patients with c.−23+1G>A or c. −23+5G>A mutations located in the first spice site are reported: 4 out of 5 suffered from a mild disease, and 3 never needed IS therapy (17, 29, 47, 58). Interestingly, laboratory studies in these patients show a variable Treg expression, two of them had normal Treg counts but a low FOXP3 expression.
Similarly, c.1044+5G>A mutation was identified in 3 patients who did not develop enteropathy, nor needed IS therapy. This genetic defect is located between exons 9 and 10, both coding for the DNA-binding FKH domain. Molecular studies showed that this mutation affects RNA splicing, inducing the formation of both a wild-type and a truncated transcript lacking exon 10. It is therefore not surprising that FOXP3 protein expression in CD4+ T cells is normal in these cases. Two other splicing mutations (c.210+1G>C and c.817-20A>C) were described in patients with late-onset enteropathy, showing that a mild phenotype is also associated with a delayed disease onset.
Mutations in Coding Regions
Nevertheless, also mutations in coding regions appear to be correlated to atypical phenotypes. Both missense and frameshift types of mutations have been clearly associated with atypical IPEX.
Above all, mild or unusual cases of IPEX caused by the missense variant c.1040G>A (p.R347H) have been repeatedly described (12, 24, 25, 28, 33, 34). Functional studies showed that mutant cells have a preserved ability to suppress cytokine production on CD4+ T cells (33), while a reduced CD25 expression has been reported only in some patients with this genotype (12, 33). Even though this mutation can also cause severe IPEX, affected patients displayed anyhow unusual clinical features [e.g., atrophic or hemorrhagic gastritis (33, 34)] or—interestingly—severe manifestations regressed at the suspension of IS in adulthood (49).
Additional reports of missense mutations causing mild phenotypes were published for c.210G>T (p.Q70H), c.340C>T (p.R114W), c.1178A>T (p.K393M), c.1222G>A (p.V408M) and c.970T>C (p.F324L) (6, 25, 27, 44, 48, 50, 59). Interestingly, the latter was described in a healthy male whose brother was affected by IPEX (24). Both patients also presented the synonymous variant c.543C>T (p.S181S), already identified in other healthy subjects (24). Laboratory studies showed normal CD25 and FOXP3 expression, and a preserved Treg suppressive ability. This defect involves a coding region, therefore a possible explanation for this behavior is that both phenylalanine and leucine are hydrophobic, and their substitution does not affect the protein's tertiary structure. Alternatively, other genetic or epigenetic features could contribute to stabilize FOXP3 structure and guarantee Treg function.
Finally, two frameshift mutations have been reported in two late-onset cases of IPEX: c.1061delC (p.P354Qfs*36) and c.264delC (p.L89Cfs*40) (17, 21). Frameshift mutations, which totally alter the protein's primary structure, can still give rise to late-onset clinical findings. c.264delC is located in exon 2, which can physiologically be alternatively spliced, generating exon 2minus transcripts (24). Molecular studies are not available for these cases, but further research could show if such exon 2-mutated Tregs could maintain their correct functionality.
Mutations Associated With a Variable Phenotype
While on one hand some genotypes have been repeatedly associated with mild clinical manifestations, another set of mutations is not clearly related to a precise phenotype and a possible genotype-phenotype correlation needs to be further elucidated. These mutations are therefore associated with variable disease phenotypes (Figure 1C and Table 2).
Mutations in the Leucine-Zipper Domain
Four mutations associated with a variable disease phenotype involve the leucine zipper domain, required for FOXP3 homodimerization and transcriptional activity; these are: c.694T>G (p.C232G), c.725T>C (p.L242P), c.736-2A>C and c.748_750del (p. K250del). Therefore, impaired formation of FOXP3 homo-/heterodimers in Treg cells both diminishes FOXP3 functions and destabilizes its expression. This has been particularly elucidated for the intronic mutation c.736-2A>C (52), responsible for exon 7 skipping. All three patients with this mutation displayed membranous nephropathy or MCN. Indeed, FOXP3 transcripts lacking exon 7 have been associated with a Th17 differentiation, as described in patients with multifactorial autoimmune diseases such as rheumatoid arthritis and Crohn's disease (60). Further research is needed to ascertain if autoimmune manifestation (e.g., nephropathy) in IPEX patients carrying this mutation are driven by Th17 cells.
Several cases of c.694T>G (p.C232G) are reported in literature. Among these is a family cluster displaying a Common Variable Immunodeficiency-like (CVID-like) phenotype, which has rarely been described in IPEX (18). On the other hand, the same mutation has been reported in other 3 patients, who presented a more severe phenotype, though still displaying susceptibility to infections (51). Laboratory tests revealed low FOXP3+ Tregs in the first family, while FOXP3 expression was normal in the patients reported in the second manuscript.
Mutations in the DNA-Binding Domain
c.1150G>A (p.A384T) and c.1190G>A (p.R397Q) are the only two mutations involving the DNA-binding domain and determining a variable disease phenotype (26). The former has been shown to impair suppressive Treg function, while preserving FOXP3 ability to repress the production of inflammatory cytokines. This is possibly due to disruption of FOXP3 binding to histone acetyltransferase (61). On the other hand, a severe IPEX phenotype has once been reported for p.A384T (24), therefore its association with a mild disease phenotype is still debated and needs to be confirmed by further reports.
Mutations in PolyA Sequence
Several cases of mutations in the mRNA polyadenylation (PolyA) signal sequence have been described (3, 24, 53), though still underestimated, since such region is frequently neglected in usual sequencing approaches. PolyA sequence protects transcripts from degradation, and patients with these defects have low-level expression of FOXP3 (3). On the other hand, a high variability characterizes these cases, since phenotypes range from an incomplete triad to severe or unusual manifestations (e.g., adrenal insufficiency and pneumopathy). Such clinical diversity could be related to a variable amount of transcript degradation and mRNA stability in the cytoplasm (24).
Mutations Associated With Peculiar Clinical Features
Some FOXP3 mutations seem to be associated with precise organ involvement (i.e., nephropathy), specific laboratory alterations (i.e., hypogammaglobulinemia) or other peculiar clinical findings (Figure 1C and Table 3).
Nephropathy-Correlated Mutations
c.210+1G>A and c.736-2A>G splicing mutations are repeatedly associated with nephropathy. As previously described, kidney involvement is not rare in IPEX and could be due both to autoimmunity and to IS drugs side effects (29). In detail, three patients with c.736-2A>G displayed either membranous glomerulopathy or MCN, both of which are caused by an underlying autoimmune process. Kidney involvement should be suspected when facing patients with this splicing mutation, even though precise genotype-nephropathy correlations still need to be elucidated.
Hypogammaglobulinemia-Correlated Mutations
Hypogammaglobulinemia has been described in several IPEX cases carrying the c.694T>G (p.C232G) mutation (18, 51). Cysteine 232 is located immediately upstream of the leucine zipper domain and could therefore strongly impact on FOXP3 protein dimerization ability and its interaction with other transcription factors. Even though functional studies showed that Treg suppressive ability may be either suppressed or preserved (51), Shamriz et al. demonstrated important immunological consequences of this mutation (18). These include B cell class switching defect and Th1/Th17 skewing of cTFH (circulating T Follicular Helper) cells. Such findings may potentially explain the clinical findings displayed by patients with this defect (i.e., recurrent infections, candidiasis).
Other Peculiar Clinical Findings
Intriguingly, unusual clinical features have been repeatedly described in patients with c.1110G>A (p.M370I) lymphoproliferation) (24, 56) and c.751_753del (p. E251del, neutropenia, chronic inflammatory demyelinating polyneuropathy and other) (54). Finally, the exon 7-skipping mutations c.816+3/5/7G>C have been described in several patients. None of them had a mild or late-onset phenotype, while several unusual manifestations were reported (i.e., hypogammaglobulinemia, seizures, autoimmune pneumopathy) (24, 39, 55). As previously described, skipping exon 7 may be associated with an increased Th17 differentiation (60). Further evidence is needed to elucidate if such immunologic features underlie these unusual clinical presentations.
Conclusion
IPEX is a multisystem autoimmune disease, characterized by a universe of heterogeneous clinical manifestations that trespass the classical phenotype. Widespread use of genetic testing raised our awareness of atypical disease presentations. Such knowledge allowed us to unmask atypical IPEX cases, revealing that the real incidence of the disease might be underestimated. Herein we highlight the importance of picking up unusual signs of IPEX in order to facilitate physicians to suspect the disease even when canonical clinical findings are not fully respected (Box 1).
Box 1. Red flags for early recognition of atypical IPEX.
• Gastrointestinal
◦ Chronic/intermittent diarrhea resistant to formula switching
◦ Early-onset IBD or treatment-resistant IBD
◦ Coeliac disease not responding to gluten-free diet
• Endocrine
◦ Late onset type 1 diabetes mellitus
• Dermatitis associated with other autoimmune features
• Multiorgan autoimmunity with or without enteritis
• Do not exclude even if onset is in late childhood
Genotype-phenotype correlation is still unclear, but our analysis shed light on some mutations more frequently associated with a mild phenotype. Intronic mutations located in the first donor splice site seem to have a significant role; however, other genetic defects located in non-coding and coding regions of FOXP3 can result in atypical disease (Figure 1C). For instance, mutations in the DNA-binding site, a FOXP3 functional domain, classically associated with poorer survival (17), have also been related with moderate clinical manifestations. Nevertheless, the complexity of interactions of FOXP3 with other genes and its epigenetic regulation can also be responsible for phenotype variability, rather than FOXP3 variants themselves. Thus, further studies are needed to further elucidate other contributing mechanisms. Similarly, future focus on other actors such as Type 1 regulatory cells (Tr1) and on the plasticity of the immune system may reveal other intriguing aspects of IPEX immunopathogenesis (62).
Overall, our analysis contributes to increase awareness and to improve diagnosis and treatment intervention in IPEX. Notably, 16 reported patients are clinically stable without any IS regimen, showing that IPEX therapeutic scenarios may range from these cases to invasive options such as allogeneic HSCT. Meanwhile, novel gene therapy-based approaches are still under study but may definitively change the natural history of this disorder (5).
Author Contributions
SC and FC reviewed literature and performed analysis of data. EG supervised the work and wrote the original draft of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministry of Health Grant (Ricerca Finalizzata 2016, Ministero Salute RF-2016-02362384) and by the Jeffrey Modell Foundation Specific Defect Research Grant (EG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Powell BR, Buist NRM, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. (1982) 100:731–7. doi: 10.1016/S0022-3476(82)80573-8
2. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. (2000) 106:75–81. doi: 10.1172/JCI11679
3. Bennett CL, Yoshioka R, Kiyosawa H, Barker DF, Fain PR, Shigeoka AO, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
4. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann NY Acad Sci. (2016) 1417:1–18. doi: 10.1111/nyas.13011
5. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv. (2020) 6:eaaz0571. doi: 10.1126/sciadv.aaz0571
6. Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol. (2018) 141:1036–49.e5. doi: 10.1016/j.jaci.2017.10.041
7. Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-like syndrome. J Allergy Clin Immunol Pract. (2020) 82747–60.e7. doi: 10.1016/j.jaip.2020.04.070
8. Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. (2012) 3:1–25. doi: 10.3389/fimmu.2012.00211
9. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: Inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. (2008) 122:1105–13. doi: 10.1016/j.jaci.2008.09.027
10. Ge T, Wang Y, Che Y, Xiao Y, Zhang T. Atypical late-onset immune dysregulation, polyendocrinopathy, enteropathy, X-Linked syndrome with intractable diarrhea: a case report. Front Pediatr. (2017) 5:4–7. doi: 10.3389/fped.2017.00267
11. Luo Y, Chen J, Fang Y, Lou J, Yu J. A case of metaplastic atrophic gastritis in immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. BMC Pediatr. (2018) 18:1–5. doi: 10.1186/s12887-018-1169-9
12. Zama D, Cocchi I, Masetti R, Specchia F, Alvisi P, Gambineri E, et al. Late-onset of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) with intractable diarrhea. Ital J Pediatr. (2014) 40:1–7. doi: 10.1186/s13052-014-0068-4
13. De Benedetti F, Insalaco A, Diamanti A, Cortis E, Muratori F, Lamioni A, et al. Mechanistic associations of a mild phenotype of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Gastroenterol Hepatol. (2006) 4:653–9. doi: 10.1016/j.cgh.2005.12.014
14. Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. (2007) 38:112–21. doi: 10.1007/s12026-007-0022-2
15. Shanes E, Propst L, Ouyang DW, Ernst LM. Recurrent non immune fetal hydrops associated with IPEX syndrome. Pediatr Dev Pathol. (2019) 22:465–71. doi: 10.1177/1093526619834809
16. Carneiro-Sampaio M, Moreira-Filho CA, Bando SY, Demengeot J, Coutinho A. Intrauterine IPEX. Front Pediatr. (2020) 8:599283. doi: 10.3389/fped.2020.599283
17. Duclaux-Loras R, Charbit-Henrion F, Neven B, Nowak J, Collardeau-Frachon S, Malcus C, et al. Clinical heterogeneity of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a French multicenter retrospective study. Clin Transl Gastroenterol. (2018) 9:201. doi: 10.1038/s41424-018-0064-x
18. Shamriz O, Patel K, Marsh RA, Bleesing J, Joshi AY, Lucas L, et al. Hypogammaglobulinemia with decreased class-switched B-cells and dysregulated T-follicular-helper cells in IPEX syndrome. Clin Immunol. (2018) 197:219–23. doi: 10.1016/j.clim.2018.10.005
19. Patey-Mariaud De Serre N, Canioni D, Ganousse S, Rieux-Laucat F, Goulet O, Ruemmele F, et al. Digestive histopathological presentation of IPEX syndrome. Mod Pathol. (2009) 22:95–102. doi: 10.1038/modpathol.2008.161
20. Savova R, Arshinkova M, Houghton J, Konstantinova M, Gaydarova M, Georgieva E, et al. Clinical case of immune dysregulation, polyendocrinopaty, enteropathy, X-linked (IPEX) syndrome with severe immune deficiency and late onset of endocrinopathy and enteropathy. Case Rep Med. (2014) 2014:564926. doi: 10.1155/2014/564926
21. Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol. (2008) 28:581–7. doi: 10.1007/s10875-008-9196-1
22. Bindl L, Torgerson T, Perroni L, Youssef N, Ochs H, Goulet O, et al. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). J Pediatr. (2005) 147:256–9. doi: 10.1016/j.jpeds.2005.04.017
23. Nameirakpam J, Rikhi R, Rawat SS, Sharma J, Suri D. Genetics on early onset inflammatory bowel disease: an update. Genes Dis. (2020) 7:93–106. doi: 10.1016/j.gendis.2019.10.003
24. Gambineri E, Mannurita SC, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Clinical, immunological, and molecular heterogeneity of patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol. (2018) 9:2411. doi: 10.3389/fimmu.2018.02411
25. Hwang JL, Park SY, Ye H, Sanyoura M, Pastore AN, Carmody D, et al. FOXP3 mutations causing early-onset insulin-requiring diabetes but without other features of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Diabetes. (2018) 19:388–92. doi: 10.1111/pedi.12612
26. Fuchizawa T, Adachi Y, Ito Y, Higashiyama H, Kanegane H, Futatani T, et al. Developmental changes of FOXP3-expressing CD4+CD25+ regulatory T cells and their impairment in patients with FOXP3 gene mutations. Clin Immunol. (2007) 125:237–46. doi: 10.1016/j.clim.2007.08.004
27. Bae KW, Kim BE, Choi JH, Lee JH, Park YS, Kim GH, et al. A novel mutation and unusual clinical features in a patient with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Eur J Pediatr. (2011) 170:1611–5. doi: 10.1007/s00431-011-1588-1
28. Dogruel D, Gürbüz F, Turan I, Altintaş DU, Yilmaz M, Yüksel B. Unusual and early onset IPEX syndrome: a case report. Turk J Pediatr. (2019) 61:580–4. doi: 10.24953/turkjped.2019.04.015
29. Sheikine Y, Woda CB, Lee PY, Chatila TA, Keles S, Charbonnier LM, et al. Renal involvement in the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) disorder. Pediatr Nephrol. (2015) 30:1197–202. doi: 10.1007/s00467-015-3102-x
30. Halabi-Tawil M, Ruemmele FM, Fraitag S, Rieux-Laucat F, Neven B, Brousse N, et al. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br J Dermatol. (2009) 160:645–51. doi: 10.1111/j.1365-2133.2008.08835.x
31. Martín-Santiago A, Hervás JA, Hervás D, Rosell A, Caimari M, de Carlos JC, et al. Diagnostic value of the skin lesions in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Dermatol. (2013) 30:221–2. doi: 10.1111/pde.12126
32. Nieves DS, Phipps RP, Pollock SJ, Ochs HD, Zhu Q, Scott GA, et al. Dermatologic and immunologic findings in the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Arch Dermatol. (2004) 140:466–72. doi: 10.1001/archderm.140.4.466
33. McMurchy AN, Gillies J, Allan SE, Passerini L, Gambineri E, Roncarolo MG, et al. Point mutants of forkhead box P3 that cause immune dysregulation, polyendocrinopathy, enteropathy, X-linked have diverse abilities to reprogram T cells into regulatory T cells. J Allergy Clin Immunol. (2010) 126:1242–51. doi: 10.1016/j.jaci.2010.09.001
34. Scaillon M, Van Biervliet S, Bontems P, Dorchy H, Hanssens L, Ferster A, et al. Severe gastritis in an insulin-dependent child with an IPEX syndrome. J Pediatr Gastroenterol Nutr. (2009) 49:368–70. doi: 10.1097/MPG.0b013e3181a159de
35. Moudgil A, Perriello P, Loechelt B, Przygodzki R, Fitzerald W, Kamani N. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatr Nephrol. (2007) 22:1799–802. doi: 10.1007/s00467-007-0532-0
36. Chuva T, Pfister F, Beringer O, Felgentreff K, Büttner-Herold M, Amann K. PLA2R-positive (primary) membranous nephropathy in a child with IPEX syndrome. Pediatr Nephrol. (2017) 32:1621–4. doi: 10.1007/s00467-017-3682-8
37. Park E, Chang HJ, Shin J Il, Lim BJ, Jeong HJ, Lee KB, et al. Familial IPEX syndrome: different glomerulopathy in two siblings. Pediatr Int. (2015) 57:e59–61. doi: 10.1111/ped.12570
38. Hashimura Y, Nozu K, Kanegane H, Miyawaki T, Hayakawa A, Yoshikawa N, et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol. (2009) 24:1181–6. doi: 10.1007/s00467-009-1119-8
39. Baris S, Schulze I, Ozen A, Aydiner EK, Altuncu E, Karasu GT, et al. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. (2014) 34:601–6. doi: 10.1007/s10875-014-0059-7
40. Ghosh S, Seidel MG. Editorial: current challenges in immune and other acquired cytopenias of childhood. Front Pediatr. (2016) 4:4–6. doi: 10.3389/fped.2016.00003
41. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magerus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
42. Consonni F, Dotta L, Todaro F, Vairo D, Badolato R. Signal transducer and activator of transcription gain-of-function primary immunodeficiency/immunodysregulation disorders. Curr Opin Pediatr. (2017) 29:1. doi: 10.1097/MOP.0000000000000551
43. Schwab C, Gabrysch A, Olbrich P, Patino V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
44. Tsuda M, Torgerson TR, Selmi C, Gambineri E, Carneiro-Sampaio M, Mannurita SC, et al. The spectrum of autoantibodies in IPEX syndrome is broad and includes anti-mitochondrial autoantibodies. J Autoimmun. (2010) 35:265–8. doi: 10.1016/j.jaut.2010.06.017
45. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. (2002) 39:537–45. doi: 10.1136/jmg.39.8.537
46. Torgerson TR, Linane A, Moes N, Anover S, Mateo V, Rieux-Laucat F, et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. (2007) 132:1705–17. doi: 10.1053/j.gastro.2007.02.044
47. Otsubo K, Kanegane H, Kamachi Y, Kobayashi I, Tsuge I, Imaizumi M, et al. Identification of FOXP3-negative regulatory T-like (CD4 +CD25 +CD127 low) cells in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Immunol. (2011) 141:111–20. doi: 10.1016/j.clim.2011.06.006
48. Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. (2006) 116:1713–22. doi: 10.1172/JCI25112
49. Seidel MG, Boztug K, Haas OA. Immune dysregulation syndromes (IPEX, CD27 deficiency, and others): always doomed from the start? J Clin Immunol. (2016) 36:6–7. doi: 10.1007/s10875-015-0218-5
50. Rubio-Cabezas O, Minton JAL, Caswell R, Shield JP, Deiss D, Sumnik Z, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. (2009) 32:111–6. doi: 10.2337/dc08-1188
51. Okou DT, Mondal K, Faubion WA, Kobrynski LJ, Denson LA, Mulle JG, et al. Exome sequencing identifies a novel FOXP3 mutation in a 2-generation family with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. (2014) 58:561–8. doi: 10.1097/MPG.0000000000000302
52. Chen CA, Chung WC, Chiou YY, Yang YJ, Lin YC, Ochs HD, et al. Quantitative analysis of tissue inflammation and responses to treatment in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, and review of literature. J Microbiol Immunol Infect. (2016) 49:775–82. doi: 10.1016/j.jmii.2015.10.015
53. Dorsey MJ, Petrovic A, Morrow MR, Dishaw LJ, Sleasman JW. FOXP3 expression following bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Immunol Res. (2009) 44:179–84. doi: 10.1007/s12026-009-8112-y
54. Moes N, Rieuxlaucat F, Begue B, Verdier J, Neven B, Patey N, et al. Reduced expression of FOXP3 and regulatory T-cell function in severe forms of early-onset autoimmune enteropathy. Gastroenterology. (2010) 139:770–8. doi: 10.1053/j.gastro.2010.06.006
55. Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders J, et al. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol. (2010) 126:1000–5. doi: 10.1016/j.jaci.2010.05.021
56. An YF, Xu F, Wang M, Zhang ZY, Zhao XD. Clinical and molecular characteristics of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in China. Scand J Immunol. (2011) 74:304–9. doi: 10.1111/j.1365-3083.2011.02574.x
57. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. (2010) 463:808–12. doi: 10.1038/nature08750
58. Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet. (2001) 38:874–6. doi: 10.1136/jmg.38.12.874
59. Alkorta-Aranburu G, Carmody D, Cheng YW, Nelakuditi V, Ma L, Dickens JT, et al. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Mol Genet Metab. (2014) 113:315–20. doi: 10.1016/j.ymgme.2014.09.007
60. Mailer RKW. Alternative splicing of FOXP3-virtue and vice. Front Immunol. (2018) 9:530. doi: 10.3389/fimmu.2018.00530
61. Bin Dhuban K, D'Hennezel E, Nagai Y, Xiao Y, Shao S, Istomine R, et al. Suppression by human FOXP3(+) regulatory T cells requires FOXP3-TIP60 interactions. Sci Immunol. (2017) 2:eaai9297. doi: 10.1126/sciimmunol.aai9297
Keywords: immune dysregulation, IPEX, regulatory T cells, FOXP3, primary immunodeficiencies
Citation: Consonni F, Ciullini Mannurita S and Gambineri E (2021) Atypical Presentations of IPEX: Expect the Unexpected. Front. Pediatr. 9:643094. doi: 10.3389/fped.2021.643094
Received: 17 December 2020; Accepted: 13 January 2021;
Published: 05 February 2021.
Edited by:
Talal A. Chatila, Boston Children's Hospital and Harvard Medical School, United StatesReviewed by:
Markus G. Seidel, Medical University of Graz, AustriaSafa Baris, Marmara Üniversitesi Pendik Egitim ve Araştirma Hastanesi, Turkey
Copyright © 2021 Consonni, Ciullini Mannurita and Gambineri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Gambineri, eleonora.gambineri@unifi.it; e.gambineri@meyer.it