Cardiotoxicity of Anticancer Drugs: Molecular Mechanisms and Strategies for Cardioprotection
 Marco Bruno Morelli1†
Marco Bruno Morelli1†  Chiara Bongiovanni2,3†
Chiara Bongiovanni2,3†  Silvia Da Pra2,3†
Silvia Da Pra2,3†  Carmen Miano2†
Carmen Miano2†  Francesca Sacchi2,3†
Francesca Sacchi2,3†  Mattia Lauriola3
Mattia Lauriola3  Gabriele D’Uva2,3*
Gabriele D’Uva2,3*
- 1Scientific and Technological Pole, IRCCS MultiMedica, Milan, Italy
- 2National Laboratory of Molecular Biology and Stem Cell Engineering, National Institute of Biostructures and Biosystems (INBB), Bologna, Italy
- 3Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Bologna, Italy
Chemotherapy and targeted therapies have significantly improved the prognosis of oncology patients. However, these antineoplastic treatments may also induce adverse cardiovascular effects, which may lead to acute or delayed onset of cardiac dysfunction. These common cardiovascular complications, commonly referred to as cardiotoxicity, not only may require the modification, suspension, or withdrawal of life-saving antineoplastic therapies, with the risk of reducing their efficacy, but can also strongly impact the quality of life and overall survival, regardless of the oncological prognosis. The onset of cardiotoxicity may depend on the class, dose, route, and duration of administration of anticancer drugs, as well as on individual risk factors. Importantly, the cardiotoxic side effects may be reversible, if cardiac function is restored upon discontinuation of the therapy, or irreversible, characterized by injury and loss of cardiac muscle cells. Subclinical myocardial dysfunction induced by anticancer therapies may also subsequently evolve in symptomatic congestive heart failure. Hence, there is an urgent need for cardioprotective therapies to reduce the clinical and subclinical cardiotoxicity onset and progression and to limit the acute or chronic manifestation of cardiac damages. In this review, we summarize the knowledge regarding the cellular and molecular mechanisms contributing to the onset of cardiotoxicity associated with common classes of chemotherapy and targeted therapy drugs. Furthermore, we describe and discuss current and potential strategies to cope with the cardiotoxic side effects as well as cardioprotective preventive approaches that may be useful to flank anticancer therapies.
Introduction
The introduction of antineoplastic drugs has been a turning point for prognosis improvement in oncology patients. However, a large number of chemotherapeutic agents have adverse cardiovascular effects, leading to acute or delayed onset of cardiac dysfunction, commonly referred to as cardiotoxicity. Although the definition of cardiotoxicity is not universally accepted, in clinical practice, cardiotoxicity commonly indicates a decline in patients’ cardiac function measured as left ventricle ejection fraction (LVEF). Various organizations and clinical committees defined cardiotoxicity using different threshold changes in LVEF [reviewed in (1)]. Treatment with anthracyclines, namely the chemotherapy class of drugs that generated the most concerns about cardiotoxicity, is associated with an incidence of cardiac dysfunction ranging between 2% and 48% [reviewed in (2–7)]. The Cardiac Review and Evaluation Committee (CREC), a retrospective study aiming at the evaluation of the cardiotoxicity of the anti-HER2 agent trastuzumab with or without concomitant anthracycline treatment, defined cardiotoxicity as a reduction in LVEF of at least 5% to below 55% with concomitant signs or symptoms of congestive heart failure (CHF), or a decrease in LVEF of at least 10% to below 55% without associated signs or symptoms (8). Although the assessment of LVEF is a well-established clinical procedure for the early recognition of cardiotoxic side effects to prevent irreversible cardiac damage and heart failure (HF), a reduction in LVEF may not be an effective parameter to detect a subclinical myocardial dysfunction that subsequently evolves in a symptomatic CHF (9) [reviewed in (1, 10)].
During the last decades, the cardiotoxic effects of several classes of chemotherapy drugs (anthracyclines, fluoropyrimidines, taxanes, and alkylating agents) and targeted therapies (targeting monoclonal antibodies and kinase inhibitors) were documented, and the underlying molecular mechanisms were investigated to suggest and develop potential strategies to avoid or reduce these effects (Table 1). Based on retrospective pathophysiological analysis of cancer patients with HF after chemotherapy, cardiotoxic side effects can be defined as irreversible (type I) or reversible (type II) [reviewed in (11, 12)]. Irreversible cardiotoxicity (type I) is usually observed in anticancer regimes causing injury and loss of cardiac myocytes. These effects are mainly observed after administration of anthracyclines and alkylating drugs, and to a lesser degree with fluoropyrimidines. According to the class of anticancer agents, the underlying mechanisms may involve cardiomyocyte-intrinsic and/or indirect mechanisms. For example, anthracyclines are associated with a high incidence of HF as consequence of irreversible cardiac damages through impairment of cardiomyocyte-intrinsic mechanisms leading to cell death [reviewed in (5, 7, 13–15)]. Despite administration of alkylating drugs and fluoropyrimidines may also cause cardiomyocyte death and thus irreversible cardiac damage, the main mechanism appears to be mediated by a vasculature dysfunction and/or thromboembolic ischemia. However, anticancer agents may also impair cardiomyocyte function without inducing cell death. This type of cardiac dysfunction is typically reversible and is associated with a lower incidence of HF (type II cardiotoxicity). Mechanistically, it has been suggested that reversible cardiotoxicity may be consequent to the deregulation of cardiomyocyte-intrinsic mechanisms and/or alteration of other cardiac populations and extracellular factors, in particular paracrine factors, in turn influencing cardiomyocyte function [reviewed in (4)]. Targeting monoclonal antibodies or tyrosine kinase inhibitors (TKIs) are typically associated with reversible cardiac damages, and their adverse effects derive by the signaling impairment of cardioprotective factors for cardiomyocytes, such as Neuregulin-1 (NRG1), or for other cardiac cell populations, such as vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) [reviewed in (13, 16)].
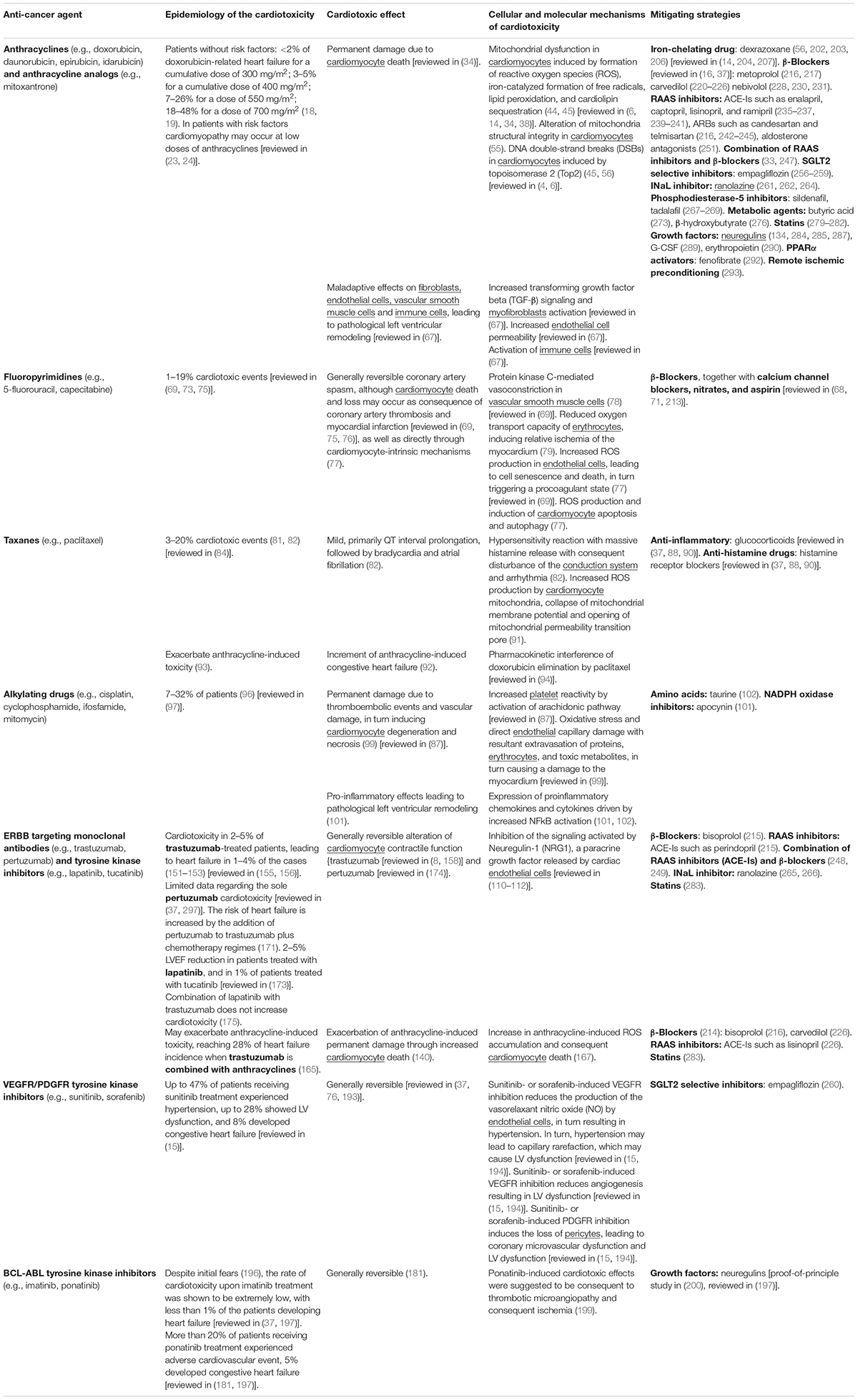
Table 1. Main features and mechanisms of cardiotoxic side effect of chemotherapies and targeted therapies along with mitigating strategies.
Importantly, the comprehension of different cellular and molecular mechanisms by which common classes of chemotherapy and targeted therapy drugs induce cardiotoxic effects is critical for developing efficient strategies for prevention, early detection, and treatment. Several therapeutical approaches have already been proposed to cope with the cardiotoxic side effects of anticancer therapies, including iron-chelating drugs, β-blockers, renin-angiotensin-aldosterone system inhibitors, sodium-glucose cotransporter-2 (SGLT2) inhibitors, late inward sodium current (INaL) selective inhibitors, phosphodiesterase-5 inhibitors, metabolic agents, statins, and growth factors. As future therapeutic goal, moving toward a protective chemoprevention approach, we need well-tolerated drugs that may flank chemotherapy to reduce clinical and subclinical cardiotoxic side effects, without interfering with the action of the antineoplastic treatments (17).
Cardiotoxicity Mechanisms Associated With Common Classes of Chemotherapy Drugs and Targeted Therapy
Chemotherapy Drugs
Anthracyclines
The anthracyclines, such as doxorubicin, daunorubicin, and epirubicin, are a class of broad-spectrum anticancer drugs extracted from Streptomyces bacterium. These compounds are used to treat different adult and pediatric hematologic cancers, such as leukemia and lymphomas, as well as many solid tumors, including breast, stomach, uterine, ovarian, bladder and lung cancers. However, anthracyclines are associated with a dose-dependent risk of cardiomyopathy and HF [reviewed in (2–7)]. Specifically, in the absence of risk factors, doxorubicin is tolerated up to a cumulative dose of 300 mg/m2, with a rate of HF of less than 2% (18). Retrospective studies have shown that an estimated 3–5% of patients, without other risk factors, would experience doxorubicin-related HF at a cumulative dose of 400 mg/m2, increasing at 7–26% and 18–48% for a dose of 550 and 700 mg/m2, respectively (18, 19). Based on these evident cardiotoxic effects, high-dose treatments with anthracycline are no longer administrated, but sub-acute and chronic cardiac effects are still a clinical problem. The use of second-generation analogs of doxorubicin, namely epirubicin or idarubicin, exhibits improvements in their therapeutic index, but the risks of inducing cardiomyopathy are not abated [reviewed in (6)]. Mitoxantrone, which is an anthracenedione, an anthracycline analog, can also damage the cardiac muscle cells, thus resulting in cardiac dysfunction (20) [reviewed in (21, 22)].
Importantly, a large body of evidence indicates that cardiomyopathy develops at lower doses of anthracyclines in the presence of risk factors, including hypertension, arrhythmias, coronary disease, combination with other anticancer agents as well as genetic predisposition to cardiotoxicity [reviewed in (17, 23, 24)]. In this regard, among the genetic factors increasing the susceptibility to anthracycline-induced cardiotoxic effects, the role of specific single-nucleotide polymorphisms (SNPs) is emerging [reviewed in (24, 25)]. Indeed, heritability analysis on multiple cell lines unveiled SNPs from 30 genes giving a greater predisposition to daunorubicin-induced cardiotoxicity (26). Specifically, several SNPs associated with anthracycline cardiotoxicity affect genes involved in anthracycline metabolism, transport, or downstream cytotoxic effects. For example, studies on pediatric cohorts enlightened polymorphisms in CBR1 and CBR3 genes (encoding for carbonyl reductases) associated with enhanced cardiotoxicity susceptibility in children with cancer (27), polymorphisms in ABCC1 and ABCC5 genes (encoding for ATP-binding cassette transporters) associated with increased anthracycline-induced cardiac dysfunction in acute lymphoblastic leukemia patients (28, 29), polymorphisms in SLC22A gene (encoding for a solute carrier) (30), as well as polymorphisms in genes playing a role in iron homeostasis (31), and others [reviewed in (25, 32)]. Oppositely, SNPs in endothelial nitric oxide synthase (NOS3) gene have been reported to be cardioprotective in patients upon a high dose of doxorubicin (29).
Cardiac injury after anthracycline administration occurs with every dose, as documented by the analysis of cardiac-biopsy specimens a few hours after a single dose of anthracycline [reviewed in (7)]. Although the vast majority (98%) of cases of anthracycline cardiotoxicity being detected within the first year after completing the treatment (33), anthracycline-induced cardiotoxicity can also manifest months to years after completing chemotherapy (33). From a pathophysiological point of view, anthracyclines were suggested to induce cardiotoxicity through cardiomyocyte-intrinsic mechanisms as well as other mechanisms involving other cardiac cell types (Figure 1). Importantly, anthracycline-induced cardiac damage may be permanent due to cardiomyocyte death through several biological processes, including apoptosis, autophagy, necrosis, necroptosis, pyroptosis, and ferroptosis [reviewed in (34)]. In this regard, the alteration of mitochondrial function and integrity emerged as a distinctive feature of anthracycline-induced cardiomyopathy [reviewed in (7, 34–39)]. Mitochondria network is well developed in the cardiac muscle, occupying 36–40% of the cardiomyocyte volume and producing around 90% of the cellular energy [reviewed in (40–43)]. Among the complex underlying molecular mechanisms involved in anthracycline-induced mitochondrial dysfunction is worth to mention the formation of reactive oxygen species (ROS), iron-catalyzed formation of free radicals, lipid peroxidation, and cardiolipin sequestering (44, 45) [reviewed in (6, 14, 34, 38)]. In this regard, in cardiac mitochondria, anthracyclines can be reduced by NAD(P)H-oxidoreductases and converted to unstable metabolites, such as doxorubicin-semiquinone radicals, which can react with molecular oxygen (O2), producing superoxide anion-free radicals and hydrogen peroxide ( and H2O2) (46) [reviewed in (37)]. ROS generated by anthracyclines affect the activity of many mitochondrial enzyme complexes, such as NOSs, NAD(P)H oxidases, catalase, and glutathione peroxidase (GPx), leading to DNA, protein, and lipid damage, and consequently to cardiomyocyte death [reviewed in (39, 47, 48)]. Moreover, anthracyclines, such as doxorubicin, have been reported to impair cardiac iron homeostasis, resulting in its overload in the cardiac tissue [reviewed in (14, 49, 50)]. Accordingly, patients with anthracycline-induced cardiac dysfunction exhibit higher iron levels in cardiac mitochondria, compared to healthy individuals or patients suffering from anthracycline-independent cardiac dysfunction (44). Doxorubicin can, in fact, chelate the free intracellular iron and form iron-doxorubicin complexes, which, in turn, are able to react with O2, further increasing the generation of ROS [reviewed in (4, 14, 49, 50)]. In addition, anthracyclines can directly interfere with the main iron-transporting/-binding proteins. For example, doxorubicin can impair cellular iron mobilization, resulting in its accumulation within ferritin (51), and can reduce the expression of the mitochondrial iron exporter ABCB8 (44). Recent studies have also focused on the detrimental role of mitochondrial iron-doxorubicin complexes triggering cardiomyocyte ferroptosis, a kind of programmed cell death dependent on iron and induced by lethal lipid peroxidation (52) [reviewed in (50)]. In this regard, doxorubicin-induced cardiotoxicity in mouse models was shown to be consequent to a decrease in the expression levels of glutathione peroxidase 4 (GPx4), which is a scavenger for lipid peroxides, in turn inducing peroxidation of unsaturated fatty acids and lipids (52). Anthracyclines are also linked to mitochondria damage because of their high affinity to cardiolipin, a mitochondrial membrane phospholipid that is involved in apoptotic pathways [reviewed in (35, 53)]. Mechanistically, doxorubicin sequesters cardiolipin avoiding its anchorage to cytochrome C or lipid-protein interfaces, thus contributing to mitochondrial dysfunction and ROS formation (54) [reviewed in (35, 53)]. Along with the impaired cardiac mitochondrial function, anthracyclines have been demonstrated to alter the structural integrity of mitochondria. Indeed, it has been reported that doxorubicin stimulates the receptor-interacting protein 3 (RIPK3)-induced activation of Ca2+-calmodulin-dependent protein kinase (CaMKII), thus triggering the opening of mitochondrial permeability transition pore (MTPT), and ultimately inducing necroptotic cardiomyocyte death (55).
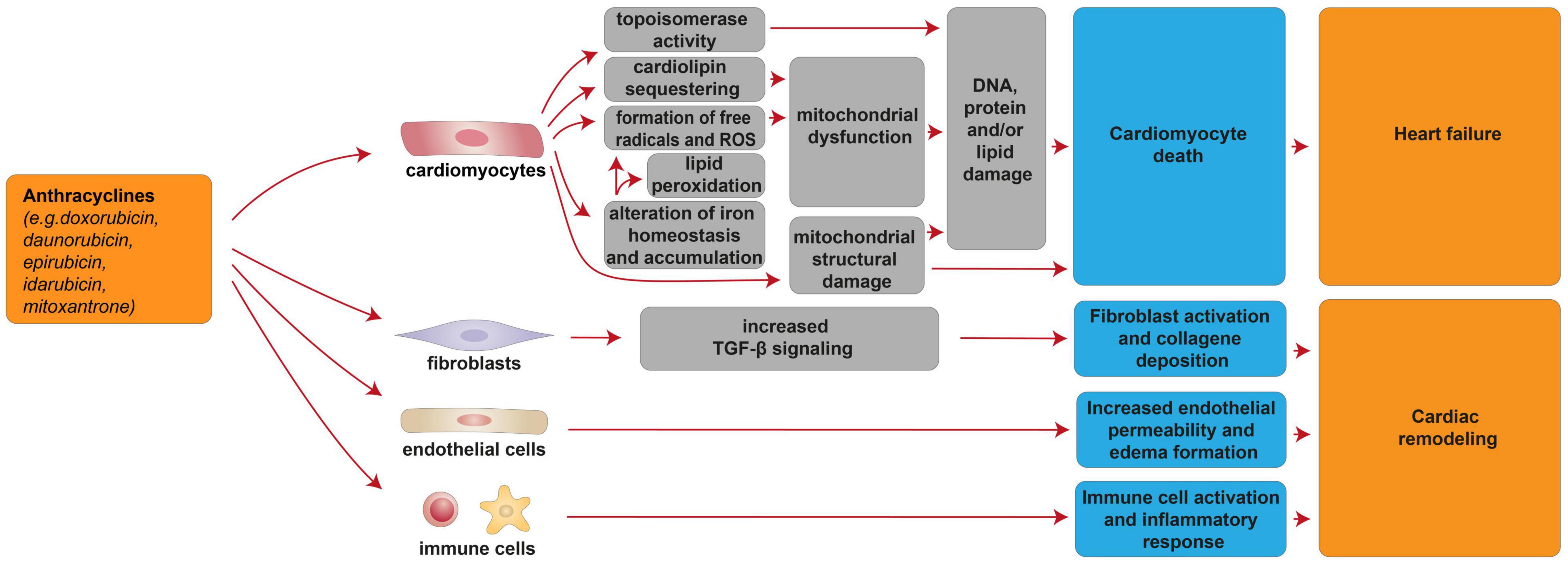
Figure 1. Cellular and molecular mechanisms of the cardiotoxic effects exerted by anthracyclines. Schematic diagram showing the impact of anthracyclines on a multitude of cardiomyocyte-intrinsic mechanisms leading to mitochondrial dysfunction and structural damage and/or DNA damage by topoisomerase activity, in turn leading to cardiomyocyte death and heart failure. Additional mechanisms of anthracycline-induced cardiotoxicity include deregulation of fibroblasts, endothelial, and immune cells, in turn concurring to cardiac remodeling.
Several lines of evidence have suggested that nuclear damage induced by topoisomerase 2 (Top2) is another pivotal event in anthracyclines’ cardiotoxic effects (45, 56) [reviewed in (4, 6)]. Specifically, doxorubicin intercalates into DNA and interacts with both Top2-alpha (Top2α) and Top2-beta (Top2β), which are enzymes responsible for managing DNA tangles and super-coils. Top2α is highly expressed in proliferating cancerous cells but not in quiescent tissues; therefore, it is considered one of the key molecular targets of anthracycline anti-tumoral effect (56). Cardiomyocyte toxicity stems from the fact that doxorubicin interacts with cardiac Top2-β, the only isoform expressed by adult mammalian cardiomyocytes. Consequently, the Top2β-doxorubicin-DNA complex induces DNA double-strand breaks (DSBs), ultimately promoting cardiomyocyte death (45, 56).
Tumor protein P53 (p53) has also been implicated in anthracyclines’ cardiotoxic response, although its involvement is currently controversial. Indeed, it has been reported that DNA breaks, induced by acute doxorubicin administration, lead to activation of the DNA damage response (DDR) network, in turn activating p53, which ultimately promotes the apoptotic cascade (57) [reviewed in (58)]. Moreover, in response to cell stress, p53 was shown to accumulate in the cytosol and to localize in mitochondria, triggering a series of death-events related to mitochondrial dysfunction, such as the permeabilization of the mitochondrial outer membrane (MOMP), the release of cytochrome C, the opening of the mitochondrial permeability transition pore (PTP), the impairment of mitochondria, and the production of ROS (59–63). Mice depleted for p53 exhibit a less impaired mitochondrial integrity and reduced cardiac dysfunction following doxorubicin treatment [reviewed in (5)]. In addition, doxorubicin-activated p53 has been shown to contribute to metabolic derangement by inhibiting mitophagy events (45) [reviewed in (4)]. As a result of cytosolic accumulation, p53 binds Parkin and abrogates its translocation to damaged mitochondria and their subsequent clearance by mitophagy (64) [reviewed in (5)]. These results support p53 as a key player in anthracycline-related cardiomyopathies (61, 62) [reviewed in (4)]. Nevertheless, other studies unveiled opposite effects depending on the dosage and timing of doxorubicin-induced cellular stress. Indeed, upon low doses of doxorubicin, which more closely recapitulate the clinical settings, it has been reported a protective role of p53, counteracting the late-onset cardiomyopathy and without activation of p53-dependent cell death cascades (65, 66).
In addition to cardiomyocyte-intrinsic mechanisms, anthracyclines exhibit a wide range of maladaptive effects on other cardiac populations, including fibroblasts, endothelial cells, vascular smooth muscle cells, and immune cells [reviewed in (67)]. In particular, doxorubicin administration was shown to increase endothelial cellular permeability, in turn causing edema formation [reviewed in (67)], to induce ROS-dependent activation of transforming growth factor beta (TGFβ) signaling, in turn triggering myofibroblast activation and collagen deposition [reviewed in (67)], and to induce the activation of the innate immune system and inflammatory response [reviewed in (67)]. Overall, these events were suggested to lead to pathological left ventricular remodeling [reviewed in (67)].
Fluoropyrimidines
Fluoropyrimidines exert the second most common cause of chemotherapy-induced cardiotoxicity [reviewed in (68–74)]. This antimetabolite drug class, which includes 5-fluorouracil (5-FU) and its prodrug capecitabine, is incorporated into DNA or RNA, thus acting as cytostatic agent for the clinical treatment of colorectal, breast, gastric, pancreatic, prostate, and bladder cancers [reviewed in (74)]. Fluoropyrimidines are generally well tolerated; nevertheless, 1–18% of the patients receiving fluoropyrimidines experiences cardiovascular toxicity [reviewed in (69–71, 73–75)]. Cardiovascular side effects associated with fluoropyrimidines include a generally reversible coronary artery spasm and myocardial ischemia, although cardiomyocyte death and loss may occur as consequence of coronary artery thrombosis and myocardial infarction [reviewed in (69, 75, 76)], as well as directly through cardiomyocyte-intrinsic mechanisms (77). These adverse effects were suggested to be mediated by vascular smooth muscle cells, erythrocytes, endothelial cells as well as directly by cardiomyocytes (Figure 2). From a molecular point of view, 5-FU was reported to induce protein kinase C-mediated vasoconstriction in vascular smooth muscle cells (78) [reviewed in (69)]. 5-FU was also shown to reduce the oxygen transport capacity of erythrocytes, inducing relative ischemia of the myocardium (79). 5-FU administration was also suggested to induce increased ROS production in endothelial cells, leading to cell senescence and death (77), in turn triggering a procoagulant state and acute thrombotic events [reviewed in (69)]. Finally, direct cardiomyocyte toxicity after fluoropyrimidine administration has also been suggested. Indeed, 5-FU has also been demonstrated to favor ROS production and to induce cardiomyocyte apoptosis and autophagy (77).
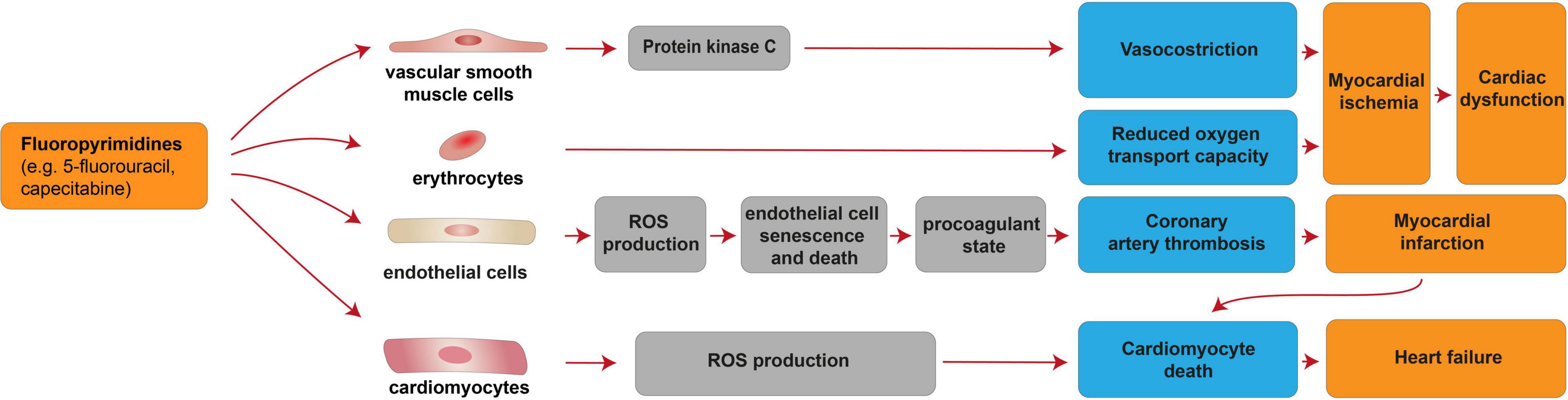
Figure 2. Cellular and molecular mechanisms of the cardiotoxic effects exerted by fluoropyrimidines. Schematic diagram showing the impact of fluoropyrimidines on cardiac dysfunction due to myocardial ischemia induced by deregulation of vascular smooth muscle cells and erythrocytes. Additional mechanisms of taxane-induced cardiotoxicity include heart failure, consequent to cardiomyocyte death induced by cardiomyocyte-intrinsic mechanisms (increased ROS production) or myocardial infarction consequent to coronary artery thrombosis caused by endothelial cell senescence and death.
Taxanes
Taxanes, such as paclitaxel, are antimitotic agents that stabilize microtubules in the mitotic spindle, thus blocking cell cycle progression. These chemotherapy drugs are widely employed in cancer treatment, including breast, lung, and ovarian cancers. However, significant toxicities limit the effectiveness of taxane-based treatment regimens (80). Taxane administration is reported to induce cardiotoxic events in 3–20% of the patients (81, 82) [reviewed in (83–85)]. Taxane-induced cardiotoxic effects include QT interval prolongation, followed by bradycardia and atrial fibrillation (82). Because taxane-induced cardiotoxicity appears to be mild in most cases and reversible upon discontinuation of the therapy, no specific agents are recommended for their management [reviewed in (86, 87)].
The underlying cellular and molecular mechanisms of taxane-induced cardiotoxicity are unclear; however, a few hypotheses have been proposed (Figure 3). Among them, hypersensitivity reaction with a massive histamine release and consequent disturbance of the conduction system and arrhythmia has been proposed (82). Hence, the administration of anti-inflammatory (glucocorticoids) and anti-histamine drugs (histamine receptor blockers), is suggested as prophylactic therapy for the management of cardiac anaphylaxis induced by taxanes [reviewed in (37, 88–90)]. Another hypothesis is cardiomyocyte damage through the drug’s actions on subcellular organelles (82). In this regard, taxanes were suggested to increase ROS production by cardiomyocyte mitochondria, the opening of mitochondrial permeability transition pore and the collapse of mitochondrial membrane potential (91).

Figure 3. Cellular and molecular mechanisms of the cardiotoxic effects exerted by taxanes. Schematic diagram showing the main cardiotoxic effects of taxanes, namely atrial fibrillation and cardiac dysfunction, as a result of the disturbance of the conduction system or cardiomyocyte dysfunction, respectively.
Among taxanes, paclitaxel has been shown to exacerbate anthracycline-induced toxicity. Indeed, combined treatment with paclitaxel and doxorubicin augmented HF events (92) and increased histopathological alterations of cardiac tissue, with extensive necrosis (93). This effect was suggested to derive from a pharmacokinetic interference of doxorubicin elimination by paclitaxel [reviewed in (94)]. No interaction between doxorubicin and other taxanes (such as docetaxel) has been reported; in line, docetaxel showed no increase in cardiac toxicity when combined with doxorubicin [reviewed in (94)].
Alkylating Drugs
Alkylating drugs, such as cisplatin, cyclophosphamide, ifosfamide, mitomycin, are crosslinking agents inducing ROS production, DNA damage and apoptosis in cancer cells [reviewed in (95)]. Cisplatin is mostly used in combination with other chemotherapy drugs to overcome drug-resistance and reduce toxicity [reviewed in (95)]. Cisplatin-based chemotherapy has been reported to cause cardiovascular diseases, particularly myocardial infarction and angina, in a range of 7–32% of patients (96) [reviewed in (97)]. In patients treated with cisplatin, a long-term unfavorable cardiovascular risk profile was observed, with hypercholesterolemia, hypertriglyceridemia, hypertension and insulin-resistance evaluated after more than 10 years from remission (98). The cardiotoxic effects of alkylating agents may be permanent and a few cellular and molecular mechanisms were suggested to contribute to these processes (Figure 4). Indeed, cisplatin administration has been linked with thromboembolic events associated with platelet aggregation and vascular damage (99) [reviewed in (87)], in turn resulting in cardiomyocyte degeneration and necrosis. The increased platelet aggregation was suggested as a direct consequence of cisplatin on the activation of the arachidonic pathway in platelets [reviewed in (87)]. The endothelial capillary damage was suggested to derive from a cisplatin-dependent increase in oxidative stress (99). Indeed, cisplatin has also been shown to induce oxidative stress in myocardial tissue, with decreased activity of glutathione and antioxidant enzymes (100, 101). The consequence of cisplatin-induced endothelial injury was suggested to be the extravasation of proteins, erythrocytes, and toxic metabolites, in turn causing damage to the myocardium (99). Finally, cisplatin has also been suggested to activate NF-κB in the cardiac tissue (101), in turn increasing the expression of proinflammatory chemokines and cytokines (102). This mechanism was proposed to result in cardiac remodeling (101), and extensive degeneration and fragmentation of cardiac muscle fibers (102).
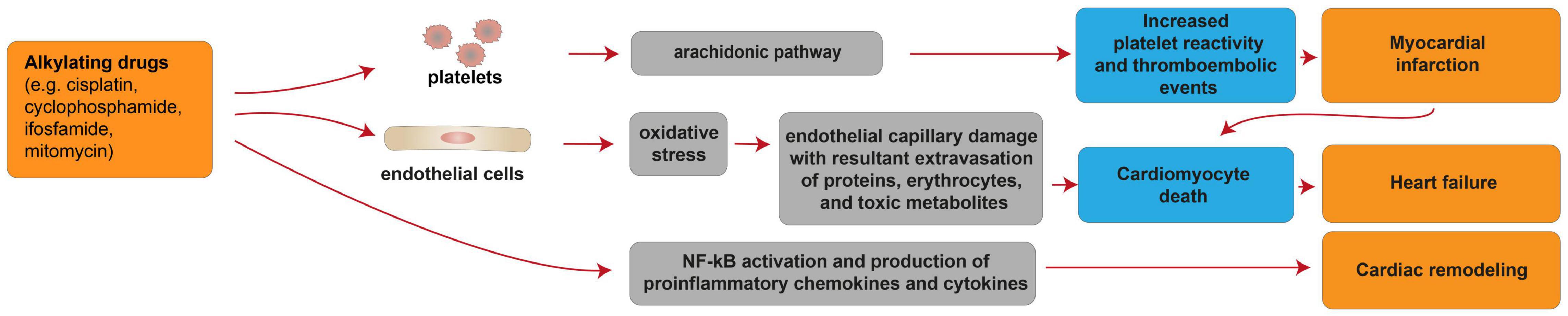
Figure 4. Cellular and molecular mechanisms of the cardiotoxic effects exerted by alkylating drugs. Schematic diagram showing the impact of alkylating agents in promoting heart failure due to cardiomyocyte death consequent to myocardial infarction. Additional mechanisms of alkylating drug-induced cardiotoxicity include heart failure consequent to cardiomyocyte death induced by oxidative stress and cardiac remodeling following activation of pro-inflammatory pathways.
The alkylating agent cyclophosphamide at high doses can cause hemorrhagic cell necrosis and may lead to HF; however, with the lower doses currently used, these side effects are infrequent (103).
Targeted Therapy
ERBB Targeted Therapies
Growth factor receptors of the ERBB family (EGFR/ERBB1, ERBB2, ERBB3, and ERBB4) play a key role in the development and progression of a variety of solid cancers [reviewed in (104–106)]. After the binding of soluble ligands, ERBB kinase receptors arrange in homo- or heterodimer complexes, which activate the tyrosine kinase activity and the consequent signaling events leading to the modulation of cell survival, proliferation, migration, and differentiation [reviewed in (104–107)]. ERBB2 (also known as HER2) receptor is a proto-oncogene frequently amplified and overexpressed in many human cancers. Unlike the other ERBB receptors, ERBB2 is unable to bind ligands but heterodimerizes with other ERBB receptors, stabilizing the ligand interaction with the coupled receptors, enhancing and diversifying the ligand-induced receptor signaling (108) [reviewed in (107)]. Several strategies have been developed to target the key role of ERBB2 signaling in tumor development and progression. Successful approaches are represented by treatment with humanized ERBB2-targeting antibodies (e.g., trastuzumab and pertuzumab) and tyrosine kinase multi-HER inhibitors (e.g., lapatinib, tucatinib, afatinib, neratinib, and dacomitinib), which effectively showed ERBB2 inhibition and tumor regression, particularly in the treatment of mammary carcinomas [reviewed in (109)].
The cardiotoxicity of ERBB2-directed therapeutics is consequent to the inhibition of the signaling activated by Neuregulin-1 (NRG1), a paracrine growth factor released by cardiac endothelial cells featuring pivotal functions in the heart (Figure 5) [reviewed in (110–112)]. NRG1, together with its tyrosine kinase receptors ERBB4, ERBB3, and ERBB2, is essential for heart development (113–115) [reviewed in (110, 116, 117)] and tunes heart regenerative, inflammatory, fibrotic, and metabolic processes (118, 119) [reviewed in (110, 117, 120–122)]. In cardiomyocytes, the most prominently expressed NRG1 receptors are ERBB4 and ERBB2 (123) and NRG1 stimulates fetal/neonatal cardiomyocyte proliferation, hypertrophy, sarcomerogenesis, and survival (114, 115, 124–127) [reviewed in (110, 116, 117, 120, 121)]. ERBB2 forms heterodimers with ERBB4 and is necessary for NRG1-elicited cardiomyocyte proliferation during embryonic and neonatal stages (122, 124). However, cardiac ERBB2 expression levels decline soon after birth in mice, as part of the mechanism leading to cardiomyocyte terminal differentiation, cell cycle withdrawal and loss of cardiac regenerative ability (124) [reviewed in (122)].

Figure 5. Cellular and molecular mechanisms of the cardiotoxic effect exerted by ERBB targeting monoclonal antibodies and tyrosine kinase inhibitors. Schematic diagram showing the impact of ERBB targeting therapies on cardiomyocyte dysfunction caused by the impairment of Neuregulin-1 signaling. However, in combination with anthracyclines, anti-HER2 monoclonal antibody trastuzumab may also induce heart failure as a consequence of cardiomyocyte death induced by ROS accumulation.
Despite low levels described in adulthood, ERBB2 appears to play a role in the prevention of dilated cardiomyopathy. Indeed, mice with ventricular-restricted deletion of ERBB2 exhibited multiple independent parameters of dilated cardiomyopathy, such as chamber dilatation, wall thinning, and decreased contractility (128). Decreased NRG1 signaling in postnatal life is associated with adverse cardiac function and susceptibility to stress [reviewed in (110, 116)]. The expression and activation of ERBB4 and ERBB2 receptors were found lower in myocardium from HF patients (129). In mice subjected to pressure overload, ERBB4 and ERBB2 undergo relevant reduction at mRNA and protein levels with the progression to HF (130).
Conversely, enhanced activity of NRG1 counteracts cardiac remodeling and HF progression [reviewed in (110, 116)]. Systemic administration of NRG1 improves cardiac function following various types of cardiac injuries in adult mice (115, 127, 131, 132) [reviewed in (110, 133)] and HF patients (117, 134–136) [reviewed in (137)].
Cardiac upregulation of ERBB2 was documented upon adverse hemodynamic or other stressful or toxic stimuli, including anthracycline therapies (138, 139). This increase is required to sustain cardiomyocyte survival and cardiac function under stress conditions. Indeed, cardiomyocytes isolated from mice with ventricular-restricted deletion of ERBB2 were more susceptible to anthracycline toxicity, revealing a role for ERBB2 in cardiomyocyte survival upon chemotherapy administration (128). Conversely, cardiac-specific overexpression of ERBB2 in mice has been shown to decrease cardiomyocyte death upon doxorubicin administration (140).
EGFR (also known as ERBB1) is associated with cancer progression and its inhibition via monoclonal antibodies (such as cetuximab and panitumumab) or TKIs (such as erlotinib and gefitinib) has been the first strategy evaluated among growth factor receptors targeting therapies (141, 142). Nowadays, EGFR inhibitors are clinically used for the treatment of several solid cancers, including lung, head and neck, colorectal, and pancreatic cancers (142). Although cetuximab-associated cardiotoxicity has been reported in the clinical literature, the incidence of cardiac events in patients remains very low (143, 144).
ERBB Targeting Monoclonal Antibodies
Trastuzumab, the first ERBB2-targeting humanized monoclonal antibody, binds the extracellular domain IV of ERBB2 receptor leading to the inhibition of ligand-independent heterodimerization between ERBB2 and other ERBB family members (145, 146) [reviewed in (105, 147)]. From a clinical perspective, the cardiotoxicity of monoclonal antibodies targeting ERBB2, such as trastuzumab, is moderate and reversible [reviewed in (148–150)]. Trastuzumab monotherapy is associated with cardiotoxicity in 2–5% of patients, leading to HF in 1–4% of the cases (151–153) [reviewed in (154–157)]. The mechanism of trastuzumab-induced cardiotoxicity appears to be the alteration of cardiomyocyte contractile function without cardiomyocyte death [reviewed in (8, 158)]. Interestingly, Erbb2 gene polymorphisms that alter the ERBB2 protein sequence have been identified, and two of them (Ile 655 Val and Pro 1170 Ala) were associated with an increased risk of cardiotoxicity from trastuzumab therapy (32, 159–164). Importantly, with the concomitant association of trastuzumab and anthracyclines, HF incidence increased to 28% (165, 166). Thus, trastuzumab-mediated blockade of ERBB2 signaling increases anthracycline-induced toxicity. The molecular mechanism underlying this combinatorial phenomenon may be due to the key role of ERBB2 in the management of oxidative stress in the heart: interrupting the neuregulin/ERBB2 axis, which is responsible for the activation of the glutathione reductase system, facilitates the anthracycline-induced accumulation of ROS and subsequent calcium influx, finally leading to caspase activation and cardiomyocyte death (167). Once anti-ERBB2 agents inhibit the ERBB2 protective mechanisms in cardiomyocytes, the doxorubicin oxidative damage was reported to increase (158) [reviewed in (37)].
Pertuzumab, a new generation of ERBB2-targeting therapies, is an antibody against domain II specifically designed to inhibit ligand-induced ERBB2 heterodimerization (168, 169). The data regarding the sole pertuzumab cardiotoxicity effects are still limited. Currently, combining trastuzumab/pertuzumab and trastuzumab/lapatinib, in order to induce a dual blockade of HER2, is part of the standard of care (170). In this regard, a recent study reporting a systematic review of eight randomized controlled trials showed that the risk of HF is increased by the addition of pertuzumab to trastuzumab plus chemotherapy therapeutic regimens (171).
ERBB Kinase Inhibitors
Tyrosine kinase inhibitors selectively target and inhibit several oncogenic relevant receptor-tyrosine kinases (RTKs), inducing survival benefits in therapies for various hematological and solid cancers [reviewed in (172)]. TKIs include single-targeted and multi-targeted TKIs. A small group of small TKIs, including lapatinib (ERBB2 and EGFR inhibitor), tucatinib (ERBB2 inhibitor), erlotinib (EGFR inhibitor), gefitinib (EGFR inhibitor), afatinib (EGFR, ERBB2, and ERBB4 inhibitor), neratinib (EGFR, ERBB2, and ERBB4 inhibitor), and dacomitinib (EGFR, ERBB2, and ERBB4 inhibitor), has been developed to target ERBB receptors. However, these ERBB blockers can also exert cardiac toxicity in treated patients. In particular, about 2–5% of patients treated with lapatinib displayed a reduced LVEF, and similar effects were reported in 1% of patients treated with tucatinib [reviewed in (173)]. The decline in cardiac function is generally reversible [reviewed in (174)]. Regarding combinatorial anti-ERBB strategies, little is known about the cardiotoxic potential of ERBB2 double blockade with trastuzumab plus lapatinib. Although stronger inhibition of the HER2 pathway using two anti-HER2 drugs was initially expected to result in greater impairment of cardiomyocytes, preclinical tests suggested a possible cardioprotective mechanism exerted by lapatinib. Adjuvant Lapatinib and/or Trastuzumab Treatment Optimisation (ALTTO), a randomized, multi-center, open-label, phase III study of adjuvant lapatinib plus trastuzumab treatment in patients with HER2/ERBB2 positive primary breast cancers (ClinicalTrials.gov, identifier NCT00490139), as well as other clinical trials with double ERBB2 blockade, support the safety of lapatinib plus trastuzumab treatment, since a lower, although not statistically significant, incidence of cardiac events was detected in patients in the trastuzumab plus lapatinib arm. This evidence does not imply that lapatinib has a cardioprotective effect, nor that it should be a preferred option for patients with an increased risk of cardiotoxicity (175).
Afatinib, an ERBB family blocker, approved for the first-line treatment of advanced non-small cell lung cancer (NSCLC) with EGFR mutations, is one of the few TKIs with a low risk of cardiotoxicity [reviewed in (176)]. Finally, cardiac side effects of the irreversible pan-ERBB inhibitor neratinib [reviewed in (177)] were reported neither in phase I clinical studies in solid tumors (178, 179) nor in a phase II trial in advanced HER2-positive breast cancer (179).
Multi-Targeted Tyrosine Kinase Inhibitors
In addition to single- or multi-targeted ERBB family inhibitors (see the previous paragraph), other multi-targeted TKIs were developed to effectively block multiple pathways of intracellular signal transduction. The broad kinase-signaling inhibition of several TKIs, such as sunitinib, sorafenib, imatinib, and nilotinib, includes the vascular endothelial growth factor receptors (VEGFRs), platelet-derived growth factor receptors (PDGFRs), BCR-ABL, and c-KIT. This wide action results in a strong anti-malignancy effect of this class of drugs, although correlated with reversible myocardial dysfunctions with a wide range of severity (180) [reviewed in (37, 181–183)]. Clinical analysis of TKI anti-tumoral therapies shows that compounds with broader off-target effects as kinases inhibitors (lower selectivity in targeting a specific kinase) correlated to higher degree of cardiotoxicity, particularly in case the inhibited kinase plays a role in the maintenance of the cardiovascular system (184–186) [reviewed in (37)]. In this regard, sunitinib, which targets VEGFR/PDGFR and interferes with more than 30 tyrosine kinases; sorafenib, which targets VEGFR/PDGFR and inhibits at least 15 tyrosine kinases, including RAF/MEK/ERK pathway, and ponatinib, which targets BCR-ABL and several other RTKs, are responsible for major clinical concerns related to cardiotoxicity (172) [reviewed in (37, 187, 188)]. Of note, these three compounds (sunitinib, sorafenib, and sonatinib) target VEGF, PDGFR, and c-Kit, namely three tyrosine kinase receptors involved in multiple key functions in the cardiovascular system, whose inhibition is likely the cause of the observed cardiotoxic effects (Figure 6). Particularly, sunitinib, which presents an effective multi-targeted inhibition of growth-factor receptors able to reduce the angiogenesis and tumor cell survival/proliferation (182, 189), is considered more cardiotoxic than other anti-angiogenic and TKI drugs (182). Based on clinical studies, 47% of patients receiving sunitinib treatment exhibited hypertension, up to 28% showed LV dysfunction, and 8% developed CHF [reviewed in (15)]. Patients with pre-existing cardiovascular diseases or previous cardio-toxicant exposure show even higher risks [reviewed in (190–192)]. However, cardiac dysfunctions induced by sunitinib and other inhibitors of tyrosine kinases have shown high reversibility; after treatment withdrawal, hypertension and cardiac dysfunction were alleviated or wholly restored (193) [reviewed in (37)]. Indeed, the majority of sunitinib-treated patients were able to carry on with sunitinib therapy following the resolution of cardiovascular events (193). Similarly, reversible cardiotoxicity has been reported upon sorafenib treatment [reviewed in (76)].
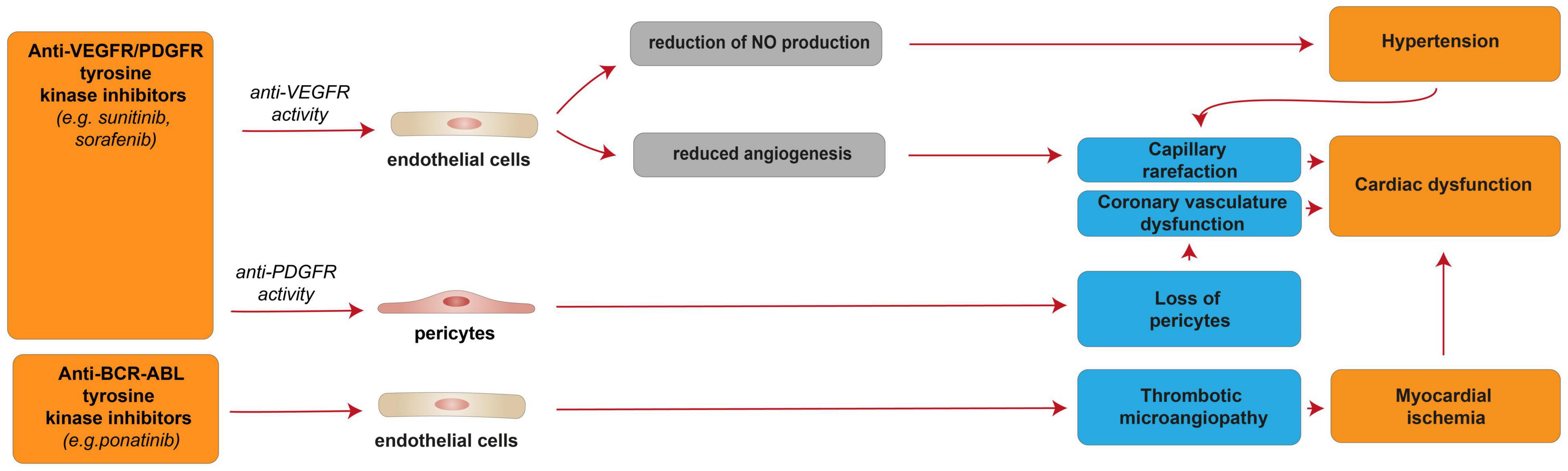
Figure 6. Cellular and molecular mechanisms of the cardiotoxic effects exerted by VEGFR/PDGFR and BCR-ABL and tyrosine kinase inhibitors. Schematic diagram showing the impact of VEGFR/PDGFR and BCR-ABL inhibition, resulting in a reversible cardiac disfunction. Anti-VEFGR activity impairs cardiac function by inducing capillary rarefaction consequent to reduced angiogenesis or hypertension derived from reduced NO production. Anti-PDGFR activity induces cardiac dysfunction by promoting the loss of pericytes, which in turn impairs the coronary vasculature. Anti-BCR-ABL inhibition may results in myocardial ischemia and cardiac dysfunction consequent to thrombotic microangiopathy.
The cellular and molecular details of the observed elevated blood pressure and cardiac dysfunction in patients treated with anti-VEGF/PDGFR drugs, such as sunitinib and sorafenib, are not fully understood. Nevertheless, sunitinib- and sorafenib-induced VEGFR inhibition was suggested to reduce the production of the vasodilator nitric oxide (NO) by endothelial cells, in turn resulting in hypertension [reviewed in (15, 194)]. Hypertension is known to lead to capillary rarefaction, which may be responsible for the cardiac dysfunction observed in sunitinib and sorafenib-treated patients [reviewed in (15)]. Indeed, given the high energy dependency, the heart is usually highly vulnerable to any altered blood supply. However, the capillary rarefaction potentially responsible for cardiac dysfunction may also be a direct consequence of reduced angiogenesis following sunitinib- or sorafenib-induced VEGFR inhibition [reviewed in (15, 194)]. Further, sunitinib- or sorafenib-induced PDGFR inhibition was suggested to induce the loss of pericytes, in turn leading to coronary microvascular dysfunction (195) [reviewed in (15, 194)]. Sunitinib, as an off-target effect, has also been suggested to inhibit AMPK activity, in turn inducing energy depletion in cardiomyocytes (184). However, another study found that sunitinib treatment in cardiomyocytes does not affect cellular ATP levels and that myocytes are not protected from sunitinib by pre-treatment with AMPK-activating drug metformin (189).
Imatinib, a TKI that inhibits BCR-ABL fusion protein, c-KIT, and PDGFR, is used to treat chronic myeloid leukemia and gastrointestinal stromal cancers. Despite initial fears (196), the rate of cardiotoxicity upon imatinib treatment was shown to be very low, with less than 1% of the patients developing HF [reviewed in (37, 197)]. Nevertheless, the inhibition of CaMKII in adult rat cardiac fibroblasts was shown to reduce the production of mitochondrial superoxide triggered by sunitinib and imatinib treatments (198).
Interestingly, ponatinib, a BCR-ABL kinase inhibitor developed to treat patients with imatinib resistance driven by T315I “gatekeeper” mutation, has been associated with a high rate of cardiovascular adverse events. Indeed, more than 20% of patients receiving ponatinib treatment experienced adverse cardiovascular events, and 5% developed CHF [reviewed in (181, 197)]. Of note, these cardiotoxic effects are often reversible with interruption of the therapy (181). The mechanisms of ponatinib-induced cardiotoxic effects are unclear; however, they were suggested to be consequent to thrombotic microangiopathy and consequent ischemia (Figure 6) (199), although cardiomyocyte death was also reported to occur in the zebrafish model (200).
Strategies to Reduce Anticancer Drug-Associated Cardiovascular Toxicity
Several therapeutical approaches already known in clinical usage have been proposed to reduce cardiotoxicities, such as iron-chelating drugs, β-blockers, renin-angiotensin-aldosterone system (RAAS) inhibitors, SGLT2 inhibitors, late inward sodium current (INaL) selective inhibitors, phosphodiesterase-5 inhibitors, metabolic agents, statins as well as growth factors and hormones [previously reviewed in (201)]. Here we will discuss these classes of drugs, focusing on their mechanisms of action and the therapeutic validity and effectiveness.
Iron-Chelating Drugs
The iron-chelating drug dexrazoxane has been identified as one of the most promising cardioprotective therapies in these last years and represents the only FDA-approved drug specific for anthracycline-induced cardiotoxicity (202, 203). Dexrazoxane is a pro-drug that rapidly turns into its active form after entering in cardiomyocytes, in turn counteracting the formation of anthracyclines-iron complexes and the subsequent adverse cardiac effects [reviewed in (204)]. Importantly, the development of iron-chelating drugs to prevent anthracycline-induced cardiotoxicity has emerged as an approach of relevant clinical importance in the context of the genetic predisposition of patients suffering from iron-related genetic disorders, such as hereditary hemochromatosis (31, 205). Initially, the cardioprotective functions of this iron chelator were ascribed majorly to its ability to affect iron regulatory proteins and reduce iron accumulation (206) [reviewed in (14, 207)]. However, additional mechanisms have been suggested to drive the cardioprotective activity exerted by dexrazoxane following anthracycline administration. Specifically, dexrazoxane has been shown to modify the topoisomerase 2 (Top2β) configuration preventing its interface with anthracyclines, thereby avoiding the Top2-DNA cleavage complexes (56, 208). Close derivatives of dexrazoxane lacking the interaction with Top2β were found not to be protective in relevant chronic anthracycline cardiotoxicity models (206, 209). Thus, cardioprotective effects of dexrazoxane in chronic anthracycline cardiotoxicity were suggested to derive from the inhibition of the interaction between anthracyclines and Top2β, rather than to its metal-chelating action (209) [reviewed in (210)].
Recently, a study on the cardioprotective effects of dexrazoxane, based on seven randomized trials and two retrospective trials for a total of 2177 patients with breast cancer receiving anthracyclines with or without trastuzumab reported that dexrazoxane reduces the risk of clinical HF and cardiac events in these patients without significantly impacting cancer outcomes (203). Thus, dexrazoxane represents a therapeutical strategy to limit anthracycline cardiotoxicity.
β-Blockers
β-blockers, also known as beta-adrenergic blocking agents, are a class of drugs that blocks the effects of the hormone epinephrine (adrenaline), causing the heart to beat more slowly and with less force, thus lowering blood pressure. These drugs are predominantly used to manage the reduction in left ventricular ejection fraction (LVEF), preventing symptomatic HF and protecting the heart from a second heart attack event after the first one (secondary prevention) [reviewed in (211, 212)]. The choice of β-blockers as a therapy for cardiac dysfunctions associated with anticancer drugs is mostly based on the dual cardioprotective role exerted by antihypertensive or antiarrhythmic drugs, which preserve cardiovascular function while inhibiting tumor angiogenesis [reviewed in (212)]. β-blockers, together with calcium channel blockers, nitrates, and aspirin are recommended for the management of fluoropyrimidines-induced cardiotoxicity as therapies for angina chest pain, albeit the absence of randomized controlled trials to support their efficacy [reviewed in (68, 71, 213)]. Furthermore, a large number of observations indicate β-adrenergic receptor signaling alterations as a feature of anthracycline-induced cardiomyopathy and in other forms of dilated cardiomyopathies [reviewed in (16, 37)]. A retrospective survey between 2005 and 2010 on 920 breast cancer patients who received anthracyclines and trastuzumab showed an association of continuous β-blocker treatment with a significantly lower incidence of HF events (214). Bisoprolol, another second-generation β-blocker, showed stronger efficacy compared to angiotensin-converting-enzyme inhibitor (ACE-I) perindopril in attenuating the LVEF decline in patients who received trastuzumab, even though it was unable to avoid left ventricular remodeling (215). However, administration of metoprolol, a second-generation β-blocker, did not affect LVEF decline determined by adjuvant, anthracycline-containing regimens with or without trastuzumab and radiation (216) and showed a non-statistically significant reduction in the incidence of anthracycline-induced HF events (217).
In in vitro and ex vivo set-up, carvedilol, a non-selective β- and α1-AR antagonist with strong antioxidant properties, reduced doxorubicin-induced ROS release and cardiomyocyte apoptosis (218) as well as mitochondrial respiration dysfunctions and calcium overloading (219). In rat models of doxorubicin-induced cardiomyopathy, carvedilol showed a significant cardioprotective effect, while atenolol, a β-blocker selective for β1-AR and without antioxidant properties, did not, thus suggesting that carvedilol cardioprotective efficacy relies more on its antioxidant properties than on the β-AR blocking action (220). In clinical trials of patients undergoing anthracycline chemotherapy, the prophylactic use of carvedilol decreased the ventricular dysfunction (221–223). In children receiving anthracyclines for acute lymphocytic leukemia, pre-treatment with carvedilol reduced troponin, diastolic dysfunction, and lactate dehydrogenase levels (224, 225). Furthermore, a randomized trial on 468 breast cancer patients treated with anthracyclines with/without trastuzumab showed reduced cardiotoxicity upon carvedilol administration, hence recommending carvedilol as a strategy to reduce trastuzumab interruptions (226).
Nebivolol is a cardio-selective β-blocker with mild vasodilating effects due to its interaction with the arginine/NO pathway [reviewed in (227)]. In isolated perfused rat hearts model of anthracycline-induced cardiotoxicity, treatment with nebivolol increased NO levels and significantly reduced oxidative stress, and improved cardiac function (228). Mechanistically, experiments in the rat model suggested that nebivolol administration reduces alterations in cardiomyocyte histomorphometry induced by doxorubicin through modulation of caspase-3, NO synthase (NOS), and TNF-α (229). In randomized placebo-controlled studies, the prophylactic use of nebivolol preserved the cardiac diastolic and systolic function from anthracycline-induced toxicity (230, 231).
To date, the cardioprotective efficacy of β-blockers needs to be further validated in large clinical trials. In addition, in clinical practice, the usage of β-blockers is hampered by their adverse effects in fragile patients, indicating their possible application only in patients with a high cardiotoxicity risk.
Renin-Angiotensin-Aldosterone System Inhibitors
Several studies showed that alteration of the RAAS has a crucial role in modulating anthracycline-induced cardiotoxicity [reviewed in (232)]. Therefore, the development of RAAS inhibitors, including ACE-Is, angiotensin receptor type 1 blockers (ARBs), as well as aldosterone antagonists, may be effective in the prevention and treatment of anthracycline-induced cardiotoxicity [reviewed in (232, 233)].
Angiotensin-converting-enzyme inhibitors, such as enalapril, captopril, lisinopril, and ramipril, impair the conversion of angiotensin I to angiotensin II, with a consequent decrease of angiotensin II receptor type 1 (AT1R) stimulation and its downstream signaling. These compounds have been demonstrated effective in the treatment of hypertension, as well as in reducing mortality in left ventricular dysfunction after myocardial infarction and CHF (234). Preclinical studies in animal models have demonstrated that ACE-Is, such as enalapril, captopril, and lisinopril, can effectively counteract the cardiotoxic effects after single high-dose, multiple low-doses or chronic exposure of anthracyclines (235–238). Mechanistically, ACE-Is’ therapy has been shown to result in the neutralization of ROS damage, reduction of interstitial fibrosis, limitation of intracellular calcium overload, along with improvement of mitochondrial respiration and cardiomyocyte metabolism (235, 236) [reviewed in (232)]. In retrospective clinical analysis, enalapril administration to doxorubicin-induced HF children increased cardiac hemodynamic parameters; however, these parameters declined after a few years (239). ACE-I therapy with ramipril or enalapril was also shown to induce the recovery of cardiac parameters in patients with doxorubicin-induced cardiac function decline (240). However, no significant improvement in exercise ability or contractile state of pediatric cancer patients receiving anthracyclines was also reported upon enalapril administration, albeit with reduction of left ventricular end-systolic wall stress (241). Clinical trials on HER2-positive breast cancer patients under anthracycline-trastuzumab therapy enlightened cardioprotective effects upon the administration of the ACE-I lisinopril (226).
Angiotensin receptor type 1 blockers, such as candesartan and telmisartan, inhibit angiotensin II binding to AT1R. In preclinical rat models, candesartan significantly reversed the daunorubicin-induced myocardial pathological changes and cardiac dysfunction (242). Candesartan administration was shown to significantly alleviate the decline in LVEF occurring during adjuvant, anthracycline-containing regimens with or without trastuzumab and radiation (216). Furthermore, in a small prospective study of 49 patients free from cardiovascular diseases and affected by solid cancers, telmisartan treatment starting before chemotherapy was able to reduce epirubicin-induced ROS damage by antagonizing the pro-inflammatory signals and reversing the early myocardial impairment (243). Telmisartan administration was also associated with long-lasting (up to 18 months) protection from early and acute myocardial dysfunction in patients treated with epirubicin (244, 245). In contrast, the administration of candesartan was unable to protect against the decrease in left ventricular ejection fraction during or shortly after trastuzumab treatment (246).
Importantly, clinical trials have also shown that a combination of ACE-Is or ARBs and β-blockers has beneficial effects in treating cardiotoxicity induced by anthracyclines and/or anti-HER2 agents. Indeed, the combination of ACE-Is (enalapril) and β-blockers significantly reduced the incidence of cardiac dysfunction along with prevention of the onset of late cardiotoxicity in patients receiving anthracyclines (33, 247). A small phase I trial conducted on 20 women suffering from breast cancer assessed the safety of continuing trastuzumab treatment despite cardiotoxicity onset if patients received ACE-Is and β-blockers following a staggered protocol (248). Another study unveiled the combination of ACE-Is, β-blockers and close cardiac monitoring as an effective strategy for cardioprotection in patients receiving HER2-targeted therapies (249).
Further studies focused on the cardioprotective role of aldosterone antagonists, which inhibit the last step of the RAAS and are already known for their beneficial effects on injury-induced cardiac remodeling and fibrosis [reviewed in (250)]. In a small clinical trial, spironolactone has been reported to prevent anthracycline-related cardiac dysfunction in breast cancer patients (251).
Sodium-Glucose Cotransporter-2 Inhibitors
Sodium-glucose cotransporter-2 selective inhibitors (empagliflozin, canagliflozin, and dapagliflozin) are a group of compounds that have been shown to have protective effects on the progression of HF [reviewed in (252)]. Indeed, EMPA-REG OUTCOME trial demonstrated that empagliflozin reduces major adverse cardiovascular events, cardiovascular death, and hospitalization rates for HF (253). Similarly, EMPEROR-Preserved trials found a reduced risk of HF hospitalization for 9718 patients with HF treated with empagliflozin (254). In a new systematic review meta-analysis of seven studies, for a total of 5,150 HF patients, empagliflozin was effective in reducing cardiovascular death or hospitalization for worsening HF condition (255). Therefore, SGLT2 inhibitors represent a promising treatment for chronic HF patients.
Recently, the potentially protective effects of SGLT2 inhibitors on the cardiac dysfunction induced by chemotherapies and targeted therapies were also investigated in preclinical studies in animal models. In this regard, protective effect by empagliflozin against anthracycline-induced cardiac impairment, diastolic dysfunction, and maladaptive cardiac remodeling has been documented (256–259). Mechanistically, empagliflozin was suggested to reduce ferroptosis, inflammatory response (NF-κB signaling), apoptosis, and fibrosis induced by doxorubicin through the involvement of NLRP3 and MyD88-related pathway (257, 258). A recent pre-clinical study reported that empagliflozin can also improve the cardiac dysfunction induced by anti-VEGFR/PDGFR multi-TKI sunitinib, via regulation of cardiomyocyte autophagy, in turn mediated by the AMPK-mTOR signaling pathway (260).
Late Inward Sodium Current Inhibitors
Selective inhibitors of late inward sodium current (INaL), such as ranolazine, have proven effective in treating experimental HF in several experimental models of cardiac dysfunction given its antiarrhythmic, anti-ischemic, and ATP-sparing features. Experimental evidence suggests that anthracyclines indirectly induce INaL hyperactivation, resulting in cytosolic calcium overload (261–263). INaL hyperactivation contributes to mitochondrial calcium depletion and dysregulation that, in turn, triggers mitochondrial ROS generation (oxidative stress), as well as NAD(P)H and ATP depletion (energetic stress); as a result, these events lead to cardiomyocyte impairment, diastolic dysfunction, and HF progression (261, 262, 264). Importantly, in animal models of doxorubicin-induced cardiotoxicity, ranolazine administration attenuated diastolic cardiac dysfunction and prevented worsening of systolic function by reducing oxidative stress and cardiomyocyte functional derangements (261, 262, 264). Moreover, ranolazine limited trastuzumab-induced cardiac dysfunction in mice by acting as a regulator of cardiac redox balance (265). In a very small randomized clinical study on 24 low-risk patients with diastolic dysfunction induced by anthracycline-based or fluoropyrimidine-/platinum-based therapies, patients were treated for 5 weeks with ranolazine or standard therapy, observing a complete recovery from diastolic dysfunction in all subjects in ranolazine group (12 patients) (266). Thus, the therapeutic use of this drug is promising, although needs validation in large clinical trials specific for each type of chemotherapy.
Phosphodiesterase-5 Inhibitors
Phosphodiesterase-5 inhibitors, such as sildenafil and tadalafil, were demonstrated to induce cardioprotective effects in animal models affected by doxorubicin cardiac toxicity (267–269). Sildenafil demonstrated cardioprotective activity against anthracycline-induced cardiac dysfunction by inducing the opening of mitochondrial KATP channels, leading to preserving mitochondrial potential and functions, myofibrillar integrity, and preventing cardiomyocyte apoptosis (267). The cardiac effects of sildenafil were also suggested to be dependent on the NO-signaling pathway since its protective activity was abolished by both L-NAME (inhibitor of NOS) and 5-hydroxydecanoate (inhibitor of ATP-sensitive K+ channels) (270). Tadalafil’s effects on cardiotoxicity reduction, instead, were suggested to be mainly due to NO-mediated increases of protein kinase G (PKG) activity and cGMP signaling, which is significantly reduced by doxorubicin administration (268, 269).
Metabolic Agents
Butyric acid, a short-chain fatty acid produced daily by the gut microbiota, has proven beneficial in models of cardiovascular diseases (271) [reviewed in (272)]. A novel butyric acid derivative, phenylalanine-butyramide (FBA), has been shown to protect animal models from doxorubicin-induced cardiotoxicity by decreasing oxidative stress and improving mitochondrial function (273). Of note, FBA prevented doxorubicin-induced cardiomyocyte apoptosis, left ventricular dilatation, and fibrosis (273).
Another metabolic agent, β-hydroxybutyrate (BHB), produced by fatty-acid oxidation in the liver under the fasting state, was shown to play a cardioprotective role in diabetic and HF with preserved ejection fraction (HFpEF) mouse models, when administrated as a dietary supplement or directly injected (274, 275). Interestingly, BHB was also reported to induce protection against anthracycline-induced cardiac function decline and partially reverted the maladaptive remodeling, characterized by increased cardiomyocyte size and decreased fibrosis (276). In vitro, BHB administration reduces oxidative stress and ameliorates mitochondrial functions, decreasing cardiomyocyte cell injury and apoptosis (276).
Statins
Statins reduce cholesterol synthesis by inhibiting the enzyme HMG CoA reductase. However, statins have emerged as pleiotropic factors playing a positive role on the cardiovascular system, including ROS production and oxidative stress, and the consequent cardiac mitochondrial dysfunction [reviewed in (277, 278)]. Importantly, the treatment of breast cancer patients undergoing anthracycline-based therapy with statins has been reported to be associated with a lower risk for HF and to prevent the decrease of the left ventricular ejection fraction (279–282). A similar cardioprotective activity of statins was reported for trastuzumab-based therapies (283).
Growth Factors
Administration of the growth factor Neuregulin-1 (NRG1β) has been shown to improve cardiac function following injury in adult mice (127, 134) [reviewed in (110, 133)] and in HF patients (135, 136) [reviewed in (137)]. Importantly, administration of NRG1β has also been shown to protect cardiac myocytes from anthracycline-induced apoptosis (134, 167, 284, 285) [reviewed in (286)]. Further, NRG1 administration in the zebrafish model was reported to reduce cardiomyocyte apoptosis induced by the multi-TKI ponatinib (200) [reviewed in (197)]. However, NRG1β is not clinically relevant as a therapy for cardiomyopathy induced by anticancer drugs because of its well-established cancer-promoting role. To solve this issue, an engineered bivalent NRG1 (NN), which preferentially induces ERBB4 homo-dimer formation in cardiomyocytes, has been developed and shown to protect against doxorubicin-induced cardiotoxicity, maintaining the same cardioprotective properties of NRG1 but with reduced pro-neoplastic potential (287). Nevertheless, although up to now there is no evidence in the literature about detrimental side effects in response to bivalent NRG1, NN has not been recruited into a clinical trial yet. Further studies are therefore recommended to assess if this combinatorial treatment is sufficient to mitigate the cardiotoxic side effects of chemotherapeutic agents.
Granulocyte colony-stimulating factor (G-CSF) is a hematopoietic growth factor that affects proliferation and differentiation, especially of progenitors of the neutrophil and granulocyte lineages, therefore it is currently used clinically in combination with doxorubicin to counteract doxorubicin-induced myelosuppression (288). Interestingly, a role for G-CSF has also been suggested in doxorubicin-induced cardiomyopathy. Indeed, an attenuation of cardiomyocyte atrophic degeneration and a decrease of myocardial fibrosis have been reported after G-CSF administration in doxorubicin-treated mice (289). Intriguingly, G-CSF was suggested to exert an anti-atrophic and anti-inflammatory activity directly on cardiomyocytes (289).
Among stromal cells, the beneficial role of endothelial progenitor cells (EPCs) has emerged to counteract the cardiotoxicity of cancer therapies. For example, erythropoietin (EPO) has been shown to promote angiogenesis by increasing the number of EPCs, thereby improving cardiac function after doxorubicin treatment (290).
Other Strategies
A few other strategies were suggested to reduce the adverse cardiovascular side effects of common chemotherapies and targeted therapies. In this regard, the sulfur-containing amino acid taurine (2-aminoethanesulfonic acid) has been shown to exert beneficial effects in CHF, ischemic heart disease, hypertension, atherosclerosis, and diabetic cardiomyopathy (291). Intriguingly, taurine was also shown to reduce cisplatin-induced cardiotoxicity by suppressing the generation of ROS, ER stress, and inflammation (102). Apocynin, a specific NADPH oxidase inhibitor, has been shown to reduce cisplatin-induced oxidative stress, inflammation and apoptosis (101).
Preclinical studies demonstrated that fenofibrate, a PPARα activator, counteracted doxorubicin-induced cardiotoxicity in mice by increasing circulating EPCs, stimulating cardiac NO activation and inducing the production of pro-angiogenic factors such as SDF-1 and VEGF (292).
Besides molecular strategies, remote ischemic preconditioning (RIPC), which consists of reversible repetitive interruptions in blood flow, ischemia, and reperfusion, seems a good approach to reduce anthracycline-induced cardiotoxicity (293). Indeed, large animals, subjected to RIPC before each doxorubicin injection, have shown a preserved cardiac contractility and mitochondrial integrity, concomitantly with a higher cardiac performance and reduced fibrosis (293).
Conclusion and Future Perspectives
Although anticancer therapies greatly improve survival and quality of life of oncological patients, their negative impact on cardiac well-being is a very critical issue. In addition to common risk factors, such as age, hypertension, arrhythmias, and coronary disease, it has emerged the identification of genetic variants related to an increased predisposition to cardiotoxicity of chemotherapies and targeted therapies, in particular for anthracyclines and anti-HER2 therapies. Thus, the development of individualized treatments, based on the forecast of the cardiotoxic side effects, may acquire a considerable clinical relevance for the future perspective. Importantly, the cellular and molecular mechanisms mediating the cardiotoxicity of common classes of chemotherapy and targeted therapy drugs are emerging, providing a rationale for the development of novel strategies for cardioprotection. Recent clinical trials have tested multiple cardioprotective drugs, highlighting the ability of some of them in counteracting or limiting the cardiotoxic effects of anticancer treatments. However, many of these therapeutic strategies still have certain limits and need some precautions. Among them, the lack of validation in large clinical trials, the underlying molecular mechanisms still not fully understood, as well as the risk-benefit controversies. In this regard, it is extremely important to take into account the tolerability of the adverse effects that these therapies may entail, including fatigue and dizziness, in patients already fatigued by antitumoral therapy.
Despite multiple cellular and molecular mechanisms being suggested to mediate the cardiotoxic effect of anti-cancer drugs, cardiomyocyte death has emerged as the major cause of long-term irreversible cardiac disfunction. These important side effects have been documented for anthracyclines, fluoropyrimidines, and alkylating drugs. This is because lost cardiomyocytes cannot be efficiently regenerated due to the very low ability of the adult mammalian heart to produce new cardiomyocytes (294, 295) [reviewed in (296)]. Although the cytotoxic effect of anticancer treatments resides on a wide range of biological mechanisms, the development of strategies aiming at increasing cardiomyocyte survival is thus encouraged to reduce anticancer drug-induced cardiomyocyte death and the consequent permanent damage. In the future, the administration of cardiomyocyte survival factors flanking chemotherapy and targeted therapies should be further explored. In this regard, a plethora of factors and signaling pathways has been shown to trigger endogenous cardiomyocyte proliferation for cardiac regenerative strategies [reviewed in (296)], thus their modulation may be also explored for cancer patients with permanent damage by anticancer drugs. Some of these factors also regulate cardiomyocyte survival, thus their modulation may be tested as a preventive strategy to reduce permanent cardiotoxic effect of anticancer drugs. Obviously, the potential interfering with the action of the antineoplastic treatments should be carefully evaluated.
In conclusion, cardiovascular adverse effects resulting from antineoplastic therapies are important concerns for the health of cancer patients and could question the choice of undertaking or interrupting treatments. Nowadays, some drugs have been clinically tested to counteract the cardiotoxicity related to anticancer care, and we here propose a further evaluation of factors that up to now are mainly known for their role in cardiomyocyte proliferation and survival, as promising strategies for protection and/or regeneration of the cardiac tissue. Moreover, an increasing synergistic effort would be required for the oncologic and cardiologic research fields to assure cancer patients a long-term relapse-free survival and high-quality cardiovascular health.
Author Contributions
All authors significantly contributed to the writing of the manuscript.
Funding
This project was supported by the Fondazione Cariplo to GD’U and ML (Grant Number: GR 2017-0800) and by Ministry of Health – Ricerca Corrente – IRCCS MultiMedica.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lambert J, Thavendiranathan P. Controversies in the definition of cardiotoxicity: do we care? – American college of cardiology. Am Coll Cardiol. (2016). Available online at: https://www.acc.org/latest-in-cardiology/articles/2016/07/07/14/59/controversies-in-the-definition-of-cardiotoxicity (accessed February 13, 2022).
2. Cardinale D, Iacopo F, Cipolla CM. Cardiotoxicity of anthracyclines. Front Cardiovasc Med. (2020) 7:26. doi: 10.3389/fcvm.2020.00026
3. Menna P, Salvatorelli E. Primary prevention strategies for anthracycline cardiotoxicity: a brief overview. Chemotherapy. (2017) 62:159–68. doi: 10.1159/000455823
4. Mercurio V, Pirozzi F, Lazzarini E, Marone G, Rizzo P, Agnetti G, et al. Models of heart failure based on the cardiotoxicity of anticancer drugs. J Card Fail. (2016) 22:449–58. doi: 10.1016/j.cardfail.2016.04.008
5. Menna P, Paz OG, Chello M, Covino E, Salvatorelli E, Minotti G. Anthracycline cardiotoxicity. Expert Opin Drug Saf. (2012) 11(Suppl. 1):S21–36. doi: 10.1517/14740338.2011.589834
6. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. (2004) 56:185–229. doi: 10.1124/pr.56.2.6
7. Sawyer DB. Anthracyclines and heart failure. N Engl J Med. (2013) 368:1154–6. doi: 10.1056/NEJMCIBR1214975
8. Seidman A, Hudis C, Pierri MK, Shak S, Paton V, Ashby M, et al. Cardiac dysfunction in the trastuzumab clinical trials experience. J Clin Oncol. (2002) 20:1215–21. doi: 10.1200/JCO.2002.20.5.1215
9. Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American society of echocardiography and the European association of cardiovascular imaging. Eur Hear J Cardiovasc Imaging. (2014) 15:1063–93. doi: 10.1093/ehjci/jeu192
10. Vaduganathan M, Prasad V. Cardiovascular risk assessment in oncological clinical trials: is there a role for centralized events adjudication? Eur J Heart Fail. (2016) 18:128–32. doi: 10.1002/ejhf.457
11. Ewer MS, Lippman SM. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J Clin Oncol. (2005) 23:2900–2. doi: 10.1200/JCO.2005.05.827
12. Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J. (2013) 34:1102–11. doi: 10.1093/eurheartj/ehs181
13. Ky B, Vejpongsa P, Yeh ETH, Force T, Moslehi JJ. Emerging paradigms in cardiomyopathies associated with cancer therapies. Circ Res. (2013) 113:754–64. doi: 10.1161/CIRCRESAHA.113.300218
14. Gammella E, Maccarinelli F, Buratti P, Recalcati S, Cairo G. The role of iron in anthracycline cardiotoxicity. Front Pharmacol. (2014) 5:25. doi: 10.3389/fphar.2014.00025
15. Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. (2014) 3:e000665. doi: 10.1161/JAHA.113.000665
16. Molinaro M, Ameri P, Marone G, Petretta M, Abete P, Di Lisa F, et al. Recent advances on pathophysiology, diagnostic and therapeutic insights in cardiac dysfunction induced by antineoplastic drugs. Biomed Res Int. (2015) 2015:138148. doi: 10.1155/2015/138148
17. Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. (2010) 102:14–25. doi: 10.1093/jnci/djp440
18. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79. doi: 10.1002/cncr.11407
19. Von Hoff DD, Layard MW, Basa P, Davis HL, Von Hoff AL, Rozencweig M, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. (1979) 91:710–7. doi: 10.7326/0003-4819-91-5-710
20. Shaikh AY, Suryadevara S, Tripathi A, Ahmed M, Kane JL, Escobar J, et al. Mitoxantrone-induced cardiotoxicity in acute myeloid leukemia-a velocity vector imaging analysis. Echocardiography. (2016) 33:1166–77. doi: 10.1111/echo.13245
21. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JAP, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. (2016) 90:2063–76. doi: 10.1007/s00204-016-1759-y
22. Saletan S. Mitoxantrone: an active, new antitumor agent with an improved therapeutic index. Cancer Treat Rev. (1987) 14:297–303. doi: 10.1016/0305-7372(87)90021-1
23. Qiu S, Zhou T, Qiu B, Zhang Y, Zhou Y, Yu H, et al. Risk factors for anthracycline-induced cardiotoxicity. Front Cardiovasc Med. (2021) 8:736854. doi: 10.3389/fcvm.2021.736854
24. Tromp J, Steggink LC, Van Veldhuisen DJ, Gietema JA, van der Meer P. Cardio-oncology: progress in diagnosis and treatment of cardiac dysfunction. Clin Pharmacol Ther. (2017) 101:481–90. doi: 10.1002/cpt.614
25. Chang VY, Wang JJ. Pharmacogenetics of chemotherapy-induced cardiotoxicity. Curr Oncol Rep. (2018) 20:52. doi: 10.1007/s11912-018-0696-8
26. Duan S, Bleibel WK, Huang RS, Shukla SJ, Wu X, Badner JA, et al. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. (2007) 67:5425–33. doi: 10.1158/0008-5472.CAN-06-4431
27. Blanco JG, Sun C-L, Landier W, Chen L, Esparza-Duran D, Leisenring W, et al. Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes–a report from the children’s oncology group. J Clin Oncol. (2012) 30:1415–21. doi: 10.1200/JCO.2011.34.8987
28. Semsei AF, Erdelyi DJ, Ungvari I, Csagoly E, Hegyi MZ, Kiszel PS, et al. ABCC1 polymorphisms in anthracycline-induced cardiotoxicity in childhood acute lymphoblastic leukaemia. Cell Biol Int. (2012) 36:79–86. doi: 10.1042/CBI20110264
29. Krajinovic M, Elbared J, Drouin S, Bertout L, Rezgui A, Ansari M, et al. Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharmacogenomics J. (2016) 16:530–5. doi: 10.1038/tpj.2015.63
30. Visscher H, Rassekh SR, Sandor GS, Caron HN, van Dalen EC, Kremer LC, et al. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics. (2015) 16:1065–76. doi: 10.2217/pgs.15.61
31. Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. (2003) 102:2574–80. doi: 10.1182/blood-2003-03-0869
32. Yang X, Li G, Guan M, Bapat A, Dai Q, Zhong C, et al. Potential gene association studies of chemotherapy-induced cardiotoxicity: a systematic review and meta-analysis. Front Cardiovasc Med. (2021) 8:651269. doi: 10.3389/fcvm.2021.651269
33. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
34. Renu K, V G A, P B TP, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy – an update. Eur J Pharmacol. (2018) 818:241–53. doi: 10.1016/j.ejphar.2017.10.043
35. Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. (2020) 126:926–41. doi: 10.1161/CIRCRESAHA.119.314681
36. Murabito A, Hirsch E, Ghigo A. Mechanisms of anthracycline-induced cardiotoxicity: is mitochondrial dysfunction the answer? Front Cardiovasc Med. (2020) 7:35. doi: 10.3389/fcvm.2020.00035
37. Tocchetti CG, Cadeddu C, Di Lisi D, Femminò S, Madonna R, Mele D, et al. From molecular mechanisms to clinical management of antineoplastic drug-induced cardiovascular toxicity: a translational overview. Antioxid Redox Signal. (2019) 30:2110–53. doi: 10.1089/ars.2016.6930
38. Ghigo A, Li M, Hirsch E. New signal transduction paradigms in anthracycline-induced cardiotoxicity. Biochim Biophys Acta. (2016) 1863:1916–25. doi: 10.1016/j.bbamcr.2016.01.021
39. Pagliaro P, Penna C. Redox signalling and cardioprotection: translatability and mechanism. Br J Pharmacol. (2015) 172:1974–95. doi: 10.1111/bph.12975
40. Bonora M, Wieckowski MR, Sinclair DA, Kroemer G, Pinton P, Galluzzi L. Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat Rev Cardiol. (2019) 16:33–55. doi: 10.1038/s41569-018-0074-0
41. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–50. doi: 10.1038/nrcardio.2016.203
42. Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the american heart association. Circ Res. (2016) 118:1960–91. doi: 10.1161/RES.0000000000000104
43. Aon MA, Cortassa S. Mitochondrial network energetics in the heart. Wiley Interdiscip Rev Syst Biol Med. (2012) 4:599–613. doi: 10.1002/wsbm.1188
44. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. (2014) 124:617–30. doi: 10.1172/JCI72931
45. Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
46. Davies KJA, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. (1986) 261:3060–7. doi: 10.1016/s0021-9258(17)35746-0
47. Tullio F, Angotti C, Perrelli M-G, Penna C, Pagliaro P. Redox balance and cardioprotection. Basic Res Cardiol. (2013) 108:392. doi: 10.1007/s00395-013-0392-7
48. Penna C, Mancardi D, Rastaldo R, Pagliaro P. Cardioprotection: a radical view free radicals in pre and postconditioning. Biochim Biophys Acta. (2009) 1787:781–93. doi: 10.1016/j.bbabio.2009.02.008
49. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. (2021) 12:339. doi: 10.1038/s41419-021-03614-x
50. Qin Y, Guo T, Wang Z, Zhao Y. The role of iron in doxorubicin-induced cardiotoxicity: recent advances and implication for drug delivery. J Mater Chem B. (2021) 9:4793–803. doi: 10.1039/d1tb00551k
51. Kwok JC, Richardson DR. Anthracyclines induce accumulation of iron in ferritin in myocardial and neoplastic cells: inhibition of the ferritin iron mobilization pathway. Mol Pharmacol. (2003) 63:849–61. doi: 10.1124/mol.63.4.849
52. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI insight. (2020) 5:e132747. doi: 10.1172/jci.insight.132747
53. Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. (2009) 46:1439–53. doi: 10.1016/j.freeradbiomed.2009.03.004
54. Parker MA, King V, Howard KP. Nuclear magnetic resonance study of doxorubicin binding to cardiolipin containing magnetically oriented phospholipid bilayers. Biochim Biophys Acta. (2001) 1514:206–16. doi: 10.1016/s0005-2736(01)00371-6
55. Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. (2016) 22:175–82. doi: 10.1038/nm.4017
56. Lyu YL, Kerrigan JE, Lin C-P, Azarova AM, Tsai Y-C, Ban Y, et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. (2007) 67:8839–46. doi: 10.1158/0008-5472.CAN-07-1649
57. McSweeney KM, Bozza WP, Alterovitz W-L, Zhang B. Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell Death Discov. (2019) 5:102. doi: 10.1038/s41420-019-0182-6
58. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
59. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. (2000) 275:16202–12. doi: 10.1074/jbc.275.21.16202
60. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. (2005) 434:652–8. doi: 10.1038/nature03317
61. Baumann K. Cell death: multitasking p53 promotes necrosis. Nat Rev Mol Cell Biol. (2012) 13:480–1. doi: 10.1038/nrm3401
62. Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. (2012) 149:1536–48. doi: 10.1016/j.cell.2012.05.014
63. Blandino G, Valenti F, Sacconi A, Di Agostino S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: emerging insights in cancer disease. Semin Cell Dev Biol. (2020) 98:105–17. doi: 10.1016/j.semcdb.2019.05.011
64. Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ. Inhibition of mitochondrial p53 accumulation by PFT-μ prevents cisplatin-induced peripheral neuropathy. Front Mol Neurosci. (2017) 10:108. doi: 10.3389/fnmol.2017.00108
65. Li J, Wang P-Y, Long NA, Zhuang J, Springer DA, Zou J, et al. p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc Natl Acad Sci USA. (2019) 116:19626–34. doi: 10.1073/pnas.1904979116
66. Nishi M, Wang P-Y, Hwang PM. Protective role of p53 in doxorubicin-induced cardiomyopathy as a mitochondrial disease. Mol Cell Oncol. (2020) 7:1724598. doi: 10.1080/23723556.2020.1724598
67. Gambardella J, Trimarco B, Iaccarino G, Sorriento D. Cardiac nonmyocyte cell functions and crosstalks in response to cardiotoxic drugs. Oxid Med Cell Longev. (2017) 2017:1089359. doi: 10.1155/2017/1089359
68. Jurczyk M, Król M, Midro A, Kurnik-Łucka M, Poniatowski A, Gil K. Cardiotoxicity of fluoropyrimidines: epidemiology, mechanisms, diagnosis, and management. J Clin Med. (2021) 10:4426. doi: 10.3390/jcm10194426
69. Shiga T, Hiraide M. Cardiotoxicities of 5-fluorouracil and other fluoropyrimidines. Curr Treat Options Oncol. (2020) 21:27. doi: 10.1007/s11864-020-0719-1
70. Deac AL, Burz CC, Bocsan IC, Buzoianu AD. Fluoropyrimidine-induced cardiotoxicity. World J Clin Oncol. (2020) 11:1008–17. doi: 10.5306/wjco.v11.i12.1008
71. Depetris I, Marino D, Bonzano A, Cagnazzo C, Filippi R, Aglietta M, et al. Fluoropyrimidine-induced cardiotoxicity. Crit Rev Oncol Hematol. (2018) 124:1–10. doi: 10.1016/j.critrevonc.2018.02.002
72. Sorrentino MF, Kim J, Foderaro AE, Truesdell AG. 5-fluorouracil induced cardiotoxicity: review of the literature. Cardiol J. (2012) 19:453–8. doi: 10.5603/cj.2012.0084
73. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. (2009) 8:191–202. doi: 10.1517/14740330902733961
74. Alter P, Herzum M, Soufi M, Schaefer JR, Maisch B. Cardiotoxicity of 5-fluorouracil. Cardiovasc Hematol Agents Med Chem. (2006) 4:1–5. doi: 10.2174/187152506775268785
75. Madeddu C, Deidda M, Piras A, Cadeddu C, Demurtas L, Puzzoni M, et al. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. J Cardiovasc Med (Hagerstown). (2016) 17(Suppl. 1):S12–8. doi: 10.2459/JCM.0000000000000376
76. Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. (2015) 12:547–58. doi: 10.1038/nrcardio.2015.65
77. Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One. (2015) 10:e0115686. doi: 10.1371/journal.pone.0115686
78. Mosseri M, Fingert HJ, Varticovski L, Chokshi S, Isner JM. In vitro evidence that myocardial ischemia resulting from 5-fluorouracil chemotherapy is due to protein kinase C-mediated vasoconstriction of vascular smooth muscle. Cancer Res. (1993) 53:3028–33.
79. Spasojević I, Jelić S, Zakrzewska J, Bacić G. Decreased oxygen transfer capacity of erythrocytes as a cause of 5-fluorouracil related ischemia. Molecules. (2008) 14:53–67. doi: 10.3390/molecules14010053
80. Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H. Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf. (2007) 6:609–21. doi: 10.1517/14740338.6.5.609
81. Osman M, Elkady MA. Prospective study to evaluate the effect of paclitaxel on cardiac ejection fraction. Breast Care (Basel). (2017) 12:255–9. doi: 10.1159/000471759
82. Rowinsky EK, McGuire WP, Guarnieri T, Fisherman JS, Christian MC, Donehower RC. Cardiac disturbances during the administration of taxol. J Clin Oncol. (1991) 9:1704–12. doi: 10.1200/JCO.1991.9.9.1704
83. Batra A, Patel B, Addison D, Baldassarre LA, Desai N, Weintraub N, et al. Cardiovascular safety profile of taxanes and vinca alkaloids: 30 years FDA registry experience. Open Hear. (2021) 8:e001849. doi: 10.1136/openhrt-2021-001849
84. Schlitt A, Jordan K, Vordermark D, Schwamborn J, Langer T, Thomssen C. Cardiotoxicity and oncological treatments. Dtsch Arztebl Int. (2014) 111:161–8. doi: 10.3238/arztebl.2014.0161
85. Arbuck SG, Strauss H, Rowinsky E, Christian M, Suffness M, Adams J, et al. A reassessment of cardiac toxicity associated with taxol. J Natl Cancer Inst Monogr. (1993) 117–30. Available online at: https://pubmed.ncbi.nlm.nih.gov/7912518/ (accessed February 24, 2022).
86. Rosa GM, Gigli L, Tagliasacchi MI, Di Iorio C, Carbone F, Nencioni A, et al. Update on cardiotoxicity of anti-cancer treatments. Eur J Clin Invest. (2016) 46:264–84. doi: 10.1111/eci.12589
87. Schimmel KJM, Richel DJ, van den Brink RBA, Guchelaar H-J. Cardiotoxicity of cytotoxic drugs. Cancer Treat Rev. (2004) 30:181–91. doi: 10.1016/j.ctrv.2003.07.003
88. Geiger S, Lange V, Suhl P, Heinemann V, Stemmler H-J. Anticancer therapy induced cardiotoxicity: review of the literature. Anticancer Drugs. (2010) 21:578–90. doi: 10.1097/CAD.0b013e3283394624
89. Finley RS, Rowinsky EK. Patient care issues: the management of paclitaxel-related toxicities. Ann Pharmacother. (1994) 28:S27–30. doi: 10.1177/10600280940280S507
90. Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC. Clinical toxicities encountered with paclitaxel (taxol). Semin Oncol. (1993) 20:1–15.
91. Varbiro G, Veres B, Gallyas F, Sumegi B. Direct effect of taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med. (2001) 31:548–58. doi: 10.1016/s0891-5849(01)00616-5
92. Gianni L, Viganò L, Locatelli A, Capri G, Giani A, Tarenzi E, et al. Human pharmacokinetic characterization and in vitro study of the interaction between doxorubicin and paclitaxel in patients with breast cancer. J Clin Oncol. (1997) 15:1906–15. doi: 10.1200/JCO.1997.15.5.1906
93. Saad SY, Najjar TAO, Alashari M. Cardiotoxicity of doxorubicin/paclitaxel combination in rats: effect of sequence and timing of administration. J Biochem Mol Toxicol. (2004) 18:78–86. doi: 10.1002/jbt.20012
94. Sparano JA. Doxorubicin/taxane combinations: cardiac toxicity and pharmacokinetics. Semin Oncol. (1999) 26:14–9.
95. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. (2014) 740:364–78. doi: 10.1016/j.ejphar.2014.07.025
96. Haugnes HS, Wethal T, Aass N, Dahl O, Klepp O, Langberg CW, et al. Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: a 20-year follow-up study. J Clin Oncol. (2010) 28:4649–57. doi: 10.1200/JCO.2010.29.9362
97. Alexandre J, Moslehi JJ, Bersell KR, Funck-Brentano C, Roden DM, Salem J-E. Anticancer drug-induced cardiac rhythm disorders: current knowledge and basic underlying mechanisms. Pharmacol Ther. (2018) 189:89–103. doi: 10.1016/j.pharmthera.2018.04.009
98. Meinardi MT, Gietema JA, van der Graaf WTA, van Veldhuisen DJ, Runne MA, Sluiter WJ, et al. Cardiovascular morbidity in long-term survivors of metastatic testicular cancer. J Clin Oncol. (2000) 18:1725–32. doi: 10.1200/JCO.2000.18.8.1725
99. Al-Majed AA, Sayed-Ahmed MM, Al-Yahya AA, Aleisa AM, Al-Rejaie SS, Al-Shabanah OA. Propionyl-L-carnitine prevents the progression of cisplatin-induced cardiomyopathy in a carnitine-depleted rat model. Pharmacol Res. (2006) 53:278–86. doi: 10.1016/j.phrs.2005.12.005
100. El-Awady E-SE, Moustafa YM, Abo-Elmatty DM, Radwan A. Cisplatin-induced cardiotoxicity: mechanisms and cardioprotective strategies. Eur J Pharmacol. (2011) 650:335–41. doi: 10.1016/j.ejphar.2010.09.085
101. El-Sawalhi MM, Ahmed LA. Exploring the protective role of apocynin, a specific NADPH oxidase inhibitor, in cisplatin-induced cardiotoxicity in rats. Chem Biol Interact. (2014) 207:58–66. doi: 10.1016/j.cbi.2013.11.008
102. Chowdhury S, Sinha K, Banerjee S, Sil PC. Taurine protects cisplatin induced cardiotoxicity by modulating inflammatory and endoplasmic reticulum stress responses. Biofactors. (2016) 42:647–64. doi: 10.1002/biof.1301
103. Braverman AC, Antin JH, Plappert MT, Cook EF, Lee RT. Cyclophosphamide cardiotoxicity in bone marrow transplantation: a prospective evaluation of new dosing regimens. J Clin Oncol. (1991) 9:1215–23. doi: 10.1200/JCO.1991.9.7.1215
104. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
105. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. (2012) 12:553–63. doi: 10.1038/nrc3309
106. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
107. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. (2006) 7:505–16. doi: 10.1038/nrm1962
108. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. (1996) 16:5276–87. doi: 10.1128/MCB.16.10.5276
109. Wang J, Xu B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct Target Ther. (2019) 4:34. doi: 10.1038/s41392-019-0069-2
110. Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. (2012) 111:1376–85. doi: 10.1161/CIRCRESAHA.112.267286
111. De Keulenaer GW, Doggen K, Lemmens K. The vulnerability of the heart as a pluricellular paracrine organ: lessons from unexpected triggers of heart failure in targeted ErbB2 anticancer therapy. Circ Res. (2010) 106:35–46. doi: 10.1161/CIRCRESAHA.109.205906
112. Fuller SJ, Sivarajah K, Sugden PH. ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J Mol Cell Cardiol. (2008) 44:831–54. doi: 10.1016/j.yjmcc.2008.02.278
113. Del Monte-Nieto G, Ramialison M, Adam AAS, Wu B, Aharonov A, D’Uva G, et al. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature. (2018) 557:439–71. doi: 10.1038/s41586-018-0110-6
114. Lai D, Liu X, Forrai A, Wolstein O, Michalicek J, Ahmed I, et al. Neuregulin 1 sustains the gene regulatory network in both trabecular and nontrabecular myocardium. Circ Res. (2010) 107:715–27. doi: 10.1161/CIRCRESAHA.110.218693
115. Liu J, Bressan M, Hassel D, Huisken J, Staudt D, Kikuchi K, et al. A dual role for ErbB2 signaling in cardiac trabeculation. Development. (2010) 137:3867–75. doi: 10.1242/dev.053736
116. Wadugu B, Kuhn B. The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am J Physiol Hear Circ Physiol. (2012) 302:H2139–47. doi: 10.1152/ajpheart.00063.2012ajpheart.00063.2012
117. Pentassuglia L, Sawyer DB. The role of neuregulin-1beta/ErbB signaling in the heart. Exp Cell Res. (2009) 315:627–37. doi: 10.1016/j.yexcr.2008.08.015
118. Hedhli N, Huang Q, Kalinowski A, Palmeri M, Hu X, Russell RR, et al. Endothelium-derived neuregulin protects the heart against ischemic injury. Circulation. (2011) 123:2254–62. doi: 10.1161/CIRCULATIONAHA.110.991125
119. Hedhli N, Dobrucki LW, Kalinowski A, Zhuang ZW, Wu X, Russell RR, et al. Endothelial-derived neuregulin is an important mediator of ischaemia-induced angiogenesis and arteriogenesis. Cardiovasc Res. (2012) 93:516–24. doi: 10.1093/cvr/cvr352
120. De Keulenaer GW, Feyen E, Dugaucquier L, Shakeri H, Shchendrygina A, Belenkov YN, et al. Mechanisms of the multitasking endothelial protein NRG-1 as a compensatory factor during chronic heart failure. Circ Heart Fail. (2019) 12:e006288. doi: 10.1161/CIRCHEARTFAILURE.119.006288
121. Harvey RP, Wystub-Lis K, del Monte-Nieto G, Graham RM, Tzahor E. Cardiac regeneration therapies – targeting neuregulin 1 signalling. Hear Lung Circ. (2016) 25:4–7. doi: 10.1016/j.hlc.2015.08.014
122. D’Uva G, Tzahor E. The key roles of ERBB2 in cardiac regeneration. Cell Cycle. (2015) 14:2383–4. doi: 10.1080/15384101.2015.1063292
123. Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. (1998) 273:10261–9. doi: 10.1074/jbc.273.17.10261
124. D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. (2015) 17:627–38. doi: 10.1038/ncb3149
125. Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, et al. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. (2015) 7:281ra45. doi: 10.1126/scitranslmed.aaa5171
126. Gemberling M, Karra R, Dickson AL, Poss KD. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife. (2015) 4:e05871. doi: 10.7554/eLife.05871
127. Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. (2009) 138:257–70. doi: 10.1016/j.cell.2009.04.060S0092-8674(09)00522-4
128. Crone SA, Zhao Y-YY, Fan L, Gu Y, Minamisawa S, Liu Y, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. (2002) 8:459–65. doi: 10.1038/nm0502-459nm0502-459
129. Rohrbach S, Niemann B, Silber RE, Holtz J. Neuregulin receptors erbB2 and erbB4 in failing human myocardium – depressed expression and attenuated activation. Basic Res Cardiol. (2005) 100:240–9. doi: 10.1007/s00395-005-0514-4
130. Rohrbach S, Yan X, Weinberg EO, Hasan F, Bartunek J, Marchionni MA, et al. Neuregulin in cardiac hypertrophy in rats with aortic stenosis. Differential expression of erbB2 and erbB4 receptors. Circulation. (1999) 100:407–12. doi: 10.1161/01.cir.100.4.407
131. Mendes-Ferreira P, Maia-Rocha C, Adão R, Mendes MJ, Santos-Ribeiro D, Alves BS, et al. Neuregulin-1 improves right ventricular function and attenuates experimental pulmonary arterial hypertension. Cardiovasc Res. (2016) 109:44–54. doi: 10.1093/cvr/cvv244
132. Gu A, Jie Y, Sun L, Zhao S, Mingyan E, You Q. RhNRG-1β protects the myocardium against irradiation-induced damage via the ErbB2-ERK-SIRT1 signaling pathway. PLoS One. (2015) 10:e0137337. doi: 10.1371/journal.pone.0137337
133. Xu Y, Li X, Liu X, Zhou M. Neuregulin-1/ErbB signaling and chronic heart failure. Adv Pharmacol. (2010) 59:31–51. doi: 10.1016/S1054-3589(10)59002-1
134. Liu X, Gu X, Li Z, Li X, Li H, Chang J, et al. Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J Am Coll Cardiol. (2006) 48:1438–47. doi: 10.1016/j.jacc.2006.05.057
135. Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, et al. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. (2010) 55:1907–14. doi: 10.1016/j.jacc.2009.12.044
136. Lenihan DJ, Anderson SA, Lenneman CG, Brittain E, Muldowney JAS, Mendes L, et al. A phase I, single ascending dose study of cimaglermin alfa (neuregulin 1β3) in patients with systolic dysfunction and heart failure. JACC Basic to Transl Sci. (2016) 1:576–86. doi: 10.1016/j.jacbts.2016.09.005
137. Sawyer DB, Caggiano A. Neuregulin-1beta for the treatment of systolic heart failure. J Mol Cell Cardiol. (2011) 51:501–5. doi: 10.1016/j.yjmcc.2011.06.016
138. de Korte MA, de Vries EG, Lub-de Hooge MN, Jager PL, Gietema JA, van der Graaf WT, et al. 111Indium-trastuzumab visualises myocardial human epidermal growth factor receptor 2 expression shortly after anthracycline treatment but not during heart failure: a clue to uncover the mechanisms of trastuzumab-related cardiotoxicity. Eur J Cancer. (2007) 43:2046–51. doi: 10.1016/j.ejca.2007.06.024
139. Gabrielson K, Bedja D, Pin S, Tsao A, Gama L, Yuan B, et al. Heat shock protein 90 and ErbB2 in the cardiac response to doxorubicin injury. Cancer Res. (2007) 67:1436–41. doi: 10.1158/0008-5472.CAN-06-3721
140. Belmonte F, Das S, Sysa-Shah P, Sivakumaran V, Stanley B, Guo X, et al. ErbB2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am J Physiol Heart Circ Physiol. (2015) 309:H1271–80. doi: 10.1152/ajpheart.00517.2014
141. Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chimeric human-murine monoclonal antibody. Drugs Today. (2005) 41:107–27. doi: 10.1358/dot.2005.41.2.882662
142. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. (2008) 358:1160–74. doi: 10.1056/NEJMra0707704
143. Ishiguro M, Watanabe T, Yamaguchi K, Satoh T, Ito H, Seriu T, et al. Japanese post-marketing surveillance of cetuximab (Erbitux®) in patients with metastatic colorectal cancer. Jpn J Clin Oncol. (2012) 42:287–94. doi: 10.1093/jjco/hys005
144. Huang W-T, Chen H-H, Yeh C-H, Lu Y-C, Hwang W-S, Huang J-S, et al. A postmarketing surveillance study on erbitux (cetuximab) in patients with metastatic colorectal cancer refractory to irinotecan-containing treatment. J Investig Med. (2013) 61:1108–14. doi: 10.2310/JIM.0b013e3182a6799d
145. Cho H-S, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin fab. Nature. (2003) 421:756–60. doi: 10.1038/nature01392
146. Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. (2009) 15:429–40. doi: 10.1016/j.ccr.2009.03.020
147. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. (2005) 5:341–54. doi: 10.1038/nrc1609
148. Perez EA. Cardiac issues related to trastuzumab. Breast. (2004) 13:171–2. doi: 10.1016/j.breast.2004.02.008
149. Perez EA, Rodeheffer R. Clinical cardiac tolerability of trastuzumab. J Clin Oncol. (2004) 22:322–9. doi: 10.1200/JCO.2004.01.120
150. Keefe DL. Trastuzumab-associated cardiotoxicity. Cancer. (2002) 95:1592–600. doi: 10.1002/cncr.10854
151. Yavas O, Yazici M, Eren O, Oyan B. The acute effect of trastuzumab infusion on ECG parameters in metastatic breast cancer patients. Swiss Med Wkly. (2007) 137:556–8. doi: 10.4414/smw.2007.11899
152. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. (2002) 20:719–26. doi: 10.1200/JCO.2002.20.3.719
153. Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. (1999) 17:2639–48. doi: 10.1200/JCO.1999.17.9.2639
154. Lin M, Xiong W, Wang S, Li Y, Hou C, Li C, et al. The research progress of trastuzumab-induced cardiotoxicity in HER-2-positive breast cancer treatment. Front Cardiovasc Med. (2021) 8:821663. doi: 10.3389/fcvm.2021.821663
155. Healey Bird BRJ, Swain SM. Cardiac toxicity in breast cancer survivors: review of potential cardiac problems. Clin Cancer Res. (2008) 14:14–24. doi: 10.1158/1078-0432.CCR-07-1033
156. Hudis CA, Clifford H. Trastuzumab – mechanism of action and use in clinical practice. N Engl J Med. (2007) 357:39–51. doi: 10.1056/nejmra043186
157. Morris PG, Hudis CA. Trastuzumab-related cardiotoxicity following anthracycline-based adjuvant chemotherapy: how worried should we be? J Clin Oncol. (2010) 28:3407–10. doi: 10.1200/JCO.2009.26.0125
158. Ewer MS, Ewer SM. Troponin I provides insight into cardiotoxicity and the anthracycline-trastuzumab interaction. J Clin Oncol. (2010) 28:3901–4. doi: 10.1200/JCO.2010.30.6274
159. Beauclair S, Formento P, Fischel JL, Lescaut W, Largillier R, Chamorey E, et al. Role of the HER2 [Ile655Val] genetic polymorphism in tumorogenesis and in the risk of trastuzumab-related cardiotoxicity. Ann Oncol Off J Eur Soc Med Oncol. (2007) 18:1335–41. doi: 10.1093/annonc/mdm181
160. Roca L, Diéras V, Roché H, Lappartient E, Kerbrat P, Cany L, et al. Correlation of HER2, FCGR2A, and FCGR3A gene polymorphisms with trastuzumab related cardiac toxicity and efficacy in a subgroup of patients from UNICANCER-PACS 04 trial. Breast Cancer Res Treat. (2013) 139:789–800. doi: 10.1007/s10549-013-2587-x
161. Han X, Diao L, Xu Y, Xue W, Ouyang T, Li J, et al. Association between the HER2 Ile655Val polymorphism and response to trastuzumab in women with operable primary breast cancer. Ann Oncol Off J Eur Soc Med Oncol. (2014) 25:1158–64. doi: 10.1093/annonc/mdu111
162. Gómez Peña C, Dávila-Fajardo CL, Martínez-González LJ, Carmona-Sáez P, Soto Pino MJ, Sánchez Ramos J, et al. Influence of the HER2 Ile655Val polymorphism on trastuzumab-induced cardiotoxicity in HER2-positive breast cancer patients: a meta-analysis. Pharmacogenet Genomics. (2015) 25:388–93. doi: 10.1097/FPC.0000000000000149
163. Stanton SE, Ward MM, Christos P, Sanford R, Lam C, Cobham MV, et al. Pro1170 Ala polymorphism in HER2-neu is associated with risk of trastuzumab cardiotoxicity. BMC Cancer. (2015) 15:267. doi: 10.1186/s12885-015-1298-6
164. Vazdar L, Gabrić ID, Kruljac I, Pintarić H, Šeparović R, Kirigin Biloš LS, et al. Influence of Ile655Val polymorphism on trastuzumab-induced cardiotoxicity in early-stage HER2 positive breast cancer. Sci Rep. (2021) 11:14395. doi: 10.1038/s41598-021-93634-6
165. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. (2001) 344:783–92. doi: 10.1056/nejm200103153441101
166. Cardinale D, Colombo A, Torrisi R, Sandri MT, Civelli M, Salvatici M, et al. Trastuzumab-induced cardiotoxicity: clinical and prognostic implications of troponin I evaluation. J Clin Oncol. (2010) 28:3910–6. doi: 10.1200/JCO.2009.27.3615
167. Timolati F, Ott D, Pentassuglia L, Giraud MN, Perriard JC, Suter TM, et al. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. (2006) 41:845–54. doi: 10.1016/j.yjmcc.2006.08.002
168. Capelan M, Pugliano L, De Azambuja E, Bozovic I, Saini KS, Sotiriou C, et al. Pertuzumab: new hope for patients with HER2-positive breast cancer. Ann Oncol. (2013) 24:273–82. doi: 10.1093/annonc/mds328
169. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, De Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. (2004) 5:317–28. doi: 10.1016/S1535-6108(04)00083-2
170. von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer. N Engl J Med. (2017) 377:122–31. doi: 10.1056/NEJMoa1703643
171. Alhussein MM, Mokbel A, Cosman T, Aghel N, Yang EH, Mukherjee SD, et al. Pertuzumab cardiotoxicity in patients with HER2-positive cancer: a systematic review and meta-analysis. CJC Open. (2021) 3:1372–82. doi: 10.1016/j.cjco.2021.06.019
172. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. (2007) 7:332–44. doi: 10.1038/nrc2106
173. Schlam I, Swain SM. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now. NPJ Breast Cancer. (2021) 7:56. doi: 10.1038/s41523-021-00265-1
174. Perez EA, Koehler M, Byrne J, Preston AJ, Rappold E, Ewer MS. Cardiac safety of lapatinib: pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin Proc. (2008) 83:679–86. doi: 10.4065/83.6.679
175. Eiger D, Pondé NF, Agbor-Tarh D, Moreno-Aspitia A, Piccart M, Hilbers FS, et al. Long-term cardiac outcomes of patients with HER2-positive breast cancer treated in the adjuvant lapatinib and/or trastuzumab treatment optimization trial. Br J Cancer. (2020) 122:1453–60. doi: 10.1038/s41416-020-0786-x
176. Ewer MS, Patel K, O’Brien D, Lorence RM. Cardiac safety of afatinib: a review of data from clinical trials. Cardio Oncol. (2015) 1:3. doi: 10.1186/s40959-015-0006-7
177. Sendur MAN, Aksoy S, Altundag K. Cardiotoxicity of novel HER2-targeted therapies. Curr Med Res Opin. (2013) 29:1015–24. doi: 10.1185/03007995.2013.807232
178. Wong K-K, Fracasso PM, Bukowski RM, Lynch TJ, Munster PN, Shapiro GI, et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. (2009) 15:2552–8. doi: 10.1158/1078-0432.CCR-08-1978
179. Ito Y, Suenaga M, Hatake K, Takahashi S, Yokoyama M, Onozawa Y, et al. Safety, efficacy and pharmacokinetics of neratinib (HKI-272) in Japanese patients with advanced solid tumors: a phase 1 dose-escalation study. Jpn J Clin Oncol. (2012) 42:278–86. doi: 10.1093/jjco/hys012
180. Novo G, Di Lisi D, Bronte E, Macaione F, Accurso V, Badalamenti G, et al. Cardiovascular toxicity in cancer patients treated with tyrosine kinase inhibitors: a real-world single-center experience. Oncology. (2020) 98:445–51. doi: 10.1159/000505486
181. Singh AP, Umbarkar P, Tousif S, Lal H. Cardiotoxicity of the BCR-ABL1 tyrosine kinase inhibitors: emphasis on ponatinib. Int J Cardiol. (2020) 316:214–21. doi: 10.1016/j.ijcard.2020.05.077
182. Yang Y, Bu P. Progress on the cardiotoxicity of sunitinib: prognostic significance, mechanism and protective therapies. Chem Biol Interact. (2016) 257:125–31. doi: 10.1016/j.cbi.2016.08.006
183. Yang B, Papoian T. Preclinical approaches to assess potential kinase inhibitor-induced cardiac toxicity: past, present and future. J Appl Toxicol. (2018) 38:790–800. doi: 10.1002/jat.3584
184. Hasinoff BB, Patel D, O’Hara KA. Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor sunitinib. Mol Pharmacol. (2008) 74:1722–8. doi: 10.1124/mol.108.050104
185. Hasinoff BB, Patel D. Mechanisms of myocyte cytotoxicity induced by the multikinase inhibitor sorafenib. Cardiovasc Toxicol. (2010) 10:1–8. doi: 10.1007/s12012-009-9056-0
186. Hasinoff BB, Patel D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol Appl Pharmacol. (2010) 249:132–9. doi: 10.1016/j.taap.2010.08.026
187. Cheng H, Force T. Molecular mechanisms of cardiovascular toxicity of targeted cancer therapeutics. Circ Res. (2010) 106:21–34. doi: 10.1161/CIRCRESAHA.109.206920
188. Green MR, Newton MD, Fancher KM. Off-target effects of BCR-ABL and JAK2 inhibitors. Am J Clin Oncol. (2016) 39:76–84. doi: 10.1097/COC.0000000000000023
189. Kerkela R, Woulfe KC, Durand J-B, Vagnozzi R, Kramer D, Chu TF, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. (2009) 2:15–25. doi: 10.1111/j.1752-8062.2008.00090.x
190. Zhu X, Stergiopoulos K, Wu S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: systematic review and meta-analysis. Acta Oncol. (2009) 48:9–17. doi: 10.1080/02841860802314720
191. Wu S, Chen JJ, Kudelka A, Lu J, Zhu X. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. (2008) 9:117–23. doi: 10.1016/S1470-2045(08)70003-2
192. Curigliano G, Mayer EL, Burstein HJ, Winer EP, Goldhirsch A. Cardiac toxicity from systemic cancer therapy: a comprehensive review. Prog Cardiovasc Dis. (2010) 53:94–104. doi: 10.1016/j.pcad.2010.05.006
193. Ewer MS, Suter TM, Lenihan DJ, Niculescu L, Breazna A, Demetri GD, et al. Cardiovascular events among 1090 cancer patients treated with sunitinib, interferon, or placebo: a comprehensive adjudicated database analysis demonstrating clinically meaningful reversibility of cardiac events. Eur J Cancer. (2014) 50:2162–70. doi: 10.1016/j.ejca.2014.05.013
194. Mourad J-J, Levy BI. Mechanisms of antiangiogenic-induced arterial hypertension. Curr Hypertens Rep. (2011) 13:289–93. doi: 10.1007/s11906-011-0206-y
195. Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med. (2013) 5:187ra69. doi: 10.1126/scitranslmed.3005066
196. Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. (2006) 12:908–16. doi: 10.1038/nm1446
197. Choksey A, Timm KN. Cancer therapy-induced cardiotoxicity-a metabolic perspective on pathogenesis, diagnosis and therapy. Int J Mol Sci. (2021) 23:441. doi: 10.3390/ijms23010441
198. McMullen CJ, Chalmers S, Wood R, Cunningham MR, Currie S. Sunitinib and imatinib display differential cardiotoxicity in adult rat cardiac fibroblasts that involves a role for calcium/calmodulin dependent protein kinase II. Front Cardiovasc Med. (2020) 7:630480. doi: 10.3389/fcvm.2020.630480
199. Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood. (2019) 133:1597–606. doi: 10.1182/blood-2018-10-881557
200. Singh AP, Glennon MS, Umbarkar P, Gupte M, Galindo CL, Zhang Q, et al. Ponatinib-induced cardiotoxicity: delineating the signalling mechanisms and potential rescue strategies. Cardiovasc Res. (2019) 115:966–77. doi: 10.1093/cvr/cvz006
201. Kalam K, Marwick TH. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur J Cancer. (2013) 49:2900–9. doi: 10.1016/j.ejca.2013.04.030
202. Ganatra S, Nohria A, Shah S, Groarke JD, Sharma A, Venesy D, et al. Upfront dexrazoxane for the reduction of anthracycline-induced cardiotoxicity in adults with preexisting cardiomyopathy and cancer: a consecutive case series. Cardio Oncol. (2019) 5:1. doi: 10.1186/s40959-019-0036-7
203. Macedo AVS, Hajjar LA, Lyon AR, Nascimento BR, Putzu A, Rossi L, et al. Efficacy of dexrazoxane in preventing anthracycline cardiotoxicity in breast cancer. JACC CardioOncol. (2019) 1:68–79. doi: 10.1016/j.jaccao.2019.08.003
204. Simůnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. (2009) 61:154–71. doi: 10.1016/s1734-1140(09)70018-0
205. Lipshultz SE, Lipsitz SR, Kutok JL, Miller TL, Colan SD, Neuberg DS, et al. Impact of hemochromatosis gene mutations on cardiac status in doxorubicin-treated survivors of childhood high-risk leukemia. Cancer. (2013) 119:3555–62. doi: 10.1002/cncr.28256
206. Hasinoff BB, Schroeder PE, Patel D. The metabolites of the cardioprotective drug dexrazoxane do not protect myocytes from doxorubicin-induced cytotoxicity. Mol Pharmacol. (2003) 64:670–8. doi: 10.1124/mol.64.3.670
207. Cascales A, Sánchez-Vega B, Navarro N, Pastor-Quirante F, Corral J, Vicente V, et al. Clinical and genetic determinants of anthracycline-induced cardiac iron accumulation. Int J Cardiol. (2012) 154:282–6. doi: 10.1016/j.ijcard.2010.09.046
208. Lenèová-Popelová O, Jirkovskı E, Jansová H, Jirkovská-Vávrová A, Vostatková-Tichotová L, Mazurová Y, et al. Cardioprotective effects of inorganic nitrate/nitrite in chronic anthracycline cardiotoxicity: comparison with dexrazoxane. J Mol Cell Cardiol. (2016) 91:92–103. doi: 10.1016/j.yjmcc.2015.12.021
209. Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, Hasinoff BB, et al. Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology. (2009) 255:72–9. doi: 10.1016/J.TOX.2008.10.011
210. Stìrba M, Popelová O, Vávrový A, Jirkovskı E, Kovaříková P, Geršl V, et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal. (2013) 18:899–929. doi: 10.1089/ars.2012.4795
211. Joseph P, Swedberg K, Leong DP, Yusuf S. The evolution of β-blockers in coronary artery disease and heart failure (part 1/5). J Am Coll Cardiol. (2019) 74:672–82. doi: 10.1016/J.JACC.2019.04.067
212. Freemantle N, Cleland J, Young P, Mason J, Harrison J. beta Blockade after myocardial infarction: systematic review and meta regression analysis. BMJ. (1999) 318:1730–7. doi: 10.1136/bmj.318.7200.1730
213. Kanduri J, More LA, Godishala A, Asnani A. Fluoropyrimidine-associated cardiotoxicity. Cardiol Clin. (2019) 37:399–405. doi: 10.1016/j.ccl.2019.07.004
214. Seicean S, Seicean A, Alan N, Plana JC, Budd GT, Marwick TH. Cardioprotective effect of β-adrenoceptor blockade in patients with breast cancer undergoing chemotherapy: follow-up study of heart failure. Circ Heart Fail. (2013) 6:420–6. doi: 10.1161/CIRCHEARTFAILURE.112.000055
215. Pituskin E, Mackey JR, Koshman S, Jassal D, Pitz M, Haykowsky MJ, et al. Multidisciplinary approach to novel therapies in cardio-oncology research (MANTICORE 101-breast): a randomized trial for the prevention of trastuzumab-associated cardiotoxicity. J Clin Oncol. (2017) 35:870–7. doi: 10.1200/JCO.2016.68.7830
216. Gulati G, Heck SL, Ree AH, Hoffmann P, Schulz-Menger J, Fagerland MW, et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): a 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur Heart J. (2016) 37:1671–80. doi: 10.1093/eurheartj/ehw022
217. Georgakopoulos P, Roussou P, Matsakas E, Karavidas A, Anagnostopoulos N, Marinakis T, et al. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: a prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am J Hematol. (2010) 85:894–6. doi: 10.1002/ajh.21840
218. Spallarossa P, Garibaldi S, Altieri P, Fabbi P, Manca V, Nasti S, et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J Mol Cell Cardiol. (2004) 37:837–46. doi: 10.1016/j.yjmcc.2004.05.024
219. Santos DL, Moreno AJM, Leino RL, Froberg MK, Wallace KB. Carvedilol protects against doxorubicin-induced mitochondrial cardiomyopathy. Toxicol Appl Pharmacol. (2002) 185:218–27. doi: 10.1006/taap.2002.9532
220. Matsui H, Morishima I, Numaguchi Y, Toki Y, Okumura K, Hayakawa T. Protective effects of carvedilol against doxorubicin-induced cardiomyopathy in rats. Life Sci. (1999) 65:1265–74. doi: 10.1016/s0024-3205(99)00362-8
221. Kalay N, Basar E, Ozdogru I, Er O, Cetinkaya Y, Dogan A, et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2006) 48:2258–62. doi: 10.1016/j.jacc.2006.07.052
222. Tashakori Beheshti A, Mostafavi Toroghi H, Hosseini G, Zarifian A, Homaei Shandiz F, Fazlinezhad A. Carvedilol administration can prevent doxorubicin-induced cardiotoxicity: a double-blind randomized trial. Cardiology. (2016) 134:47–53. doi: 10.1159/000442722
223. Nabati M, Janbabai G, Baghyari S, Esmaili K, Yazdani J. Cardioprotective effects of carvedilol in inhibiting doxorubicin-induced cardiotoxicity. J Cardiovasc Pharmacol. (2017) 69:279–85. doi: 10.1097/FJC.0000000000000470
224. Avila MS, Ayub-Ferreira SM, de Barros Wanderley MR, das Dores Cruz F, Gonçalves Brandão SM, Rigaud VOC, et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY trial. J Am Coll Cardiol. (2018) 71:2281–90. doi: 10.1016/j.jacc.2018.02.049
225. El-Shitany NA, Tolba OA, El-Shanshory MR, El-Hawary EE. Protective effect of carvedilol on adriamycin-induced left ventricular dysfunction in children with acute lymphoblastic leukemia. J Card Fail. (2012) 18:607–13. doi: 10.1016/j.cardfail.2012.06.416
226. Guglin M, Krischer J, Tamura R, Fink A, Bello-Matricaria L, McCaskill-Stevens W, et al. Randomized trial of lisinopril versus carvedilol to prevent trastuzumab cardiotoxicity in patients with breast cancer. J Am Coll Cardiol. (2019) 73:2859–68. doi: 10.1016/J.JACC.2019.03.495
227. Fongemie J, Felix-Getzik EA. Review of nebivolol pharmacology and clinical evidence. Drugs. (2015) 75:1349–71. doi: 10.1007/s40265-015-0435-5
228. de Nigris F, Rienzo M, Schiano C, Fiorito C, Casamassimi A, Napoli C. Prominent cardioprotective effects of third generation beta blocker nebivolol against anthracycline-induced cardiotoxicity using the model of isolated perfused rat heart. Eur J Cancer. (2008) 44:334–40. doi: 10.1016/j.ejca.2007.12.010
229. Mohamed EA, Kassem HH. Protective effect of nebivolol on doxorubicin-induced cardiotoxicity in rats. Arch Med Sci. (2018) 14:1450–8. doi: 10.5114/aoms.2018.79008
230. Kaya MG, Ozkan M, Gunebakmaz O, Akkaya H, Kaya EG, Akpek M, et al. Protective effects of nebivolol against anthracycline-induced cardiomyopathy: a randomized control study. Int J Cardiol. (2013) 167:2306–10. doi: 10.1016/j.ijcard.2012.06.023
231. Cochera F, Dinca D, Bordejevic DA, Citu IM, Mavrea AM, Andor M, et al. Nebivolol effect on doxorubicin-induced cardiotoxicity in breast cancer. Cancer Manag Res. (2018) 10:2071–81. doi: 10.2147/CMAR.S166481
232. Sobczuk P, Czerwińska M, Kleibert M, Cudnoch-Jêdrzejewska A. Anthracycline-induced cardiotoxicity and renin-angiotensin-aldosterone system-from molecular mechanisms to therapeutic applications. Heart Fail Rev. (2022) 27:295–319. doi: 10.1007/s10741-020-09977-1
233. Ayuna A, Abidin N. The role of neurohormonal blockers in the primary prevention of acute-, early-, and late-onset anthracycline-induced cardiotoxicity. Egypt Hear J. (2020) 72:59. doi: 10.1186/s43044-020-00090-0
234. Hanif K, Bid HK, Konwar R. Reinventing the ACE inhibitors: some old and new implications of ACE inhibition. Hypertens Res. (2010) 33:11–21. doi: 10.1038/hr.2009.184
235. Abd El-Aziz MA, Othman AI, Amer M, El-Missiry MA. Potential protective role of angiotensin-converting enzyme inhibitors captopril and enalapril against adriamycin-induced acute cardiac and hepatic toxicity in rats. J Appl Toxicol. (2001) 21:469–73. doi: 10.1002/jat.782
236. Boucek RJ, Steele A, Miracle A, Atkinson J. Effects of angiotensin-converting enzyme inhibitor on delayed-onset doxorubicin-induced cardiotoxicity. Cardiovasc Toxicol. (2003) 3:319–29. doi: 10.1385/ct:3:4:319
237. Hullin R, Métrich M, Sarre A, Basquin D, Maillard M, Regamey J, et al. Diverging effects of enalapril or eplerenone in primary prevention against doxorubicin-induced cardiotoxicity. Cardiovasc Res. (2018) 114:272–81. doi: 10.1093/cvr/cvx162
238. Okumura K, Jin D, Takai S, Miyazaki M. Beneficial effects of angiotensin-converting enzyme inhibition in adriamycin-induced cardiomyopathy in hamsters. Jpn J Pharmacol. (2002) 88:183–8. doi: 10.1254/jjp.88.183
239. Lipshultz SE, Lipsitz SR, Sallan SE, Simbre VC, Shaikh SL, Mone SM, et al. Long-term enalapril therapy for left ventricular dysfunction in doxorubicin-treated survivors of childhood cancer. J Clin Oncol. (2002) 20:4517–22. doi: 10.1200/JCO.2002.12.102
240. Jensen BV, Skovsgaard T, Nielsen SL. Functional monitoring of anthracycline cardiotoxicity: a prospective, blinded, long-term observational study of outcome in 120 patients. Ann Oncol. (2002) 13:699–709. doi: 10.1093/annonc/mdf132
241. Silber JH, Cnaan A, Clark BJ, Paridon SM, Chin AJ, Rychik J, et al. Enalapril to prevent cardiac function decline in long-term survivors of pediatric cancer exposed to anthracyclines. J Clin Oncol. (2004) 22:820–8. doi: 10.1200/JCO.2004.06.022
242. Soga M, Kamal FA, Watanabe K, Ma M, Palaniyandi S, Prakash P, et al. Effects of angiotensin II receptor blocker (candesartan) in daunorubicin-induced cardiomyopathic rats. Int J Cardiol. (2006) 110:378–85. doi: 10.1016/j.ijcard.2005.08.061
243. Cadeddu C, Piras A, Mantovani G, Deidda M, Dessì M, Madeddu C, et al. Protective effects of the angiotensin II receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress, and early ventricular impairment. Am Heart J. (2010) 160:487.e1–7. doi: 10.1016/j.ahj.2010.05.037
244. Dessì M, Madeddu C, Piras A, Cadeddu C, Antoni G, Mercuro G, et al. Long-term, up to 18 months, protective effects of the angiotensin II receptor blocker telmisartan on epirubin-induced inflammation and oxidative stress assessed by serial strain rate. Springerplus. (2013) 2:198. doi: 10.1186/2193-1801-2-198
245. Dessì M, Piras A, Madeddu C, Cadeddu C, Deidda M, Massa E, et al. Long-term protective effects of the angiotensin receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress and myocardial dysfunction. Exp Ther Med. (2011) 2:1003–9. doi: 10.3892/etm.2011.305
246. Boekhout AH, Gietema JA, Milojkovic Kerklaan B, van Werkhoven ED, Altena R, Honkoop A, et al. Angiotensin II-receptor inhibition with candesartan to prevent trastuzumab-related cardiotoxic effects in patients with early breast cancer: a randomized clinical trial. JAMA Oncol. (2016) 2:1030–7. doi: 10.1001/jamaoncol.2016.1726
247. Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. (2010) 55:213–20. doi: 10.1016/j.jacc.2009.03.095
248. Leong DP, Cosman T, Alhussein MM, Kumar Tyagi N, Karampatos S, Barron CC, et al. Safety of continuing trastuzumab despite mild cardiotoxicity: a phase I trial. JACC CardioOncol. (2019) 1:1–10. doi: 10.1016/j.jaccao.2019.06.004
249. Lynce F, Barac A, Geng X, Dang C, Yu AF, Smith KL, et al. Prospective evaluation of the cardiac safety of HER2-targeted therapies in patients with HER2-positive breast cancer and compromised heart function: the SAFE-HEaRt study. Breast Cancer Res Treat. (2019) 175:595–603. doi: 10.1007/s10549-019-05191-2
250. Ezekowitz JA, McAlister FA. Aldosterone blockade and left ventricular dysfunction: a systematic review of randomized clinical trials. Eur Heart J. (2009) 30:469–77. doi: 10.1093/eurheartj/ehn543
251. Akpek M, Ozdogru I, Sahin O, Inanc M, Dogan A, Yazici C, et al. Protective effects of spironolactone against anthracycline-induced cardiomyopathy. Eur J Heart Fail. (2015) 17:81–9. doi: 10.1002/ejhf.196
252. Berliner D, Bauersachs J. Current drug therapy in chronic heart failure: the new guidelines of the european society of cardiology (ESC). Korean Circ J. (2017) 47:543–54. doi: 10.4070/kcj.2017.0030
253. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
254. Butler J, Packer M, Filippatos G, Ferreira JP, Zeller C, Schnee J, et al. Effect of empagliflozin in patients with heart failure across the spectrum of left ventricular ejection fraction. Eur Heart J. (2022) 43:416–26. doi: 10.1093/eurheartj/ehab798
255. Pan D, Xu L, Chen P, Jiang H, Shi D, Guo M. Empagliflozin in patients with heart failure: a systematic review and meta-analysis of randomized controlled trials. Front Cardiovasc Med. (2021) 8:683281. doi: 10.3389/fcvm.2021.683281
256. Barış VÖ, Dinçsoy AB, Gedikli E, Zırh S, Müftüoğlu S, Erdem A. Empagliflozin significantly prevents the doxorubicin-induced acute cardiotoxicity via non-antioxidant pathways. Cardiovasc Toxicol. (2021) 21:747–58. doi: 10.1007/s12012-021-09665-y
257. Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. (2021) 20:150. doi: 10.1186/s12933-021-01346-y
258. Sabatino J, De Rosa S, Tammè L, Iaconetti C, Sorrentino S, Polimeni A, et al. Empagliflozin prevents doxorubicin-induced myocardial dysfunction. Cardiovasc Diabetol. (2020) 19:66. doi: 10.1186/s12933-020-01040-5
259. Oh CM, Cho S, Jang JY, Kim H, Chun S, Choi M, et al. Cardioprotective potential of an SGLT2 inhibitor against doxorubicin-induced heart failure. Korean Circ J. (2019) 49:1183–95. doi: 10.4070/kcj.2019.0180
260. Ren C, Sun K, Zhang Y, Hu Y, Hu B, Zhao J, et al. Sodium-Glucose cotransporter-2 inhibitor empagliflozin ameliorates sunitinib-induced cardiac dysfunction via regulation of AMPK-mTOR signaling pathway-mediated autophagy. Front Pharmacol. (2021) 12:664181. doi: 10.3389/fphar.2021.664181
261. Corradi F, Paolini L, De Caterina R. Ranolazine in the prevention of anthracycline cardiotoxicity. Pharmacol Res. (2014) 79:88–102. doi: 10.1016/j.phrs.2013.11.001
262. Cappetta D, Esposito G, Coppini R, Piegari E, Russo R, Ciuffreda LP, et al. Effects of ranolazine in a model of doxorubicin-induced left ventricle diastolic dysfunction. Br J Pharmacol. (2017) 174:3696–712. doi: 10.1111/bph.13791
263. Wang YX, Korth M. Effects of doxorubicin on excitation-contraction coupling in guinea pig ventricular myocardium. Circ Res. (1995) 76:645–53. doi: 10.1161/01.res.76.4.645
264. Tocchetti CG, Carpi A, Coppola C, Quintavalle C, Rea D, Campesan M, et al. Ranolazine protects from doxorubicin-induced oxidative stress and cardiac dysfunction. Eur J Heart Fail. (2014) 16:358–66. doi: 10.1002/ejhf.50
265. Riccio G, Antonucci S, Coppola C, D’Avino C, Piscopo G, Fiore D, et al. Ranolazine attenuates trastuzumab-induced heart dysfunction by modulating ROS production. Front Physiol. (2018) 9:38. doi: 10.3389/fphys.2018.00038
266. Minotti G, Menna P, Calabrese V, Greco C, Armento G, Annibali O, et al. Pharmacology of ranolazine versus common cardiovascular drugs in patients with early diastolic dysfunction induced by anthracyclines or nonanthracycline chemotherapeutics: a phase 2b minitrial. J Pharmacol Exp Ther. (2019) 370:197–205. doi: 10.1124/jpet.119.258178
267. Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. (2005) 111:1601–10. doi: 10.1161/01.CIR.0000160359.49478.C2
268. Koka S, Das A, Zhu S-G, Durrant D, Xi L, Kukreja RC. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther. (2010) 334:1023–30. doi: 10.1124/jpet.110.170191
269. Jin Z, Zhang J, Zhi H, Hong B, Zhang S, Guo H, et al. Beneficial effects of tadalafil on left ventricular dysfunction in doxorubicin-induced cardiomyopathy. J Cardiol. (2013) 62:110–6. doi: 10.1016/j.jjcc.2013.03.018
270. Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. (2005) 280:12944–55. doi: 10.1074/JBC.M404706200
271. Bartolomaeus H, Balogh A, Yakoub M, Homann S, Markó L, Höges S, et al. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. (2019) 139:1407–21. doi: 10.1161/CIRCULATIONAHA.118.036652
272. Chen X-F, Chen X, Tang X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin Sci (Lond). (2020) 134:657–76. doi: 10.1042/CS20200128
273. Russo M, Guida F, Paparo L, Trinchese G, Aitoro R, Avagliano C, et al. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur J Heart Fail. (2019) 21:519–28. doi: 10.1002/ejhf.1439
274. Thai PN, Miller CV, King MT, Schaefer S, Veech RL, Chiamvimonvat N, et al. Ketone ester D-β-hydroxybutyrate-(R)-1,3 butanediol prevents decline in cardiac function in type 2 diabetic mice. J Am Heart Assoc. (2021) 10:e020729. doi: 10.1161/JAHA.120.020729
275. Liao S, Tang Y, Yue X, Gao R, Yao W, Zhou Y, et al. β-hydroxybutyrate mitigated heart failure with preserved ejection fraction by increasing treg cells via Nox2/GSK-3β. J Inflamm Res. (2021) 14:4697–706. doi: 10.2147/JIR.S331320
276. Liu Y, Wei X, Wu M, Xu J, Xu B, Kang L. Cardioprotective roles of β-hydroxybutyrate against doxorubicin induced cardiotoxicity. Front Pharmacol. (2020) 11:603596. doi: 10.3389/fphar.2020.603596
277. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. (2017) 120:229–43. doi: 10.1161/CIRCRESAHA.116.308537
278. Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. (2004) 109:III39–43. doi: 10.1161/01.CIR.0000131517.20177.5a
279. Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2011) 58:988–9. doi: 10.1016/J.JACC.2011.05.025
280. Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational clinical cohort study. J Am Coll Cardiol. (2012) 60:2384–90. doi: 10.1016/j.jacc.2012.07.067
281. Chotenimitkhun R, D’Agostino R, Lawrence JA, Hamilton CA, Jordan JH, Vasu S, et al. Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. Can J Cardiol. (2015) 31:302–7. doi: 10.1016/j.cjca.2014.11.020
282. Abdel-Qadir H, Bobrowski D, Zhou L, Austin PC, Calvillo-Argüelles O, Amir E, et al. Statin exposure and risk of heart failure after anthracycline- or trastuzumab-based chemotherapy for early breast cancer: a propensity score—matched cohort study. J Am Heart Assoc. (2021) 10:e018393. doi: 10.1161/JAHA.119.018393
283. Calvillo-Argüelles O, Abdel-Qadir H, Michalowska M, Billia F, Suntheralingam S, Amir E, et al. Cardioprotective effect of statins in patients with HER2-positive breast cancer receiving trastuzumab therapy. Can J Cardiol. (2019) 35:153–9. doi: 10.1016/j.cjca.2018.11.028
284. Fukazawa R, Miller TA, Kuramochi Y, Frantz S, Kim YD, Marchionni MA, et al. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardiol. (2003) 35:1473–9. doi: 10.1016/j.yjmcc.2003.09.012
285. Bian Y, Sun M, Silver M, Ho KKL, Marchionni MA, Caggiano AO, et al. Neuregulin-1 attenuated doxorubicin-induced decrease in cardiac troponins. AJP Hear Circ Physiol. (2009) 297:H1974–83. doi: 10.1152/ajpheart.01010.2008
286. Vasti C. Neuregulin-1/erbB activities with focus on the susceptibility of the heart to anthracyclines. World J Cardiol. (2014) 6:653. doi: 10.4330/wjc.v6.i7.653
287. Jay SM, Murthy AC, Hawkins JF, Wortzel JR, Steinhauser ML, Alvarez LM, et al. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation. (2013) 128:152–61. doi: 10.1161/CIRCULATIONAHA.113.002203
288. Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. (1991) 78:2791–808. doi: 10.1016/S0083-6729(01)63006-1
289. Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, et al. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest. (2007) 87:440–55. doi: 10.1038/labinvest.3700530
290. Hamed S, Barshack I, Luboshits G, Wexler D, Deutsch V, Keren G, et al. Erythropoietin improves myocardial performance in doxorubicin-induced cardiomyopathy. Eur Heart J. (2006) 27:1876–83. doi: 10.1093/eurheartj/ehl044
291. Xu Y-J, Arneja AS, Tappia PS, Dhalla NS. The potential health benefits of taurine in cardiovascular disease. Exp Clin Cardiol. (2008) 13:57–65.
292. Huang W-P, Yin W-H, Chen J-S, Huang P-H, Chen J-W, Lin S-J. Fenofibrate attenuates doxorubicin-induced cardiac dysfunction in mice via activating the eNOS/EPC pathway. Sci Rep. (2021) 11:1159. doi: 10.1038/s41598-021-80984-4
293. Galán-Arriola C, Villena-Gutiérrez R, Higuero-Verdejo MI, Díaz-Rengifo IA, Pizarro G, López GJ, et al. Remote ischaemic preconditioning ameliorates anthracycline-induced cardiotoxicity and preserves mitochondrial integrity. Cardiovasc Res. (2021) 117:1132–43. doi: 10.1093/cvr/cvaa181
294. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell. (2015) 161:1566–75. doi: 10.1016/j.cell.2015.05.026
295. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. (2009) 324:98–102. doi: 10.1126/science.1164680
296. Bongiovanni C, Sacchi F, Da Pra S, Pantano E, Miano C, Morelli MB, et al. Reawakening the intrinsic cardiac regenerative potential: molecular strategies to boost dedifferentiation and proliferation of endogenous cardiomyocytes. Front Cardiovasc Med. (2021) 8:750604. doi: 10.3389/fcvm.2021.750604
Keywords: cardiotoxicity, cardioncology, cardiomyocyte death, cardiomyocyte dysfunction, cardiomyocyte survival, chemotherapy, targeted therapy, cardioprotection
Citation: Morelli MB, Bongiovanni C, Da Pra S, Miano C, Sacchi F, Lauriola M and D’Uva G (2022) Cardiotoxicity of Anticancer Drugs: Molecular Mechanisms and Strategies for Cardioprotection. Front. Cardiovasc. Med. 9:847012. doi: 10.3389/fcvm.2022.847012
Received: 31 December 2021; Accepted: 03 March 2022;
Published: 15 April 2022.
Edited by:
Chun Liu, Stanford University, United StatesReviewed by:
Lichao Liu, Stanford University, United StatesShane Rui Zhao, Stanford University, United States
Copyright © 2022 Morelli, Bongiovanni, Da Pra, Miano, Sacchi, Lauriola and D’Uva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriele D’Uva, gabrielematteo.duva2@unibo.it
†These authors have contributed equally to this work