Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)—multiple rather than a single mechanism
 Boris Gole
Boris Gole Lisa Wiesmüller
Lisa Wiesmüller- Division of Gynecological Oncology, Department of Obstetrics and Gynecology, Ulm University, Ulm, Germany
Despite manifold efforts to achieve reduced-intensity and -toxicity regimens, secondary leukemia has remained the most severe side effect of chemotherapeutic cancer treatment. Rearrangements involving a short telomeric <1 kb region of the mixed lineage leukemia (MLL) gene are the most frequently observed molecular changes in secondary as well as infant acute leukemia. Due to the mode-of-action of epipodophyllotoxins and anthracyclines, which have widely been used in cancer therapy, and support from in vitro experiments, cleavage of this MLL breakpoint cluster hotspot by poisoned topoisomerase II was proposed to trigger the molecular events leading to malignant transformation. Later on, clinical patient data and cell-based studies addressing a wider spectrum of stimuli identified cellular stress signaling pathways, which create secondary DNA structures, provide chromatin accessibility, and activate nucleases other than topoisomerase II at the MLL. The MLL destabilizing signaling pathways under discussion, namely early apoptotic DNA fragmentation, transcription stalling, and replication stalling, may all act in concert upon infection-, transplantation-, or therapy-induced cell cycle entry of hematopoietic stem and progenitor cells (HSPCs), to permit misguided cleavage and error-prone DNA repair in the cell-of-leukemia-origin.
Introduction
The KMT2A gene, better known as MLL (mixed lineage or myeloid/lymphoid leukemia) encodes a lysine (K)-specific histone methyltransferase 2A, which functions as an epigenetic regulator of transcription (Daniel and Nussenzweig, 2012; Takeda et al., 2013). The enzyme is primarily connected to hematopoietic and embryonic development (Hess et al., 1997), but was also described to contribute to the S-phase DNA damage checkpoint (Liu et al., 2010). The gene spans the breakpoint cluster region at chromosomal position 11q23, frequently rearranged in acute leukemia, especially in therapy-related and infant cases (Ziemin-van der Poel et al., 1991; Emerenciano et al., 2007; Cowell and Austin, 2012). Despite over two decades of efforts, the reasons underlying the exceptionally high breakability of the KMT2A/MLL locus are still not clear. The same is true also for the mechanisms leading to its rearrangements. In this review we summarize the facts and hypotheses which in our view are relevant for understanding of KMT2A/MLL breakage and rearrangements, with a focus on therapy-related cases.
Incidence and Risk of MLL Rearrangements
Rearrangements of the MLL gene were found in 5.2% of all the acute myeloid leukemia (AML) cases and in 22% of all the acute lymphoid leukemia (ALL) cases (De Braekeleer et al., 2005). Patients with MLL rearrangements have poorer prognosis than the ones without, with shorter event free and overall survival rates (Tamai et al., 2008; Cerveira et al., 2012; Chen et al., 2013). Interestingly, myelodsyplastic syndrome (MDS) patients with the 11q23 rearrangement t(2; 11)(p21; q23) which does not affect the MLL gene but rather upregulates a downstream lying miRNA MIR125B1 resulting in inhibited primary human CD34+ cell differentiation, have a favorable prognosis (Bousquet et al., 2008; Dvorak et al., 2014).
In childhood ALL, MLL rearrangements are found in 44–85% of the infants (<1 year old), which decreases down to 3% in the elder (1–10 years) patients (Emerenciano et al., 2007, 2013; Al-Sudairy et al., 2014). Data from siblings revealed that both genetic and environmental risk factors are implied in childhood ALL (Schmiegelow et al., 2012). As MLL rearrangements in these patients can arise during in utero fetal hematopoiesis (Gale et al., 1997), prenatal exposure and consequently lifestyle of the mother are highly relevant for development of this type of leukemia. Increased MLL rearrangements were indeed observed in amniocytes from long-term smokers in a small prospective study (de la Chica et al., 2011), as well as a statistically significant association between intake of hormones during pregnancy and risk of in utero MLL rearrangements in a study enrolling several 100 children (Pombo-de-Oliveira et al., 2006). Estrogen, in particular, was demonstrated to induce MLL breakage and rearrangements in cultured lymphoblastoid cells (Schnyder et al., 2009). The most prominent hints to exogenous MLL destabilizing sources stem from reports on the intake of dietary flavonoids during pregnancy. Bioflavonoids, such as quercetin, hydroquinone or genistein which are present in citrus, certain types of berries and root vegetables, can induce MLL cleavage and rearrangements ex vivo in human hematopoietic stem and progenitor cells (HSPCs) isolated from umbilical cord blood (Strick et al., 2000; van Waalwijk van Doorn-Khosrovani et al., 2007) and in utero in mice (Vanhees et al., 2011).
With rearrangements found in ~40% of therapy-related acute leukemia/myelodsyplastic syndrome (t-AL/MDS), MLL is the most frequently rearranged gene in t-AL (Shivakumar et al., 2008; Pullarkat et al., 2009; Shim et al., 2010; Abdulwahab et al., 2012; Cowell and Austin, 2012). The lifelong risk of t-AL/MDS in patients receiving chemo- and/or radio-therapy was found to be 0.2% among US cancer patients (Morton et al., 2013). Depending on the type of primary cancer and especially the therapy used, it varied between 0.02 and 12% (Maddams et al., 2011; Abdulwahab et al., 2012; Ezoe, 2012; Koontz et al., 2013; Morton et al., 2013). The majority of cases occur during the first 5 years after treatment of primary cancer (Shivakumar et al., 2008; Pullarkat et al., 2009; Ezoe, 2012; Koontz et al., 2013). Though causalities are often difficult to assess as patients receive complex treatments, the highest (up to 12%) incidence of t-AL was observed in patients treated with topoisomerase II inhibitors such as epipodophyllotoxins and anthracyclines (Abdulwahab et al., 2012; Ezoe, 2012). Use of topoisomerase II inhibitors also seemed connected to over 90% of all the 11q23 rearrangements in patients receiving chemotherapy (Shivakumar et al., 2008). A causal link between topoisomerase II inhibitor treatment and MLL rearrangements was supported by in vitro data on stable MLL rearrangements in human embryonic and hematopoietic stem cells (HSCs) treated with topoisomerase II inhibitor etoposide (Libura et al., 2008; Bueno et al., 2009).
Interestingly, bioflavonoids were connected to MLL rearrangements in infant ALL and were shown to inhibit human topoisomerase II (Strick et al., 2000), so that causes for infant ALL might be similar to the ones for t-AL. Both types of leukemia are characterized by short latency after the initiation event- exposure of the hematopoietic system to the MLL destabilizing agents in utero or during primary cancer treatment, respectively. To the best of our knowledge no links between bioflavonoids/topoisomerase II-inhibiting compounds and MLL rearrangements in de novo AL were described so far. Striking similarities between infant and therapy-related AL were, however, found when comparing the breakage distribution within the MLL as discussed below.
Distribution of Break Sites Leading to Leukemic Rearrangements
More than 95% of the MLL rearrangements fall within a ~7.3 kb breakpoint cluster region (MLLbcr). This was originally described to range from exon 9 to intron 11/exon 12 of the MLL gene (Reichel et al., 2001; Meyer et al., 2013). However, according to the current reference sequence (GRCh38.p2, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) this region corresponds to exon 8—intron 10/exon 11 (Figures 1A,B). For the purpose of clarity and comparability to publications cited in this review, we use the original numbering throughout this paper. Within the MLLbcr the distribution of breaks is not uniform, but rather forms two clusters which together harbor ~80% of all the breaks (Meyer et al., 2013). The centromeric cluster can be attributed mostly to cases of de novo AL in adults, while breaks from t-AL and infant ALL cluster at the telomeric part of the MLLbcr (Broeker et al., 1996; Cimino et al., 1997; Reichel et al., 2001; Cowell and Austin, 2012; Meyer et al., 2013), in particular, within a short ~600 bp translocation hotspot at the intron 11/exon 12 boundary (Mirault et al., 2006). Similar clustering to intron 11/exon 12 boundary can be found also when looking at breaks and rearrangements following topoisomerase II inhibitor and flavonoid treatments (van Waalwijk van Doorn-Khosrovani et al., 2007; Le et al., 2009), providing further support for a mechanistic connection between secondary and infant ALL.
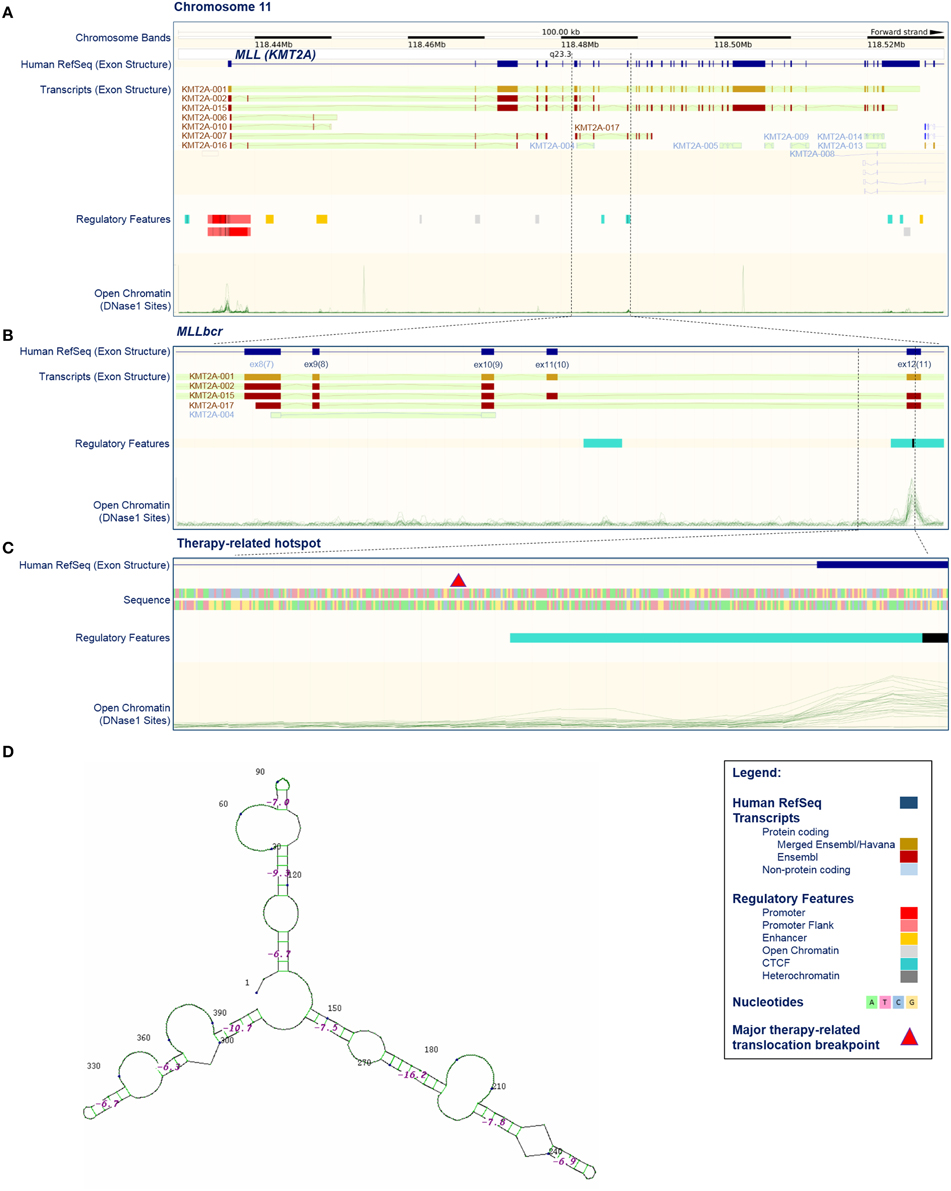
Figure 1. Position, structure, and regulatory features of MLL breakpoint cluster region. (A–C) Selected features of the KMT2A/MLL gene based on current issue of Ensembl data (release 79, March 2015; http://www.ensembl.org/index.html; Cunningham et al., 2015). (A) KMT2A/MLL position on chromosome 11, exon structure according to the current reference sequence (Human RefSeq), transcripts (protein coding and non-protein coding), regulatory features and open chromatin (DNase I sensitive) sites. Some transcripts harbor exons that are not acknowledged by the reference sequence (for example compare Human RefSeq, KMT2A-001 and KMT2A-002). Open chromatin sites are detectable in promoter-exon 1, exon 12 and exon 37. The position of the breakpoint cluster region (MLLbcr) is marked by dashed lines. (B) Enhanced view of the MLLbcr with exon structure of the reference sequence and transcripts, regulatory features and open chromatin (DNase I) sites. The position of the therapy-related hotspot is marked by dashed lines. (C) Enhanced view of a therapy-related hotspot flanking a major therapy-related translocation breakpoint described by Mirault et al. (2006), with exon structure, sequence, regulatory features and open chromatin (DNase I) sites. The major therapy-related breakpoint cluster (marked by red triangle) lies at the 5′ end of intron 11, just 3′ of the DNase I and CTCF-binding site found in 17/18 and 18/18 cell lines used in Ensembl, respectively. The indicated 399 bp segment was used in reporter studies with exogenous MLLbcr fragment (Boehden et al., 2004; Gole et al., 2014; Ireno et al., 2014). (D) Secondary structure of the 399 bp therapy-related hotspot. The structure was calculated using programs performing searches based on sequence, namely Quadruplex forming G-Rich Sequences (QGRS) Mapper (http://bioinformatics.ramapo.edu/QGRS/index.php) and M-Fold for hairpins (http://mfold.rna.albany.edu/?q=mfold).
Over 100 different rearrangements and over 60 translocation partners of MLL have been described so far, however most of them were reported only sporadically (Meyer et al., 2006, 2009). A handful of translocation partners, namely AFF1/AF4, MLLT3/AF9, MLLT1/ENL, MLLT10/AF10, and MLLT4/AF6, were found to be involved in 70–95% of the MLL translocations, with some differences in frequencies of individual partners in ALL vs. AML, infant vs. adult, and de novo vs. therapy-induced leukemia (Meyer et al., 2009, 2013; De Braekeleer et al., 2011; Cerveira et al., 2012; Emerenciano et al., 2013). Relatively narrow assortment of recurrent translocation partners most likely reflects some growth advantage of the mutant cells due to the particular rearrangement (Strick et al., 2006; Bueno et al., 2009). In line with this concept, examination of the blood samples from breast cancer and lymphoma patients undergoing topoisomerase II inhibitor treatment showed that MLL translocations are much more common than the actual t-AL cases (Le et al., 2009). Stable MLL rearrangements in human HSCs treated with etoposide did indeed increase the proliferative capacity of the cells (Libura et al., 2008; Bueno et al., 2009), though additional abnormalities seem to be needed for the completion of malignant transformation (Bueno et al., 2009). As a prominent example, the MLL-AF9 fusion gene was shown to be actively transcribed (Betti et al., 2003) and the resulting fusion protein promoted expression of Hoxa9 transcription factor which has been associated with leukemic transformation (Muntean et al., 2010). Despite the fact that reciprocal gene fusions represent the majority of stable MLL rearrangements, a plethora of additional MLL aberrations namely gene-internal duplications, inversions, deletions, insertions, and complex rearrangements were identified in leukemia patients (Meyer et al., 2013), underscoring the fact that the MLLbcr bears intrinsic features causing exceptionally high fragility.
Which Enzymes Cause MLL Breakage?
Already in the 1990s, MLLbcr breakage and rearrangements causing secondary leukemia in patients undergoing chemotherapy were linked to the action of topoisomerase II inhibitors. Thus, cleavage by topoisomerase II at consensus sequences within a centromeric MLLbcr fragment was demonstrated in vitro (Felix et al., 1995). Translocation breakpoint junctions of a t(4; 11) and a t(9; 11) secondary leukemia patients were found to coincide with topoisomerase II sensitive sites, even though not consensus sequences, in the centromeric and telomeric MLLbcr fragment, respectively (Lovett et al., 2001; Whitmarsh et al., 2003). Characterization of topoisomerase II inhibitor-induced effects on the MLLbcr in lymphoblasts from a patient undergoing chemotherapy, in ex vivo treated peripheral blood cells from healthy individuals and in vitro treated leukemia cell lines, identified breakage within a telomeric MLLbcr fragment comprising a topoisomerase II recognition sequence (Aplan et al., 1996). Topoisomerase II inhibition by either etoposide or teniposide treatment was shown to induce the same cleavage pattern in the telomeric MLLbcr region in lymphoblastoid cells, T47D breast cancer cells, and human fibroblasts (Mirault et al., 2006). Causal involvement of topoisomerase II was supported by use of cells with mutated topoisomerase II showing diminished breakage (Mirault et al., 2006; Cowell et al., 2012) and by protein expression inducing hypersensitivity to etoposide (Tamaichi et al., 2013).
Despite these well-supported links between topoisomerase II action and MLLbcr breakage, several lines of evidence indicate that topoisomerase II is not essential for this process. First, using various human leukemia and other cancer cell lines as well as mouse embryonic fibroblasts, several groups showed that MLLbcr cleavage depends on apoptosis (but not necrosis) rather than topoisomerase II, even when cells are treated with topoisomerase II inhibitors (Stanulla et al., 1997; Betti et al., 2005; Hars et al., 2006). It was hypothesized that some cells can escape the cell death process and therefore transform apoptosis-induced DNA damage into leukemic translocations (Vaughan et al., 2002). In line with this, it is possible to induce MLLbcr breakage and rearrangements in cultured cells with a wide range of compounds not targeting topoisomerase II, such as with topoisomerase I inhibitor camptothecin, DNA polymerase inhibitor aphidicolin, microtubule inhibitor vinblastine, antimetabolites (5-fluorouracil, cytosine arabinoside, methotrexate), alkylating agents (melphalan, methylmethanesulfonate), the cross-linking agent cisplatin, with ionizing irradiation, and with various other genotoxic compounds with less defined modes of action (Stanulla et al., 1997; Ploski and Aplan, 2001; Sim and Liu, 2001; Betti et al., 2005; Le et al., 2009; Gole et al., 2014; Ireno et al., 2014; Kraft et al., 2015). It has even been reported that MLLbcr breakage and rearrangements can be induced by some non-genotoxic agents such as N-methylformamide (Ploski and Aplan, 2001) or by estrogen (Le et al., 2009), though for most non-genotoxic compounds this seems not to be the case (Ireno et al., 2014). Clinical data also describe 11q23/MLL rearrangements in patients treated with chemotherapies not addressing topoisomerase II (Faller et al., 2009; Zámečníkova, 2011). Most importantly, catalytic topoisomerase II inhibition by merbarone prior to formation of the cleavable complex or RNA interference-mediated silencing of the enzyme did not abrogate MLLbcr cleavage downstream of different genotoxic treatments (Betti et al., 2005). In this context it is important to note that topoisomerase II poisoning by epipodophyllotoxins like etoposide relies on the formation of a complex composed of the drug, DNA, and topoisomerase II blocking DNA re-ligation and resulting in breakage, i.e., genotoxic stress (Felix et al., 1995).
From these observations it was proposed that signaling events downstream of topoisomerase II-induced DNA double-strand break (DSB) formation are the cause of MLLbcr breakage and/or that several distinct enzymes can be involved (Stanulla et al., 1997; Mirault et al., 2006). The most widely discussed signaling mechanism, which underlies MLLbcr breakage is apoptosis leading to apoptotic DNA fragmentation (Stanulla et al., 1997; Betti et al., 2001; Sim and Liu, 2001). This two-step process of DNA degradation during apoptosis is executed by so-called apoptotic nucleases, primarily by caspase-activated DNase (CAD) but also Artemis, DNase I, DNase II, DNase γ, Endonuclease G, topoisomerase II, and cofactors such as apoptosis-inducing factor (AIF) and cyclophilins (Samejima and Earnshaw, 2005; Britton et al., 2009; Widlak and Garrard, 2009). Accordingly, MLLbcr cleavage could be part of early DNA fragmentation into high molecular weight (50–300 kb) fragments. Deregulated checkpoint control might permit repair of the resulting DSBs and thus survival, while erroneous DSB repair might rearrange MLL in some cells mediating malignant transformation (Stanulla et al., 1997; Hars et al., 2006). Indeed, genomic mapping revealed that the ~600 bp MLLbcr hotspot for therapy-related translocations contains not only topoisomerase II but also topoisomerase I and apoptotic cleavage sites (Mirault et al., 2006) and was shown to be DNase I-sensitive (Strissel et al., 1998). In further support of the concept of apoptotic fragmentation-driven MLL rearrangements, incidence of MLL fusions was decreased in etoposide-treated mouse embryo fibroblasts from CAD knockdown mice or after application of the pan-caspase inhibitor zVAD-fmk (Hars et al., 2006). Corresponding results were obtained in lymphoblastoid TK6 cells after apoptosis induction with anti-CD95-antibody and zVAD-fmk exposure (Betti et al., 2003; Le et al., 2009), indicating that induction of MLL breakage and rearrangements are observed downstream of receptor-mediated and mitochondrial pathway-dependent apoptotic signaling.
However, caspase activation seems not to be required for MLL breakage and rearrangements in each cellular setting. Etoposide-induced MLLbcr breakage was caspase-independent in CEM cells (Mirault et al., 2006). Further, irradiated MCF7 cells, which lack functional caspase-3 and thus CAD activation, show the same MLLbcr fragmentation as TK6 cells treated with anti-CD95 antibody (Betti et al., 2005). In our recent work, we used T47D cells, which are known to mimic the MLLbcr cleavage pattern of lymphoblastoid cells after genotoxic treatment (Mirault et al., 2006) and to be particularly responsive to replication stress induced by aphidicolin. Again, we found that MLLbcr cleavage is caspase-independent after aphidicolin treatment despite active apoptosis signaling (Gole et al., 2014). Moreover, in the context of enhanced replication stress, caspase-independent Endonuclease G rather than caspase-activated CAD, topoisomerase I, DNase I, DNase II, or Artemis was essential for MLLbcr rearrangements in T47D and HeLa cells (Gole et al., 2014). Replication stress is one of the outcomes of topoisomerase II-inhibitory treatments, as topoisomerase II action is required for relaxation of supercoiled DNA during replication (Felix et al., 1995). Moreover, epipodophyllotoxins generate reactive oxygen species that create various DNA lesions ultimately blocking replication (Berquist and Wilson, 2012). Therefore, erroneous responses to replication stress associated with replication fork stalling are a possible additional explanation for MLLbcr breakage and rearrangements after topoisomerase II inhibition using epipodophyllotoxins.
Properties of the MLLbcr Locus
Collective data showing that MLLbcr breaks and rearrangements most likely can be induced by topoisomerase II- or apoptosis/caspase-mediated but also independent mechanisms spurred the search for a common denominator, which could explain breakability of MLLbcr under all these different conditions. Clues might come from the intrinsic properties of the MLLbcr region itself. Soon after the discovery of the involvement of the MLL locus in leukemogenesis it was proposed that the chromatin structure of the region might play a role. Thus, the telomeric half of the MLLbcr, which harbors topoisomerase II-sensitive sites was described to have properties of a scaffold/matrix-attachment region (SAR/MAR) (Broeker et al., 1996). Later on, however, it was noticed that the MLLbcr-DNA breaks showed a higher density outside of high-affinity SAR/MAR fragments, probably because SAR/MAR-DNA is protected by a high protein content (Strissel et al., 2000; Hensel et al., 2001). Similarly, the breakpoint cluster region in the AFF1/AF4 gene, a common translocation partner of MLL, also contains MAR features but again with an inverse correlation between breakage and high-affinity SAR/MAR fragments (Hensel et al., 2001). On the other hand it was discovered that the telomeric part of the MLLbcr has characteristics of open chromatin with high DNase I sensitivity and low histone H1 content (Strissel et al., 1998; Khobta et al., 2004). DNase I sensitivity peaks in the MLLbcr, similarly as in the promoter of the MLL gene, is positioned in the 5′-half of exon 12, just 3′ to the major therapy-related translocation breakpoint (Figure 1C). The breakpoint cluster region of AFF1/AF4 also showed DNase I sensitivity (Strick et al., 2006), so that the therapy/infant-related MLLbcr breaks and rearrangements could be due to increased accessibility of this translocation hotspot for nucleases such as topoisomerase II and Endonuclease G. No such features were described for the centromeric part of the MLLbcr (Figure 1B), suggesting that the mechanisms of MLL breakage leading to de novo AL are quite different from the ones in t-AL and infant ALL.
At first glance contradictory to the proposed impact of the chromatin context on the fragility of the MLLbcr, experiments with extrachromosomal episomes containing the therapy-related MLLbcr fragment or with randomly chromosomally integrated extra MLLbcr copies showed similar cleavage and rearrangements of exogenous and the endogenous MLLbcr in situ (Stanulla et al., 2001; Boehden et al., 2004; Gole et al., 2014). Still, when investigating a randomly integrated copy of the therapy-related MLLbcr fragment, we noticed dependency of breakage and rearrangements on RNF20 (Gole et al., 2014), a histone H2B monoubiquitinase whose activity has been linked with increased chromatin accessibility (Fierz et al., 2011). This RNF20 influence was not found when analyzing extrachromosomal MLLbcr fragment stability (Gole et al., 2014). Further arguing against a mere cis-regulatory effect of the MLLbcr DNA sequence, we saw differences in the inducibility of rearrangements between cell clones with single copies of the MLLbcr integrated into different chromosomal sites in K562 myleoid leukemia cells (Ireno et al., 2014). Taken together, accessibility of naked or poorly chromatinized DNA within extrachromosomal episomes or open chromatin at intrachromosomal loci seem to significantly contribute to the t-AL/infant ALL-related MLLbcr breaks.
On the other hand, several pieces of evidence argue against chromatin accessibility as the sole reason for MLLbcr fragility, as at least two more segments of the MLL gene which are not recurrently rearranged have DNase I sensitive sites, namely within the promoter region of exon 1 and within exon 37 (Figure 1A). Interestingly, when murine Mll was introduced into human cells on an episome, it was cleaved at the site of the endogenous Mll in murine cells, which was the first hint that the sequence of MLL itself plays an important role in breakage distribution (Stanulla et al., 2001). Later on it was discovered that MLLbcr breaks at non-random positions at a sequence reminiscent of nick-forming sequences, i.e., hypersensitive regions positioned regularly at loop-size intervals in the eukaryotic chromatin, which are targeted during high-order apoptotic DNA fragmentation (Székvölgyi et al., 2006). These sequences may form secondary structures such as hairpins and these hairpins were proposed to represent the basis of MLLbcr fragility (Székvölgyi et al., 2006). Indeed, a hairpin secondary structure was described at a topoisomerase II cleavage site at the 5′-terminus of MLL exon 12, with positioning of the topoisomerase II site in the loop and the actual breakpoint at the stem of the hairpin (Le et al., 2009). A broader look at the intron 11/exon 12 boundary reveals that the whole region harboring major therapy-related translocation breakpoints is predicted to fold into a complex secondary structure with hairpins (Figure 1D). Such non-B DNA structures can form during DNA replication when longer runs of single-stranded DNA are transiently formed as well as in a replication-independent manner (Wang et al., 2013). When left unresolved, such structures can obstruct progression of the DNA replication and/or RNA transcription machinery (Le et al., 2009; Wang et al., 2013), both of which could contribute to MLLbcr breakage and further rearrangements (Le et al., 2009) as discussed in more in detail below.
Replication Stalling or Transcription Stalling?
Replication Stalling
Breakage of the therapy-related MLLbcr hotspot in response to treatment with aphidicolin, an inhibitor of polymerase α, δ, and ε (Ozeri-Galai et al., 2011), indicated involvement of replication stress in MLLbcr destabilization, particularly as apoptosis induction was not necessarily coupled with treatment (Gole et al., 2014). Together with formation of a stable secondary structure predicted to halt the progression of replication forks (Figure 1D), these MLLbcr features were reminiscent of so-called fragile sites (Durkin and Glover, 2007; Ozeri-Galai et al., 2011; Debatisse et al., 2012). Fragile sites by definition are loci with frequent breaks under replication stress. While Rare Fragile Sites (RFS) are found only in few individuals such as with micro- or mini-satellite repeat expansion-associated inheritable diseases, Common fragile sites (CFS) are population-wide hotspots for chromosomal rearrangements, whereby >200 were mapped in humans (Letessier et al., 2011; Debatisse et al., 2012). CFS were discovered as gaps and constrictions in metaphase chromosomes of cells grown under mild replication stress conditions following treatment of cells with aphidicolin (Glover et al., 1984). Further work showed that CFS are also responsive to replication impediments such as due to secondary structures, decelerated replication such as due to depletion of nucleotide pools, perturbations of replication due to failure of origin firing or deregulated checkpoint control mechanisms, and interference with transcription (Letessier et al., 2011; Ozeri-Galai et al., 2011; Debatisse et al., 2012). Genomic analysis revealed that CFS are prone to sister chromatid exchange, loss of heterozygosity, deletions, and tandem segmental genomic duplications. Intriguingly, CFS correlate with chromosomal breakpoints in tumors. Altogether, even though not formally defined as CFS, the therapy-related MLLbcr hotspot shares crucial characteristics with these fragile sites.
Depending on the cause, severity, and persistence of DNA replication stress it emerges as stalling and/or collapse of DNA replication forks. DNA lesions represent an obstacle to continued fork progression and therefore are a major source of replication stress. Replication stalling can directly create DSBs, when the fork encounters a single-stranded DNA break. However, DSBs may also arise during replication reactivation, when damage bypass mechanisms transiently introduce breaks such as during inter-strand DNA cross-link repair or when strand exchange intermediates are created that are vulnerable to incisions by structure specific nucleases such as upon replication fork reversal (Atkinson and McGlynn, 2009; Thompson and Hinz, 2009). Endogenous and exogenous reactive oxygen species are a prominent source of a wide spectrum of lesions ranging from base modifications to inter-strand DNA cross-links (Berquist and Wilson, 2012; Ensminger et al., 2014). Other replication blocking insults may arise from environmental, nutritional, and life-style risk factors, as well as from therapeutic interventions such as chemo- and radiotherapies, i.e., the well-described cause of secondary leukemia associated with MLLbcr rearrangements (Shivakumar et al., 2008; Pullarkat et al., 2009; Shim et al., 2010; Abdulwahab et al., 2012; Cowell and Austin, 2012). The combination of deregulated replication and genotoxic stress such as in case of oncogene- or hormone-induced proliferation together with reactive oxygen species and reactive metabolite formation, respectively, seems to be particularly detrimental to the integrity of the genome (Bolton and Thatcher, 2008; Macheret and Halazonetis, 2015).
Replication stress-induced rearrangements could also be the common trigger for rearrangements within the telomeric MLL fragment causing both therapy-related and infant ALL (Broeker et al., 1996; Cimino et al., 1997; Reichel et al., 2001; Cowell and Austin, 2012; Meyer et al., 2013). HSCs and progenitor cells, i.e., the cells-of-leukemia-origin, are known to exit from quiescence into an active cycle upon infection, enforced self-renewal due to bone marrow transplantation, and in response to genotoxic insults. A recent publication provided in vivo evidence in mice for the appearance of DSBs and single-stranded breaks as a direct consequence of HSC cell cycle entry under conditions mimicking viral infection (Walter et al., 2015). Serial transplantation of HSCs into immunodeficient mice as well as clinical HSC transplantation were shown to trigger replication stress and persistent DNA damage (Yahata et al., 2011). The authors identified mitochondrial reactive oxygen species as mediator of replication stress-induced DNA damage in both cases. Proliferative stress further correlated with genomic instability in bone marrow transplants during repopulation of recipients favoring secondary tumor formation (Hertenstein et al., 2005; Flynn and Kaufman, 2007). Murine HSCs, when pushed out of quiescence to reconstitute mature blood cells by massive self-renewal following irradiation or chemotherapeutic treatment show hypersensitivity to further genotoxic treatment and proneness to lymphomagenesis (Cheshier et al., 1999; Labi et al., 2010; Trumpp et al., 2010; Desai et al., 2014). MLLbcr rearrangements in t-AL and infant ALL associated with radio-/chemotherapy and fetal hematopoiesis, respectively, are thus compatible with HSC exit from quiescence and hypersensitivity to replication-associated damage. Further clues to an involvement of replication stress in MLLbcr rearrangements came from Fanconi anemia (FA) patients, as the FA pathway is central to the stabilization and reactivation of replication forks (Thompson and Hinz, 2009). The integrity of the FA pathway was reported essential to prevent high rates of HSC death upon replication stress (Ceccaldi et al., 2012; Walter et al., 2015). Supporting a causal role of replication-associated damage in MLLbcr rearrangements, therapy-related t(11; 16)-AML with a MLL-CBP fusion was observed in a pre-B-cell ALL pediatric patient with FA (Sugita et al., 2000), and MLL partial tandem duplications (MLL-PTD) were reported in bone marrow samples from FA patients (Quentin et al., 2011).
Transcription Stalling
As with replication stalling, various hurdles ranging from repetitive sequences to chromatin changes may also cause transcription stalling and several pieces of evidence suggested involvement of transcription stalling in MLLbcr destabilization. Using chromatin immunoprecipitation (ChIP) in Jurkat immortalized human T-lymphocytes and human CD34+ HSPCs, the telomeric MLLbcr region encompassing the therapy-related hotspot at intron 11/exon 12 was characterized by chromatin marks reminiscent of promoters with the lowest histone H1 content and H4-acetylated islands (Khobta et al., 2004). In 2007, Marschalek and co-workers (Scharf et al., 2007) confirmed a chromatin structure that supports active transcription. Even further, they proposed that this telomeric MLLbcr fragment colocalizes with a gene-internal promoter resulting in a truncated MLL protein. Interestingly, the authors detected a similar putative gene-internal promoter in the homologous region in the murine Mll. Intriguingly, etoposide-induced break sites were then mapped at the RNA polymerase II binding and transcription initiation site within the putative gene-internal promoter in Jurkat and REH human cell lines as well as peripheral blood mononuclear cells (Scharf et al., 2007). However, no such information on a gene-internal promoter at intron 11/exon 12 is included in current Ensembl data (release 79, March 2015, Cunningham et al., 2015) which assembles information from 18 different human cell lines (including K562, DND-41 leukemic T-cells and GM12878 B-lymphocytes). Nevertheless, a DNase I sensitive open chromatin structure was detectable in the critical region. It further coincides with binding sites for CTCF that can function as a transcription factor, demarcate chromatin domains, and mediate chromosomal looping interactions (Figures 1B,C). One possibility to reconcile the different observations could be cell type-dependent regulatory effects. Importantly, replication and transcription cannot be looked upon separately, because collisions between transcription and replication machineries or RNA-DNA hybrid (R-loops) formed during transcription are known to impede replication fork progression (Macheret and Halazonetis, 2015). Conflicts between transcription and replication can stem from deregulated checkpoint control mechanisms changing the length of cell cycle phases and the program of origin firing during malignant transformation processes.
Different views exist also regarding the order of events leading to DNA breakage and subsequent rearrangements at transcriptionally active sites. The two major hypotheses imply either that rearrangements occur after DNA damage at the juxtaposed sequences/genes (contact first) or due to increased mobility of distant sequences/genes after DNA damage (breakage first). The contact first hypothesis implies transcription stalling as the basis of DNA break formation. In support of such a model regarding MLL rearrangements, it was noticed that MLL transcripts can be founded in close proximity to actively transcribed translocation partners AF4 and AF9 in KG1 myeloid leukemia cells and Nalm-6 pre-B leukemia cells and AF4 and AF9 transcripts in close proximity to actively transcribed MLL (Cowell et al., 2012). This suggests that these genes could be part of common transcription factories, where simultaneous DNA breaks on translocation partners in close proximity are possible. Supporting the common transcription factory/contact first hypothesis are also observations of MLL-AF4 mRNA fusion transcripts in the absence of the corresponding gene fusion in both tumor and normal hematopoietic cells from infant ALL patients (Uckun et al., 1998) and in peripheral blood mononuclear cells from healthy individuals (Kowarz et al., 2011). These observations were interpreted such that premature termination of transcription may trigger intra- and to a lesser extent inter-genic trans-splicing events resulting in fusion transcripts at common transcription factories. Due to the intriguing coincidence of trans-splicing and translocation events involving the same gene pairs, and inspired by findings from yeast on RNA-templated DNA repair (Storici et al., 2007), the authors proposed that fusion transcripts may in fact guide error-prone repair of DNA lesions arising in one of the translocating genes (Kowarz et al., 2011, 2012). In a recent study addressing the role of prenatal hormone exposure in MLL translocations leading to infant ALL, estradiol-induced MLL and MLLT3/AF9 colocalization as well as fusion transcript formation were detected. Interestingly, this process required the protein activation-induced cytidine deaminase, AID (Wright et al., 2014). Even though primarily known to be involved in targeted Ig gene maturation processes, AID has also been shown to exert unspecific genotoxic effects upon activation by estrogen (Pauklin et al., 2009). ChIP experiments revealed localization of AID in MLLbcr intron 11 (Wright et al., 2014), to where it may become recruited by stalled RNA polymerase in analogy to the situation during Ig gene switching (Pavri et al., 2010). Given that aberrantly activated AID can cleave at 150 target sites outside of Ig genes including well-described translocation hotspots (Hakim et al., 2012), it represents a key candidate for involvement in genome rearrangements following transcription stalling.
The breakage first hypothesis positions contact formation as an event secondary to DNA break formation. One of the reasons arguing against the contact first hypothesis was that 3D-FISH analysis indicated closer spatial proximity of MLL and ENL genes in interphase nuclei of myeloid (AML-193, PLB-985) and lymphoid cells (Nalm-6, IM-9) as compared with MLL and AF4 genes, even though AF4 represents the most frequent partner in MLL translocations (Gué et al., 2006). Evidence supporting breakage as the primary event leading to chromosome rearrangements was obtained also with the 3D time-lapse microscopy in living U2OS osteosarcoma cells using the fluorescently labeled 53BP1-GFP protein as a DSB marker. These experiments revealed enhanced mobility of damaged as compared with intact chromatin domains and susceptibility of only DSB containing chromatin to retardation of the movements by agents that affect chromatin organization (Krawczyk et al., 2012). Glukhov and colleagues (Glukhov et al., 2013) more specifically addressed the mobility of the MLLbcr in etoposide-treated Jurkat cells. The authors observed that the telomeric MLLbcr DNA end moved out of the chromosome 11 territory within 1 h following cleavage, which could increase the probability to meet and erroneously merge with a translocation partner.
Observations made on the frequency of Igh translocations in murine B lymphocytes may reconcile the two models in that they showed that site-directed DNA break formation is strictly associated with translocations, whereas in the absence of targeted DNA breaks rearrangements are related to the contact frequency with the partner genes, i.e., reflect the nuclear architecture (Hakim et al., 2012). Thus, targeted cleavage seems to govern downstream events. Notably, MLLbcr cleavage and at least intramolecular rearrangements do not require higher-order chromatin structures, take place on non-replicating episomes and independently of the MLLbcr sequence orientation with respect to a neighboring transcriptional promoter arguing against an essential role of replication or transcription stalling immediately within the MLLbcr (Stanulla et al., 2001; Boehden et al., 2004; Gole et al., 2014). However, chromosomal structures and processes promote MLLbcr breakage in cis and/or influence cleavage in trans such as via early apoptotic signaling and activation of Endonuclease G (Gole et al., 2014) (Figure 2). Intriguingly, damaged DNA and complex DNA structures, such as R-loops, all of which can promote replication and/or transcription stalling are preferential targets of Endonuclease G-mediated cleavage (Ruiz-Carrillo and Renaud, 1987; Ohsato et al., 2002; Kalinowska et al., 2005) making it a strong candidate for the nuclease responsible for MLLbcr cleavage.
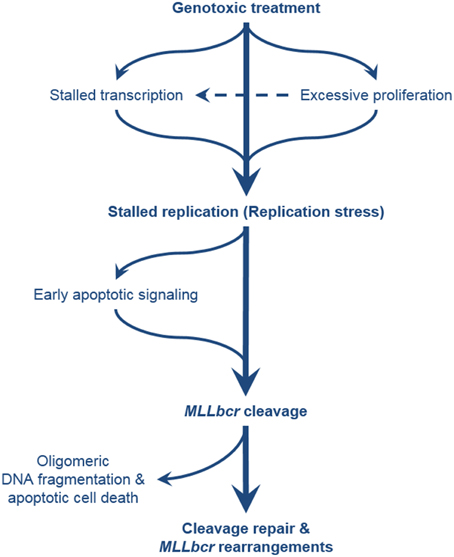
Figure 2. Comprehensive overview of the pathways leading to MLLbcr rearrangements. Exposure of the cells to genotoxic agents can directly or indirectly cause stalling of DNA replication, e.g., by antimetabolite treatment, and by formation of DNA adducts and downstream repair intermediates (e.g., by treatment with alkylating agents and downstream excision repair) or transcription stalling, respectively. Excessive proliferation of the hematopoietic stem/progenitor cells (cells-of-leukemia-origin) due to enforced self-renewal after bone marrow transplantation or genotoxic insults also results in replication stress. MLLbcr in turn is cleaved either as part of the attempt to rescue stalled forks or as part of DNA damage-induced early apoptotic high order DNA fragmentation. DSBs at the MLLbcr can be repaired through NHEJ, MMEJ, or homology-directed repair which can lead to leukemogenic rearrangements preventing further oligomeric DNA fragmentation and cell death.
How is Broken MLLbcr Repaired?
The striking disparity in the distribution of breakpoints associated with de novo vs. therapy-related AL clearly involves differential targeting of DNA breaks but may additionally be influenced by the involvement of specific DNA repair mechanisms (Broeker et al., 1996). So far, most of the information on the involvement of DNA repair pathways originated from the mapping of fusion points and the analysis of rearranged sequences. In this way, Alu repeat-mediated homology-directed repair was shown to create partial tandem MLL duplications in a group of de novo AML patients (So et al., 1997; Strout et al., 1998). The corresponding Alu elements are present in MLL intron 1 and in the centromeric part of the MLLbcr, and consequently are not involved in therapy-associated rearrangements mostly affecting the telomeric part of the MLLbcr. On the other hand, collective data from several groups were interpreted such that non-homologous end-joining (NHEJ) is the major mechanism for the creation of translocations. Sequence analysis of intergenic fusions frequently lacked overlapping homologies or revealed micro-homologies (mini-direct repeats) rather than extended homologies, which pointed to misrepair by error-prone classical NHEJ or alternative microhomology-mediated end joining (MMEJ) mechanisms (Reichel et al., 2001; Whitmarsh et al., 2003; Le et al., 2009). These features were detected in infant and adult ALL as well as t-AL patients and consequently in centromeric and telomeric MLLbcr regions. More specifically, MLL-AF4 fusions in 40 ALL patients diagnosed with t(4;11) showed filler DNA of ≤21 nucleotides and mini-direct repeats of ≤7 nucleotides at the inter-chromosomal junctions in more than half of the cases. Additionally, duplications of ≤463 nucleotides of parental sequences, inversions of ≤267 nucleotides, and/or deletions of ≤4413 nucleotides were found (Reichel et al., 2001). Analysis of a t-AML case diagnosed with t(9;11) revealed a MLL-AF9 translocation fusing a micro-homologous TATTA sequence without gain or loss of any further nucleotides (Whitmarsh et al., 2003). Analysis of blood samples from breast cancer and lymphoma patients undergoing chemotherapeutic treatments identified MLL rearrangements with micro-homologous sequences of 2–8 bp in most of the junction sequences (Le et al., 2009). Even ex vivo flavonoid exposure of primary human CD34+ HSPCs was demonstrated to induce DSBs and translocations in the MLL intron 11/exon 12 region mostly via error-prone repair involving micro-homologies but in fewer cases via homology-directed repair at Alu-like sequences (van Waalwijk van Doorn-Khosrovani et al., 2007). Interestingly, in almost all presumed MMEJ cases repetitive LINE/L1 or SINE/Alu-like elements were detected adjacent to the breakpoints on at least one side and short palindromic sequences predicted to be formed. When Zinc Finger Nuclease technology was used to introduce cuts precisely within the MLL, detrimental genetic changes were only observed upon additional inhibition of the key NHEJ factor DNA-PK, which was accompanied by a rise in homology-directed activities (Do et al., 2012). This result suggested that clean breaks within the MLLbcr less frequently cause leukemogenic rearrangements, because they are quickly removed by classical NHEJ. Biochemical analysis indeed demonstrated association of DNA-PK with the MLLbcr following ionizing irradiation-induced breakage (Betti et al., 2001).
With the advance of technologies—starting from the analysis of cytogenetically defined karyotypes toward long-distance inverse PCR-based sequence analysis—detection of genomic changes has seen a dramatic sensitivity rise. Reflecting higher resolution, the spectrum of newly described MLLbcr rearrangements became larger in recent years. Previously, MLL was known to predominantly participate in reciprocal chromosomal translocations. More recently, non-reciprocal rearrangements resulting in insertions, inversions, and deletions were identified to account for a significant fraction of MLLbcr rearrangements. Complex rearrangements requiring at least tree DSBs and involving at least two chromosomes were observed to account for a fraction of up to 26% of MLL rearrangements. Complex rearrangements may result in three-way translocations, chromosomal translocations associated with deletions or with fragment insertions. In the most extreme case of complex chromosomal translocations many fusion alleles are generated during a single cell division in a process called chromothripsis (De Braekeleer et al., 2011; Cerveira et al., 2012; Meyer et al., 2013).
A major source of non-reciprocal complex rearrangements is break-induced replication, whereby replication fork collapse can be induced by ionizing irradiation or oncogenes followed by processing into one-ended DSBs, which are repaired by break-induced replication, a form of homology-directed repair, which involves replication template switching (Macheret and Halazonetis, 2015). Desai et al. (2014) demonstrated reliance of HSPCs, the cells-of-leukemia-origin, on homology-directed repair for DSB processing upon exit from quiescence. Thus, pushing HSPCs of homology-directed repair-deficient Exo1 mutant mice into the cell cycle by treatment with 5-Fluorouracil or poly-IC caused hypersensitivity to ionizing radiation. There is also evidence for MLLbcr rearrangements by the homology-directed repair mechanism homologous recombination, i.e., the DSB repair pathway considered to be most error-free. First, sequence analysis of partial tandem duplications of the MLL gene in AML patients suggested recombination between imperfectly homologous Alu sequences through generation of a heteroduplex fusion (Strout et al., 1998). Second, knockdown experiments of replication stress-induced MLLbcr rearrangements indicated involvement of RAD51 (Gole et al., 2014). Third, a pathogenic role of altered ATM function was proposed, when a germline missense ATM mutation was detected in the phosphatidylinositol-3 (PI-3) kinase coding region of a pediatric leukemia patient with MLL rearrangement (Oguchi et al., 2003). ATM deficiency was further shown to result in excessive binding of the DNA recombination protein RAD51 at the MLLbcr translocation breakpoint hotspot of 11q23 chromosome translocation after etoposide exposure. Binding of Replication protein A (RPA) and the chromatin remodeler INO80, which facilitate RAD51 loading on damaged DNA, to the hotspot were also increased by ATM deficiency. Thus, in addition to activating DNA damage signaling, ATM may avert chromosome translocations by preventing excessive loading of recombinational repair proteins onto translocation breakpoint hotspots (Sun et al., 2010). In this context it is of interest that reduced ATM levels were found in cycling human HSPCs as compared with cycling mature human peripheral blood lymphocytes (Kraft et al., 2015), providing one possible explanation for HSPC proneness to aberrant homologous recombination events at the MLLbcr.
Conclusions
Therapy-induced leukemia represents the most severe side effect of chemotherapy after successful treatment of the primary tumor. Evaluation of population-based data for the years 1975–2008 from nine US cancer registries revealed a t-AML incidence of 5.7% among the 29% of cancer patients who had received initial chemotherapy (Morton et al., 2013). During the observation period the t-AML risk rose among some cancer patients like non-Hodgkin lymphoma (NHL) patients, while it declined among others like ovarian carcinoma patients. Changes were explained by more frequent administration of subsequent therapies for persistent or relapsed NHL and a shift from an alkylating (melphalan) to a crosslinking (platinum) drug-based chemotherapy of ovarian carcinoma, documenting the influence of cumulative dose and the mode-of-action of the drug. Accordingly, attempts to optimize treatment modalities have focused on dose- and drug-adaptation in analogy to reduced-intensity conditioning regimens for patients requiring bone marrow transplantation (Myers and Davies, 2009). However, a recent study showed that the risk of secondary malignancies did not decline with reduced-intensity and reduced-toxicity conditioning regimens (Shimoni et al., 2013). Moreover, the benefits from modern medicine with a 3% annual increase of successfully treated cancer survivors, call our attention to the predicted rise in secondary malignancy risk (Maddams et al., 2011; Dong and Chen, 2014). Knowing that age is the most prominent cancer risk factor, the worldwide rise in longevity will further increase cancer incidences. Additionally, the hematopoietic system accumulates replication stress damage with age (Flach et al., 2014), which may rise the vulnerability for chemotherapy-induced MLLbcr rearrangements (Gole et al., 2014). So far, the benefits of chemotherapy significantly outweigh the risk of adverse effects (Dong and Chen, 2014). Novel targeted approaches such as PARP or mTOR inhibitor therapies inducing replication stress and EndoG nuclear translocation, respectively, may also bear a risk of MLLbcr destabilization (Gole et al., 2014). Hopes for better survival of patients with MLL-rearranged leukemia come from novel treatment regimens such as a triple immunotherapy targeting tumor-associated antigen and natural killer cell resistance (Chan et al., 2012). The challenge for the future will, however, be to identify markers for secondary leukemia risk and to develop compounds preventing t-AML for rational design of personalized combination therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We cordially thank Karen M. Vasquez, University of Texas, Austin, USA, for generating and sharing MLL DNA primary structure prediction data.
References
Abdulwahab, A., Sykes, J., Kamel-Reid, S., Chang, H., and Brandwein, J. M. (2012). Therapy-related acute lymphoblastic leukemia is more frequent than previously recognized and has a poor prognosis. Cancer 118, 3962–3967. doi: 10.1002/cncr.26735
Al-Sudairy, R., Al-Nasser, A., Alsultan, A., Al Ahmari, A., Abosoudah, I., Al-Hayek, R., et al. (2014). Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia in Saudi Arabia: a multi-institutional retrospective national collaborative study. Pediatr. Blood Cancer 61, 74–80. doi: 10.1002/pbc.24584
Aplan, P. D., Chervinsky, D. S., Stanulla, M., and Burhans, W. C. (1996). Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood 87, 2649–2658.
Atkinson, J., and McGlynn, P. (2009). Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492. doi: 10.1093/nar/gkp244
Berquist, B. R., and Wilson, D. M. III. (2012). Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 327, 61–72. doi: 10.1016/j.canlet.2012.02.001
Betti, C. J., Villalobos, M. J., Diaz, M. O., and Vaughan, A. T. (2001). Apoptotic triggers initiate translocations within the MLL gene involving the nonhomologous end joining repair system. Cancer Res. 61, 4550–4555.
Betti, C. J., Villalobos, M. J., Diaz, M. O., and Vaughan, A. T. (2003). Apoptotic stimuli initiate MLL-AF9 translocations that are transcribed in cells capable of division. Cancer Res. 63, 1377–1381.
Betti, C. J., Villalobos, M. J., Jiang, Q., Cline, E., Diaz, M. O., Loredo, G., et al. (2005). Cleavage of the MLL gene by activators of apoptosis is independent of topoisomerase II activity. Leukemia 19, 2289–2295. doi: 10.1038/sj.leu.2403966
Boehden, G. S., Restle, A., Marschalek, R., Stocking, C., and Wiesmüller, L. (2004). Recombination at chromosomal sequences involved in leukaemogenic rearrangements is differentially regulated by p53. Carcinogenesis 25, 1305–1313. doi: 10.1093/carcin/bgh092
Bolton, J. L., and Thatcher, G. R. (2008). Potential mechanisms of estrogen quinone carcinogenesis. Chem. Res. Toxicol. 21, 93–101. doi: 10.1021/tx700191p
Bousquet, M., Quelen, C., Rosati, R., Mansat-De Mas, V., La Starza, R., Bastard, C., et al. (2008). Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J. Exp. Med. 205, 2499–2506. doi: 10.1084/jem.20080285
Britton, S., Frit, P., Biard, D., Salles, B., and Calsou, P. (2009). ARTEMIS nuclease facilitates apoptotic chromatin cleavage. Cancer Res. 69, 8120–8126. doi: 10.1158/0008-5472.CAN-08-4400
Broeker, P. L., Super, H. G., Thirman, M. J., Pomykala, H., Yonebayashi, Y., Tanabe, S., et al. (1996). Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 87, 1912–1922.
Bueno, C., Catalina, P., Melen, G. J., Montes, R., Sánchez, L., Ligero, G., et al. (2009). Etoposide induces MLL rearrangements and other chromosomal abnormalities in human embryonic stem cells. Carcinogenesis 30, 1628–1637. doi: 10.1093/carcin/bgp169
Ceccaldi, R., Parmar, K., Mouly, E., Delord, M., Kim, J. M., Regairaz, M., et al. (2012). Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 11, 36–49. doi: 10.1016/j.stem.2012.05.013
Cerveira, N., Lisboa, S., Correia, C., Bizarro, S., Santos, J., Torres, L., et al. (2012). Genetic and clinical characterization of 45 acute leukemia patients with MLL gene rearrangements from a single institution. Mol. Oncol. 6, 553–564. doi: 10.1016/j.molonc.2012.06.004
Chan, W. K., Kung Sutherland, M., Li, Y., Zalevsky, J., Schell, S., and Leung, W. (2012). Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin. Cancer Res. 18, 6296–6305. doi: 10.1158/1078-0432.CCR-12-0668
Chen, Y., Kantarjian, H., Pierce, S., Faderl, S., O'Brien, S., Qiao, W., et al. (2013). Prognostic significance of 11q23 aberrations in adult acute myeloid leukemia and the role of allogeneic stem cell transplantation. Leukemia 27, 836–842. doi: 10.1038/leu.2012.319
Cheshier, S. H., Morrison, S. J., Liao, X., and Weissman, I. L. (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A. 96, 3120–3125. doi: 10.1073/pnas.96.6.3120
Cimino, G., Rapanotti, M. C., Biondi, A., Elia, L., Lo Coco, F., Price, C., et al. (1997). Infant acute leukemias show the same biased distribution of ALL1 gene breaks as topoisomerase II related secondary acute leukemias. Cancer Res. 57, 2879–2883.
Cowell, I. G., and Austin, C. A. (2012). Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health 9, 2075–2091. doi: 10.3390/ijerph9062075
Cowell, I. G., Sondka, Z., Smith, K., Lee, K. C., Manville, C. M., Sidorczuk-Lesthuruge, M., et al. (2012). Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. U.S.A. 109, 8989–8994. doi: 10.1073/pnas.1204406109
Cunningham, F., Amode, M. R., Barrell, D., Beal, K., Billis, K., Brent, S., et al. (2015). Ensembl 2015. Nucleic Acids Res. 43, D662-D669. doi: 10.1093/nar/gku1010
Daniel, J. A., and Nussenzweig, A. (2012). Roles for histone H3K4 methyltransferase activities during immunoglobulin class-switch recombination. Biochim. Biophys. Acta 1819, 733–738. doi: 10.1016/j.bbagrm.2012.01.019
Debatisse, M., Le Tallec, B., Letessier, A., Dutrillaux, B., and Brison, O. (2012). Common fragile sites: mechanisms of instability revisited. Trends Genet. 28, 22–32. doi: 10.1016/j.tig.2011.10.003
De Braekeleer, E., Meyer, C., Douet-Guilbert, N., Basinko, A., Le Bris, M. J., Morel, F., et al. (2011). Identification of MLL partner genes in 27 patients with acute leukemia from a single cytogenetic laboratory. Mol. Oncol. 5, 555–563. doi: 10.1016/j.molonc.2011.08.003
De Braekeleer, M., Morel, F., Le Bris, M. J., Herry, A., and Douet-Guilbert, N. (2005). The MLL gene and translocations involving chromosomal band 11q23 in acute leukemia. Anticancer Res. 25, 1931–1944.
de la Chica, R. A., Mediano, C., Salido, M., Espinet, B., Manresa, J. M., and Solé, F. (2011). Increased MLL gene rearrangements in amniocytes from fetuses of mothers who smoke. Leuk. Res. 35, 1066–1069. doi: 10.1016/j.leukres.2011.04.010
Desai, A., Qing, Y., and Gerson, S. L. (2014). Exonuclease 1 is a critical mediator of survival during DNA double strand break repair in nonquiescent hematopoietic stem and progenitor cells. Stem Cells 32, 582–593. doi: 10.1002/stem.1596
Do, T. U., Ho, B., Shih, S. J., and Vaughan, A. (2012). Zinc Finger Nuclease induced DNA double stranded breaks and rearrangements in MLL. Mutat. Res. 740, 34–42. doi: 10.1016/j.mrfmmm.2012.12.006
Dong, C., and Chen, L. (2014). Second malignancies after breast cancer: the impact of adjuvant therapy. Mol. Clin. Oncol. 2, 331–336. doi: 10.3892/mco.2014.250
Durkin, S. G., and Glover, T. W. (2007). Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192. doi: 10.1146/annurev.genet.41.042007.165900
Dvorak, P., Lysak, D., Vokurka, S., Michalova, K., Sarova, I., Jonasova, A., et al. (2014). The translocation t(2;11)(p21;q23) without MLL gene rearrangement–a possible marker of good prognosis in myelodysplastic syndrome patients. Hematol. Oncol. 32, 82–86. doi: 10.1002/hon.2089
Emerenciano, M., Koifman, S., and Pombo-de-Oliveira, M. S. (2007). Acute leukemia in early childhood. Braz. J. Med. Biol. Res. 40, 749–760. doi: 10.1590/S0100-879X2007000600002
Emerenciano, M., Meyer, C., Mansur, M. B., Marschalek, R., and Pombo-de-Oliveira, M. S. (2013). The distribution of MLL breakpoin ts correlates with outcome in infant acute leukaemia. Br. J. Haematol. 161, 224–236. doi: 10.1111/bjh.12250
Ensminger, M., Iloff, L., Ebel, C., Nikolova, T., Kaina, B., and Löbrich, M. (2014). DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 206, 29–43. doi: 10.1083/jcb.201312078
Ezoe, S. (2012). Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Environ. Res. Public Health 9, 2444–2453. doi: 10.3390/ijerph9072444
Faller, B. A., Robu, V. G., and Borghaei, H. (2009). Therapy-related acute myelogenous leukemia with an 11q23/MLL translocation following adjuvant cisplatin and vinorelbine for non-small-cell lung cancer. Clin. Lung Cancer 10, 438–440. doi: 10.3816/CLC.2009.n.082
Felix, C. A., Lange, B. J., Hosler, M. R., Fertala, J., and Bjornsti, M. A. (1995). Chromosome band 11q23 translocation breakpoints are DNA topoisomerase II cleavage sites. Cancer Res. 55, 4287–4292.
Fierz, B., Chatterjee, C., McGinty, R. K., Bar-Dagan, M., Raleigh, D. P., and Muir, T. W. (2011). Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 7, 113–119. doi: 10.1038/nchembio.501
Flach, J., Bakker, S. T., Mohrin, M., Conroy, P. C., Pietras, E. M., Reynaud, D., et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. doi: 10.1038/nature13619
Flynn, C. M., and Kaufman, D. S. (2007). Donor cell leukemia: insight into cancer stem cells and the stem cell niche. Blood 109, 2688–2691. doi: 10.1182/blood-2006-07-021980
Gale, K. B., Ford, A. M., Repp, R., Borkhardt, A., Keller, C., Eden, O. B., et al. (1997). Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl. Acad. Sci. U.S.A. 94, 13950–13954. doi: 10.1073/pnas.94.25.13950
Glover, T. W., Berger, C., Coyle, J., and Echo, B. (1984). DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 67, 136–142. doi: 10.1007/BF00272988
Glukhov, S. I., Rubtsov, M. A., Alexeyevsky, D. A., Alexeevski, A. V., Razin, S. V., and Iarovaia, O. V. (2013). The broken MLL gene is frequently located outside the inherent chromosome territory in human lymphoid cells treated with DNA topoisomerase II poison etoposide. PLoS ONE 8:e75871. doi: 10.1371/journal.pone.0075871
Gole, B., Baumann, C., Mian, E., Ireno, C. I., and Wiesmüller, L. (2014). Endonuclease G initiates DNA rearrangements at the MLL breakpoint cluster upon replication stress. Oncogene. doi: 10.1038/onc.2014.268. [Epub ahead of print].
Gué, M., Sun, J. S., and Boudier, T. (2006). Simultaneous localization of MLL, AF4 and ENL genes in interphase nuclei by 3D-FISH: MLL translocation revisited. BMC Cancer 6:20. doi: 10.1186/1471-2407-6-20
Hakim, O., Resch, W., Yamane, A., Klein, I., Kieffer-Kwon, K. R., Jankovic, M., et al. (2012). DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484, 69–74. doi: 10.1038/nature10909
Hars, E. S., Lyu, Y. L., Lin, C. P., and Liu, L. F. (2006). Role of apoptotic nuclease caspase-activated DNase in etoposide-induced treatment-related acute myelogenous leukemia. Cancer Res. 66, 8975–8979. doi: 10.1158/0008-5472.CAN-06-1724
Hensel, J. P., Gillert, E., Fey, G. H., and Marschalek, R. (2001). Breakpoints of t(4;11) translocations in the human MLL and AF4 genes in ALL patients are preferentially clustered outside of high-affinity matrix attachment regions. J. Cell. Biochem. 82, 299–309. doi: 10.1002/jcb.1161
Hertenstein, B., Hambach, L., Bacigalupo, A., Schmitz, N., McCann, S., Slavin, S., et al. (2005). Development of leukemia in donor cells after allogenic transplantation-a survey of the European Group of Blood and Marrow Transplantation (EBMT). Haematologica 90, 969–975.
Hess, J. L., Yu, B. D., Li, B., Hanson, R., and Korsmeyer, S. J. (1997). Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 90, 1799–1806.
Ireno, I. C., Baumann, C., Stöber, R., Hengstler, J. G., and Wiesmüller, L. (2014). Fluorescence-based recombination assay for sensitive and specific detection of genotoxic carcinogens in human cells. Arch. Toxicol. 88, 1141–1159. doi: 10.1007/s00204-014-1229-3
Kalinowska, M., Garncarz, W., Pietrowska, M., Garrard, W. T., and Widlak, P. (2005). Regulation of the human apoptotic DNase/RNase endonuclease G: involvement of Hsp70 and ATP. Apoptosis 10, 821–830. doi: 10.1007/s10495-005-0410-9
Khobta, A., Carlo-Stella, C., and Capranico, G. (2004). Specific histone patterns and acetylase/deacetylase activity at the breakpoint-cluster region of the human MLL gene. Cancer Res. 64, 2656–2662. doi: 10.1158/0008-5472.CAN-03-1126
Koontz, M. Z., Horning, S. J., Balise, R., Greenberg, P. L., Rosenberg, S. A., Hoppe, R. T., et al. (2013). Risk of therapy-related secondary leukemia in Hodgkin lymphoma: the Stanford University experience over three generations of clinical trials. J. Clin. Oncol. 31, 592–598. doi: 10.1200/JCO.2012.44.5791
Kowarz, E., Dingermann, T., and Marschalek, R. (2012). Do non-genomically encoded fusion transcripts cause recurrent chromosomal translocations? Cancers (Basel). 4, 1036–1049. doi: 10.3390/cancers4041036
Kowarz, E., Merkens, J., Karas, M., Dingermann, T., and Marschalek, R. (2011). Premature transcript termination, trans-splicing and DNA repair: a vicious path to cancer. Am. J. Blood Res. 1, 1–12.
Kraft, D., Rall, M., Volcic, M., Metzler, E., Groo, A., Stahl, A., et al. (2015). NF-κB-dependent DNA damage-signaling differentially regulates DNA double-strand break repair mechanisms in immature and mature human hematopoietic cells. Leukemia. doi: 10.1038/leu.2015.28. [Epub ahead of print].
Krawczyk, P. M., Borovski, T., Stap, J., Cijsouw, T., ten Cate, R., Medema, J. P., et al. (2012). Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 125, 2127–2133. doi: 10.1242/jcs.089847
Labi, V., Erlacher, M., Krumschnabel, G., Manzl, C., Tzankov, A., Pinon, J., et al. (2010). Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 24, 1602–1607. doi: 10.1101/gad.1940210
Le, H., Singh, S., Shih, S. J., Du, N., Schnyder, S., Loredo, G. A., et al. (2009). Rearrangements of the MLL gene are influenced by DNA secondary structure, potentially mediated by topoisomerase II binding. Genes Chromosomes Cancer 48, 806–815. doi: 10.1002/gcc.20685
Letessier, A., Millot, G. A., Koundrioukoff, S., Lachagès, A. M., Vogt, N., Hansen, R. S., et al. (2011). Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123. doi: 10.1038/nature09745
Libura, J., Ward, M., Solecka, J., and Richardson, C. (2008). Etoposide-initiated MLL rearrangements detected at high frequency in human primitive hematopoietic stem cells with in vitro and in vivo long-term repopulating potential. Eur. J. Haematol. 81, 185–195. doi: 10.1111/j.1600-0609.2008.01103.x
Liu, H., Takeda, S., Kumar, R., Westergard, T. D., Brown, E. J., Pandita, T. K., et al. (2010). Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 467, 343–346. doi: 10.1038/nature09350
Lovett, B. D., Lo Nigro, L., Rappaport, E. F., Blair, I. A., Osheroff, N., Zheng, N., et al. (2001). Near-precise interchromosomal recombination and functional DNA topoisomerase II cleavage sites at MLL and AF-4 genomic breakpoints in treatment-related acute lymphoblastic leukemia with t(4;11) translocation. Proc. Natl. Acad. Sci. U.S.A. 98, 9802–9807. doi: 10.1073/pnas.171309898
Macheret, M., and Halazonetis, T. D. (2015). DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 10, 425–448. doi: 10.1146/annurev-pathol-012414-040424
Maddams, J., Parkin, D. M., and Darby, S. C. (2011). The cancer burden in the United Kingdom in 2007 due to radiotherapy. Int. J. Cancer 129, 2885–2893. doi: 10.1002/ijc.26240
Meyer, C., Hofmann, J., Burmeister, T., Gröger, D., Park, T. S., Emerenciano, M., et al. (2013). The MLL recombinome of acute leukemias in 2013. Leukemia 27, 2165–2176. doi: 10.1038/leu.2013.135
Meyer, C., Kowarz, E., Hofmann, J., Renneville, A., Zuna, J., Trka, J., et al. (2009). New insights to the MLL recombinome of acute leukemias. Leukemia 23, 1490–1499. doi: 10.1038/leu.2009.33
Meyer, C., Kowarz, E., Schneider, B., Oehm, C., Klingebiel, T., Dingermann, T., et al. (2006). Genomic DNA of leukemic patients: target for clinical diagnosis of MLL rearrangements. Biotechnol. J. 1, 656–663. doi: 10.1002/biot.200600037
Mirault, M. E., Boucher, P., and Tremblay, A. (2006). Nucleotide-resolution mapping of topoisomerase-mediated and apoptotic DNA strand scissions at or near an MLL translocation hotspot. Am. J. Hum. Genet. 79, 779–791. doi: 10.1086/507791
Morton, L. M., Dores, G. M., Tucker, M. A., Kim, C. J., Onel, K., Gilbert, E. S., et al. (2013). Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975-2008. Blood 121, 2996–3004. doi: 10.1182/blood-2012-08-448068
Muntean, A. G., Tan, J., Sitwala, K., Huang, Y., Bronstein, J., Connelly, J. A., et al. (2010). The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 17, 609–621. doi: 10.1016/j.ccr.2010.04.012
Myers, K. C., and Davies, S. M. (2009). Hematopoietic stem cell transplantation for bone marrow failure syndromes in children. Biol. Blood Marrow Transplant. 15, 279–292. doi: 10.1016/j.bbmt.2008.11.037
Oguchi, K., Takagi, M., Tsuchida, R., Taya, Y., Ito, E., Isoyama, K., et al. (2003). Missense mutation and defective function of ATM in a childhood acute leukemia patient with MLL gene rearrangement. Blood 101, 3622–3627. doi: 10.1182/blood-2002-02-0570
Ohsato, T., Ishihara, N., Muta, T., Umeda, S., Ikeda, S., Mihara, K., et al. (2002). Mammalian mitochondrial endonuclease G. Digestion of R-loops and localization in intermembrane space. Eur. J. Biochem. 269, 5765–5770. doi: 10.1046/j.1432-1033.2002.03238.x
Ozeri-Galai, E., Lebofsky, R., Rahat, A., Bester, A. C., Bensimon, A., and Kerem, B. (2011). Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 43, 122–131. doi: 10.1016/j.molcel.2011.05.019
Pauklin, S., Sernández, I. V., Bachmann, G., Ramiro, A. R., and Petersen-Mahrt, S. K. (2009). Estrogen directly activates AID transcription and function. J. Exp. Med. 206, 99–111. doi: 10.1084/jem.20080521
Pavri, R., Gazumyan, A., Jankovic, M., Di Virgilio, M., Klein, I., Ansarah-Sobrinho, C., et al. (2010). Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143, 122–133. doi: 10.1016/j.cell.2010.09.017
Ploski, J. E., and Aplan, P. D. (2001). Characterization of DNA fragmentation events caused by genotoxic and non-genotoxic agents. Mutat. Res. 473, 169–180. doi: 10.1016/S0027-5107(00)00147-0
Pombo-de-Oliveira, M. S., Koifman, S., and Brazilian Collaborative Study Group of Infant Acute Leukemia. (2006). Infant acute leukemia and maternal exposures during pregnancy. Cancer Epidemiol. Biomarkers Prev. 15, 2336–2341. doi: 10.1158/1055-9965.EPI-06-0031
Pullarkat, V., Slovak, M. L., Dagis, A., Bedell, V., Somlo, G., Nakamura, R., et al. (2009). Acute leukemia and myelodysplasia after adjuvant chemotherapy for breast cancer: durable remissions after hematopoietic stem cell transplantation. Ann. Oncol. 20, 2000–2006. doi: 10.1093/annonc/mdp232
Quentin, S., Cuccuini, W., Ceccaldi, R., Nibourel, O., Pondarre, C., Pagès, M. P., et al. (2011). Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 117, e161–e170. doi: 10.1182/blood-2010-09-308726
Reichel, M., Gillert, E., Angermüller, S., Hensel, J. P., Heidel, F., Lode, M., et al. (2001). Biased distribution of chromosomal breakpoints involving the MLL gene in infants versus children and adults with t(4;11) ALL. Oncogene 20, 2900–2907. doi: 10.1038/sj.onc.1204401
Ruiz-Carrillo, A., and Renaud, J. (1987). Endonuclease G: a (dG)n X (dC)n-specific DNase from higher eukaryotes. EMBO J. 6, 401–407.
Samejima, K., and Earnshaw, W. C. (2005). Trashing the genome: the role of nucleases during apoptosis. Nat. Rev. Mol. Cell Biol. 6, 677–688. doi: 10.1038/nrm1715
Scharf, S., Zech, J., Bursen, A., Schraets, D., Oliver, P. L., Kliem, S., et al. (2007). Transcription linked to recombination: a gene-internal promoter coincides with the recombination hot spot II of the human MLL gene. Oncogene 26, 1361–1371. doi: 10.1038/sj.onc.1209948
Schmiegelow, K., Lausten Thomsen, U., Baruchel, A., Pacheco, C. E., Pieters, R., Pombo-de-Oliveira, M. S., et al. (2012). High concordance of subtypes of childhood acute lymphoblastic leukemia within families: lessons from sibships with multiple cases of leukemia. Leukemia 26, 675–681. doi: 10.1038/leu.2011.274
Schnyder, S., Du, N. T., Le, H. B., Singh, S., Loredo, G. A., and Vaughan, A. T. (2009). Estrogen treatment induces MLL aberrations in human lymphoblastoid cells. Leuk. Res. 33, 1400–1404. doi: 10.1016/j.leukres.2009.01.023
Shim, H., Chi, H. S., Jang, S., Seo, E. J., Park, C. J., Lee, J. H., et al. (2010). Therapy-related acute leukemia in breast cancer patients: twelve cases treated with a topoisomerase inhibitor. Korean J. Hematol. 45, 177–182. doi: 10.5045/kjh.2010.45.3.177
Shimoni, A., Shem-Tov, N., Chetrit, A., Volchek, Y., Tallis, E., Avigdor, A., et al. (2013). Secondary malignancies after allogeneic stem-cell transplantation in the era of reduced-intensity conditioning; the incidence is not reduced. Leukemia 27, 829–835. doi: 10.1038/leu.2012.299
Shivakumar, R., Tan, W., Wilding, G. E., Wang, E. S., and Wetzler, M. (2008). Biologic features and treatment outcome of secondary acute lymphoblastic leukemia–a review of 101 cases. Ann. Oncol. 19, 1634–1638. doi: 10.1093/annonc/mdn182
Sim, S. P., and Liu, L. F. (2001). Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. J. Biol. Chem. 276, 31590–31595. doi: 10.1074/jbc.M103962200
So, C. W., Ma, Z. G., Price, C. M., Dong, S., Chen, S. J., Gu, L. J., et al. (1997). MLL self fusion mediated by Alu repeat homologous recombination and prognosis of AML-M4/M5 subtypes. Cancer Res. 57, 117–122.
Stanulla, M., Chhalliyil, P., Wang, J., Jani-Sait, S. N., and Aplan, P. D. (2001). Mechanisms of MLL gene rearrangement: site-specific DNA cleavage within the breakpoint cluster region is independent of chromosomal context. Hum. Mol. Genet. 10, 2481–2491. doi: 10.1093/hmg/10.22.2481
Stanulla, M., Wang, J., Chervinsky, D. S., Thandla, S., and Aplan, P. D. (1997). DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Mol. Cell. Biol. 17, 4070–4079.
Storici, F., Bebenek, K., Kunkel, T. A., Gordenin, D. A., and Resnick, M. A. (2007). RNA-templated DNA repair. Nature 447, 338–341. doi: 10.1038/nature05720
Strick, R., Strissel, P. L., Borgers, S., Smith, S. L., and Rowley, J. D. (2000). Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. U.S.A. 97, 4790–4795. doi: 10.1073/pnas.070061297
Strick, R., Zhang, Y., Emmanuel, N., and Strissel, P. L. (2006). Common chromatin structures at breakpoint cluster regions may lead to chromosomal translocations found in chronic and acute leukemias. Hum. Genet. 119, 479–495. doi: 10.1007/s00439-006-0146-9
Strissel, P. L., Strick, R., Rowley, J. D., and Zeleznik-Le, N. J. (1998). An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood 92, 3793–3803.
Strissel, P. L., Strick, R., Tomek, R. J., Roe, B. A., Rowley, J. D., and Zeleznik-Le, N. J. (2000). DNA structural properties of AF9 are similar to MLL and could act as recombination hot spots resulting in MLL/AF9 translocations and leukemogenesis. Hum. Mol. Genet. 9, 1671–1679. doi: 10.1093/hmg/9.11.1671
Strout, M. P., Marcucci, G., Bloomfield, C. D., and Caligiuri, M. A. (1998). The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. U.S.A. 95, 2390–2395. doi: 10.1073/pnas.95.5.2390
Sugita, K., Taki, T., Hayashi, Y., Shimaoka, H., Kumazaki, H., Inoue, H., et al. (2000). MLL-CBP fusion transcript in a therapy-related acute myeloid leukemia with the t(11;16)(q23;p13) which developed in an acute lymphoblastic leukemia patient with Fanconi anemia. Genes Chromosomes Cancer 27, 264–269. doi: 10.1002/(SICI)1098-2264(200003)27:3<264::AID-GCC6>3.0.CO;2-#
Sun, J., Oma, Y., Harata, M., Kono, K., Shima, H., Kinomura, A., et al. (2010). ATM modulates the loading of recombination proteins onto a chromosomal translocation breakpoint hotspot. PLoS ONE 5:e13554. doi: 10.1371/journal.pone.0013554
Székvölgyi, L., Hegedüs, E., Molnár, M., Bacsó, Z., Szarka, K., Beck, Z., et al. (2006). Nick-forming sequences may be involved in the organization of eukaryotic chromatin into approximately 50 kbp loops. Histochem. Cell Biol. 125, 63–73. doi: 10.1007/s00418-005-0073-1
Takeda, S., Liu, H., Sasagawa, S., Dong, Y., Trainor, P. A., Cheng, E. H., et al. (2013). HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Invest. 123, 3154–3165. doi: 10.1172/JCI65566
Tamai, H., Yamaguchi, H., Hamaguchi, H., Yagasaki, F., Bessho, M., Kobayashi, T., et al. (2008). Clinical features of adult acute leukemia with 11q23 abnormalities in Japan: a co-operative multicenter study. Int. J. Hematol. 87, 195–202. doi: 10.1007/s12185-008-0034-2
Tamaichi, H., Sato, M., Porter, A. C., Shimizu, T., Mizutani, S., and Takagi, M. (2013). Ataxia telangiectasia mutated-dependent regulation of topoisomerase II alpha expression and sensitivity to topoisomerase II inhibitor. Cancer Sci. 104, 178–184. doi: 10.1111/cas.12067
Thompson, L. H., and Hinz, J. M. (2009). Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat. Res. 668, 54–72. doi: 10.1016/j.mrfmmm.2009.02.003
Trumpp, A., Essers, M., and Wilson, A. (2010). Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10, 201–209. doi: 10.1038/nri2726
Uckun, F. M., Herman-Hatten, K., Crotty, M. L., Sensel, M. G., Sather, H. N., Tuel-Ahlgren, L., et al. (1998). Clinical significance of MLL-AF4 fusion transcript expression in the absence of a cytogenetically detectable t(4;11)(q21;q23) chromosomal translocation. Blood 92, 810–821.
Vanhees, K., de Bock, L., Godschalk, R. W., van Schooten, F. J., and van Waalwijk van Doorn-Khosrovani, S. B. (2011). Prenatal exposure to flavonoids: implication for cancer risk. Toxicol. Sci. 120, 59–67. doi: 10.1093/toxsci/kfq388
van Waalwijk van Doorn-Khosrovani, B. S., Janssen, J., Maas, L. M., Godschalk, R. W., Nijhuis, J. G., and van Schooten, F. J. (2007). Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis 28, 1703–1709. doi: 10.1093/carcin/bgm102
Vaughan, A. T., Betti, C. J., and Villalobos, M. J. (2002). Surviving apoptosis. Apoptosis 7, 173–177. doi: 10.1023/A:1014374717773
Walter, D., Lier, A., Geiselhart, A., Thalheimer, F. B., Huntscha, S., Sobotta, M. C., et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552. doi: 10.1038/nature14131
Wang, G., Gaddis, S., and Vasquez, K. M. (2013). Methods to detect replication-dependent and replication-independent DNA structure-induced genetic instability. Methods 64, 67–72. doi: 10.1016/j.ymeth.2013.08.004
Whitmarsh, R. J., Saginario, C., Zhuo, Y., Hilgenfeld, E., Rappaport, E. F., Megonigal, M. D., et al. (2003). Reciprocal DNA topoisomerase II cleavage events at 5′-TATTA-3′ sequences in MLL and AF-9 create homologous single-stranded overhangs that anneal to form der(11) and der(9) genomic breakpoint junctions in treatment-related AML without further processing. Oncogene 22, 8448–8459. doi: 10.1038/sj.onc.1207052
Widlak, P., and Garrard, W. T. (2009). Roles of the major apoptotic nuclease-DNA fragmentation factor-in biology and disease. Cell. Mol. Life Sci. 66, 263–274. doi: 10.1007/s00018-008-8472-9
Wright, R. L., Slemmons, K. K., and Vaughan, A. T. (2014). Estradiol induces gene proximity and MLL-MLLT3 fusion in an AICDA-mediated pathway. Leuk. Lymphoma 17, 1–24. doi: 10.3109/10428194.2014.954112
Yahata, T., Takanashi, T., Muguruma, Y., Ibrahim, A. A., Matsuzawa, H., Uno, T., et al. (2011). Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 118, 2941–2950. doi: 10.1182/blood-2011-01-330050
Zámečníkova, A. (2011). Acquisition of mixed lineage leukemia rearrangement in a chronic myeloid leukemia patient while on imatinib. Hematol. Rep. 3:e13. doi: 10.4081/hr.2011.e13
Keywords: MLL breakpoint cluster region, infant acute leukemia, therapy-related leukemia, apoptosis, replication stress, error-prone DNA repair
Citation: Gole B and Wiesmüller L (2015) Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)—multiple rather than a single mechanism. Front. Cell Dev. Biol. 3:41. doi: 10.3389/fcell.2015.00041
Received: 24 April 2015; Accepted: 12 June 2015;
Published: 25 June 2015.
Edited by:
Ashok Kumar, University of Louisville, USAReviewed by:
Wen-Shu Wu, University of Illinois at Chicago, USAKaren M. Vasquez, The University of Texas at Austin, USA
Copyright © 2015 Gole and Wiesmüller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Wiesmüller, Division of Gynecological Oncology, Department of Obstetrics and Gynecology, Ulm University, Prittwitzstrasse 43, 89075 Ulm, Germany, lisa.wiesmueller@uni-ulm.de