
- 1 Cell Signalling Research Group, School of Biomedical Sciences, The Medical School, Queen’s Medical Centre, University of Nottingham, Nottingham, UK
- 2 CVGI iMED, AstraZeneca UK Ltd., Macclesfield, Cheshire, UK
Discovery of G protein coupled receptors for long chain free fatty acids (FFAs), FFA1 (GPR40) and GPR120, has expanded our understanding of these nutrients as signaling molecules. These receptors have emerged as important sensors for FFA levels in the circulation or the gut lumen, based on evidence from in vitro and rodent models, and an increasing number of human studies. Here we consider their promise as therapeutic targets for metabolic disease, including type 2 diabetes and obesity. FFA1 directly mediates acute FFA-induced glucose-stimulated insulin secretion in pancreatic beta-cells, while GPR120 and FFA1 trigger release of incretins from intestinal endocrine cells, and so indirectly enhance insulin secretion and promote satiety. GPR120 signaling in adipocytes and macrophages also results in insulin sensitizing and beneficial anti-inflammatory effects. Drug discovery has focused on agonists to replicate acute benefits of FFA receptor signaling, with promising early results for FFA1 agonists in man. Controversy surrounding chronic effects of FFA1 on beta-cells illustrates that long term benefits of antagonists also need exploring. It has proved challenging to generate highly selective potent ligands for FFA1 or GPR120 subtypes, given that both receptors have hydrophobic orthosteric binding sites, which are not completely defined and have modest ligand affinity. Structure activity relationships are also reliant on functional read outs, in the absence of robust binding assays to provide direct affinity estimates. Nevertheless synthetic ligands have already helped dissect specific contributions of FFA1 and GPR120 signaling from the many possible cellular effects of FFAs. Approaches including use of fluorescent ligand binding assays, and targeting allosteric receptor sites, may improve further pre-clinical ligand development at these receptors, to exploit their unique potential to target multiple facets of diabetes.
Introduction
Free fatty acids (FFAs) have traditionally been viewed as nutrients and metabolic substrates (Yaney and Corkey, 2003). However there is emerging evidence for direct signaling pathways activated by FFAs with key roles in physiology and pathology, particularly with respect to metabolic diseases such as type 2 diabetes. In common with many other lipid mediators, long chain FFAs are also ligands for the transcription factor family of peroxisome proliferator-activated receptors (PPARs), which for example regulate expression of genes involved in lipid metabolism (Varga et al., 2011). Within the last decade, deorphanization of several FFA binding G protein coupled receptors (GPCRs) has enhanced the spectrum of short and long term signaling pathways activated by these molecules (Stoddart et al., 2008; Hudson et al., 2011; Talukdar et al., 2011). As the largest family of cell surface receptors in man, GPCRs have proved tractable drug targets in the past (Overington et al., 2006). However it is perhaps less recognized that this clinical exploitation is currently restricted to around 5% of the known receptor proteins. The FFA GPCRs provide good illustrative examples of receptors whose therapeutic targeting might yield real benefits for patients, but for which there are also obstacles to characterizing their pre-clinical pharmacology and developing selective high affinity synthetic ligands and translating these findings through to the clinic. Chiefly these reside around the lipophilicity of both the ligands and receptor binding sites, and also understanding the complex interplay between the pleiotropic effects of FFAs – encompassing influences on cell membrane composition, metabolism, and actions on various receptors.
Here we illustrate these challenges by focusing on two GPCRs responsive to saturated and unsaturated FFAs with long (C12–C22) aliphatic chains (Figure 1). FFA1 (also known as GPR40) is a receptor which is related in amino sequence to medium and short chain FFA GPCRs FFA2 and FFA3 (Briscoe et al., 2003; Itoh et al., 2003; Stoddart et al., 2008; Hudson et al., 2011). GPR120 is a distant phylogenetic relative of FFA1 (Fredriksson et al., 2003; Ichimura et al., 2009), but interestingly has co-evolved a similar specificity for endogenous FFA ligands (Hirasawa et al., 2005; Talukdar et al., 2011). We review how the distribution and function of these receptors highlights them as novel targets, particularly for treatment of type 2 diabetes in which prevalence worldwide is set to escalate to 366 million by 2030 (Wild et al., 2004; Rennie and Jebb, 2005; Ogden et al., 2006). With some caveats, significant progress has been made in elucidating the signaling roles of these receptors in pancreatic β-cells, adipocytes, intestinal enteroendocrine cells, and elsewhere. In part this has been achieved by successful development of synthetic agonists and antagonists, and there are further opportunities for improved ligand development at these receptors in the future.
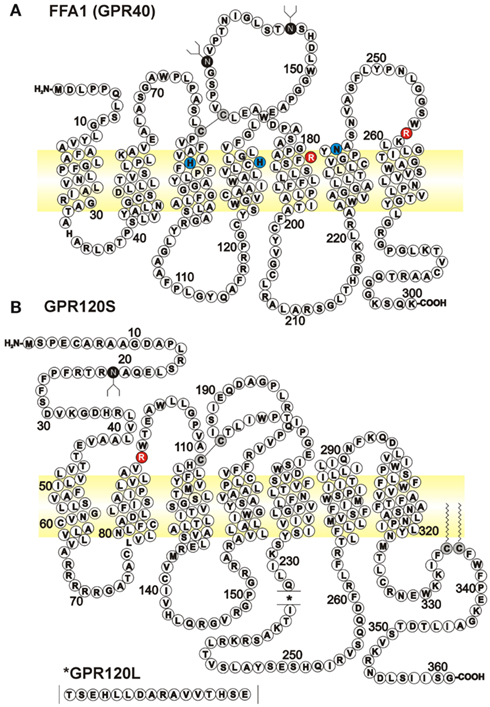
Figure 1. Amino acid sequences for human long chain FFA GPCRs. Diagrams represent FFA1 (A), GenBank NM_005303) and the short isoform of GPR120S [(B), BC101175] with the position of the 16 amino acid insert in GPR120L (NM_181745) indicated in the inset. Putative glycosylation sites are indicated on the extracellular Asn residues by white on black, while the conserved disulfide bridge from extracellular loop 2, and two unproven palmitoylation sites in GPR120, are also indicated by black on gray Cys residues. Basic Arg residues (white on red) have been implicated in recognizing the carboxylate anions of FFA and other agonists (Sum et al., 2007; Suzuki et al., 2008). A number of other residues have been identified as important for GW9508 binding to FFA1, some of which are indicated here (black on blue; Sum et al., 2007); however the specificity with which these mutations alter GW9508 binding, compared to more general effects on FFA1 activation has since been questioned (Smith et al., 2009).
Long Chain FFA GPCRs as Therapeutic Targets
FFA1 – A FFA Sensing Receptor in the Pancreatic β-Cell
FFA1 was the first long FFA receptor cloned and described in 2003, as a predominantly Gq/11 coupled receptor which responds to both saturated (e.g., palmitic acid, C16:0), mono-unsaturated (e.g., oleic acid, C18:1) and poly unsaturated long chain FFAs (e.g., docosahexaenoic acid, DHA), C22:6; Briscoe et al., 2003; Itoh et al., 2003; Kotarsky et al., 2003). It is also the FFA receptor for which the widest range of synthetic ligands are now available (Table 1; Figure 2), including both agonists (such as GW9508) and antagonists (e.g., GW1100, pKb 6.0; Briscoe et al., 2006; Garrido et al., 2006; Pfizer compound 15i; Humphries et al., 2009). In part the screening for novel FFA1 compounds was aided by the early discovery that certain thiazolidinedione (TZD) PPARγ ligands, such as rosiglitazone, were also agonists at FFA GPCRs (Kotarsky et al., 2003; Tan et al., 2008; Smith et al., 2009). Some of the novel FFA1 agonists (e.g., Tan et al., 2008; Zhou et al., 2010) are thus structurally based on the TZD backbone, while others arose from conformational restriction of the aliphatic FFA carbon backbone (e.g., Christiansen et al., 2008). Recent studies have also explored ways to improve the pharmacokinetic properties of both FFA1 agonist (Christiansen et al., 2011) and antagonist series (Humphries et al., 2009).
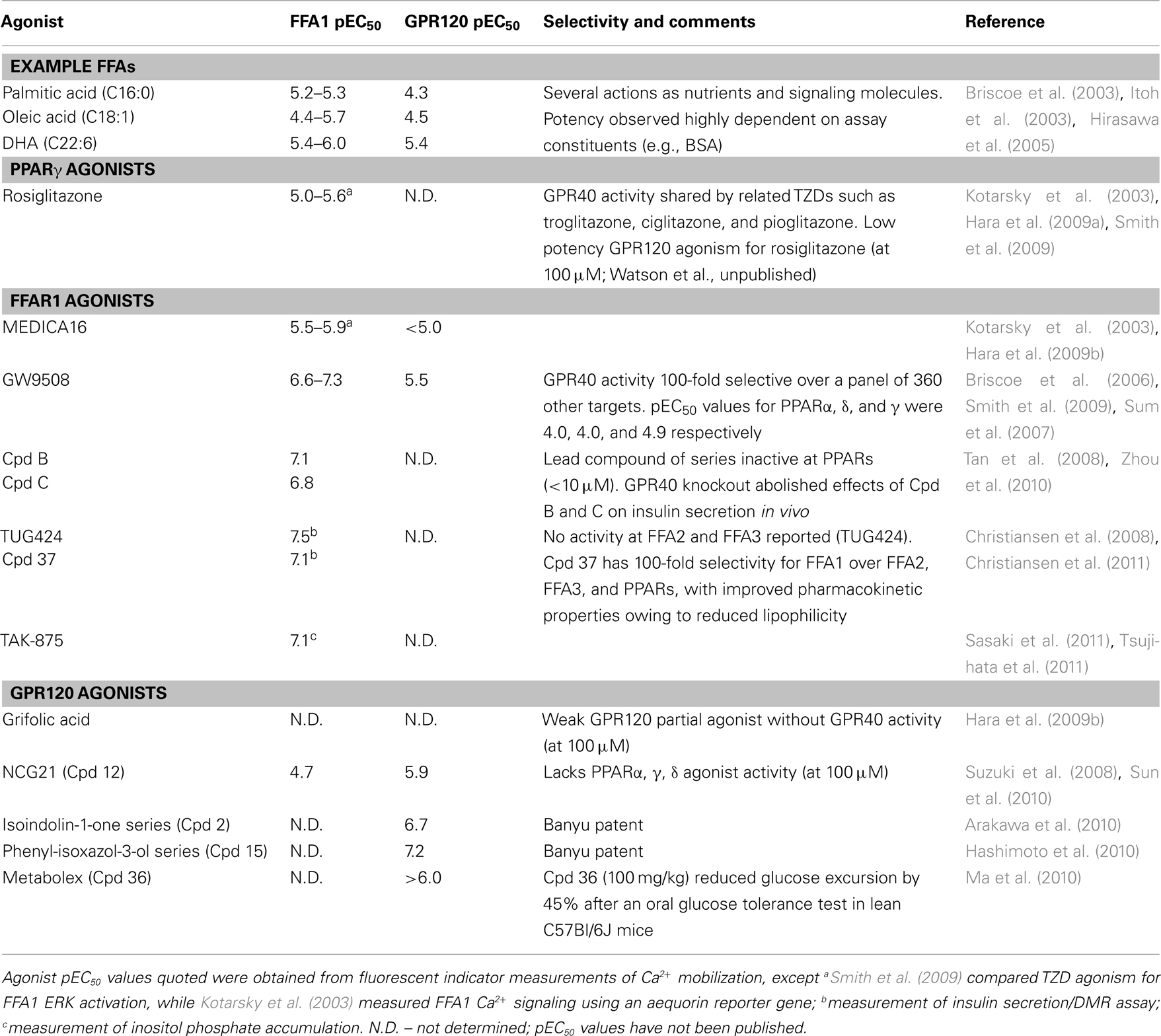
Table 1. Summary of long chain FFA GPCR agonist pharmacology.
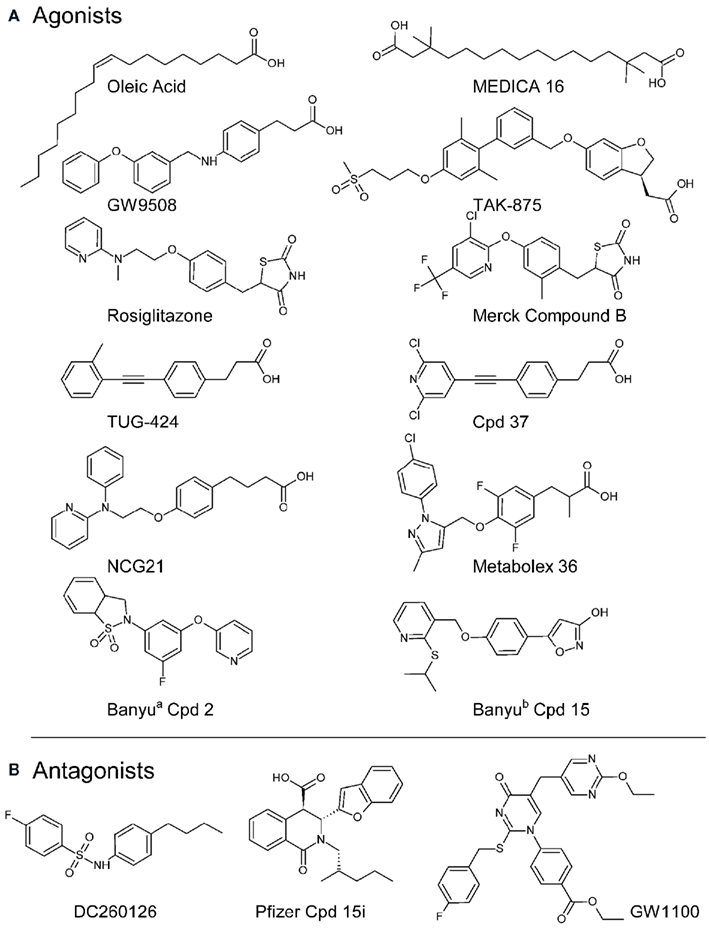
Figure 2. Chemical structures of example FFA GPCR agonists (A) and three reported FFA1 antagonists (B). Information on agonist pharmacology, with references, is provided in Table 1. The structure of Metabolex example 36 is reproduced from the relevant GPR120 agonist patent (Ma et al., 2010) and two Banyu compounds are shown from isoindolin-1-one derivativesa (cpd 2; Arakawa et al., 2010) and the phenyl-isoxazol-3-ol seriesb (cpd 15; Hashimoto et al., 2010). Both GW1100 (Briscoe et al., 2006), Pfizer compound 15i (Humphries et al., 2009), and DC260126 (Hu et al., 2009) inhibited agonist stimulated FFA1 receptor calcium responses in transfected cells with respective pIC50 values of 6.0, 7.7, and 6.0.
The predominant (though not exclusive) expression of FFA1 is in the pancreas and in particular β-cells (Briscoe et al., 2003; Itoh et al., 2003; Tomita et al., 2005, 2006). Several investigations have confirmed that FFA1 plays a crucial role in the short term stimulation of insulin secretion from the β-cell, including pharmacological, genetic knockout, or RNA interference knockdown approaches (Itoh et al., 2003; Steneberg et al., 2005; Briscoe et al., 2006; Latour et al., 2007; Tan et al., 2008; Alquier et al., 2009; Wu et al., 2010). Thus development of FFA1 agonists might be expected to be beneficial in type 2 diabetes treatment. However this contrasts with the well known relationship between long term circulating FFA levels and β-cell dysfunction (Yaney and Corkey, 2003). Elevated plasma FFA are common in type 2 diabetes and are linked with the onset of peripheral and hepatic insulin resistance and are also suggested to represent a critical link between insulin resistance and β-cell dysfunction (Boden and Shulman, 2002). The contribution of FFA1 to such chronic deleterious effects of long chain FFAs has largely been addressed by knockout and transgenic animal models, and has proved somewhat controversial. Using FFA1 knockout mice, Steneberg et al. (2005) demonstrated the importance of FFA1 for acute insulin secretion, but also showed that the absence of FFA1 protected mice on a high fat diet (HFD) from a number of characteristics associated with type 2 diabetic phenotype, such as obesity-induced hyperinsulinemia, glucose intolerance, and hypertriglyceridemia. Conversely islet cell specific transgene expression of FFA1 led to a number of indicators of β-cell dysfunction (Steneberg et al., 2005). Recent in vivo data using a small molecule antagonist DC260126 at least in part supports these observations, and makes the case for FFA1 antagonists, rather than agonists, as clinically relevant ligands for type 2 diabetes treatment (Hu et al., 2009; Zhang et al., 2010). However, other studies on FFA1 knockout mice have failed to replicate the original findings, suggesting that these animals are as susceptible as wild type littermates to the adverse consequences of a HFD (Latour et al., 2007; Kebede et al., 2008; Lan et al., 2008; Tan et al., 2008). Indeed one investigation reported a beneficial result of transgenic FFA1 overexpression in improved glucose tolerance (Nagasumi et al., 2009). Some of the discrepancies in these results may be down to the precise experimental conditions used, but they are also indicative of the complex multiple mechanisms, even at the cellular level, which generate FFA responses. Given our current understanding of FFA1 signaling, in which many different FFAs appear equi-effective agonists (Briscoe et al., 2003; Itoh et al., 2003), it is unlikely that this receptor can be solely responsible for long term mechanisms which differ substantially between pro-apoptotic effects of saturated FFAs and protective actions of unsaturated FFAs (Dhayal et al., 2008). Moreover constitutive knockout and transgenic mouse studies cannot report the effects of FFA1 in complete isolation from other FFA metabolic and nuclear receptor signaling cascades. For example, cross talk between FFA1 and other receptor pathways is clearly evident from the changes in PPAR and lipid handling gene transcription resulting from FFA1 overexpression (Steneberg et al., 2005). Knockout studies also involve wider loss of FFA1 from other tissues, where its physiological functions are currently less clear – for example in glucagon producing pancreatic cells (Flodgren et al., 2007), intestinal enteroendocrine cells (Edfalk et al., 2008), taste buds (Cartoni et al., 2010), and neurons (Briscoe et al., 2003; Ma et al., 2007; see GPR120 agonists – a multi pronged attack on type 2 diabetes? below).
The vast majority of FFA1 studies have used in vitro or in vivo animal models, but its expression and function in human isolated islets suggests applicability of these findings to man (Tomita et al., 2005, 2006; Vettor et al., 2008). There are also interesting links between the prevalence of FFA1 coding polymorphisms with insulin secretory capacity in a male cohort (R211H; Ogawa et al., 2005), or with obese individuals at risk from diabetes (G180S; Vettor et al., 2008). However thus far there is little published information on GPR40 ligands from clinical trials. Pre-clinically a number of pharmaceutical companies have reported effective compounds in various models of T2DM (Negoro et al., 2010; Herling et al., 2011; Jagannath et al., 2011; Sasaki et al., 2011; Tsujihata et al., 2011). Clinically the most advanced GPR40 agonists are developed by Takeda. Initial PK data in healthy volunteers on the Takeda pharmaceuticals agonist TAK-875 demonstrated good tolerability and pharmacokinetic properties suitable for a once-daily regimen (Naik et al., 2011). Excitingly recent data presented at the American Diabetes Association conference demonstrated a dose-dependent decrease in HbA1c in patients treated with TAK-875 for 12 weeks with an efficacy similar to glimepiride but a significantly reduced incidence of hypoglycemia (Viswanathan et al., 2011). Therefore whilst a requirement for antagonists to block the pancreatic effects of chronically elevated FFAs still remains an untested hypothesis, there is mounting evidence that an FFA1 agonist will produce significant benefits to type 2 diabetes patients.
GPR120 Agonists – A Multi Pronged Attack on Type 2 Diabetes?
The second long chain FFA receptor, GPR120, demonstrates a similar ligand specificity to FFA1 in its activation by C12–C22 saturated and unsaturated FFAs (Hirasawa et al., 2005) – though ω3 polyunsaturated FFAs, such as DHA, have often been the focus as endogenous ligands (Oh et al., 2010; Talukdar et al., 2011). GPR120 is activated by some synthetic agonists (Table 1; Figure 2) designed for PPAR or FFA1 receptors, but with lower potency, such as GW9508 (Briscoe et al., 2006) or MEDICA16 (Kotarsky et al., 2003; Hara et al., 2009b). GW1100 is relatively selective as an antagonist for FFA1 over GPR120 (Briscoe et al., 2006). Conversely members of the compound series from a TZD derivative reported by Suzuki et al. (2008) are ∼10-fold more potent at GPR120 than FFA1 and display little PPAR activity (e.g., NCG21; Table 1); other compounds from the same group have yet to be fully characterized (Sun et al., 2010). Grifolic acid has also been suggested as a low potency, and relatively low efficacy, GPR120 partial agonist (Hara et al., 2009b). Finally, the structures of additional GPR120 agonists, without a full description of their pharmacology, have entered the public domain through patent applications (Arakawa et al., 2010; Hashimoto et al., 2010; Ma et al., 2010; Figure 2; Table 1). Interestingly both compound series revealed in the Banyu patents indicate that a carboxylic acid group is dispensable for generating GPR120 agonists with sub micromolar potency (Table 1; Arakawa et al., 2010; Hashimoto et al., 2010).
Like FFA1, GPR120 appears mainly coupled to Gq/11 proteins leading to intracellular Ca2+ mobilization, and also stimulates protein kinases such as extracellular signal related kinase (ERK) and Akt (Hirasawa et al., 2005; Katsuma et al., 2005; Oh et al., 2010). However GPR120 is also phosphorylated after agonist stimulation, and recruits β-arrestin adaptors responsible for desensitization, internalization, and G protein independent signaling pathways (Hirasawa et al., 2005; Burns and Moniri, 2010; Oh et al., 2010). A further complication, compared to FFA1, is that human GPR120 has two splice variants which differ in their coding regions (Fredriksson et al., 2003) – the “short” isoform (GPR120S; GenBank accession number BC101175) contains 361 residues, whilst the “long” isoform (GPR120L; NM_181745) contains 16 additional amino acid residues between positions 231 and 247 in intracellular loop 3 (ICL3; Figure 1). Early studies cloned and examined the pharmacology of GPR120L (Hirasawa et al., 2005; Briscoe et al., 2006), but more recent investigations suggest that the short isoform homolog may predominate in other primate and rodent species (Tanaka et al., 2008b; Moore et al., 2009). Data from our laboratories (Watson et al., in preparation) suggest clear differences in the intracellular signaling properties of the two isoforms, with human GPR120L impaired in G protein dependent but not arrestin dependent signaling. Thus it will be important in future to confirm the GPR120 splice variant involved in the cell type specific responses described below.
In common with FFA1, GPR120 is an anti-diabetic drug target but the focus has been on its indirect effects – on insulin secretion and insulin resistance – through its expression in enteroendocrine cells, adipocytes, and immune cells (Miyauchi et al., 2009; Talukdar et al., 2011). GPR120 is localized to intestinal enteroendocrine cells, such as colonic L cells, and its stimulation releases incretin hormones such as glucagon like peptide 1 (GLP1) and cholecystokinin (CCK) into the circulation (Hirasawa et al., 2005; Tanaka et al., 2008a). These peptides have beneficial actions in stimulating insulin secretion, and in the context of obesity related diabetes, they also promote satiety. From a drug discovery point of view, targeting colonic endocrine cells with GPR120 agonist may have practical advantages, in that the receptors in these cells sense luminal, rather than circulating FFAs. Thus theoretically, orally administered GPR120 drugs would not require absorption into the systemic circulation for this therapeutic action although this does depend on the luminal/apical distribution of the receptors.
Although incretin hormone secretion was originally described as a GPR120 specific effect (Hirasawa et al., 2005), it is clear that FFA1 is also co-expressed in several types of insulin endocrine cell and can exert similar actions in vitro and in vivo (Edfalk et al., 2008; Liou et al., 2011). RT-PCR studies have identified both GPR120 and FFA1 mRNAs in intestinal endocrine cell lines such as STC-1 (Hirasawa et al., 2005). While not demonstrating co-localization of the receptors at the single cell level, this complicates the assignment of a particular effect to one FFA receptor in the absence of highly selective tool compounds. Notably the majority of studies that have indicated the significance of FFA1 in β-cells, or GPR120 in colonic L cells, do so by genetic or knockdown approaches which manipulate receptor expression – in general such evidence suggests the target receptor is necessary, rather than sufficient, for a particular response. This evidence leaves open the possibility that both receptors act in concert to produce their physiological effects in these cell types, through signaling cross talk or perhaps closer association as a GPCR heterodimer (Pin et al., 2007). FFA1 and GPR120 are also both detected by RT-PCR in taste buds, and may act thus act as “preference” sensors for dietary FFAs (Matsumura et al., 2009; Cartoni et al., 2010). Though yet to be fully tested, this raises a possibility of anti-obesity drugs which target such chemosensors to modulate appetite for fat containing diets (Dramane et al., 2011). This is of course an area that could be further exploited by the food industry and if this hypothesis is proved correct one could envisage a future application for FFA receptor ligands as a satiety factor in food with limited fat content.
More recently independent roles for GPR120, not shared by FFA1, have been proposed due to its expression in differentiated adipocytes and macrophages (Gotoh et al., 2007; Oh et al., 2010). In adipocytes, GPR120 activation increased glucose uptake and adipogenesis, for example in response to circulating ω3 polyunsaturated fatty acids, and GPR120 signaling in macrophages exerted anti-inflammatory actions (Gotoh et al., 2007; Oh et al., 2010; Talukdar et al., 2011). Similar in vivo dual effects of ω3 FFAs, acting via GPR120, were confirmed through comparison of wild type and GPR120 knockout mice (Oh et al., 2010). Thus as novel treatments for type 2 diabetes, GPR120 agonists might be unique in improving insulin sensitivity while also reducing the “metabolic” inflammation implicated in the disease pathogenesis (Talukdar et al., 2011). Oh et al. (2010) also revealed that GPR120 responses in adipocytes and macrophages relied on distinct cell signaling mechanisms. In adipocytes, its enhancement of glucose uptake was wholly dependent on Gq/11 protein activation, while its anti-inflammatory effect required β-arrestin adaptor proteins. This requirement for different GPR120 signaling cascades raises interesting questions about whether such pathways can be activated selectively by agonist-GPR120 complexes in different cell types – through selective expression of different receptor splice variants, or therapeutically, through the use of functionally “biased” GPR120 ligands (Rajagopal et al., 2010).
Our knowledge of GPR120 pharmacology, and availability of tool compounds to dissect it, is thus less advanced compared to FFA1, particularly given the lack of reported antagonists for GPR120. However the expression pattern and known functions of this receptor suggest that GPR120 agonists may have distinct synergistic actions in different tissues that offer a multi faceted approach to mitigate the causes and symptoms of type 2 diabetes. Indeed it can be argued that such agonists are already marketed as nutritional supplements. For example the case has been made for orally administered fish oil poly unsaturated fatty acids (PUFAs), such as DHA, acting physiologically at GPR120 (Oh et al., 2010; Talukdar et al., 2011). Other beneficial dietary FFAs, such as conjugated linoleic acid, are also FFA GPCR agonists (Schmidt et al., 2011). Given the apparent lack of selectivity of FFA GPCRs for a range of “good” and “bad” long chain FFAs, it will be necessary to test the extent to which the specific benefits of PUFAs derive from binding FFA1 or GPR120 receptors. As an alternative, the possibility of targeting GPR120 with synthetic ligands is now supported by emerging data from patents filed by Banyu and Metabolex (Arakawa et al., 2010; Hashimoto et al., 2010; Ma et al., 2010). These companies have described potent small molecule GPR120 agonists (Table 1; Figure 2) with good oral bioavailability, which increased glucose-stimulated insulin secretion in mouse models representing both lean and disease model states (Hashimoto et al., 2010; Ma et al., 2010). A more thorough characterization of GPR120 function elsewhere – in taste buds, lung tissue (Miyauchi et al., 2009), and bone osteoblasts and osteoclasts (Cornish et al., 2008) – may also lead to new therapeutic avenues, together with an increased awareness of potential side effects.
Experimental Challenges in Understanding Long Chain FFA Receptor Pharmacology
Pleiotropic Actions of FFAs
As illustrated in the previous sections, one of the key controls for FFA1 or GPR120 investigations is to demonstrate that the response is specific for that receptor. There is potential for the involvement of additional FFA GPCRs, other fatty acid binding proteins and PPAR nuclear receptors, and effects as metabolism substrates (Yaney and Corkey, 2003; Hudson et al., 2011; Varga et al., 2011). As their rapid metabolism by β-oxidation indicates, the lifespan of endogenous FFA ligands during incubations is relatively short, and their metabolites may have their own independent actions. In addition chronic changes in the quality and quantity of FFAs in the plasma lead to changes plasma membrane composition and fluidity. This has the capacity to influence several membrane signaling pathways indirectly (Calder, 2008; Gawrisch et al., 2008), including those of other GPCRs such as the ghrelin receptor (Delhanty et al., 2010). Polyunsaturated FFAs in particular (e.g., arachidonic acid, DHA) are themselves converted into new signaling mediators such as the prostaglandins and the resolvins, with their own receptors (Serhan et al., 2011).
The increased pharmacological toolbox of synthetic ligands, which lack some of the endogenous FFA effects, can now help in isolating FFA GPCR actions. Limited specificity (e.g., GW9508 for FFA1 versus GPR120), relatively low affinity, and possible unknown pharmacological actions (e.g., at PPARs; Table 1) of these compounds must still be borne in mind when assessing their effects. Isolating responses mediated by FFA GPCRs might also be aided by consideration that rapid signaling events are expected from these cell surface receptors (e.g., intracellular calcium mobilization), though fast non-genomic actions of PPARs have also been reported, prior to real recognition of the possible existence of FFA GPCRs (Gardner et al., 2005). In recombinant cell lines, clean systems for assessing FFA receptor pharmacology can be demonstrated by comparison with non-transfected cells, or by receptor expression through an inducible promoter system such as that controlled by the tetracycline repressor protein (Stoddart et al., 2007; Smith et al., 2009). In endogenously expressing cell lines or primary cells siRNA or shRNA transfection can perform a similar function, with careful controls to demonstrate the specificity and magnitude of the receptor knockdown achieved (Itoh et al., 2003; Hirasawa et al., 2005; Tanaka et al., 2008a; Wu et al., 2010). In future the delineation of FFA1 and GPR120 effects in vivo using receptor knockout mice (Katsuma et al., 2005; Steneberg et al., 2005; Latour et al., 2007; Lan et al., 2008; Oh et al., 2010), might be improved by conditional or tissue-specific gene deletions – for example to avoid developmental compensation through alternative signaling pathways.
Lipophilicity and Endogenous FFAs
Poor aqueous solubility of FFAs, together with the requirement for relatively high concentrations (μM) to fully define FFA1 and GPR120 concentration response relationships(Briscoe et al., 2003; Itoh et al., 2003; Hirasawa et al., 2005), presents practical problems in in vitro signaling assays. Solubility can be aided by inclusion of solvents such as DMSO, but the solvent concentration required often leads to non-specific effects and cellular toxicity. Alternatively fatty acid free bovine serum albumin (BSA) can be included in the assay buffer, which replicates its role as a fatty acid carrier protein in plasma. While improving solubility, this has a disadvantage in reducing the availability of FFAs and thus the observed potency in stimulating FFA1 or GPR120 signaling (Itoh et al., 2003; Hirasawa et al., 2005; Stoddart et al., 2007). The affinity of BSA for saturated and unsaturated FFAs differs, which may also generate indirect differences in FFA GPCR activity. Consequently apparent changes in potency for FFA at their cognate receptors could be due to a complex relationship between affinity differences for BSA and the receptor. If BSA is included its potential ability to bind synthetic ligands under investigation must also be considered. More widely, the high lipophilicity of most current FFA GPCR drugs can lead to problematic pharmacokinetic properties when taking these compounds forward in vivo. However a recent investigation has shown for one FFA1 agonist series, this issue can be addressed by reducing lipophilicity whilst retaining in vitro activity (Christiansen et al., 2011).
A second consideration for in vitro assays is the likelihood that long chain FFAs are present in most standard serum-containing cell culture media, and may also be produced endogenously by the cell lines themselves. This tone may influence FFA GPCR expression (e.g., in transfected cells) and signaling behavior in the long term, and potentially the assay activity of “competing” exogenously added ligands. The demonstration that “constitutive” (i.e., agonist-independent) activity of FFA1 in fact derives from endogenous FFA stimulation provides a notable illustration of this problem (Stoddart et al., 2007). Such effects can be minimized by the use of inducible expressing cell lines, the addition of fatty acid free BSA as a sink for endogenous FFAs (Stoddart et al., 2007) or dialysis of cell culture serum.
Hydrophobic FFA Receptor Binding Sites
The FFA carboxylate anion is essential for endogenous agonist activity at FFA1 and GPR120, with corresponding FFA esters being inactive (Itoh et al., 2003; Hirasawa et al., 2005). This negative charge is a shared characteristic of most synthetic agonists, and may be substituted in TZD derivatives by the 2,4-dione moiety (Figure 2). Mutagenesis studies have identified co-ordinating basic residues at the top of transmembrane domains V (Arg 183) and VII (Arg 258) essential for FFA1 activation by FFAs, GW9508, and TZDs (Sum et al., 2007; Smith et al., 2009). An equivalent amino acid in upper transmembrane domain II of GPR120 (Arg 99) has also been suggested (Suzuki et al., 2008; Figure 1). The location of these residues suggests that for both receptors, the lipophilic portions of endogenous FFAs and synthetic agonists extend into a hydrophobic binding pocket within the transmembrane domain bundle (see modeling studies Sum et al., 2007 and Sun et al., 2010). Some additional contacts (His98, His137, Asn244), present only in FFA1 (Figure 1), have been suggested to contribute to GW9508 binding (Sum et al., 2007; Tikhonova et al., 2007), and this may provide one explanation for its selectivity over GPR120 (∼100-fold) as an agonist. However a more recent investigation questions whether His137 and Asn244 mutations are specific for particular FFA agonists (Smith et al., 2009). Such discrepancies highlight the problems of interpreting the effects of binding mutations from FFA1 functional assays (see Determining Ligand Affinity at FFA1 and GPR120 and Functional Assessment of FFA1 and GPR120 Pharmacology below), which may represent convolved influences on ligand affinity, efficacy, or indeed a general ability of receptors to undergo conformational change to an active state. Thus the overall agonist binding pocket is rather ill defined for both receptors in molecular terms, other than knowledge of the ligand size (e.g., chain length) required to occupy it. Its reliance on hydrophobic contacts presents potential challenges for generating high affinity selective ligands that distinguish FFA1 from GPR120, or indeed as the dual activity of TZDs demonstrates, from similar binding sites in PPAR receptors.
As an alternative it may be preferable to consider the development of allosteric modulators for these receptors as a strategy. Such compounds use alternative binding sites to influence receptor activity in the presence or absence of the endogenous ligand. They are released from the structural constraints imposed by the orthosteric site, and because allosteric binding pockets are receptor specific, they may also offer improved selectivity (May et al., 2007; Smith and Milligan, 2010). Such compounds have been already identified for short chain FFA receptor FFA2 (Lee et al., 2008; Smith and Milligan, 2010; Smith et al., 2011). A recent US patent filed by Amgen Inc (Brown et al., 2010) suggests that similar allosteric ligands may also have the potential to impact the biology and future drug development possibilities for FFA1 receptors.
Determining Ligand Affinity at FFA1 and GPR120
The moderate (high nM–μM) perceived affinity of FFA receptor ligands, their lipophilic nature, and potential lack of target selectivity, have all impeded the development of successful probes for radioligand binding assays – the staple screening assay for pharmacologists to estimate compound affinity, in terms of its equilibrium dissociation constant Kd, and thus build basic structure activity relationships (SARs). There is one published radioligand binding report (using a tritiated ligand from the TAK-875 compound series; Negoro et al., 2010), which together with Amgen patent information (Brown et al., 2010) indicates that FFA1 binding assays may be achievable with the right probe. In our hands the high level of non-specific binding present with radiolabeled lipophilic ligands presents a key challenge (Brown et al., unpublished observations). Two other binding techniques have developed enhanced specificity for the ligand–receptor interaction under study. Bartoschek et al. (2010) generated FFAR1 binding data from saturation transfer difference 1H nuclear magnetic resonance (NMR) measurements, in which NMR spectra are obtained only when ligands bind a macromolecular complex, such as a receptor protein. They showed that this method allowed estimation of agonist IC50 values from competition binding experiments, which displayed close correspondence with functional potencies. Second a flow cytometry based assay used the fluorescent FFA BODIPY-C12 as a binding probe to immunoprecipitated FFA1 (Hara et al., 2009a). TZDs and GW9508 (with lower than expected affinity) were able to displace BODIPY-C12 binding specifically. As a more widespread screening assay it is likely that this approach would still require improvements in the ratio of specific to non-specific binding, and confirmation that FFAs bound the inside, rather than the exterior of the solubilized FFA1 protein. However this study does highlight the opportunities for using fluorescent FFA GPCR ligands in future, perhaps improving the specificity for the detection of ligand–receptor interaction using techniques such as TR-FRET (Zwier et al., 2010).
In the absence of robust binding techniques, an alternative approach, employed for GPR120, has been to model binding interactions in silico by molecular docking simulations (Sun et al., 2010). The success of such homology modeling depends on the structural match between the crystal template (in this case, of bovine rhodopsin) and the receptor under study, in the confirmation required (e.g., agonist active state). Differences between the limited crystal structures available for closely related rhodopsin-like GPCRs suggests that this relationship cannot always be guaranteed (Kobilka, 2011). Nevertheless, Sun et al. (2010) were able to use homology modeling to predict the binding energies and SAR of a novel compound series of GPR120 agonists – predictions which were borne out by functional measurements of agonist activity.
Functional Assessment of FFA1 and GPR120 Pharmacology
Pharmacological analysis of FFA receptors must currently do without direct assessments of ligand affinity, but a wide range of functional signaling assays is possible in cells, as for other GPCRs (see overview in Figure 3). Consideration should first be given to the species variant of FFA receptor under investigation and the impact this may have on ligand pharmacology. For example the two splice variants of human GPR120 have already been discussed above (GPR120 agonists – a multi pronged attack on type 2 diabetes?), and it is currently unknown whether the longer isoform is present in other primates or rodents (Tanaka et al., 2008b; Moore et al., 2009). Initial investigations derived similar agonist pharmacology for human, rat, and mouse FFA1 receptors (Itoh et al., 2003). However heterologous expression of mouse FFA1 in Xenopus oocytes suggests that this species variant is also responsive to short and medium chain FFAs (greater than C4; Stewart et al., 2006). Potentially this might imply unique features for the mouse FFA1 orthosteric binding site. Thus far, synthetic agonist pharmacology derived from murine cell lines (e.g., insulin secreting MIN-6 cells, incretin secreting STC-1 cells; Hirasawa et al., 2005; Katsuma et al., 2005; Briscoe et al., 2006) or in vivo models has provided broadly consistent with human receptor data.
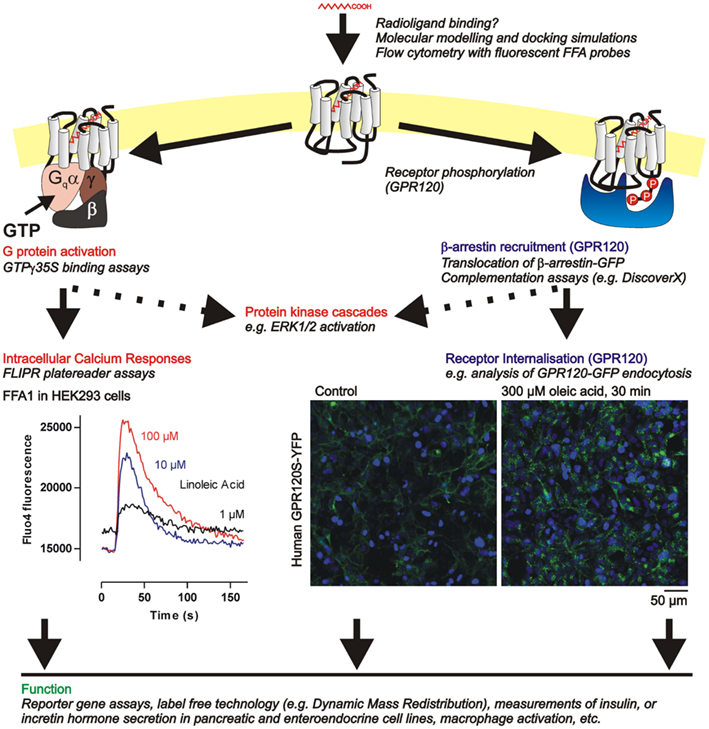
Figure 3. Summary of functional assays available to investigate FFA G protein coupled receptor pharmacology in cell lines. The indirect approaches to assessing ligand binding indicated here are discussed further in Section “Determining Ligand Affinity at FFA1 and GPR120.” Several signaling endpoints related to the G protein (FFA1, GPR120) or β-arrestin pathways (GPR120) can be measured using the example assays in italics (see text, Functional Assessment of FFA1 and GPR120 Pharmacology) culminating in assessment of function at the cellular level. Example data from our own laboratory illustrates (i) calcium responses in HEK293 cells expressing human GPR40 and stimulated with the C18:2 FFA linoleic acid, and (ii) internalization of human GPR120S–YFP receptors (green), following vehicle or oleic acid treatment, also in transfected HEK293 cells. In the image panels nuclei are counterstained with the dye H33342 (blue).
Functional assays provide SAR data for agonists in terms of their potency (e.g., pEC50 values) and maximal responses (Emax). It should be noted that these are empirical parameters that depend both on the affinity and efficacy of the agonist ligands, together with assay dependent parameters such receptor density and the extent of signal amplification at the endpoint measured. Null methods (e.g., Schild analysis) do provide opportunities to estimate antagonist dissociation constants, and determine whether their mode of binding is competitive and reversible – for example for GW1100 (Briscoe et al., 2006). However it is still more common to quote inhibitory IC50 values for antagonists, which will also depend on the agonist concentration and the assay system chosen (Briscoe et al., 2006; Hu et al., 2009; Humphries et al., 2009).
GTPγ35S binding assays have been successfully employed to investigate FFA1 pharmacology, with the proviso that receptor-Gq/11α fusion proteins and immunoprecipitation of the GTPγ35S bound complexes are required to enhance the signal (Stoddart et al., 2007; Smith et al., 2009). This technique has two distinct advantages for assessing pharmacology. Measurement is at the first stage of the receptor signaling pathway (activation of the G protein), resulting in less signal amplification and a better discrimination, through changes in Rmax, of agonists with differing efficacy. Second ligands are allowed to bind in a pre-incubation before G protein activation is assessed by addition of GTPγ35S, and ensures equilibrium conditions in competition experiments, for example when assessing antagonist or allosteric modulator function (Stoddart et al., 2007).
Both FFA1 and GPR120 couple to calcium mobilization, allowing routine high throughput assessment of function through fluorescence based measurements of intracellular calcium concentration (Briscoe et al., 2003, 2006; Itoh et al., 2003; Hirasawa et al., 2005; Sun et al., 2010). It should be borne in mind that these are non-equilibrium kinetic assays and thus they can be influenced by the rates of ligand association and dissociation (which can be slow for FFAs), as well as agonist affinity and efficacy. Early investigations using human GPR120 reported that calcium mobilization required fusion to a promiscuous G16α (Hirasawa et al., 2005) or chimeric Gqi5α protein (Ma et al., 2010), or high efficiency viral transfection (Briscoe et al., 2006), but in retrospect this may be a particular consequence of reduced G protein coupling efficiency of the long, compared to the short isoform (Tanaka et al., 2008b; Moore et al., 2009; Watson et al., in preparation). Responses assessed via ERK stimulation (Itoh et al., 2003; Hirasawa et al., 2005; Smith et al., 2009) or reporter gene activation (Briscoe et al., 2003, 2006) should also be feasible given the current understanding of effector coupling of these receptors.
Given the specific involvement of the β-arrestin signaling pathway in GPR120 anti-inflammatory responses, inclusion of related readouts may be advisable. Alternative β-arrestin recruitment assays measure interaction between modified receptor and arrestin proteins using bioluminescence resonance energy transfer (BRET), protease based systems (Tango, Invitrogen Life Technologies, Grand Island, NY, USA) or β-galactosidase based complementation assay (DiscoverX, Fremont, CA, USA). Direct analysis of β-arrestin-GFP translocation or downstream receptor-GFP internalization is also possible (Hirasawa et al., 2005; Oh et al., 2010). Increasingly it is recognized that dual assessment of both G protein and β-arrestin dependent responses has the potential to reveal functionally selective ligands that discriminate between these pathways (Rajagopal et al., 2010). Our own data (Watson et al., in preparation), based on quantitative imaging assessment of arrestin recruitment and internalization (Kilpatrick et al., 2010), indicates that both GPR120 isoforms, but not FFA1, engage this pathway effectively.
Finally measurements of function in cells (e.g., insulin, or incretin hormone secretion; Itoh et al., 2003; Hirasawa et al., 2005; Briscoe et al., 2006) allow closer correspondence with the physiological actions expected in subsequent in vivo assays, for particular ligands. This is exemplified by recent patent data supporting a role for GPR120 small molecule agonists in glucose-stimulated insulin secretion in isolated rat islets (Ma et al., 2010). Within this category label free technologies, such as dynamic mass redistribution (DMR), also offer sensitive measurements that can be applied to endogenous receptors in living cell lines or primary cells, without modification by labeling reagents or reporter transfections – and in a high throughput 384 well plate format. DMR technology uses an optical biosensor that measures the perpendicular movement of cellular constituents. It directs broadband light into the bottom of the plate, and ligand induced movement of cellular contents results in a shift in the wavelength of the light that is directed back into the sensor (Fang et al., 2006). This shift in wavelength is measured, and in the case of GPCRs, gives a readout that is thought to be dependent upon the G protein coupling of the receptor in question (Schröder et al., 2010). Schröder et al. (2010) demonstrated the use of DMR to determine agonist pharmacology and Gq/11 coupling signature of FFA1, and other studies have now characterized novel FFA1 agonists (TUG424, conjugated linoleic acid isomers) by this technique (Christiansen et al., 2008; Schmidt et al., 2011). We have shown that this system is also applicable to study GPR120 activation (Watson et al., unpublished observations).
Conclusion
FFA1 and GPR120 have emerged as receptors with key roles to play in the sensing of circulating and intestinal long chain FFAs in pancreatic β-cells, intestinal endocrine cells and elsewhere. In vitro and animal model data highlight a unique potential for co-ordinated benefits of synthetic ligands at these receptors, by counteracting multiple inflammatory and metabolic factors contributing to the development of type 2 diabetes. Promising initial data suggests that FFA1 agonists may be effective in man, and we await similar human studies, predicted from the recent patent applications, which examine GPR120 agonists. There is some uncertainty over the long term effects of FFA1 and GPR120 signaling, and this still leaves open the best choice of therapeutic ligand – agonist or antagonist. As drug targets, the lipophilic orthosteric binding sites of FFA1 and GPR120 present challenges for developing high affinity, selective orthosteric ligands, using the functional assays currently available. However future drug discovery at these receptors will be aided by expanding knowledge of receptor structure and the advent of novel fluorescent ligand binding approaches. Perhaps there will also be possibilities for improved ligand specificity at these receptors, using allosteric modulators or “biased” ligands which functionally select particular FFA signaling pathways.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Leigh Stoddart for constructive comments on the manuscript. Sarah-Jane Watson is a postgraduate student sponsored by the UK Engineering and Physical Sciences Research Council, and work in NDHs laboratory on FFA receptors has been funded by the Royal Society UK and AstraZeneca.
References
Alquier, T., Peyot, M. L., Latour, M. G., Kebede, M., Sorensen, C. M., Gesta, S., Ronald Kahn, C., Smith, R. D., Jetton, T. L., Metz, T. O., Prentki, M., and Poitout, V. (2009). Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 58, 2607–2615.
Arakawa, K., Nishimura, T., Sugimoto, Y., Takahashi, H., and Shimamura, T. (2010). Novel isoindolin-1-one derivative. International Patent WO 2010/104195.
Bartoschek, S., Klabunde, T., Defossa, E., Dietrich, V., Stengelin, S., Griesinger, C., Carlomagno, T., Focken, I., and Wendt, K. U. (2010). Drug design for G-protein-coupled receptors by a ligand-based NMR method. Angew. Chem. Int. Ed. Engl. 49, 1426–1429.
Boden, G., and Shulman, G. I. (2002). Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and β-cell dysfunction. Eur. J. Clin. Invest. 32(Suppl. 3), 14–23.
Briscoe, C. P., Peat, A. J., Mckeown, S. C., Corbett, D. F., Goetz, A. S., Littleton, T. R., Mccoy, D. C., Kenakin, T. P., Andrews, J. L., Ammala, C., Fornwald, J. A., Ignar, D. M., and Jenkinson, S. (2006). Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br. J. Pharmacol. 148, 619–628.
Briscoe, C. P., Tadayyon, M., Andrews, J. L., Benson, W. G., Chambers, J. K., Eilert, M. M., Ellis, C., Elshourbagy, N. A., Goetz, A. S., Minnick, D. T., Murdock, P. R., Sauls, H. R. Jr., Shabon, U., Spinage, L. D., Strum, J. C., Szekeres, P. G., Tan, K. B., Way, J. M., Ignar, D. M., Wilson, S., and Muir, A. I. (2003). The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303–11311.
Brown, S. P., Dransfield, P. J., Heuze, J., Kohn, T. J., Liu, J., Medina, J., Pateropomg, V., Shen, W., Vimolratana, M., Wang, Y., Lu, M., and Zhu, L. (2010). Conformationally constrained carboxylic acid derivatives useful for treating metabolic disorders. US Patent 2010/0298367A1.
Burns, R. N., and Moniri, N. H. (2010). Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem. Biophys. Res. Commun. 396, 1030–1035.
Calder, P. C. (2008). The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot. Essent. Fatty Acids 79, 101–108
Cartoni, C., Yasumatsu, K., Ohkuri, T., Shigemura, N., Yoshida, R., Godinot, N., Le Coutre, J., Ninomiya, Y., and Damak, S. (2010). Taste preference for fatty acids is mediated by GPR40 and GPR120. J. Neurosci. 30, 8376–8382.
Christiansen, E., Urban, C., Grundmann, M., Due-Hansen, M. E., Hagesaether, E., Schmidt, J., Pardo, L., Ullrich, S., Kostenis, E., Kassack, M. U., and Ulven, T. (2011). Identification of a potent and selective free fatty acid receptor 1 (FFA1/GPR40) agonist with favorable physicochemical and in vitro ADME properties. J. Med. Chem. 54, 6691–6703.
Christiansen, E., Urban, C., Merten, N., Liebscher, K., Karlsen, K. K., Hamacher, A., Spinrath, A., Bond, A. D., Drewke, C., Ullrich, S., Kassack, M. U., Kostenis, E., and Ulven, T. (2008). Discovery of potent and selective agonists for the free fatty acid receptor 1 (FFA(1)/GPR40), a potential target for the treatment of type II diabetes. J. Med. Chem. 51, 7061–7064.
Cornish, J., MacGibbon, A., Lin, J. M., Watson, M., Callon, K. E., Tong, P. C., Dunford, J. E., van der Does, Y., Williams, G. A., Grey, A. B., Naot, D., and Reid, I. R. (2008). Modulation of osteoclastogenesis by fatty acids. Endocrinology 149, 5688–5695.
Delhanty, P. J., van Kerkwijk, A., Huisman, M., van de Zande, B., Verhoef-Post, M., Gauna, C., Hofland, L., Themmen, A. P., and van der Lely, A. J. (2010). Unsaturated fatty acids prevent desensitization of the human growth hormone secretagogue receptor by blocking its internalization. Am. J. Physiol. Endocrinol. Metab. 299, E497–E505.
Dhayal, S., Welters, H. J., and Morgan, N. G. (2008). Structural requirements for the cytoprotective actions of mono-unsaturated fatty acids in the pancreatic beta-cell line, BRIN-BD11. Br. J. Pharmacol. 153, 1718–1727.
Dramane, G., Akpona, S., Simonin, A. M., Besnard, P., and Khan, N. A. (2011). Cell signaling mechanisms of gustatory perception of lipids: can the taste cells be the target of anti-obesity agents? Curr. Med. Chem. 18, 3417–3422.
Edfalk, S., Steneberg, P., and Edlund, H. (2008). Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57, 2280–2287.
Fang, Y., Ferrie, A. M., Fontaine, N. H., Mauro, J., and Balakrishnan, J. (2006). Resonant waveguide grating biosensor for living cell sensing. Biophys. J. 91, 1925–1940.
Flodgren, E., Olde, B., Meidute-Abaraviciene, S., Winzell, M. S., Ahren, B., and Salehi, A. (2007). GPR40 is expressed in glucagon producing cells and affects glucagon secretion. Biochem. Biophys. Res. Commun. 354, 240–245.
Fredriksson, R., Hoglund, P. J., Gloriam, D. E., Lagerstrom, M. C., and Schioth, H. B. (2003). Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 554, 381–388.
Gardner, O. S., Dewar, B. J., and Graves, L. M. (2005). Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signaling. Mol. Pharmacol. 68, 933–941.
Garrido, D. M., Corbett, D. F., Dwornik, K. A., Goetz, A. S., Littleton, T. R., Mckeown, S. C., Mills, W. Y., Smalley, T. L. Jr., Briscoe, C. P., and Peat, A. J. (2006). Synthesis and activity of small molecule GPR40 agonists. Bioorg. Med. Chem. Lett. 16, 1840–1845.
Gawrisch, K., Soubias, O., and Mihailescu, M. (2008). Insights from biophysical studies on the role of polyunsaturated fatty acids for function of G-protein coupled membrane receptors. Prostaglandins Leukot. Essent. Fatty Acids 79, 131–134.
Gotoh, C., Hong, Y. H., Iga, T., Hishikawa, D., Suzuki, Y., Song, S. H., Choi, K. C., Adachi, T., Hirasawa, A., Tsujimoto, G., Sasaki, S., and Roh, S. G. (2007). The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun. 354, 591–597.
Hara, T., Hirasawa, A., Sun, Q., Koshimizu, T. A., Itsubo, C., Sadakane, K., Awaji, T., and Tsujimoto, G. (2009a). Flow cytometry-based binding assay for GPR40 (FFAR1; free fatty acid receptor 1). Mol. Pharmacol. 75, 85–91.
Hara, T., Hirasawa, A., Sun, Q., Sadakane, K., Itsubo, C., Iga, T., Adachi, T., Koshimizu, T. A., Hashimoto, T., Asakawa, Y., and Tsujimoto, G. (2009b). Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch. Pharmacol. 380, 247–255.
Hashimoto, N., Sasaki, Y., Nakama, C., and Ishikawa, M. (2010). Novel phenyl-isoxazol-3-ol derivative. US Patent 2010/0130559.
Herling, A. W., Defossa, E., Haschke, G., Dietrich, V., Stengelin, S., Keil, S., Dudda, A., Wagner, M., Ruus, P., and Ruetten, H. (2011). “Pharmacological activation of GPR40 reduces increased blood glucose levels by modulated insulin secretion in a glucose-dependent manner,” in 71st American Diabetes Association Meeting (Poster Presentation), San Diego, CA, 0452–PP.
Hirasawa, A., Tsumaya, K., Awaji, T., Katsuma, S., Adachi, T., Yamada, M., Sugimoto, Y., Miyazaki, S., and Tsujimoto, G. (2005). Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11, 90–94.
Hu, H., He, L. Y., Gong, Z., Li, N., Lu, Y. N., Zhai, Q. W., Liu, H., Jiang, H. L., Zhu, W. L., and Wang, H. Y. (2009). A novel class of antagonists for the FFAs receptor GPR40. Biochem. Biophys. Res. Commun. 390, 557–563.
Hudson, B. D., Smith, N., and Milligan, G. (2011). Experimental challenges to targeting poorly characterized GPCRs: uncovering the therapeutic potential for free fatty acid receptors Adv. Pharmacol. 62, 175–218.
Humphries, P. S., Benbow, J. W., Bonin, P. D., Boyer, D., Doran, S. D., Frisbie, R. K., Piotrowski, D. W., Balan, G., Bechle, B. M., Conn, E. L., Dirico, K. J., Oliver, R. M., Soeller, W. C., Southers, J. A., and Yang, X. (2009). Synthesis and SAR of 1,2,3,4-tetrahydroisoquinolin-1-ones as novel G-protein-coupled receptor 40 (GPR40) antagonists. Bioorg. Med. Chem. Lett. 19, 2400–2403.
Ichimura, A., Hirasawa, A., Hara, T., and Tsujimoto, G. (2009). Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediat. 89, 82–88.
Itoh, Y., Kawamata, Y., Harada, M., Kobayashi, M., Fujii, R., Fukusumi, S., Ogi, K., Hosoya, M., Tanaka, Y., Uejima, H., Tanaka, H., Maruyama, M., Satoh, R., Okubo, S., Kizawa, H., Komatsu, H., Matsumura, F., Noguchi, Y., Shinohara, T., Hinuma, S., Fujisawa, Y., and Fujino, M. (2003). Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176.
Jagannath, M. M., Somesh, B. P., Venkataranganna, M. R., Anup, O., Anilkumar, D., Verma, M. K., Sanghamitra, B., Bhawna, C., Manojkumar, S., Sowmya, R., Jayalakshmi, S., and Sunil, V. (2011). “CNX-011-67, a novel orally available GPR40 agonist, enhances glucose stimulated insulin secretion and significantly reduces fasting and non-fasting hyperglycemia – studies in vitro and in vivo in a preclinical model of type 2 diabetes,” in 71st American Diabetes Association Meeting (Poster Presentation), San Diego, CA, 0031–LB.
Katsuma, S., Hatae, N., Yano, T., Ruike, Y., Kimura, M., Hirasawa, A., and Tsujimoto, G. (2005). Free fatty acids inhibit serum deprivation-induced apoptosis through GPR120 in a murine enteroendocrine cell line STC-1. J. Biol. Chem. 280, 19507–19515.
Kebede, M., Alquier, T., Latour, M. G., Semache, M., Tremblay, C., and Poitout, V. (2008). The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes 57, 2432–2437.
Kilpatrick, L. E., Briddon, S. J., Hill, S. J., and Holliday, N. D. (2010). Quantitative analysis of neuropeptide Y receptor association with beta-arrestin2 measured by bimolecular fluorescence complementation. Br. J. Pharmacol. 160, 892–906.
Kobilka, B. K. (2011). Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 32, 213–218.
Kotarsky, K., Nilsson, N. E., Flodgren, E., Owman, C., and Olde, B. (2003). A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 301, 406–410.
Lan, H., Hoos, L. M., Liu, L., Tetzloff, G., Hu, W., Abbondanzo, S. J., Vassileva, G., Gustafson, E. L., Hedrick, J. A., and Davis, H. R. (2008). Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes 57, 2999–3006.
Latour, M. G., Alquier, T., Oseid, E., Tremblay, C., Jetton, T. L., Luo, J., Lin, D. C., and Poitout, V. (2007). GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 56, 1087–1094.
Lee, T., Schwandner, R., Swaminath, G., Weiszmann, J., Cardozo, M., Greenberg, J., Jaeckel, P., Ge, H., Wang, Y., Jiao, X., Liu, J., Kayser, F., Tian, H., and Li, Y. (2008). Identification and functional characterization of allosteric agonists for the G protein-coupled receptor FFA2. Mol. Pharmacol. 74, 1599–1609.
Liou, A. P., Lu, X., Sei, Y., Zhao, X., Pechhold, S., Carrero, R. J., Raybould, H. E., and Wank, S. (2011). The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140, 903–912.
Ma, D., Tao, B., Warashina, S., Kotani, S., Lu, L., Kaplamadzhiev, D. B., Mori, Y., Tonchev, A. B., and Yamashima, T. (2007). Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neurosci. Res. 58, 394–401.
Ma, J., Novack, A., Nashashibi, I., Pham, P., Rabbat, C. J., Song, J., Shi, D. F., Zhao, Z., Choi, Y. J., and Chen, X. (2010). Aryl GPR120 receptor agonists and uses thereof. International Patent WO 2010/048207.
Matsumura, S., Eguchi, A., Mizushige, T., Kitabayashi, N., Tsuzuki, S., Inoue, K., and Fushiki, T. (2009). Colocalization of GPR120 with phospholipase-Cbeta2 and alpha-gustducin in the taste bud cells in mice. Neurosci. Lett. 450, 186–190.
May, L. T., Leach, K., Sexton, P. M., and Christopoulos, A. (2007). Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51.
Miyauchi, S., Hirasawa, A., Iga, T., Liu, N., Itsubo, C., Sadakane, K., Hara, T., and Tsujimoto, G. (2009). Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn Schmiedebergs Arch. Pharmacol. 379, 427–434.
Moore, K., Zhang, Q., Murgolo, N., Hosted, T., and Duffy, R. (2009). Cloning, expression, and pharmacological characterization of the GPR120 free fatty acid receptor from cynomolgus monkey: comparison with human GPR120 splice variants. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 154, 419–426.
Nagasumi, K., Esaki, R., Iwachidow, K., Yasuhara, Y., Ogi, K., Tanaka, H., Nakata, M., Yano, T., Shimakawa, K., Taketomi, S., Takeuchi, K., Odaka, H., and Kaisho, Y. (2009). Overexpression of GPR40 in pancreatic {beta}-cells augments glucose stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 58, 1067–1076.
Naik, H., Vakilynejad, M., Wu, J., Viswanathan, P., Dote, N., Higuchi, T., and Leifke, E. (2011). Safety, tolerability, pharmacokinetics, and pharmacodynamic properties of the GPR40 agonist TAK-875: results from a double-blind, placebo-controlled single oral dose rising study in healthy volunteers. J. Clin. Pharmacol. (in press). doi:10.1177/0091270011409230
Negoro, N., Sasaki, S., Mikami, S., Ito, M., Suzuki, M., Tsujihata, Y., Ito, R., Harada, A., Takeuchi, K., Suzuki, N., Miyazaki, J., Santou, T., Odani, T., Kanzaki, N., Funami, M., Tanaka, T., Kogame, A., Matsunaga, S., Yasuma, T., and Momose, Y. (2010). Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist. ACS Med. Chem. Lett. 1, 290–294.
Ogawa, T., Hirose, H., Miyashita, K., Saito, I., and Saruta, T. (2005). GPR40 gene Arg211His polymorphism may contribute to the variation of insulin secretory capacity in Japanese men. Metab. Clin. Exp. 54, 296–299.
Ogden, C. L., Carroll, M. D., Curtin, L. R., Mcdowell, M. A., Tabak, C. J., and Flegal, K. M. (2006). Prevalence of overweight and obesity in the United States, 1999-2004. J. Am. Med. Assoc. 295, 1549–1555.
Oh, D. Y., Talukdar, S., Bae, E. J., Imamura, T., Morinaga, H., Fan, W., Li, P., Lu, W. J., Watkins, S. M., and Olefsky, J. M. (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698.
Overington, J. P., Al-Lazikani, B., and Hopkins, A. L. (2006). How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996.
Pin, J. P., Neubig, R., Bouvier, M., Devi, L., Filizola, M., Javitch, J. A., Lohse, M. J., Milligan, G., Palczewski, K., Parmentier, M., and Spedding, M. (2007). International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol. Rev. 59, 5–13.
Rajagopal, S., Rajagopal, K., and Lefkowitz, R. J. (2010). Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386.
Sasaki, S., Kitamura, S., Negoro, N., Suzuki, M., Tsujihata, Y., Suzuki, N., Santou, T., Kanzaki, N., Harada, M., Tanaka, Y., Kobayashi, M., Tada, N., Funami, M., Tanaka, T., Yamamoto, Y., Fukatsu, K., Yasuma, T., and Momose, Y. (2011). Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J. Med. Chem. 54, 1365–1378.
Schmidt, J., Liebscher, K., Merten, N., Grundmann, M., Mielenz, M., Sauerwein, H., Christiansen, E., Due-Hansen, M. E., Ulven, T., Ullrich, S., Gomeza, J., Drewke, C., and Kostenis, E. (2011). Conjugated linoleic acids mediate insulin release through islet G protein-coupled receptor FFA1/GPR40. J. Biol. Chem. 286, 11890–11894.
Schröder, R., Janssen, N., Schmidt, J., Kebig, A., Merten, N., Hennen, S., Muller, A., Blattermann, S., Mohr-Andra, M., Zahn, S., Wenzel, J., Smith, N. J., Gomeza, J., Drewke, C., Milligan, G., Mohr, K., and Kostenis, E. (2010). Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat. Biotechnol. 28, 943–949.
Serhan, C. N., Krishnamoorthy, S., Recchiuti, A., and Chiang, N. (2011). Novel anti-inflammatory – pro-resolving mediators and their receptors. Curr. Top. Med. Chem. 11, 629–647.
Smith, N. J., and Milligan, G. (2010). Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol. Rev. 62, 701–725.
Smith, N. J., Stoddart, L. A., Devine, N. M., Jenkins, L., and Milligan, G. (2009). The action and mode of binding of thiazolidinedione ligands at free fatty acid receptor 1. J. Biol. Chem. 284, 17527–17539.
Smith, N. J., Ward, R. J., Stoddart, L. A., Hudson, B. D., Kostenis, E., Ulven, T., Morris, J. C., Trankle, C., Tikhonova, I. G., Adams, D. R., and Milligan, G. (2011). Extracellular loop 2 of the free fatty acid receptor 2 mediates allosterism of a phenylacetamide ago-allosteric modulator. Mol. Pharmacol. 80, 163–173.
Steneberg, P., Rubins, N., Bartoov-Shifman, R., Walker, M. D., and Edlund, H. (2005). The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 1, 245–258.
Stewart, G., Hira, T., Higgins, A., Smith, C. P., and Mclaughlin, J. T. (2006). Mouse GPR40 heterologously expressed in Xenopus oocytes is activated by short-, medium-, and long-chain fatty acids. Am. J. Physiol. Cell Physiol. 290, C785–C792.
Stoddart, L. A., Brown, A. J., and Milligan, G. (2007). Uncovering the pharmacology of the G protein-coupled receptor GPR40: high apparent constitutive activity in guanosine 5′-O-(3-[35S]thio)triphosphate binding studies reflects binding of an endogenous agonist. Mol. Pharmacol. 71, 994–1005.
Stoddart, L. A., Smith, N. J., and Milligan, G. (2008). International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol. Rev. 60, 405–417.
Sum, C. S., Tikhonova, I. G., Neumann, S., Engel, S., Raaka, B. M., Costanzi, S., and Gershengorn, M. C. (2007). Identification of residues important for agonist recognition and activation in GPR40. J. Biol. Chem. 282, 29248–29255.
Sun, Q., Hirasawa, A., Hara, T., Kimura, I., Adachi, T., Awaji, T., Ishiguro, M., Suzuki, T., Miyata, N., and Tsujimoto, G. (2010). Structure-activity relationships of GPR120 agonists based on a docking simulation. Mol. Pharmacol. 78, 804–810.
Suzuki, T., Igari, S., Hirasawa, A., Hata, M., Ishiguro, M., Fujieda, H., Itoh, Y., Hirano, T., Nakagawa, H., Ogura, M., Makishima, M., Tsujimoto, G., and Miyata, N. (2008). Identification of G protein-coupled receptor 120-selective agonists derived from PPARgamma agonists. J. Med. Chem. 51, 7640–7644.
Talukdar, S., Olefsky, J. M., and Osborn, O. (2011). Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 32, 543–550.
Tan, C. P., Feng, Y., Zhou, Y. P., Eiermann, G. J., Petrov, A., Zhou, C., Lin, S., Salituro, G., Meinke, P., Mosley, R., Akiyama, T. E., Einstein, M., Kumar, S., Berger, J. P., Mills, S. G., Thornberry, N. A., Yang, L., and Howard, A. D. (2008). Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 57, 2211–2219.
Tanaka, T., Katsuma, S., Adachi, T., Koshimizu, T. A., Hirasawa, A., and Tsujimoto, G. (2008a). Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedebergs Arch. Pharmacol. 377, 523–527.
Tanaka, T., Yano, T., Adachi, T., Koshimizu, T. A., Hirasawa, A., and Tsujimoto, G. (2008b). Cloning and characterization of the rat free fatty acid receptor GPR120: in vivo effect of the natural ligand on GLP-1 secretion and proliferation of pancreatic beta cells. Naunyn Schmiedebergs Arch. Pharmacol. 377, 515–522.
Tikhonova, I. G., Sum, C. S., Neumann, S., Thomas, C. J., Raaka, B. M., Costanzi, S., and Gershengorn, M. C. (2007). Bidirectional, iterative approach to the structural delineation of the functional “chemoprint” in GPR40 for agonist recognition. J. Med. Chem. 50, 2981–2989.
Tomita, T., Masuzaki, H., Iwakura, H., Fujikura, J., Noguchi, M., Tanaka, T., Ebihara, K., Kawamura, J., Komoto, I., Kawaguchi, Y., Fujimoto, K., Doi, R., Shimada, Y., Hosoda, K., Imamura, M., and Nakao, K. (2006). Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: evidence for GPR40 expression in pancreatic beta cells and implications for insulin secretion. Diabetologia 49, 962–968.
Tomita, T., Masuzaki, H., Noguchi, M., Iwakura, H., Fujikura, J., Tanaka, T., Ebihara, K., Kawamura, J., Komoto, I., Kawaguchi, Y., Fujimoto, K., Doi, R., Shimada, Y., Hosoda, K., Imamura, M., and Nakao, K. (2005). GPR40 gene expression in human pancreas and insulinoma. Biochem. Biophys. Res. Commun. 338, 1788–1790.
Tsujihata, Y., Ito, R., Suzuki, M., Harada, A., Negoro, N., Yasuma, T., Momose, Y., and Takeuchi, K. (2011). TAK-875, an orally available G protein-coupled receptor 40/free fatty acid receptor 1 agonist, enhances glucose-dependent insulin secretion and improves both postprandial and fasting hyperglycemia in type 2 diabetic rats. J. Pharmacol. Exp. Ther. 339, 228–237.
Varga, T., Czimmerer, Z., and Nagy, L. (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 1812, 1007–1022.
Vettor, R., Granzotto, M., De Stefani, D., Trevellin, E., Rossato, M., Farina, M. G., Milan, G., Pilon, C., Nigro, A., Federspil, G., Vigneri, R., Vitiello, L., Rizzuto, R., Baratta, R., and Frittitta, L. (2008). Loss-of-function mutation of the GPR40 gene associates with abnormal stimulated insulin secretion by acting on intracellular calcium mobilization. J. Clin. Endocrinol. Metab. 93, 3541–3550.
Viswanathan, P., Marcinak, J., Cao, C., Xie, B., Vakilynejad, M., and Leifke, E. (2011). A randomized, double-blind, placebo- and active-controlled, dose-ranging study to determine the efficacy and safety of the novel GPR40 agonist TAK-875 in subjects with T2DM. Presented at 71st American Diabetes Association meeting (oral presentation 0134-LBOR), San Diego, CA.
Wild, S., Roglic, G., Green, A., Sicree, R., and King, H. (2004). Global prevalence of diabetes – estimates for the year 2000 and projections for 2030. Diabetes Care 27, 1047–1053.
Wu, P., Yang, L., and Shen, X. (2010). The relationship between GPR40 and lipotoxicity of the pancreatic beta-cells as well as the effect of pioglitazone. Biochem. Biophys. Res. Commun. 403, 36–39.
Yaney, G. C., and Corkey, B. E. (2003). Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia 46, 1297–1312.
Zhang, X., Yan, G., Li, Y., Zhu, W., and Wang, H. (2010). DC260126, a small-molecule antagonist of GPR40, improves insulin tolerance but not glucose tolerance in obese Zucker rats. Biomed. Pharmacother. 64, 647–651.
Zhou, C., Tang, C., Chang, E., Ge, M., Lin, S., Cline, E., Tan, C. P., Feng, Y., Zhou, Y. P., Eiermann, G. J., Petrov, A., Salituro, G., Meinke, P., Mosley, R., Akiyama, T. E., Einstein, M., Kumar, S., Berger, J., Howard, A. D., Thornberry, N., Mills, S. G., and Yang, L. (2010). Discovery of 5-aryloxy-2,4-thiazolidinediones as potent GPR40 agonists. Bioorg. Med. Chem. Lett. 20, 1298–1301.
Zwier, J. M., Roux, T., Cottet, M., Durroux, T., Douzon, S., Bdioui, S., Gregor, N., Bourrier, E., Oueslati, N., Nicolas, L., Tinel, N., Boisseau, C., Yverneau, P., Charrier-Savournin, F., Fink, M., and Trinquet, E. (2010). A fluorescent ligand-binding alternative using Tag-lite(R) technology. J. Biomol. Screen 15, 1248–1259.
Keywords: G protein coupled receptor, free fatty acid, GPR40, GPR120, FFA1, type 2 diabetes, pancreas, adipocytes
Citation: Holliday ND, Watson S-J and Brown AJH (2012) Drug discovery opportunities and challenges at G protein coupled receptors for long chain free fatty acids. Front. Endocrin. 2:112. doi: 10.3389/fendo.2011.00112
Received: 12 September 2011;
Paper pending published: 12 November 2011;
Accepted: 15 December 2011;
Published online: 03 January 2012.
Edited by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaReviewed by:
Brian Hudson, University of Glasgow, UKGuillermo Romero, University of Pittsburgh, USA
Milka Vrecl, Veterinary faculty of the University in Ljubljana, Slovenia
Copyright: © 2012 Holliday, Watson and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nicholas D. Holliday, Cell Signalling Research Group, School of Biomedical Sciences, The Medical School, Queen’s Medical Centre, University of Nottingham, Floor C, Nottingham NG7 2UH, UK. e-mail: nicholas.holliday@nottingham.ac.uk