 Xu Wang
Xu Wang Xiang Shen
Xiang Shen Fang Fang
Fang Fang Chang-Hong Ding1
Chang-Hong Ding1 Zhen-Hua Cao
Zhen-Hua Cao Dong-Yan An
Dong-Yan An- 1Department of Neurology, Beijing Children’s Hospital, National Centre for Children’s Health, Capital Medical University, Beijing, China
- 2Running Gene Inc., Beijing, China
Objective: Whole Exome Sequencing (WES) is an effective diagnostic method for complicated and multi-system involved rare diseases. However, annotation and analysis of the WES result, especially for single case analysis still remain a challenge. Here, we introduce a method called phenotype-driven designing “virtual panel” to simplify the procedure and assess the diagnostic rate of this method.
Methods: WES was performed in samples of 30 patients, core phenotypes of probands were then extracted and inputted into an in-house software, “Mingjian” to calculate and generate associated gene list of a virtual panel. Mingjian is a self-updating genetic disease computer supportive diagnostic system that based on the databases of HPO, OMIM, HGMD. The virtual panel that generated by Mingjian system was then used to filter and annotate candidate mutations. Sanger sequencing and co-segregation analysis among the family were then used to confirm the filtered mutants.
Result: We first used phenotype-driven designing “virtual panel” to analyze the WES data of a patient whose core phenotypes are ataxia, seizures, esotropia, puberty and gonadal disorders, and global developmental delay. Two mutations, c.430T > C and c.640G > C in PMM2 were identified by this method. This result was also confirmed by Sanger sequencing among the family. The same analysing method was then used in the annotation of WES data of other 29 neurological rare disease patients. The diagnostic rate was 65.52%, which is significantly higher than the diagnostic rate before.
Conclusion: Phenotype-driven designing virtual panel could achieve low-cost individualized analysis. This method may decrease the time-cost of annotation, increase the diagnostic efficiency and the diagnostic rate.
Introduction
Rare Disease is defined as disease affected less than one in 2000 citizens in Europe, or less than one in 1250 in the United States (Schieppati et al., 2008). Rare diseases often start in childhood and accompanied by multisystem disorders which affect life quality of patients (Dodge et al., 2011; Elliott and Zurynski, 2015; Wright et al., 2018). Moreover, 33% of rare disease children die before 5 years old (Wright et al., 2018). There are now approximately 10,000 rare diseases (Elliott and Zurynski, 2015), about 4 of 5 rare disease patients are thought to have a genetic base (Plaiasu et al., 2010; Dodge et al., 2011) especially monogenic disorder (Stolk et al., 2006). For some rare disorders such as tuberous sclerosis complex, phenotypes may vary among individuals due to heterogeneous manifestations. Merely diagnosis based on clinical presentations could be a great challenge (Bai et al., 2017). Hence, gene sequencing for the pathogenic genes is vital for understanding the cause of diseases.
The mainstream of gene sequencing includes genomic microarrays, Sanger sequencing and Next-Generation sequencing (NGS). Genomic microarrays are low-resolution method for detection of 50∼100 kb copy number variation (Speicher and Carter, 2005). For small insertion or deletion less than 50 kb, Sanger sequencing and NGS could fulfill the task. Sanger sequencing, due to limited throughput, is only used when a specific gene is selected. Different diseases could have similar clinical presentations such as ataxia and mental retardation. At the same time, a disease may be caused by various genes. It is difficult to determine the pathogenic gene in every patient to perform Sanger sequencing. NGS offers much higher throughput that can facilitate sequencing up to 1000s of gene once. In addition, since sheared DNA is sequenced parallelly multiple times, therefore lower error rate is achieved compared to Sanger sequencing. Moreover, the recent study showed that NGS could also be used to detect Copy Number variation that larger than 100 kb (de Ligt et al., 2013; Feng et al., 2017). Therefore, it has been increasingly used in rare disease diagnosis.
For NGS, the range of detection object could vary from multiple disease-associated genes (gene panel), whole exome (Whole-Exome Sequencing) to whole genome (Whole-Genome Sequencing). For gene panel, various genes affected several similar diseases or diseases in the same system could be detected at the same time. Since it focuses on the specific genes, the data size is generally smaller than Whole-Exome Sequencing (WES) and Whole-Genome Sequencing (WGS), the result is easy to analyze and interpret. Although convenient, the gene list of a particular panel is constant; meanwhile, the discovery of disease-associated gene is developing. The newly discovered gene on one hand cannot be added to the already made panel, and further analysis cannot be performed. On the other hand, updating gene list every day is, however, impractical, costly and with less sense. Gene panels is at present insufficient for detection and is not recommended by most of the genetics and clinicians (Biesecker and Green, 2014; Wenger et al., 2017; Ewans et al., 2018; Jin et al., 2018).
WGS, mostly based on Illumina technology, is the sequencing method covers most part of the human genome. Although easy to perform, it is costly and time consuming to analyze and interpret data. On average, 3–4 million mutations could be discovered in each individual (Ashley et al., 2010; Lupski et al., 2010; Roach et al., 2010; Sobreira et al., 2010; Bainbridge et al., 2011). In the meantime, the mutations in the intronic region except for the ones near splicing sites are hard to predict the relative risk of phenotype, since the function of the intronic gene is still mostly undiscovered, and the mutation frequency in the intron is considerably high (Tabor et al., 2002; Abecasis et al., 2010). It is hard to estimate which mutation is deleterious. Research also presented that WGS has limited significance at the present stage (Alfares et al., 2018). By contrast, the exome represents 1–2% protein-coding gene of the whole genome thus more exomes could be sequenced per run (Gilissen et al., 2012). The result of WES is more accessible to interpret since non-synonymous mutations in the coding region could directly lead to amino acid change then affect the protein structure and function. This method could also help identify not only the unknown pathological mutations but also the undiscovered mutations (Liu et al., 2012). Re-analysis of WES data was also proved to significantly increase the diagnostic rate (Alfares et al., 2018). The cost of WES is also much lower than WGS (Gilissen et al., 2012) at present. Although the number of variants is cut down to the range between 20,000 and 50,000 (Ashley et al., 2010; Lupski et al., 2010; Roach et al., 2010; Sobreira et al., 2010; Bainbridge et al., 2011; Gilissen et al., 2012), it is still difficult to analyze and identify the pathogenicity of every variant, especially for detection of single case because of lower efficiency and time consuming. Meanwhile, due to the analysis strategy with less-efficacy, the diagnostic rate of WES with unspecific analysis was relatively low, approximately 25–30% (Yang et al., 2013; Lee et al., 2014; Shashi et al., 2016).
After carrying out, investigating and studying WES in clinic for many years, the combination of clinical information and gene sequencing is increasingly suggested in disease diagnosis (Jin et al., 2018). Here, we developed a method called “Phenotype-driven designing virtual panel,” a method that concentrates in analysing the genes of diseases with related phenotypes. The gene lists of phenotype-associated diseases were generated by a system called “Mingjian.” After inputting all phenotypes of the patient, the system will automatically list the associated genes and rank the gene by the corresponding number of phenotypes. This method is proved to improve the diagnostic rate significantly in our further test.
Methods
Whole-Exome Sequencing
Proband DNA was sequenced to discover the causal gene. DNA was isolated from peripheral blood using a DNA Isolation Kit (Bioteke, AU1802). 1ug genomic DNA was fragmented into 200–300 bp length by Covaris Acoustic System. The DNA fragments were then processed by end-repairing, A-tailing and adaptor ligation, a 4-cycle pre-capture PCR amplification, targeted sequences capture. Captured DNA fragments were eluted and amplified by 15 cycle post-capture PCR. The final products were sequenced with 150 bp paired-end reads on Illumina HiSeq X platform according to the standard manual.
The raw data converted by HiSeq X were filtered and aligned against the human reference genome (hg19) using the BWA Aligner1. The single-nucleotide polymorphisms (SNPs) were called by using the GATK software (Genome Analysis ToolKit)2. Variants were annotated using ANNOVAR3. Effects of single-nucleotide variants (SNVs) were predicted by SIFT, Polyphen-2, and MutationTaster programs. All variants were interpreted according to the standards for interpretation of sequence variations recommended by ACMG and categorized to be pathogenic, likely pathogenic, variants of unknown clinical significance (VUS), likely benign and benign. The associated phenotypic features of candidate genes were analyzed against the patient’s phenotype. Core phenotypes were extracted and used to acquire a gene list of the virtual panel by OMIM database4 and Mingjian (211.149.234.157/login). Re-annotation was conducted according to the virtual panel. The whole process was shown in Figure 1.
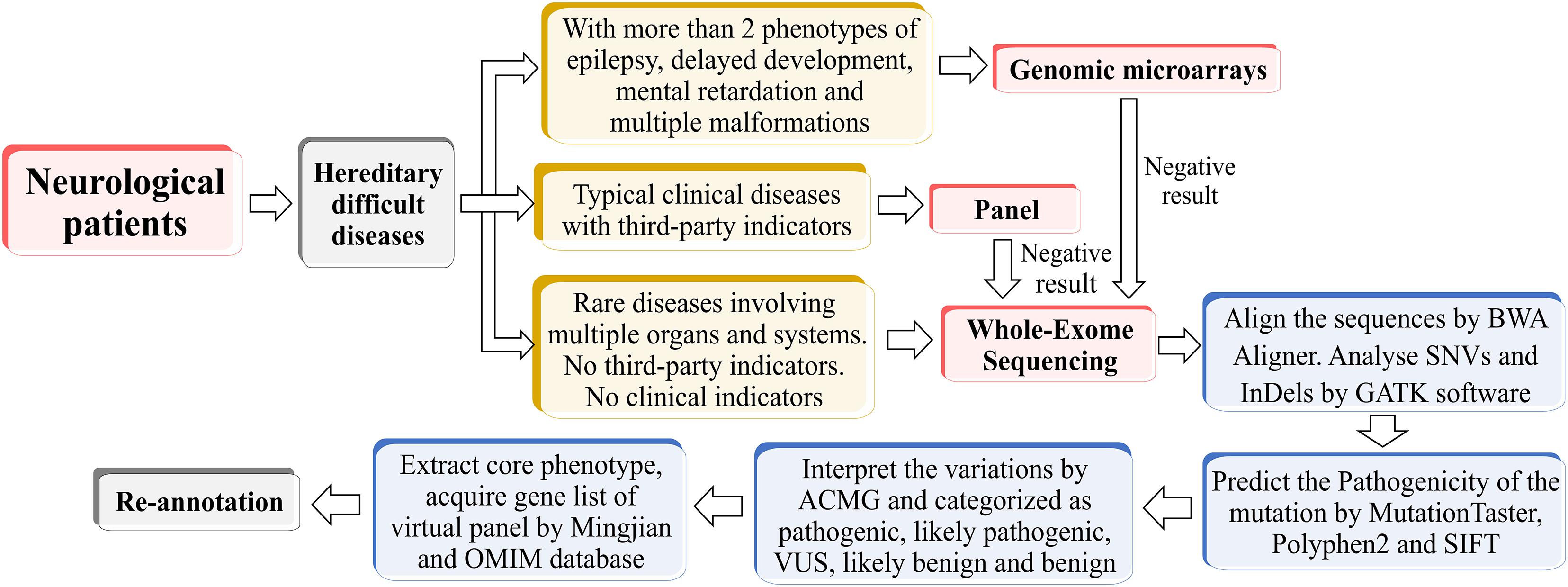
Figure 1. The flow chart of phenotype-driven designing virtual panel.
Sanger Sequencing
The candidate causal genes discovered via WES were then confirmed by Sanger sequencing, co-segregation analyses among the family were also conducted. The primers were designed using Primer Premier 5.0 (Premier Biosoft), PCR was carried out to amplify the fragments covering the mutated sites. The PCR products were further purified with Zymoclean PCR Purification Kit and then sequenced by ABI 3730 DNA Sequencer. Sanger sequencing results were analyzed by Chromas Lite v2.01 (Technelysium Pty Ltd., Tewantin, QLD, Australia).
A Case of a Diagnostic Odyssey
The patient is an 8 months old boy who was born to a normal non-consanguineous Han family by normal vaginal delivery at full-term. He had tonic seizure epilepsy with sustaining state when he first came to our hospital. His symptoms get alleviated obviously after taking levetiracetam 40 mg/kg per day. The milestone development and comprehensive development of the patient was also delayed. Physical examination: the head circumference of the patient was 41 cm, anterior fontanel was 1∗1 cm. He had internal strabismus but could chase light, he also presented large ear, low nose, inverted nipples, low muscle tension with muscle strength-4, weak tendon reflex, poor head control, round back, fat pad in buttock, bilateral cryptorchidism and short penis. His body always leaned forward when sitting (Figure 2). He could not open his mouth or speak actively. He could neither grab things initiatively. Laboratory result: MRI result presented cerebellar atrophy and delayed myelination (Figure 3); chest CT showed spine kyphosis (Figure 4); EMG result showed neurogenic damage; the LC-MS/MS result of blood (Table 1), GC-MS result of urine (Figure 5) and blood test of patient’s serum (Table 2 and Figure 6) indicated abnormal liver function.
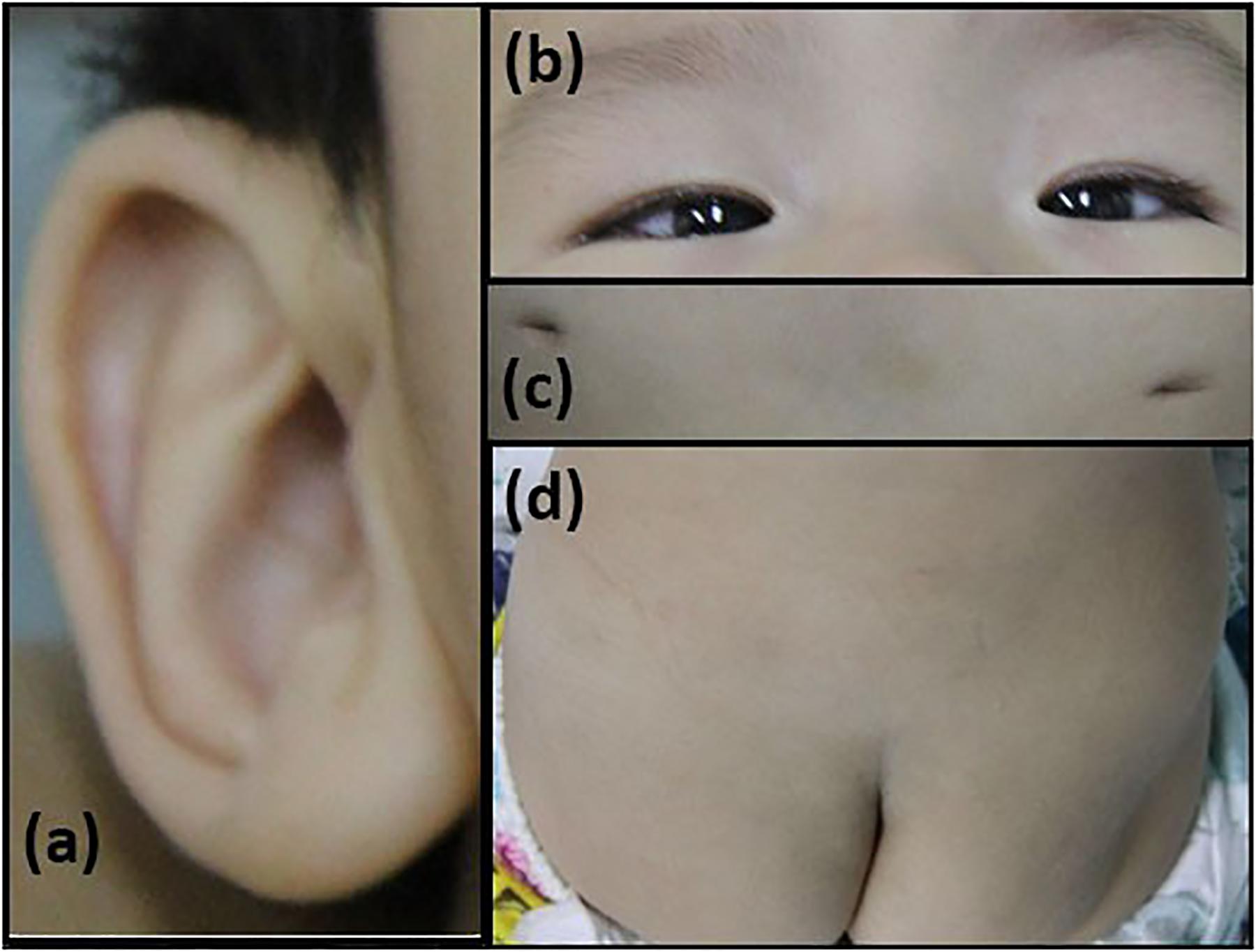
Figure 2. Phenotype of the patients. (a) large ear; (b) internal strabismus; (c) inverted nipples; (d) fat pad in the buttock.
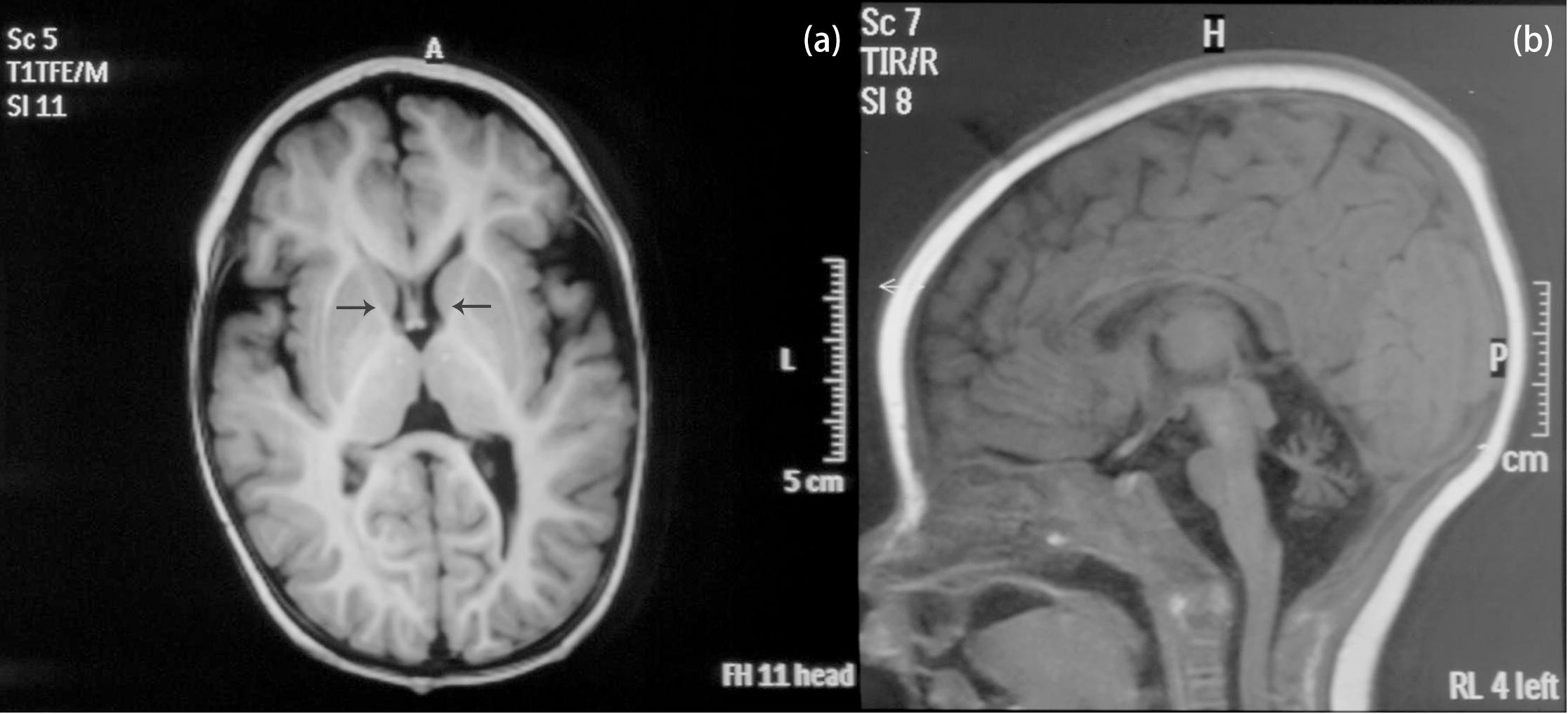
Figure 3. MRI result of the patient. (a) Coronal T1W image. Black arrows pointed the anterior limb of the internal capsule. The faint hyperintense signal of T1W indicated delayed myelination of the patient. (b) Sagittal T1W image. The patient presented cerebellar atrophy.
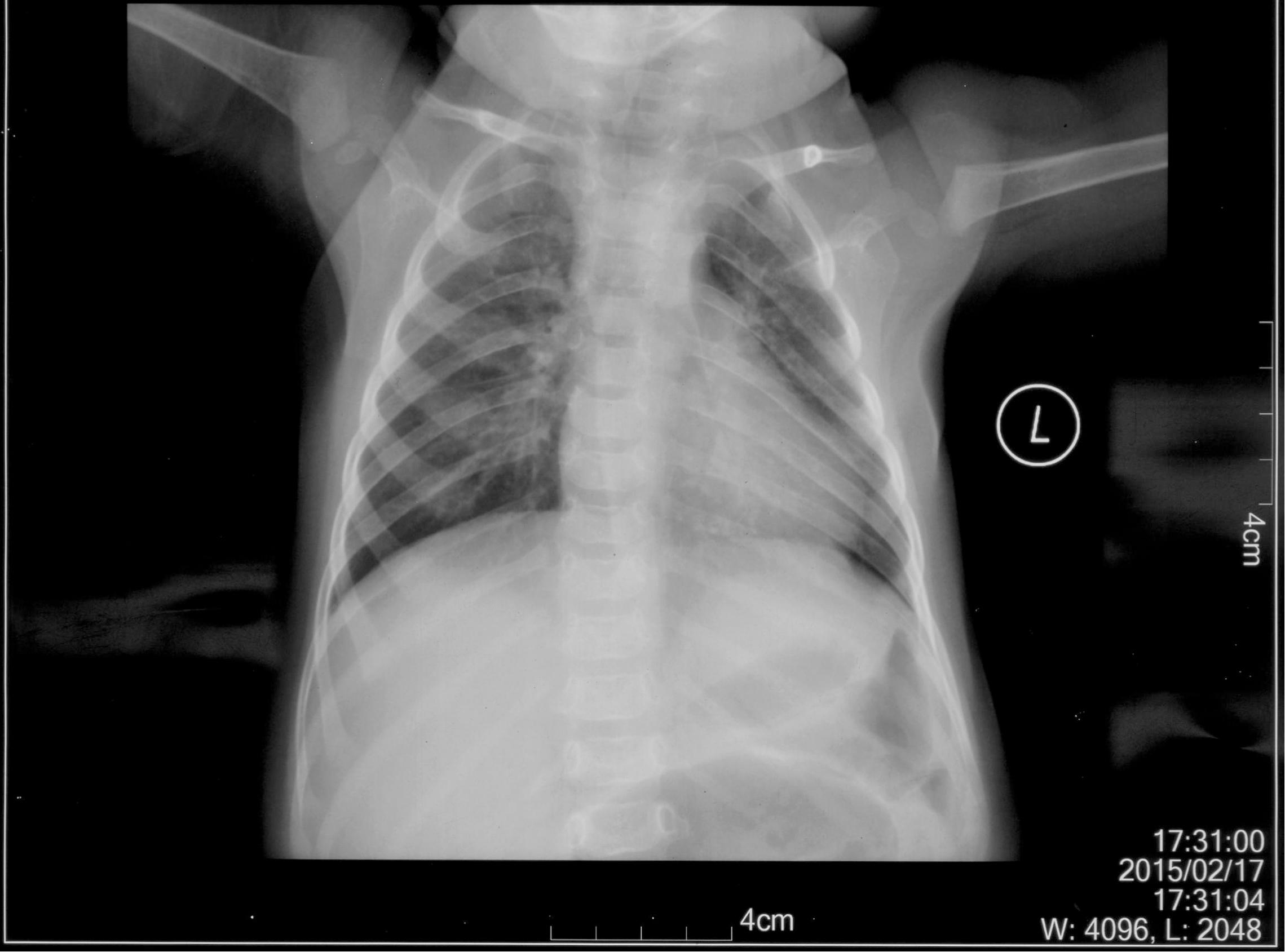
Figure 4. Chest CT result of the patient. It is shown that the patient had spine kyphosis.
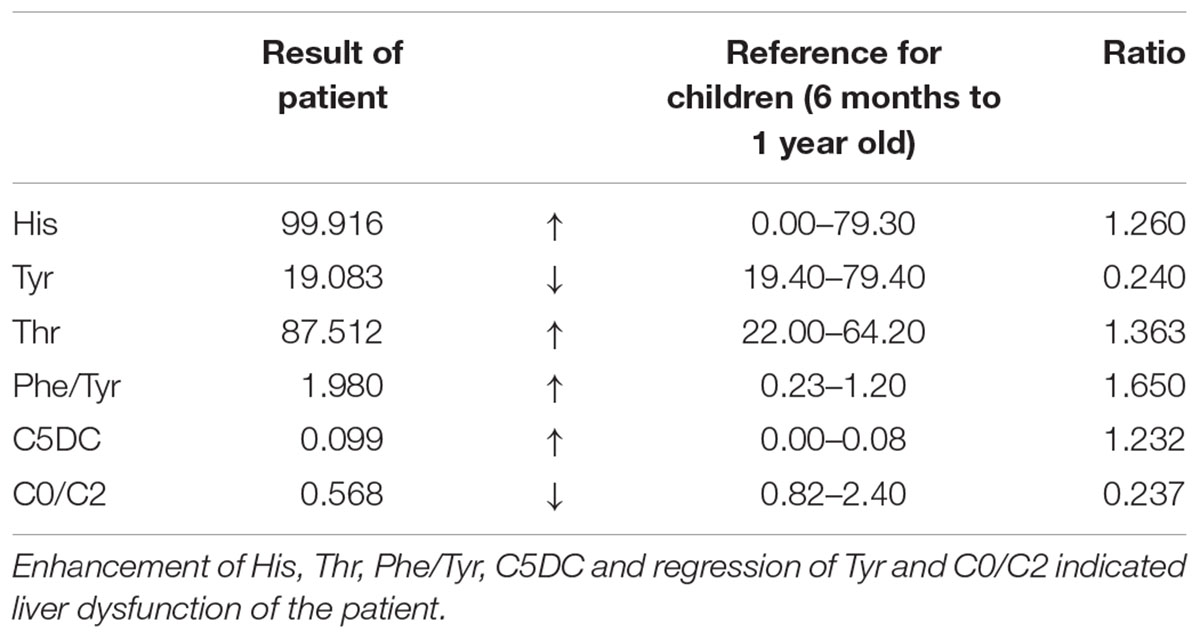
Table 1. Mass spectrum result of patient’s blood.
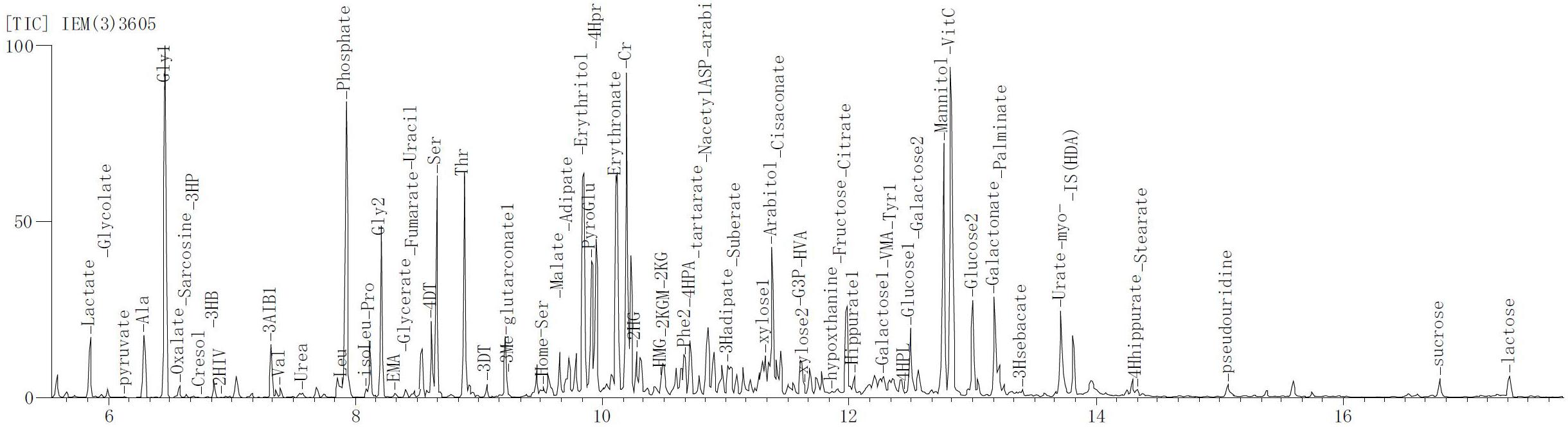
Figure 5. The mass spectrum result of the metabolite in urine. The result of the urine metabolite indicated liver abnormality of patient.
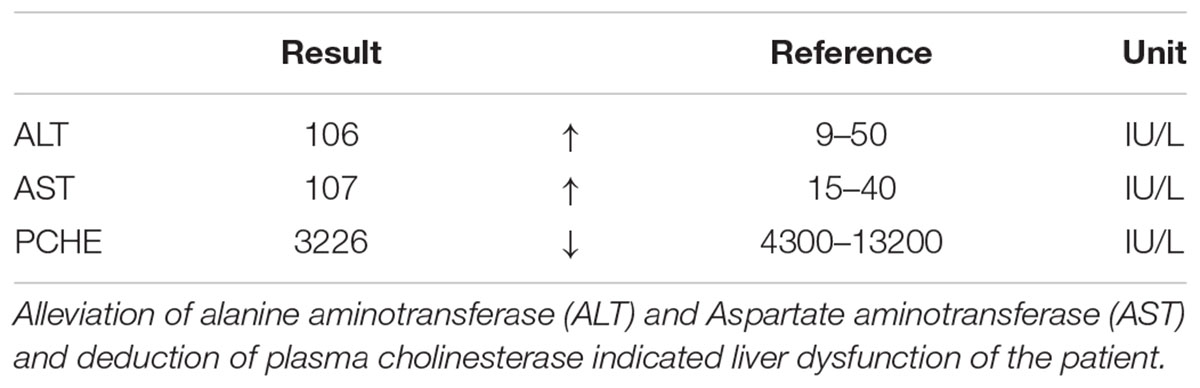
Table 2. Blood test result of the patient’s serum.
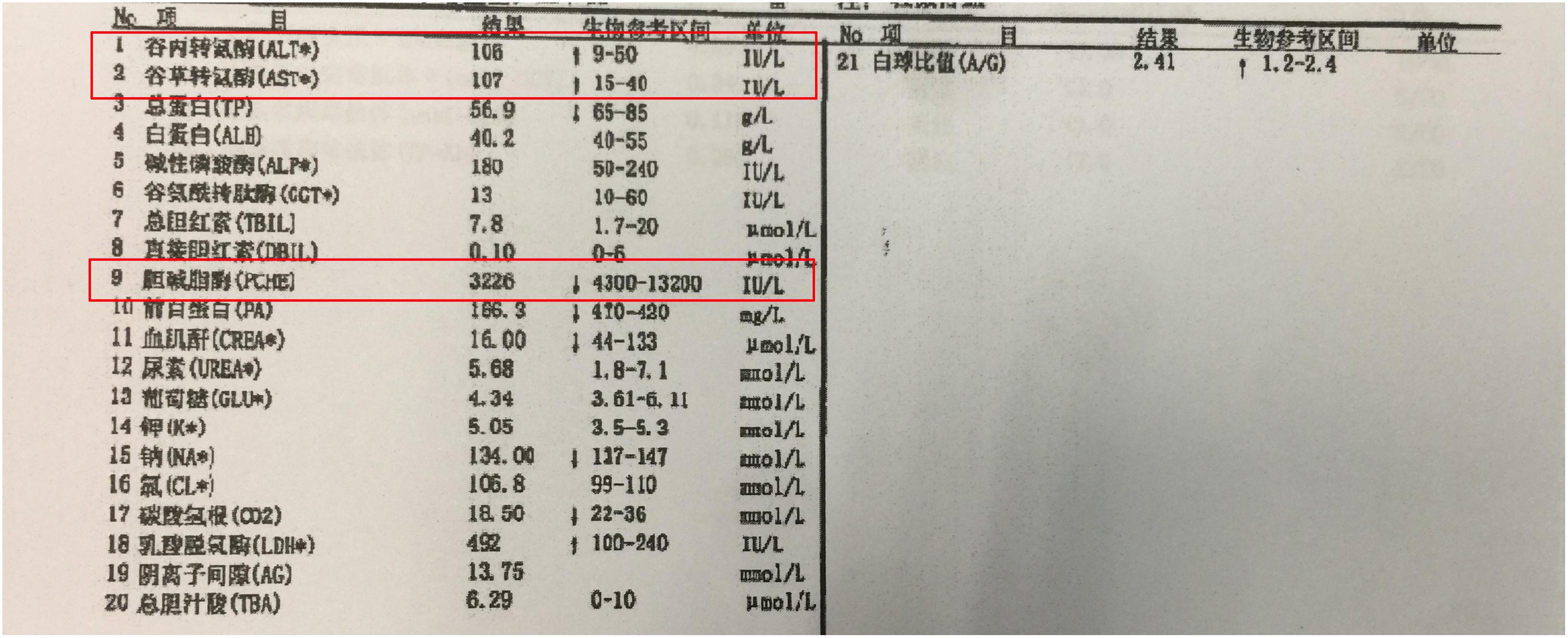
Figure 6. The blood test result of the patient’s serum. Alleviation of alanine aminotransferase (ALT) and Aspartate aminotransferase (AST) and deduction of plasma cholinesterase indicated liver dysfunction of the patient.
The elder sister of the patient, 8 years old, also shows somehow similar phenotypes. At 2 years of age, she started to have tonic epilepsy and ataxia, mental retardation, so far can only speak 2–3 words phrase. The pedigree was shown in Figure 7.
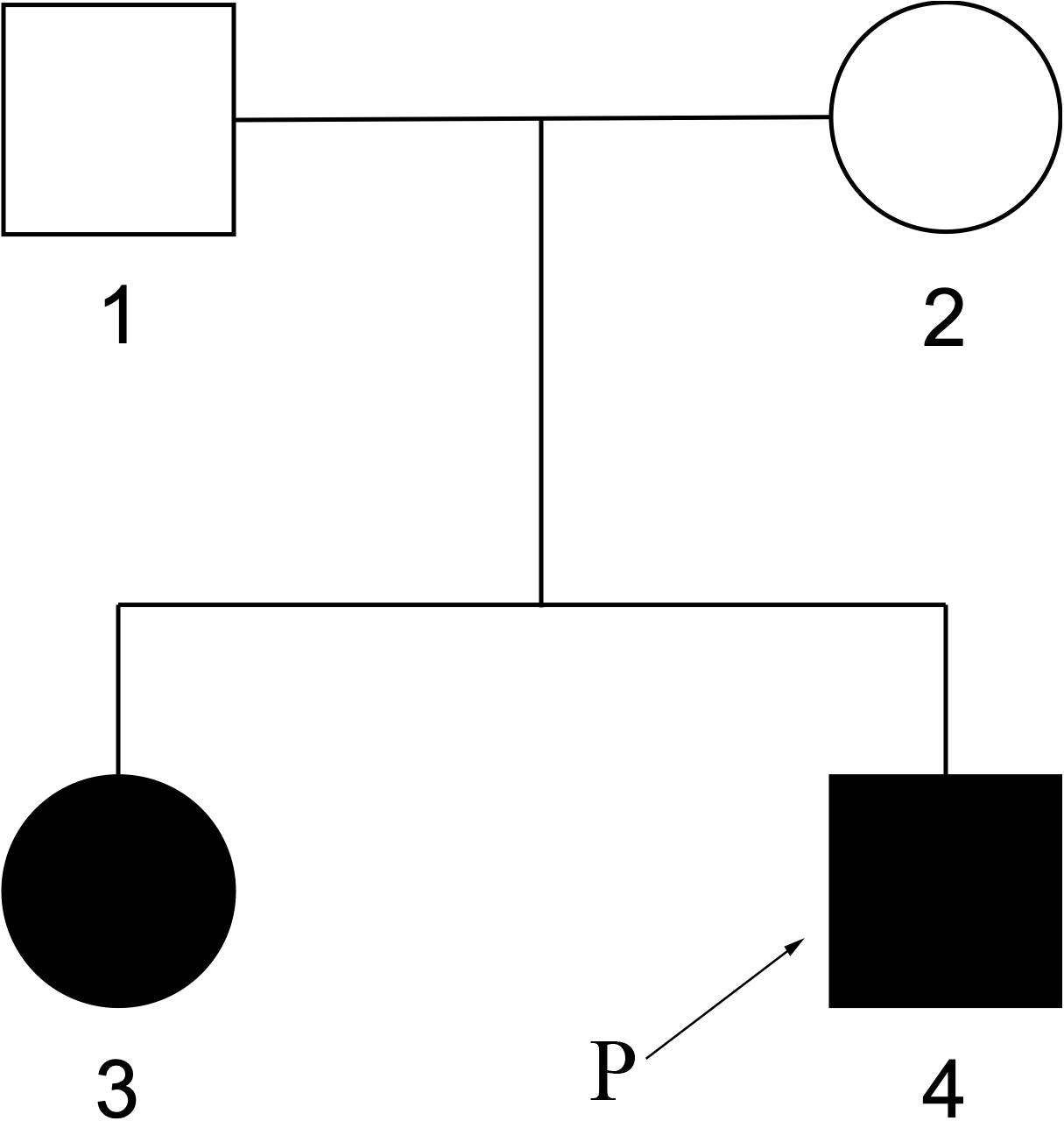
Figure 7. Pedigree of family members. The elder sister of the patient also had similar phenotypes.
The clinical presentation involved multiple systems and thus, even he has got treated at many hospitals and screened by existing detection methods, the disease was still unclear.
Results
The Gene List of Phenotype-Driven Virtual Panel
Extracting and inputting the core phenotypes: Ataxia, Seizures, Esotropia, Puberty and Gonadal disorders, Global developmental delay, Autosomal recessive (inheritance pattern). The gene list exported by Mingjian is listed in Table 3.

Table 3. Gene list exported by Software Mingjian according to the inputting core phenotypes.
Result of Whole-Exome Sequencing
Analysing the gene from gene list generated by Mingjian according to the core phenotypes, two heterozygous mutations in PMM2 gene had been found, c.430T > C in exon 5 (chr16:8905018 T > C) and c.640G > C in exon 8 (chr16:8941581G > C). These nucleotide substitutions would result in alterations in amino acid, F144L and G214R, respectively (Figure 8).
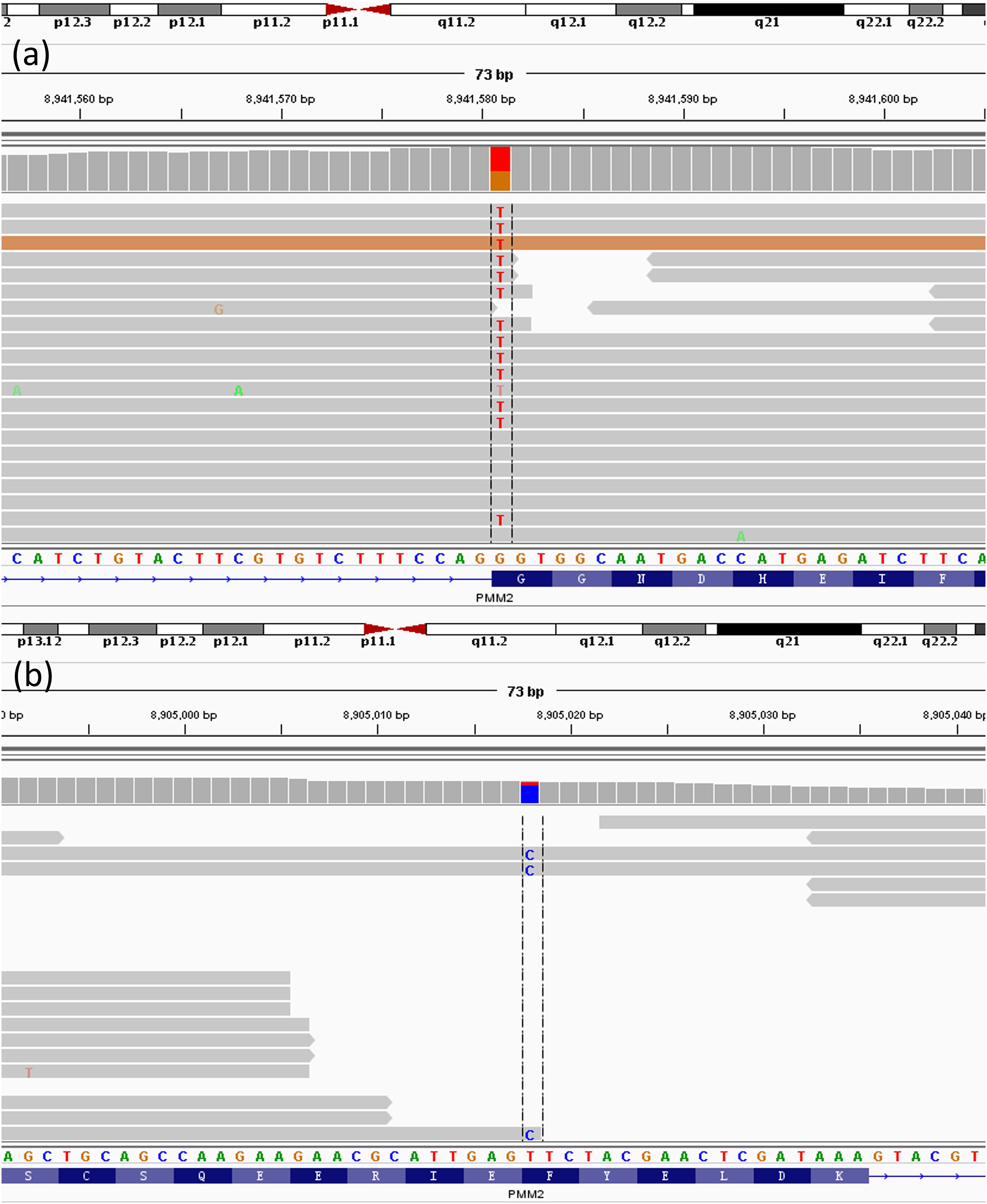
Figure 8. Next-Generation result of patient. The result shows the two heterozygous mutations in the PMM2 gene. (a) c.640G > C; (b) c.430T > G.
Further Sanger Sequencing result showed the proband’s father is the heterozygous carrier of the c.430T > C mutation, while the proband’s mother carries the c.640G > C mutation. The proband’s sister with the same clinical presentation also carries all these two mutations. Thus, the proband is the compound heterozygous for the PMM2 p.F144L/p.G214R mutations (Figure 9).
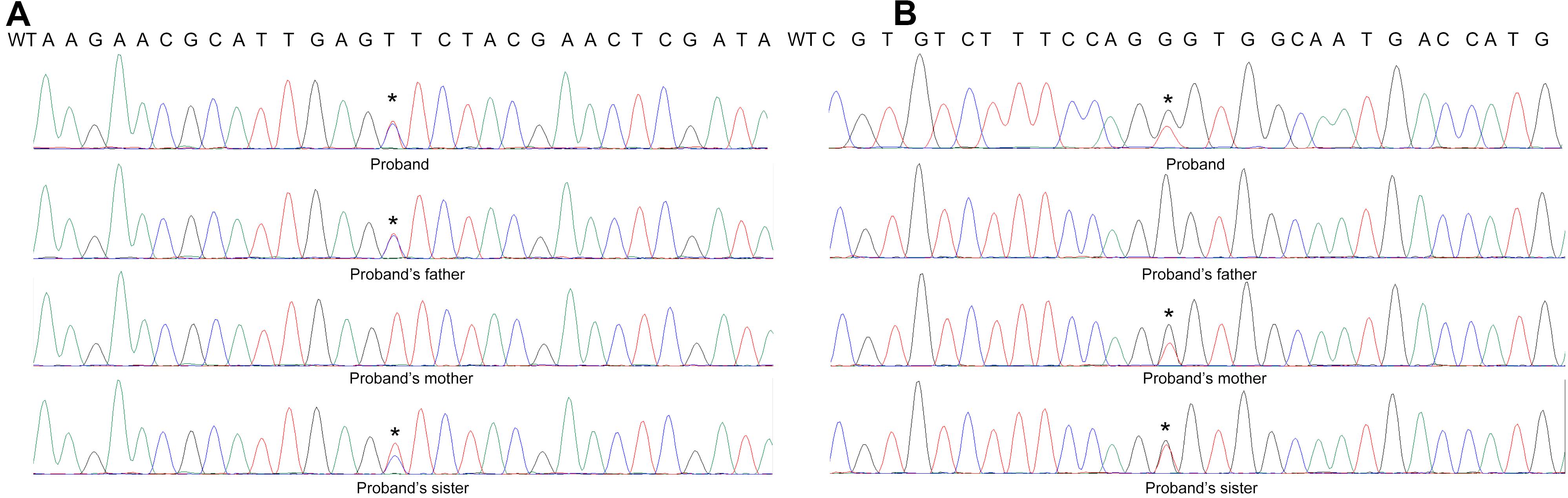
Figure 9. Sanger Sequence result of the patient’s family. The result shows that (A) the proband’s father was the heterozygous carrier of the c.430T > C mutation, while (B) the proband’s mother carried the c.640G > C mutation. The proband’s sister is also the carrier of the compound heterozygous mutations.
Mutation p.F144L is a pathologic mutation that has been reported before. This mutation could create a new site for restriction enzyme SacI causing extra splicing (Kondo et al., 1999). Another mutation p.G214R has not been reported before, however, there is another reported disease-causing mutation at the same position (c. 640G > A, G214S) (Schollen et al., 2002; Vicario et al., 2017). Since this mutation is absent from controls (PM2), detected in trans with a pathogenic variant (PM3), located at the same position with a reported pathogenic missense change (PM5), this variant was classified as “likely pathogenic” according to ACMG guidelines (Richards et al., 2015). Prediction of this mutation by MutationTaster, Provean and SIFT also turned out to be disease causing (probability > 0.99), deleterious (score = -7.66) and damaging (score = 0), respectively. The result of MutationTaster (Schwarz et al., 2014) also indicated splice site change caused by the mutation (Figure 10), however mRNA experiment was not successfully performed to prove it.
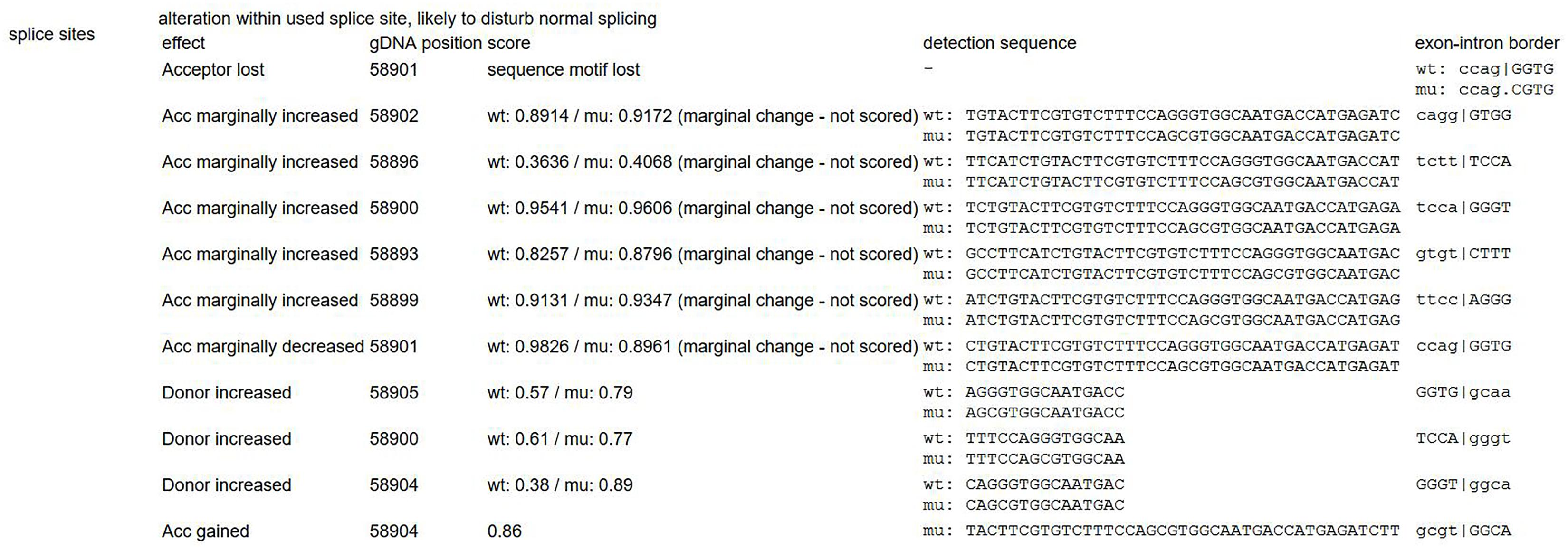
Figure 10. Indicated splicing change by MutationTaster (Schwarz et al., 2014). This mutation might disturb the exon-intron border.
Result of Other Patients
To assess the diagnostic rate of this method, “phenotype-driven virtual panel,” we decided to use the same method to analyze more neurological patients.
Clinical Information of the Patients
The clinical phenotypes of 29 patients were listed in Table 4.
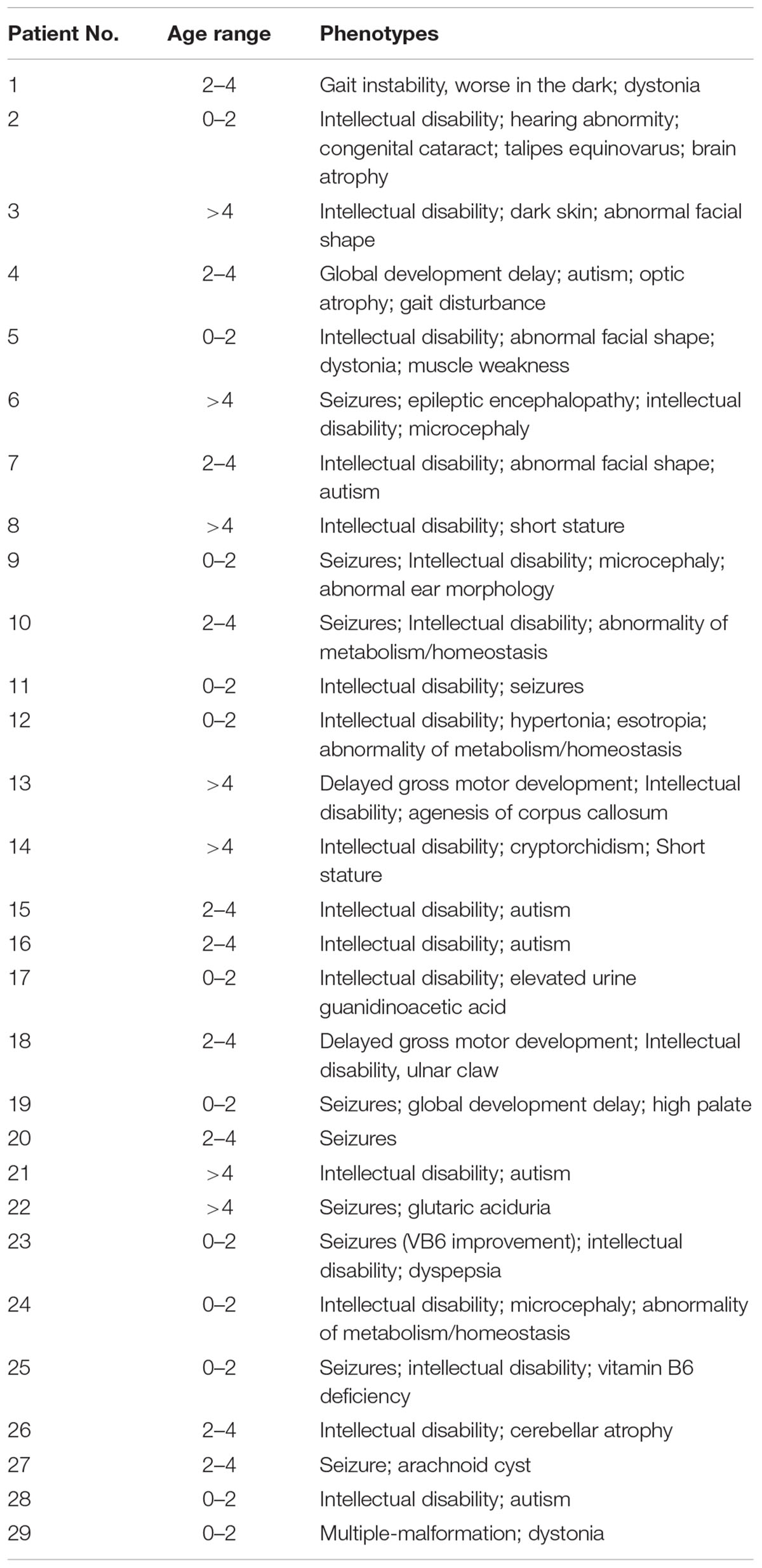
Table 4. Phenotype of 29 patients with neurological diseases.
Patients were collected from the neurology department of Beijing Children’s Hospital. Of the 29 patients, 19 patients (65%) are male, 10 patients (35%) are female. The ages range from 4 months to 17 years 6 months. Most patients have an intellectual disability. More precise clinical information, phenotypes and gene sequencing result were available in Supplementary Material.
Sequencing Results of Patients
The gene sequencing results of these 29 patients was listed in Table 5.
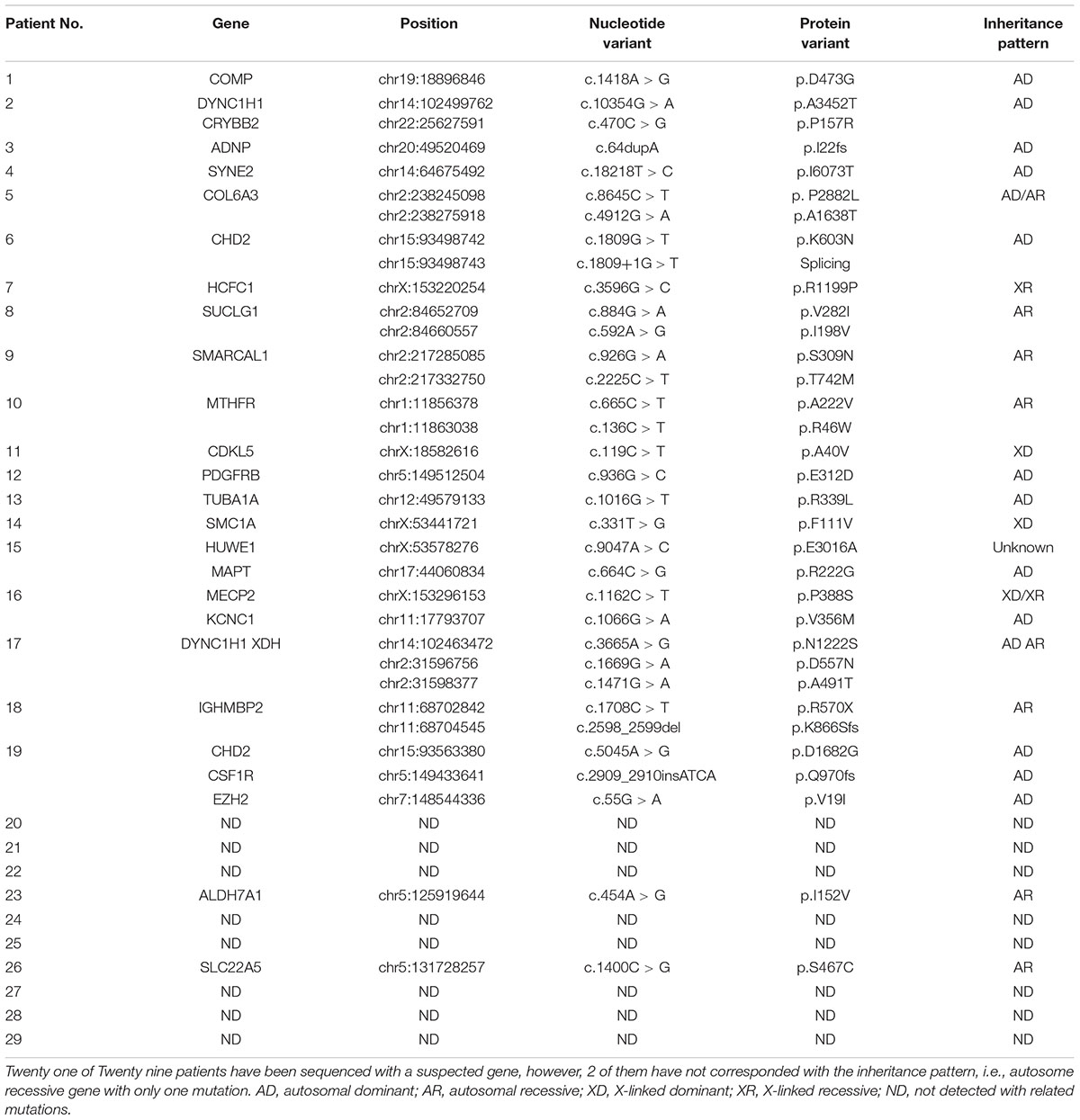
Table 5. Gene sequencing result of 29 patients with neurological diseases.
Discussion
Rare diseases, especially the ones involving multisystem are challenges for clinical diagnosis. For example, the PMM2 case described here involves not only the nervous system but also muscle, gonad, liver, spine, etc. It is hard to distinguish the fundamental factors of the pathogenesis by only examine clinical symptoms. Judging merely based on the clinical information, misdiagnosis was definitely not a rare event, especially in the generation without gene detection. A patient in our hospital who was previously diagnosed as Crouzon syndrome was finally proved to be Cytochrome P450 oxidoreductase deficiency by NGS (Hao et al., 2018). Misdiagnosis can result in a completely different treatment and might have possibility in leading deterioration. The efficacy of treatment might also be affected when the optimal treatment time is missed. Thus, gene sequencing is essential in the diagnosis of rare diseases.
Core phenotypes of patients with the neurological inherited disease are similar, i.e., ataxia, seizures, esotropia, global developmental delay, puberty and gonadal disorders in this case. It is almost impossible to only rely on clinicians’ experience to diagnose and determine candidate genes. Evaluating pathogenicity of the candidate mutations, confirming the gene function, excluding not associated mutations, choosing the clinically meaningful variants for Sanger Sequencing according to the similarity of clinical presentation is the traditional way to annotate (Jin et al., 2018). However, it is unavoidable that the function and related diseases of the redundant phenotype-unrelated mutants will be analyzed. Here, the phenotype-driven designing “virtual panel” method could automatically filter the genes that is unrelated to the patient’s symptoms, so that the analyser could only focus on the mutations in phenotype-related genes. This method can decrease the genes that should be analyzed, shorten the analysing time and make a more efficient annotation.
Moreover, designing traditional gene panel is a manual work, there might be bias occurring when selecting the gene list in the panel. Also, gene list in produced panel is constant, updating panel aligning with new discoveries is expensive and time-consuming. The virtual panel we run is designed by computer software “Mingjian,” which could avoid the bias due to personal cognition and judgement. In addition, “Mingjian” is according to the database of HPO, OMIM, and HGMD which includes all the known possible genes related to the phenotypes. Since it is actually “virtual,” updating the gene list is not an obstacle. Thus, it could contain all the present discovered, phenotype-related genes. Besides, all the undiagnosed cases can be re-analyzed when more disease-causing mutations are discovered and more linkages between disease and variations are established. Also, every patient has distinct phenotypes, a designed panel may not be applicable for every patient. Phenotype-driven “virtual panel” is based on the phenotypes of the patients, it may simply achieve low-cost individualized analysis when typical and standardized core phenotypes are extracted.
Consequently, we carried out this method in the diagnosis of more patients with neurological diseases to access the diagnostic rate. In 29 cases of patients, 21 of 29 patients were found carrying mutations in related genes. However, according to the inheritance pattern of genes, 2 heterozygous mutations of autosomal recessive genes were excluded. Other 19 of 29 patients were all confirmed with corresponding mutations by Sanger Sequencing.
For the rest of 10 patients who didn’t confirm with the relevant mutations, it may fit one of the following conditions. First, the disease-causing mutations may locate in the undefined genes or genes that have not been experimentally proved to be associated with such neurological diseases. For example, we have found that NCAM1 polymorphisms is associated with autism in a previously undiagnosed case in year 2014 (Zhang et al., 2014). This kind of cases may be solved in the future due to development of research. Secondly, some mitochondrial gene mutations may also be involved but are outside the detection range of Whole Exome Sequencing. The symptoms of most mitochondrial diseases include seizures, mental retardation, developmental delay, metabolic disorders, muscle problems and visual disorders as well (Fang et al., 2017). Both mitochondrial DNA and nuclear DNA mutations may contribute to dysfunction in mitochondria (Liu et al., 2014, 2015; Fang et al., 2017). Therefore, the disease-causing variants in these undiagnosed cases may be located in mitochondrial DNA. Moreover, insertion or deletion which is larger than 50 kb or chromosomal inversion may also cause disease. However, these mutations could not be identified by NGS due to technical limitations. This may not be a rare event since we previously diagnosed a novel DDC gene deletion in the patients who was suspected to carry mutations in DDC gene but only diagnosed with single missense variant (Dai et al., 2018).
Overall, the diagnostic rate in this study was 19/29 = 65.52%, which far exceeds the known diagnostic rate of Whole–Exome Sequencing (25–30%). Therefore, the phenotype-driven virtual panel is an effective method to analyze WES data of neurological disease.
Data Availability Statement
All the clinical and genetic data of the cases reported in this study have been submitted to the rare disease database, eRAM, at http://www.unimd.org/eram/.
Ethics Statement
This study was carried out is approved by Capital Medical University Beijing Children’s Hospital Ethics Committee (Ethics Number: 2018-k-63). The protocol was approved by the Capital Medical University Beijing Children’s Hospital Ethics Committee. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Consent for Publication
The patient’s parents gave written informed consent to studies and publication of clinical information, images and sequencing data.
Author Contributions
XW and FF designed the study. XW, FF, and C-HD collected the clinical data. XS, HZ, and Z-HC performed the WES. XS and D-YA analyzed the genetic data. XW, XS, and HZ wrote the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
XS, HZ, Z-HC, and D-YA were employed by company Running Gene Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all of the family members for their participation in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2018.01529/full#supplementary-material
Footnotes
- ^http://bio-bwa.sourceforge.net/
- ^www.broadinstitute.org/gatk
- ^annovar.openbioinformatics.org/en/latest/
- ^http://omim.org/
References
Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., Gibbs, R. A., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. doi: 10.1038/nature09534
Alfares, A., Aloraini, T., Subaie, L. A., Alissa, A., Qudsi, A. A., Alahmad, A., et al. (2018). Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 20, 1328–1333. doi: 10.1038/gim.2018.41
Ashley, E. A., Butte, A. J., Wheeler, M. T., Chen, R., Klein, T. E., Dewey, F. E., et al. (2010). Clinical assessment incorporating a personal genome. Lancet 375, 1525–1535. doi: 10.1016/S0140-6736(10)60452-7
Bai, D., Zhao, J., Li, L., Gao, J., and Wang, X. (2017). Analysis of genotypes and phenotypes in Chinese children with tuberous sclerosis complex. Sci. China Life Sci. 60, 763–771. doi: 10.1007/s11427-017-9091-x
Bainbridge, M. N., Wiszniewski, W., Murdock, D. R., Friedman, J., Gonzaga-Jauregui, C., Newsham, I., et al. (2011). Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 3:87re3. doi: 10.1126/scitranslmed.3002243
Biesecker, L. G., and Green, R. C. (2014). Diagnostic clinical genome and exome sequencing. N. Engl. J. Med. 370, 2418–2425. doi: 10.1056/NEJMra1312543
Dai, L., Ding, C., and Fang, F. (2018). A novel Ddc gene deletion mutation in two Chinese mainland siblings with aromatic l-amino acid decarboxylase deficiency. Brain Dev. doi: 10.1016/j.braindev.2018.08.003 [Epub ahead of print].
de Ligt, J., Boone, P. M., Pfundt, R., Vissers, L. E., Richmond, T., Geoghegan, J., et al. (2013). Detection of clinically relevant copy number variants with whole-exome sequencing. Hum. Mutat. 34, 1439–1448. doi: 10.1002/humu.22387
Dodge, J. A., Chigladze, T., Donadieu, J., Grossman, Z., Ramos, F., Serlicorni, A., et al. (2011). The importance of rare diseases: from the gene to society. Arch. Dis. Child. 96, 791–792. doi: 10.1136/adc.2010.193664
Elliott, E., and Zurynski, Y. (2015). Rare diseases are a ‘common’ problem for clinicians. Aust. Fam. Physician 44, 630–633.
Ewans, L. J., Schofield, D., Shrestha, R., Zhu, Y., Gayevskiy, V., Ying, K., et al. (2018). Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet. Med. doi: 10.1038/gim.2018.39 [Epub ahead of print].
Fang, F., Liu, Z., Fang, H., Wu, J., Shen, D., Sun, S., et al. (2017). The clinical and genetic characteristics in children with mitochondrial disease in China. Sci. China Life Sci. 60, 746–757. doi: 10.1007/s11427-017-9080-y
Feng, Y., Chen, D., and Wong, L.-J. C. (2017). “Detection of copy number variations (Cnvs) based on the coverage depth from the next generation sequencing data,” in Next Generation Sequencing Based Clinical Molecular Diagnosis of Human Genetic Disorders, ed. L. J. Wong (Cham: Springer),13–22.
Gilissen, C., Hoischen, A., Brunner, H. G., and Veltman, J. A. (2012). Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 20, 490–497. doi: 10.1038/ejhg.2011.258
Hao, C., Guo, J., Guo, R., Qi, Z., Li, W., and Ni, X. (2018). Compound heterozygous variants in Por gene identified by whole-exome sequencing in a Chinese pedigree with cytochrome P450 oxidoreductase deficiency. Pediatr. Invest. 2, 90–95. doi: 10.1002/ped4.12035
Jin, Y., Zhang, L., Ning, B., Hong, H., Xiao, W., Tong, W., et al. (2018). Application of genome analysis strategies in the clinical testing for pediatric diseases. Pediatr. Investig. 2, 72–81. doi: 10.1002/ped4.12044
Kondo, I., Mizugishi, K., Yoneda, Y., Hashimoto, T., Kuwajima, K., Yuasa, I., et al. (1999). Missense mutations in phosphomannomutase 2 gene in two Japanese families with carbohydrate-deficient glycoprotein syndrome type 1. Clin. Genet. 55, 50–54. doi: 10.1034/j.1399-0004.1999.550109.x
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887. doi: 10.1001/jama.2014.14604
Liu, P., Morrison, C., Wang, L., Xiong, D., Vedell, P., Cui, P., et al. (2012). Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 33, 1270–1276. doi: 10.1093/carcin/bgs148
Liu, Z., Fang, F., Ding, C., Wu, H., Lyu, J., and Wu, Y. (2014). [Sucla2-related encephalomyopathic mitochondrial Dna depletion syndrome: a case report and review of literature]. Zhonghua Er Ke Za Zhi 52, 817–821.
Liu, Z., Fang, F., Ding, C., Zhang, W., Li, J., Yang, X., et al. (2015). [Diagnosis of mitochondrial disorders in children with next generation sequencing]. Zhonghua Er Ke Za Zhi 53, 747–753.
Lupski, J. R., Reid, J. G., Gonzaga-Jauregui, C., Rio Deiros, D., Chen, D. C., Nazareth, L., et al. (2010). Whole-genome sequencing in a patient with charcot-marie-tooth neuropathy. N. Engl. J. Med. 362, 1181–1191. doi: 10.1056/NEJMoa0908094
Plaiasu, V., Nanu, M., and Matei, D. (2010). Rare Disease Day – at a glance. Maedica (Buchar) 5, 65–66.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roach, J. C., Glusman, G., Smit, A. F., Huff, C. D., Hubley, R., Shannon, P. T., et al. (2010). Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328, 636–639. doi: 10.1126/science.1186802
Schieppati, A., Henter, J. I., Daina, E., and Aperia, A. (2008). Why rare diseases are an important medical and social issue. Lancet 371, 2039–2041. doi: 10.1016/S0140-6736(08)60872-7
Schollen, E., Martens, K., Geuzens, E., and Matthijs, G. (2002). Dhplc analysis as a platform for molecular diagnosis of congenital disorders of glycosylation (Cdg). Eur. J. Hum. Genet. 10, 643–648. doi: 10.1038/sj.ejhg.5200858
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Shashi, V., Mcconkie-Rosell, A., Schoch, K., Kasturi, V., Rehder, C., Jiang, Y. H., et al. (2016). Practical considerations in the clinical application of whole-exome sequencing. Clin. Genet. 89, 173–181. doi: 10.1111/cge.12569
Sobreira, N. L., Cirulli, E. T., Avramopoulos, D., Wohler, E., Oswald, G. L., Stevens, E. L., et al. (2010). Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet. 6:e1000991. doi: 10.1371/journal.pgen.1000991
Speicher, M. R., and Carter, N. P. (2005). The new cytogenetics: blurring the boundaries with molecular biology. Nat. Rev. Genet. 6, 782–792. doi: 10.1038/nrg1692
Stolk, P., Willemen, M. J., and Leufkens, H. G. (2006). Rare essentials: drugs for rare diseases as essential medicines. Bull. World Health Organ. 84, 745–751. doi: 10.2471/BLT.06.031518
Tabor, H. K., Risch, N. J., and Myers, R. M. (2002). Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat. Rev. Genet. 3, 391–397. doi: 10.1038/nrg796
Vicario, M., Cali, T., Cieri, D., Vallese, F., Bortolotto, R., Lopreiato, R., et al. (2017). A novel Pmca3 mutation in an ataxic patient with hypomorphic phosphomannomutase 2 (Pmm2) heterozygote mutations: biochemical characterization of the pump defect. Biochim. Biophys. Acta 1863, 3303–3312. doi: 10.1016/j.bbadis.2017.08.006
Wenger, A. M., Guturu, H., Bernstein, J. A., and Bejerano, G. (2017). Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet. Med. 19, 209–214. doi: 10.1038/gim.2016.88
Wright, C. F., Fitzpatrick, D. R., and Firth, H. V. (2018). Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 19, 253–268. doi: 10.1038/nrg.2017.116
Yang, Y., Muzny, D. M., Reid, J. G., Bainbridge, M. N., Willis, A., Ward, P. A., et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 369, 1502–1511. doi: 10.1056/NEJMoa1306555
Keywords: WES, phenotype-driven, virtual panel, rare disease, annotation
Citation: Wang X, Shen X, Fang F, Ding C-H, Zhang H, Cao Z-H and An D-Y (2019) Phenotype-Driven Virtual Panel Is an Effective Method to Analyze WES Data of Neurological Disease. Front. Pharmacol. 9:1529. doi: 10.3389/fphar.2018.01529
Received: 15 July 2018; Accepted: 13 December 2018;
Published: 09 January 2019.
Edited by:
Tieliu Shi, East China Normal University, ChinaReviewed by:
Olimpia Musumeci, Università degli Studi di Messina, ItalyZhiping Liu, Augusta University, United States
Copyright © 2019 Wang, Shen, Fang, Ding, Zhang, Cao and An. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Wang, zfwx05@126.com
†These authors have contributed equally to this work