 Danielle M. A. Ratsma
Danielle M. A. Ratsma M. Carola Zillikens
M. Carola Zillikens Bram C. J. van der Eerden
Bram C. J. van der Eerden- Laboratory for Calcium and Bone Metabolism and Erasmus MC Bone Centre, Department of Internal Medicine, Erasmus University Medical Center, Rotterdam, Netherlands
Fibroblast growth factor 23 (FGF23) has been described as an important regulator of mineral homeostasis, but has lately also been linked to iron deficiency, inflammation, and erythropoiesis. FGF23 is essential for the maintenance of phosphate homeostasis in the body and activating mutations in the gene itself or inactivating mutations in its upstream regulators can result in severe chronic hypophosphatemia, where an unbalanced mineral homeostasis often leads to rickets in children and osteomalacia in adults. FGF23 can be regulated by changes in transcriptional activity or by changes at the post-translational level. The balance between O-glycosylation and phosphorylation is an important determinant of how much active intact or inactive cleaved FGF23 will be released in the circulation. In the past years, it has become evident that iron deficiency and inflammation regulate FGF23 in a way that is not associated with its classical role in mineral metabolism. These conditions will not only result in an upregulation of FGF23 transcription, but also in increased cleavage, leaving the levels of active intact FGF23 unchanged. The exact mechanisms behind and function of this process are still unclear. However, a deeper understanding of FGF23 regulation in both the classical and non-classical way is important to develop better treatment options for diseases associated with disturbed FGF23 biology. In this review, we describe how the currently known upstream regulators of FGF23 change FGF23 transcription and affect its post-translational modifications at the molecular level.
Introduction
In 1959 a first mention was made by Andrea Prader of a circulating “rachitonic” substance that was the cause of tumor-induced osteomalacia (TIO). He described a case of an 11-year-old girl, who suddenly developed severe rickets. A tumor was identified between her ribs, and resection resulted in curation of her rickets (1, 2). The first evidence of a circulating phosphaturic factor comes from experiments in hyp mice, a model for X-linked hypophosphatemic rickets (XLH), in 1989, where it was shown that a circulating factor in hyp mice could induce hypophosphatemia in wild-type mice (3). This concept was already suggested a year earlier, when a resected tumor from a TIO patient caused hypophosphatemia when transplanted into a nude mouse (4). This phosphaturic factor was first identified as fibroblast growth factor 23 (FGF23) in 2000 by the autosomal-dominant hypophosphatemic rickets (ADHR) consortium when mutations in FGF23 where found to be causing ADHR (5). Besides FGF23, other phosphatonins such as FGF7, secreted-frizzled related protein (sFRP4) and matrix extracellular phosphoglycoprotein (MEPE), have been identified but descriptions of these factors are beyond the scope of this review (6).
FGF23 is predominantly produced by osteocytes and osteoblasts in the skeleton and has a key function in regulating phosphate homeostasis (7). When serum phosphate levels rise due to bone resorption or absorption of dietary phosphate, FGF23 levels increase. FGF23 can bind to the fibroblast growth factor receptor 1 (FGFR1) in the kidneys, but needs its co-receptor, α-klotho, to execute its functions (8). Binding of FGF23 in the kidney has several effects. First of all, the expression of type II sodium-phosphate cotransporter NaPi-2a (encoded by SLC34A1), present in the cell surface of the renal proximal tubules is downregulated, resulting in decreased phosphate reabsorption (9). Secondly, FGF23 inhibits 1α-hydroxylase in the kidney, the key enzyme for the conversion of inactive 25-hydroxyvitamin D (25(OH)D) to the active 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) (10). Reduced levels of 1,25(OH)2D3 result in lower expression of NaPi-2b (SLC34A2) in the small intestine, and therefore less absorption of dietary phosphate (11, 12). The parathyroid gland is another important target for FGF23, where it is able to downregulate parathyroid hormone (PTH) synthesis and secretion in a α-klotho-dependent manner (13, 14). PTH has an important role in regulating renal phosphate reabsorption as it downregulates NaPi-2a/c (SLC34A1/SLC34A3) to reduce renal reabsorption and it activates 1α-hydroxylase (15).
FGF23 is a member of the fibroblast growth factor (FGF) family, which has a plethora of functions. The FGFs are divided into subfamilies based on their structure and phylogenic analyses (16). FGF23 is a member of the FGF19 subfamily, which consists of FGF19, FGF21, and FGF23. The FGFs in this subfamily function as circulating hormones, and are therefore also called endocrine FGFs (17, 18). Several upstream regulators inhibit or induce the expression of FGF23 in the osteocytes and late osteoblasts, which will be extensively discussed in this review. The FGF23 gene consists of three exons, that encode for a glycoprotein of 251 amino acids (19). This glycoprotein consists of three domains with the first 24 amino acids being the signal sequence, the middle part of 155 amino acids forming the core FGF homology domain and the last 72 amino acids comprising the C-terminal (20). The C-terminus of FGF23 is important for binding to klotho, while the N-terminus contains the FGFR binding domain (Figure 1) (17). The cleaved C-terminal domain competes with FGF23 for binding to klotho, and can therefore inhibit the formation of the FGF23/FGFR/Klotho complex (21). Studies performing overexpression of FGF23 in Chinese hamster ovary (CHO) cells have shown that the mature FGF23 protein lacks the signal peptide (22). Western blotting analyses of the recombinant media of these cells using an antibody against the C-terminus resulted in two products, one of ~30 kDa and a smaller one of ~10 kDa. This indicated that proteolytic processing is taking place before FGF23 is secreted (22).
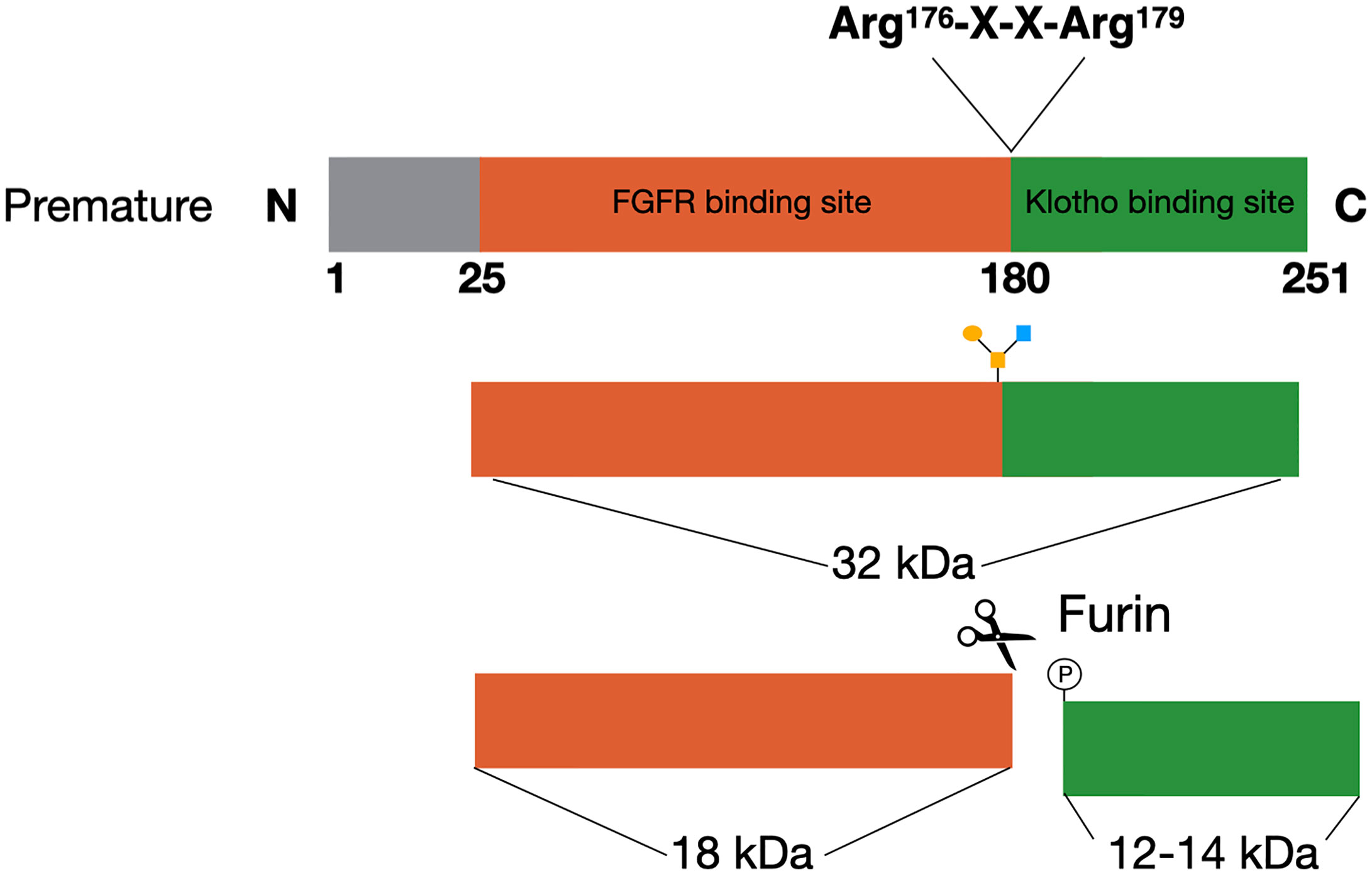
Figure 1 FGF23 processing. The premature FGF23 protein is a 251-amino acid long glycoprotein. In order to form the mature form of FGF23 the first 25 amino acids are removed through cleavage. FGF23 can be O-glycosylated at Thr178, which protects it from cleavage and results in active FGF23 in the circulation. The alternative is for FGF23 to be phosphorylated at Ser180. This results in cleavage at the consensus sequence (Arg176-X-X-Arg179) and inactive cleaved FGF23 in the circulation.
FGF23 can be cleaved by subtilisin-like proprotein convertases at a consensus sequence (Arg176-X-X-Arg179), that is unique for FGF23 as it does not appear in other members of the FGF family (Figure 1) (22, 23). Alternatively, FGF23 can be O-glycosylated at Thr178 by polypeptide N-acetylgalactosaminyltransferase 3 (Gal-NAc-T3, encoded by GalNT3), which protects FGF23 from proteolytic cleavage (24). Phosphorylation of FGF23 at Ser180 via the secretory protein kinase family with sequence similarity-20 member C (FAM20C) prevents this O-glycosylation and thereby makes FGF23 more prone to cleavage by the proprotein convertase furin (24, 25). The balance between these processes determines the ratio between active intact FGF23 (iFGF23) and inactive cleaved FGF23 (cFGF23) that will be released into the circulation (26, 27).
Although still quite limited and with many more to discover, there are a number of factors known to regulate the expression and/or cleavage of FGF23. Below we provide a current overview of both classical and non-classical regulators of FGF23.
Classical FGF23 Regulation
Local FGF23 Regulators in Osteocytes
DMP1
Dentin matrix protein 1 (DMP1) is an extracellular matrix pro-peptide that is produced by osteocytes and a member of the small integrin binding ligand N-linked glycoprotein (SIBLING) family (28). DMP1 is a suppressor of FGF23 as inactivating mutations in DMP1 result in autosomal recessive hypophosphatemic rickets (ARHR), a disease where overproduction of FGF23 results in renal phosphate wasting, osteomalacia, and rickets (29). In mouse models of Dmp1 deficiency increased Fgf23 transcription results from paracrine stimulation of Fgfr1, and downstream activation of the nuclear factor of activated T cells (Nfat) pathway (Figure 2) (30, 31). The FGF23 promotor contains an NFAT response element, which controls FGF23 expression in response to calcium and inflammatory stimuli (32). Dussold et al. showed that Nfat1 mRNA expression is increased in bone of chronic kidney disease (CKD) mice and that Dmp1 inhibits Nfat1 signaling that is activated in CKD, thereby preventing Fgf23 transcription (Figure 2) (32). This only occurs in mice with early CKD but not in more advanced stages, suggesting that other stimuli such as elevated PTH and chronic inflammation may override this effect. In DMP1-treated mice with advanced CKD, there is a small decrease in circulating Fgf23, despite transcription levels remaining stable. This might indicate that DMP1 also has a post-translational effect on FGF23 (32). However, this study only measured the total levels of serum FGF23 and to gain more insight in how DMP1 might affect FGF23 processing, levels of both iFGF23 and cFGF23 should be measured. In conclusion, although it is clear that DMP1 has an inhibitory effect on FGF23 transcription in CKD, studies are needed to describe its function in a healthy situation. Moreover, there are indications that DMP1 affects post-translational FGF23 modification, although it is unclear what the exact role of DMP1 in this process could be.
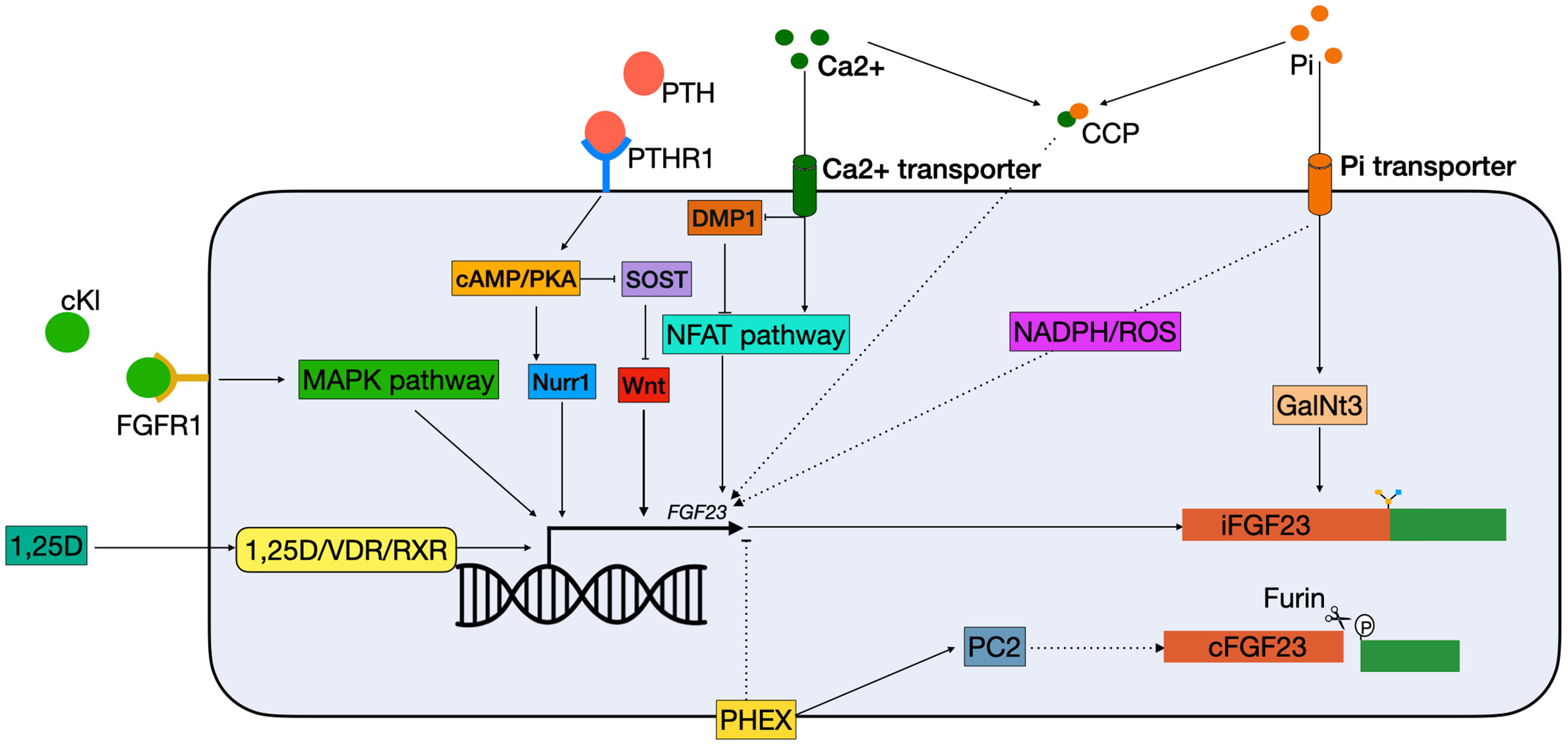
Figure 2 Schematic overview of classical FGF23 regulation. cKL binds to the FGFR1 and activates FGF23 transcription via the MAPK pathway. 1,25(OH)2D3 binds to the VDR, which heterodimerizes with the RXRs and can bind to the VDRE in the FGF23 promotor. PTH binds to the PTHR1 on the cell membrane, and activates the cAMP/PKA pathway. This has two effects: 1) NURR1 mRNA increases, resulting in increased FGF23 transcription; 2) inhibition of SOST, thereby indirectly stimulating FGF23 transcription by releasing the suppression of the WNT pathway. Both Ca2+ and Pi can activate FGF23 transcription independently. Ca2+ enters the cell via a Ca2+ transporter. In the cells it inhibits DMP1, an inhibitor of the NFAT1 pathway, thus activating the NFAT pathway, leading to FGF23 transcription stimulation. Phosphate enters the cells through phosphate transporters. In the cell it stimulates FGF23 through the production of ROS through a yet unknown mechanism. Furthermore, it increases expression of GalNT3, resulting in protection from cleavage of the full-length FGF23 protein. Also, CCPs can increase FGF23 transcription but the mechanism remains unknown. Lastly, PHEX is able to inhibit FGF23 transcription through a yet unknown mechanism and interact with PC2 to increase FGF23 cleavage. 1,25D, 1,25-dihydroxyvitamin D3; Ca2+, calcium; cAMP, Cyclic adenosine 3′,5′-monophosphate; CCP, calciprotein particles; cFGF23, cleaved fibroblast growth factor 23; cKL, cleaved klotho; DMP1, dentin matrix protein 1; iFGF23, intact fibroblast growth factor 23; FGFR1, fibroblast growth receptor 1; GalNt3, polypeptide N-acetylgalactosaminyltransferase 3; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NFAT, nuclear factor of activated T cells; Nurr1, nuclear receptor related-1 protein; PC2, proprotein convertase; PHEX, phosphate regulating endopeptidase homolog X-linked; Pi, phosphate; PKA, protein kinase A; PTH, parathyroid hormone; PTHR1, parathyroid hormone receptor 1; ROS, reactive oxygen species; RXR, retinoid X receptor; SOST, sclerostin; VDR, vitamin D receptor.
PHEX
Mutations in phosphate regulating endopeptidase homolog X-linked (PHEX) result in the most common form of chronic hypophosphatemia, named X-linked hypophosphatemia (XLH), by causing elevated levels of FGF23 (33). Hyp mice have an inactivating mutation of Phex, which mimics XLH in humans. They have a lower serum phosphate level due to high circulating levels of FGF23, which can be restored by a high phosphate diet (34). Moreover, dietary phosphate has been shown to improve bone growth and mineralization in hyp mouse, similar to what has been observed for patients with XLH (35).
PHEX is predominantly expressed by osteocytes and osteoblasts and encodes for an enzyme that degrades local SIBLINGs, in particular osteopontin (OPN) (36). Despite being an enzyme, it is thought that PHEX mostly suppresses the transcription of FGF23 rather than its degradation, though the mechanism behind this is unknown (37, 38). There is some evidence that PHEX might indirectly play a role in FGF23 cleavage. Proprotein convertase (PC2), encoded by the proprotein convertase subtilisin/kexin-type-2 (PCSK2) gene, has been shown to be upregulated by PHEX, which can result in FGF23 cleavage in vitro (Figure 2) (39). Moreover, PC2 may promote the formation of PHEX-DMP1-integrin complexes that suppress FGF23 production when they are activated by neuroendocrine protein 7B2•PC2 (40). However, despite the fact that PC2 cleaves FGF23 in vitro, there is no evidence that the two proteins are in physical contact within the osteocyte (23).
PHEX mutations are X chromosome-linked, so females with a loss of function mutation in one PHEX allele should also have an unaffected second allele. This should result in functional proteins, but nevertheless women develop XLH as well (41). Preferential X chromosome inactivation in woman has been proposed as a mechanism to explain why XLH is not a recessive disease. However, a PCR analysis of 13 female XLH patients showed no significant difference in X inactivation patterns in peripheral blood cells. compared to healthy controls (42). Moreover, a case study of identical twins, of which only one was affected with XLH, also did not reveal a difference in X inactivation patterns as analyzed in peripheral blood cells (43). Nonetheless, it should be noted that X inactivation pattern in blood cells may not be representative for other tissues, especially in twins (44). It is therefore possible that in these cases there is preferential X inactivation in other tissues, such as bone, resulting in the XLH phenotype. All in all, even though mutations in PHEX have been established to disturb FGF23 regulation, the exact regulation of FGF23 by PHEX remains largely unknown to date.
Circulating FGF23 Regulators
1,25-Dihydroxyvitamin D3
In vivo studies show that C57Bl/6 mice injected with 1,25(OH)2D3 have significantly higher Fgf23 serum levels compared to vehicle injected mice. This increase was mostly attributed to increased Fgf23 expression in the bone. When UMR106 osteosarcoma cells were treated with vitamin D, a significant upregulation of Ffg23 expression was found after 4 h. Addition of actinomycin D, a gene transcription inhibitor, completely abolished the upregulation of Fgf23, thus showing that increased Fgf23 expression by 1,25(OH)2D3 is due to a transcriptional effect (45). This effect is believed to be modulated via the vitamin D receptor (VDR) in osteocytes (46). This is also showed in patients with inactivating mutations in the VDR gene who have lower FGF23 serum levels (47). When 1,25(OH)2D3 binds to the VDR, it heterodimerizes with the retinoid X receptors (RXR), this complex can bind to the vitamin D response element (VDRE) in the promotor of target genes (Figure 2) (48). Treatment with 1,25(OH)2D3 in hypoparathyroid patients increases serum FGF23 and rapidly decreases serum phosphate, despite the fact that 1,25(OH)2D3 increases phosphate absorption in the gut (49). Moreover, Ito et al. demonstrated 1,25(OH)2D3-enhanced FGF23 promoter activity and mRNA expression in human chronic myelogenous leukemia K562 cells (46). Animal experiments have also shown that 1,25(OH)2D3 regulates Fgf23 levels. VDR null mice have low serum Fgf23 levels, which can be normalized with a rescue diet to normalize low phosphate levels and secondary hyperparathyroidism (50). However, thus far it has been unclear whether 1,25(OH)2D3 directly stimulates FGF23 expression in osteocytes via the VDR, because osteocytes have proven difficult to isolate and culture (51). Yashiro et al. have shown that 1,25(OH)2D3 treatment results in increased Fgf23 expression in the murine MC3T3-E1 osteocyte-like cell line. Knockdown of the VDR in osteocytes decreased Fgf23 expression significantly, indicating that the response of Fgf23 to 1,25(OH)2D3 is primarily mediated through the VDRE in the promoter of the Fgf23 gene (52).
It is important to note that some of the work on Fgf23 regulation by 1,25(OH)2D3 is performed using mice and rats, which are both nocturnal species. Both mice and humans use UV-exposure as a 1,25(OH)2D3 source, but as mice are nocturnal, they have less opportunity to expose themselves to UV light. Additionally, both humans and mice use their diet as a source of vitamin D (53). Mutations in the Vdr or Cyp27b1 genes in mice result in phenotypes similar to those seen in humans with these mutations (54–57). Even though vitamin D metabolism is different in mice and human, it does not preclude the use of mice as a suitable model to study the effect of 1,25(OH)2D3 on Fgf23.
PTH
Secondary hyperparathyroidism is a common complication of CKD triggered by hypocalcemia, hyperphosphatemia, and low vitamin D levels. These elevated PTH levels are usually found in combination with increased FGF23 levels (58). Moreover, it was found that PTH infusion resulted in increased FGF23 levels in healthy men, thus indicating that FGF23 is upregulated in patients with hyperparathyroidsism (59). PTH binds to the parathyroid hormone 1 receptor (PTH1R), which is expressed in numerous tissues including kidney and bone and activates several intracellular secondary messengers (Figure 2) (60, 61). In bone a major physiological role of PTH is to bind to the PTH1R on cells of the osteoblast lineage and enhancing the release of receptor activator of nuclear factor-κB ligand (RANKL), which then binds to its receptor RANK on osteoclast precursors, thereby enhancing osteoclast formation or activity (62). Data from osteoblast-like UMR-106 cells shows that PTH can also stimulate FGF23 production in osteoblasts by binding to the PTH1R and stimulating both the protein kinase A (PKA) and Wnt pathways (63). One study showed that PTH increases levels of nuclear receptor related-1 protein (NURR1) mRNA via the PKA pathway, and that overexpression of NURR1 stimulates FGF23 in the absence of PTH in UMR-106 cells. Knockdown of NURR1 prevented an increase in FGF23, indicating that NURR1 is essential for the stimulation of FGF23 by PTH in bone cells (64). Regulation of FGF23 by PTH via the Wnt pathway is through inhibition of sclerostin (SOST), a potent inhibitor of the Wnt pathway (65, 66). This effect is mediated by the PKA pathway. In adult Sabra rats, administration of Forskolin, a PKA inhibitor, resulted in decreased levels of SOST mRNA (62). Together, this indicates that activation of the PKA pathway by PTH both directly stimulates FGF23 transcription by increasing NURR1 expression, as well as indirectly by inhibition of SOST (Figure 2).
α-Klotho
α-Klotho plays an important role in phosphate regulation by FGF23 as a co-receptor for FGFR1 in the kidney. Mice lacking α-klotho have an identical phenotype as the FGF23 KO mice, as FGF23 cannot fulfill its phosphaturic function in the kidney without α-klotho (8). Moreover, in a mouse model with a 70% reduction of α-klotho expression in the distal renal tubules, significantly higher levels of serum FGF23 were found (67). This indicates that the body attempts to compensate for lower levels of α-klotho expression, by upregulating FGF23. There are several isoforms of αklotho, of which the membrane-bound klotho is important for the interaction with FGF23 in the kidney. However, the circulating or cleaved form of klotho (cKL) has also been associated with FGF23 regulation (68). This relation between cKL and upregulation of FGF23 was discovered in 2008. An infant was diagnosed with severe hypophosphatemia, but no mutations were found DMP1, PHEX, FGFR1, or FGF23. She was found to have a chromosomal translocation (t9:13) in proximity to the α-Klotho locus. Analysis of the patient’s serum revealed elevated levels of cKL and FGF23, and resulted in the hypothesis that cKL can drive FGF23 expression (69). In a mouse model mimicking this translocation, the treatment with cKl resulted in significantly higher levels of serum Fgf23 and a 150-fold increase of Fgf23 mRNA in bone (70). Moreover, c-fos and early growth response protein 1 (Egr1) were upregulated, both targets of the mitogen-activated protein kinase (MAPK) pathway, which is believed to be involved in activating Fgf23 transcription (Figure 2) (70, 71). It is hypothesized that cKl plays a role in fine-tuning the levels of iFGF23, since activity of the FGFR1c was initiated by cKl in the presence of iFGF23 (70). To investigate this relationship, the osteoblastic cell line UMR-106 was treated with FGF23, cKl or a combination. While the combination resulted in upregulated levels of EGR1 and FGF23, the single treatments were not capable of achieving this (72). Moreover, deletion of FGFR1 by CRISPR/Cas9 also ablated this response (72). This implicates a relation between circulating cKl and circulating FGF23 levels. Administration of cKl to α-klotho null mice, resulted in upregulation of Fgf23 and the prevention of hyperphosphatemia and vascular calcification (72). This may represent a mechanism to prevent hyperphosphatemia when the circulating levels of FGF23 are not adequate. In this respect, cKl might also be of clinical interest, as it is able to prevent hyperphosphatemia even in the absence of functional α-klotho. It still needs to be determined whether cKl in normal circumstances is able to change levels of FGF23 expression.
Phosphate
A major regulator of FGF23 levels is serum phosphate, which is critically maintained stable in the circulation by a combination of renal reabsorption, intestinal absorption, and bone resorption. Controlled feeding studies showed that high dietary phosphate intake results in higher levels of FGF23, while low intake results in a decrease (73–75). However, the mechanism behind phosphate-mediated FGF23 regulation remains unknown. Several studies measured phosphate levels in the serum before and after meals, but did not find a relation with diet (76, 77). Moreover, hyperphosphatemia in glial cells missing homolog 2 (Gcm2) null mice and the addition of phosphate to osteoblast cultures did not result in increased Fgf23 transcripts, a result that was also found in other studies (78–80). However, a study by Takashi et al. showed that mice fed a high phosphate diet have increased levels of serum Fgf23 compared to mice on a low phosphate diet (71). They found in UMR106 cells that this was not caused by the upregulation of Fgf23 transcription, but rather by increased expression of GalNt3, which results in a higher level of active iFGF23 in the circulation (71). Nonetheless, other studies do show that high extracellular phosphate results in enhanced FGF23 transcription (81, 82). When 4 mM phosphate was added to IDG-SW3 cells, this resulted in a 38-fold increase in Fgf23 expression. Interestingly, this effect was not found in the presence of 10 mM phosphate (82). Another study reported that phosphate enhances FGF23 transcription in UMR-106 cells. In addition, they also found that phosphate resulted in increased production of reactive oxygen species (ROS). When an inhibitor of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase was used, not only the production of ROS diminishes, but also FGF23 transcription (Figure 2) (81). There are no studies that show in vivo that ROS production is correlated with FGF23 production but it is known that patients with renal failure have increased ROS production (83). Studies assessing whether production of ROS is correlated with the risk of adverse events in the presence of high FGF23 levels are therefore warranted. It is interesting that FGF23 is mostly known as a phosphaturic hormone, but that the exact mechanism by which phosphate regulates it (production, cleavage, secretion) remains largely unclear. This underlines the importance for more research about FGF23 regulation by phosphate in both healthy individuals and patients with abnormal phosphate levels.
Calcium
A study in rats showed that increased Fgf23 transcription in response to high dietary phosphate is only possible when serum calcium levels are also high (84). This is in agreement with another study showing that it is not the increase of phosphate itself that increases Fgf23 transcription, but rather the formation of calciprotein particles (CCPs) (85). CCPs are colloidal nanoparticles that consist of calcium and phosphate circulating in the blood and are potentially harmful as they contribute to vascular calcification. UMR106 cells treated with synthesized CCPs showed increased FGF23 mRNA (85). Moreover, in C57BL/6 mice it was shown that the amount of plasma CCPs were increased after a high dietary phosphate load. In this study they also showed that the high phosphate diet also resulted in more circulating FGF23 and increased levels of mRNA Fgf23 in the bone (85). The authors hypothesize that this mechanism might constitute a negative feedback loop, since activating FGF23 results in lower levels of serum phosphate, which in turn will decrease the amount of harmful CCPs that can be formed (Figure 2). There is also evidence that calcium itself is able to upregulate FGF23 expression. In CKD, serum FGF23 levels are elevated due to hyperphosphatemia, and dietary phosphate restriction and phosphate binders are being used to keep FGF23 levels under control. Interestingly, studies showed that calcium-containing phosphate binders do not result in a decrease of serum FGF23 levels, while calcium-free phosphate binders do decrease FGF23 levels (86). Indicating that calcium itself might also modulate FGF23 in the absence of phosphate. Moreover, Cyp27b1−/− mice fed a high calcium diet had higher levels of serum FGF23. Besides, adding calcium to the culture medium of MC3T3-E1 cells resulted in higher Fgf23 expression (87). A recent study in B6 wild-type mice showed that an injection of calcium led to increased levels of circulating Fgf23 in 6 h. When an NFAT-inhibitor, 11R-VIVIT, was used, this decrease was partially inhibited. DMP1 overexpression in B6 DMPTG mice also partially prevented this increase (32). Together, this indicates that at least part of the increase of FGF23 levels can be explained by a direct induction by calcium through DMP1 inhibition and the NFAT-pathway (Figure 2). However, since inhibiting the NFAT pathway did not completely eradicate the FGF23 induction by calcium, research is needed to study whether the remaining effect is by the formation of CCPs or a tertiary mechanism.
Non-Classical FGF23 Regulation
Iron Deficiency
In 1982 a link between iron and hypophosphatemia was first described in patients receiving repeated intravenous therapy with saccharated ferric oxide (88). Later, clinical observations in ADHR patients suggested that there is a correlation between iron and FGF23 (89). In ADHR patients with low serum iron levels, higher levels of both iFGF and cFGF23 were found, while low serum iron levels in healthy individuals only result in elevated cFGF23 but not iFGF23 (90). A recent study in a mouse model of CDK showed that anemia is the primary driver of increased Fgf23 in CDK and that this can be rescued by treating the mice with erythropoiesis stimulating agents (91). These observations suggested that there is a role for iron in FGF23 regulation. In a study where wild-type mice were treated with hepcidin, a reactant that causes inflammation-induced iron deficiency, Fgf23 transcription was increased along with elevated levels of cFgf23, but not iFgf23 (92). A similar result was found in mice treated with low-iron diets and iron chelators, where mRNA levels of Fgf23 were increased, but levels of iFgf23 were only slightly elevated (Figure 3). Interestingly, the levels of iFgf23 significantly increased in CKD mice when iron deficiency was induced (92). This indicates that iron deficiency increases transcription and cleavage of FGF23 simultaneously, and that elevated circulating levels of iFGF23 are only present in situations where FGF23 cleavage is impaired, such as in CKD and ADHR patients. Paradoxically, when otherwise healthy women with iron deficiency were treated with ferric carboxymaltose, half of them developed hypophosphatemia (93). This was attributed to an acute increase in iFGF23 levels, while cFGF23 levels were decreased. When these women were treated with iron dextran the levels of cFGF23 decreased as expected but without the spike in iFGF23 (93). The mechanism by which ferric carboxymaltose increases iFGF23 levels is unknown. It could be possible that it promotes O-glycosylation of FGF23 and thereby protects it from cleavage. It should be noted that considering the fact that iron dextran administration did not cause this acute rise in iFGF23, it could be caused by the sugar molecule rather than the iron supplementation. Nevertheless, clinicians should be aware of the effect that iron supplementation can have on mineral homeostasis.
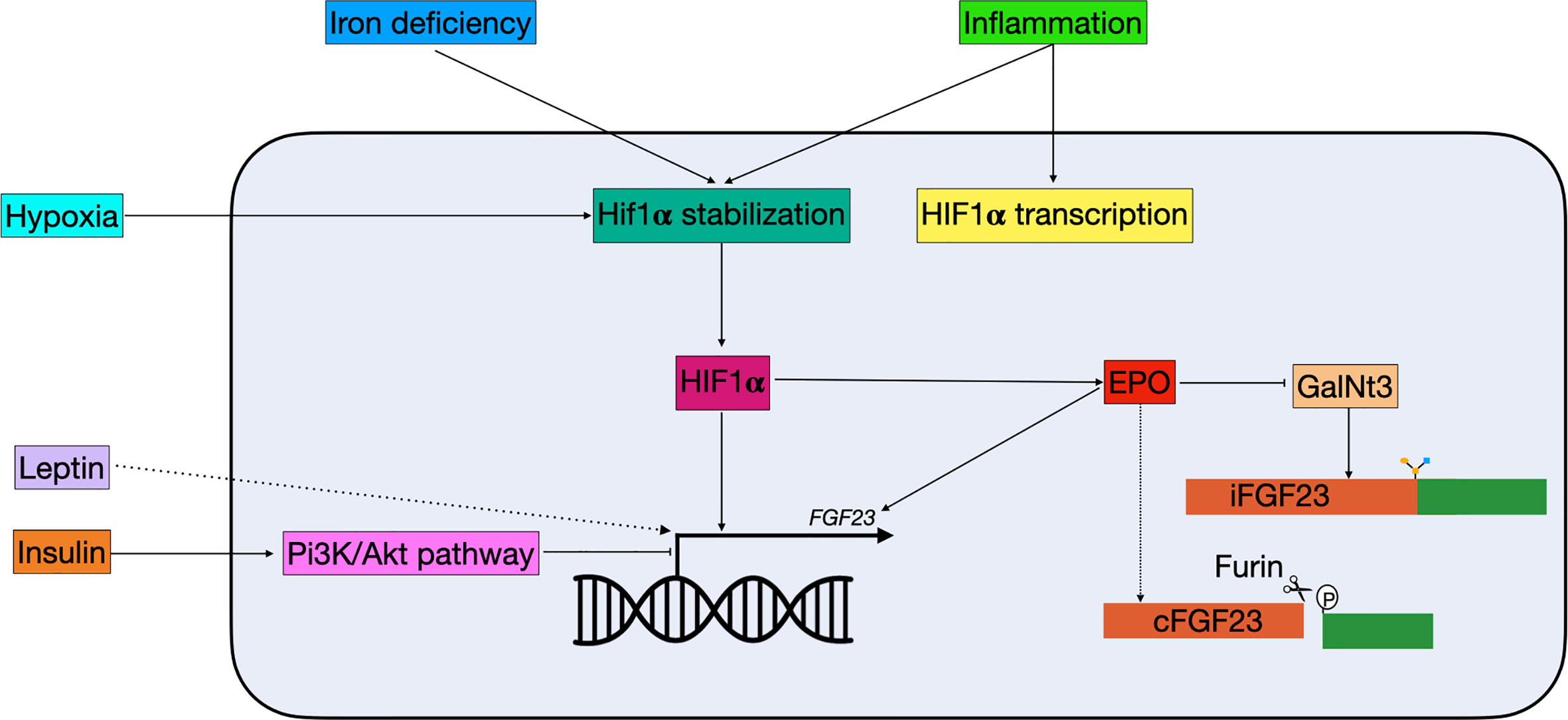
Figure 3 Schematic overview of non-classical FGF23 regulation. Hypoxia, iron deficiency, and inflammation all result in Hif1α stabilization. Hif1α, which can be stimulated by inflammation, binds to the HRE in the FGF23 promotor and stimulate expression. Hif1α can also indirectly regulate FGF23: 1) it stimulates EPO, which directly stimulates FGF23 transcription and 2) it inhibits GalNt3, resulting in increased FGF23 cleavage. Insulin is able to inhibit FGF23 transcription through the Pi3K/Akt pathway, while leptin stimulates FGF23 transcription through a yet unknown mechanism. cFGF23, cleaved fibroblast growth factor 23; EPO, erythropoietin; iFGF23, intact fibroblast growth factor 23; GalNt3, polypeptide N-acetylgalactosaminyltransferase 3; HIF1α, hypoxia inducible factor 1 α; PI3K/Akt, phosphatidylinositol 3-kinase/protein kinase B.
Inflammation
In clinical studies, it has been shown that increased inflammation markers correlate with increased serum FGF23 levels, however, the balance between its production and cleavage is maintained, as in iron deficiency (Figure 3) (94, 95). Moreover, a recent study using a mouse model of CKD has shown that production of the pro-inflammatory protein IL-1β is the driving stimulus for upregulation of Fgf23 expression in early CKD. The usage of a neutralizing antibody to IL-1β blocked the expression of Fgf23 (96). Treatment with inflammatory factors in IDG-SW3 cells also resulted in increased Fgf23 mRNA levels, but not iFgf23 in conditioned medium (97). In mice that were pretreated with a furin inhibitor, serum iFGF23 levels increased rapidly following treatment with inflammatory factors, further indicating an essential role for furin in the cleavage of intact FGF23 (92). Part of the increased Fgf23 mRNA may be mediated through an enhancer region 16 kDa upstream of the Fgf23 gene (98). Although deletion of the enhancer in a mouse model did result in lower levels of Fgf23 mRNA upon stimulation with LPS, TNF-α, and IL-1β, levels of circulating iFGF23 did not change (85).
In multiple inflammatory conditions, there is a role for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (99). Several studies have shown that NF-κB is involved in increased FGF23 transcription, via Orai1 activation (100, 101). It is therefore possible that part of the increased transcription of FGF23 can be explained by NF-κB activation.
Erythropoietin
Both inflammation and functional iron deficiency stimulate Fgf23 transcription indirectly through the production of erythropoietin (EPO) (102). Injection of recombinant human EPO in mice resulted in increased Fgf23 mRNA and cFgf23, but only marginally increased iFgf23, indicating that production and cleavage of FGF23 are directly linked, as observed in inflammation and iron deficiency (102, 103). It is currently unknown why both transcription and cleavage are upregulated during iron deficiency and inflammation. One hypothesis is that cFGF23 has a role in a hitherto undiscovered endocrine feedback loop involved in iron homeostasis and erythropoiesis, without affecting phosphate and calcium homeostasis (104). In a mouse model of acute blood loss, increased levels of cFgf23 were found while GalNt3 mRNA was decreased (105). This might be a mechanism by which changes in oxygen tension and erythropoiesis promote cFGF23 levels, without disturbing the levels of biological active iFGF23 (Figure 3) (104). Most experiments involving EPO are relatively short term, e.g., several hours or days of EPO treatment, while in a clinical setting EPO is given for months or even years. Therefore, research has been done in Tg6 mice, a model constitutively overexpressing human EPO (106). In these mice both iFgf23 and cFgf23 were increased at 6–8 weeks compared to their wild-type littermates (107). Moreover, these mice showed signs of disturbed mineral metabolism, including reduced trabecular bone mineral density, low renal αklotho, urinary calcium wasting, and reduced expression of renal and intestinal phosphate transporters (107). Together this indicates that long-term increases in EPO are able to disturb mineral metabolism, and that the relation between bone mineralization and EPO therapy should be investigated in a clinical setting.
HIF1α
Iron deficiency and inflammation are both able to alter FGF23 transcription via hypoxia inducible factor 1 α (HIF1α). HIF1α is stabilized in case of hypoxia or iron deficiency and activates downstream pathways (92). Moreover, HIF1α is believed to be involved in altered FGF23 cleavage seen in inflammation and iron deficiency. When HIF1α inhibitors were given simultaneously with the IL-1β treatment, iFGF23 levels were elevated in wild-type mice, indicating that HIF1α is directly involved in increased FGF23 cleavage caused by inflammation (92). However, HIF1α may not directly stimulate FGF23, but instead act through EPO. In a study in rats it was shown that treatment with human recombinant EPO resulted in a higher increase in cFGF23 compared to the ones treated with a HIF propyl-hydroxylase (PH) inhibitor, which stabilizes HIF (105). In rats that were pre-treated with anti-EPO antibodies, the increased cFGF caused by HIF PH inhibitor treatment was completely abolished, indicating that EPO may be the mediator in the increase of cFGF23 in response to HIF stabilization (108). Nonetheless there are some studies that suggest that HIF1α stabilization directly influences FGF23 transcription (Figure 3). Fluorescent-tagged antibodies for both HIF1α and FGF23 co-localize in perivascular cells in resected tumors of patients with TIO and chromatin immunoprecipitation assays showed that HIF1α might bind to a HIF-binding site within the FGF23 promotor (109).
For a deeper understanding of the interplay between iron, inflammation, EPO and HIF1α more research to FGF23 biology is needed. The cFGF23 fragments that are increased during iron deficiency and inflammation, do not seem to represent the classical role of FGF23 in maintaining mineral homeostasis. It is therefore important to study how exactly these processes are able to increase both FGF23 transcription and cleavage.
Insulin and Diabetes
In clinical studies diabetes was found to be associated with higher serum levels of FGF23 (110, 111). Both diabetes type 1 and 2 are associated with high levels of inflammation, which is positively correlated with high levels of FGF23 (112, 113). It is therefore possible that the relation between diabetes and high FGF23 levels is mostly explained by inflammation. However, Bär et al. showed in UMR106 cells that insulin treatment is able to suppress FGF23 gene expression in the absence of inflammatory factors (114). Most of the downstream effects of insulin are mediated through the PI3K/PKB/Akt signaling pathway, and inhibition of this pathway or its downstream transcription factor, FOXO1, ablated the effect of insulin on FGF23 expression (Figure 3) (114, 115). Moreover, in a diabetic mouse model, where use of streptozocin ablates insulin production through pancreatic β-cell destruction, a spike in serum Fgf23 was seen, which could be prevented by giving the mice daily injections of insulin (114). Finally, the authors looked at a correlation between FGF23 levels and fasted insulin concentration in healthy human volunteers. They found that the baseline insulin concentration was inversely correlated with FGF23 levels. When the volunteers were subjected to a glucose challenge, the increase in insulin was also inversely correlated with FGF23 levels (114). Together these results indicate that insulin is an inhibitor of FGF23, at least partially independent of the inflammation associated with diabetes.
Leptin
Leptin (encoded by LEP) is a hormone mainly produced by adipocytes and secreted in response to a change in body energy stores (116). Leptin deficient ob/ob mice overexpress 25(OH)D, 1,25(OH)2D3, and 1α-hydroxylase and a lower expression of Fgf23 (117, 118). Administration of leptin to these mice corrects for the overexpression of 25(OH)D, 1,25(OH)2D3, and 1α-hydroxylase (117). However, leptin does not inhibit 1α-hydroxylase in primary cell cultures, suggesting the involvement of another factor (118). Moreover, when leptin is administrated to ob/ob mice this results in an increase of serum Fgf23 levels, while decreasing serum phosphate, calcium and 1,25(OH)2D3, an effect that was not found in leptin receptor-deficient db/db mice (118). The authors also investigated whether administrating Fgf23 to ob/ob mice had similar effects as leptin, which resulted in a normalization of 1α-hydroxylase expression (118). Together this indicates that leptin is able to directly stimulate Fgf23 expression in ob/ob mice (Figure 3). In a clinical study, higher levels of leptin have also been associated with higher levels of FGF23. However, this association was not found to be significant when leptin was included in a multivariate-adjusted model (119). Even though there are some strong indications that leptin indeed stimulates FGF23, the mechanism behind it is unknown. Additional work should reveal the interaction between these hormones in order to add leptin to the definitive list of FGF23 regulators.
Discussion
Since the discovery of FGF23 in the year 2000, much knowledge has been gathered about the upstream regulators of this hormone. In this review, we distinguish the classical regulation of FGF23 involved in phosphate metabolism and the non-classical regulation affected by inflammation and iron or insulin levels. Irregularity in these processes underlie disorders in bone and mineral metabolism and insights in FGF23 regulation should provide insights with potential therapeutic consequences. Even though insights in FGF23 biology are advancing, many of the mechanisms behind it are still unclear.
Considerations When Studying FGF23 Regulation
FGF23 is primarily produced by osteocytes, which are challenging cells to study (120). Osteocytes are terminally differentiated cells and are hard to isolate in sufficient quantities. In the past years several cell lines have been developed from mouse osteocytes, which are also used in a large portion of the studies described in this review (121). However, these cell models have their limitations, as they have to be modified in order to dedifferentiate and proliferate, while osteocytes in vivo do not (122–124). Much of the research discussed in this review was performed in osteoblast cell lines (e.g., MC3T-E1), but it still remains controversial whether osteoblasts are truly a source of FGF23, as it is mostly described as a late osteocyte marker (123, 125, 126). Moreover, the osteosarcoma cell line UMR106 is often used, but as this is a cancerous cell line, FGF23 regulation might be altered/deranged (127). Because all these cell lines differ from the osteocytes found in vivo it is hard to state with certainty that mechanism found in these cell lines are representative of the situation in humans or animals.
Secondly, osteocytes are embedded in the mineralized bone matrix, and therefore might behave differently than when they are cultured on a flat surface. Research progress regarding 3D culture models might be a solution for this problem. One recent study showed that primary murine osteoblasts gain a more osteocyte-like pattern when cultured in 3D and dedifferentiated to a more osteoblast-like pattern when placed back in 2D (128). For OCY454 cells it has been shown that culturing in a 3D scaffold resulted in a more robust increase in Fgf23 transcription following PTH treatment (124). Another study showed that primary human osteocytes had a higher expression of SOST and FGF23 when they were cultured in 3D in hypoxic conditions (129). Together these results indicate that optimizing the culture conditions for osteocytes is important to study FGF23 as it would function in vivo.
The most promising option to study FGF23 regulation might be the culture of human bone chips, as they will mimic the in vivo situation more closely (130). However, limited availability and donor variation makes it difficult to produce consistent results. There is a long way to go before we can study human FGF23 biology in its natural human environment, but with the currently available models it will be possible to gain valuable insights in FGF23 regulation.
Biological Role for cFGF23
The intent of classical FGF23 regulation is unambiguous: maintaining phosphate homeostasis in the body. For non-classical FGF23 regulation this is not as straightforward, as inflammation or iron deficiency will not only result in higher FGF23 expression but also in increased cleavage. It is currently unclear whether this is a protective mechanism to maintain mineral homeostasis when HIF1α is stabilized or that the cFGF23 fragments have a yet undefined function. Based on findings in mice where cFGF23 inhibited FGF23 signaling, it can be hypothesized that cFGF23 acts as a competitive inhibitor to the FGF-receptor (21). Still, healthy humans and animals with iron deficiency often have normal serum phosphate levels (90, 131, 132). This makes it unlikely that cFGF23 would have a role in FGF23 inhibition in healthy individuals, as they would be expected to present with hyperphosphatemia. It is also possible that cFGF23 plays a role in a yet to be discovered feedback loop in maintaining iron homeostasis and erythropoiesis. Further research needs to be done to identify the precise effect of cFGF23 fragments, as they seem to have no role in classical FGF23 regulation but might have unique properties instead.
FGF23 Cleavage Machinery
The discovery of genetic mutations affecting FGF23 cleavage elegantly showed that the intracellular FGF23 cleavage machinery is essential for its functioning. Although it is clear that proteases such as furin are involved in cleaving FGF23, and that phosphorylation by FAM20C and O-glycosylation by Gal-NAc-T3 regulate this process, the exact mechanisms of FGF23 cleavage are unknown (24). The mechanisms by which FAM20C is transcriptionally and functionally regulated remains to be elucidated, though studies suggest that changes in Gal-NAc-T3 and FAM20C activity or transcription seem to be regulated by iron, iron deficiency and elevated serum phosphate (24, 133, 134). It is likely that FGF23 cleavage is far more complicated than this but whether and how other upstream regulators affect cleavage and which other proteins might be involved remain unanswered questions. For deeper understanding of FGF23 regulation it is therefore important to study how this process exactly works and what effect upstream regulators have.
Emerging Regulators
Apart from the factors described here, some other upstream regulators of FGF23 have emerged in the past years and undoubtedly there are more to be discovered. Factors that were found to cause an upregulation of FGF23 expression are glycerol-3-phosphate, cytoskeleton reorganization, NF-κB/Orai1 signaling, aldosterone, and cytosolic calcium activity (100, 101, 135, 136). These factors are mostly described in single in vitro studies or small clinical studies, and it is therefore not yet fully understood why and how they affect FGF23 transcription. This emphasizes how much there is still to uncover about FGF23 and all of its potential upstream regulators. Additionally, recent advances in RNA sequencing and bioinformatics have revealed hitherto unknown roles of circular RNAs, long non-coding RNAs (lncRNAs), Piwi-interacting (pi)RNAs, and micro (mi)RNAs in various signaling pathways (137, 138). However, the role of these non-coding RNAs in osteocyte function is still unexplored (139). Studies to the role these non-coding RNAs in osteocyte function and FGF23 signaling, could therefore unravel a whole new class of FGF23 regulators.
Therapeutic Therapies for FGF23-Mediated Pathologies
Conventional treatment for children with XLH consists of 1,25(OH)2D3 and phosphate supplementation, which results in improved growth and less rachitic deformities. However, outcomes are variable and the treatment comes with adverse effects such as hypercalcemic hyperparathyroidism and nephrocalcinosis (140). A new promising therapy is the monoclonal FGF23 antibody burosumab, which has been shown to normalize phosphate levels in both adults and children. Burosumab is currently FDA approved for the treatment of XLH and future studies need to show whether this drug can also be used in other FGF23-mediated pathologies, such as TIO (140, 141). Besides these established therapies, experimental treatments included the use of growth hormone, which seemed to improve growth during the therapy but did not result in a significant difference in adult height (142). Some new treatment options are currently being tested in hyp mice. These include 1) the use of hexa-D-arginine which increases the expression of 7B2, which indirectly suppresses FGF23 expression (39) and and 2) methods to block the FGFR in hyp mouse, which results in improved growth (143). Many of the current therapies focus on blocking FGF23 activity or managing the effect of high FGF23 levels. This underlines the importance of developing therapies that will target the upstream regulators of FGF23 directly, instead of managing the downstream consequences.
Conclusion
Advances over the past 20 years have provided knowledge regarding several regulators of FGF23 both involved in mineral metabolism and possible alternative processes. Moreover, it has become clear that FGF23 transcription and cleavage are independently regulated processes. The way upstream regulators affect these actions is not always clear. It is also yet to be discovered why the osteocyte would increase FGF23 production in response to some of these factors, only to immediately cleave them. Future research answering these questions might not only provide valuable insights in the molecular mechanisms regulating FGF23, but may also be of use to develop new therapeutic strategies for patients with FGF23-associated disorders.
Author Contributions
DR performed the literature research, wrote the manuscript and designed the figures. DR, MZ, and BE revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
DR is supported by a grant from Health Holland (PhosphoNorm; LSHM18029).
Conflict of Interest
The authors declare that the research was conducted without any commercial or financial relationships that may be regarded as a potential conflict of interest.
References
1. Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer (2011) 18:R53–77. doi: 10.1530/ERC-11-0006
2. Prader A, Illig R, Uehlinger E, Stalder G. Rickets following bone tumor. Helv Paediatr Acta (1959) 14:554–65.
3. Meyer RA, Meyer MH, Gray RW. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res Off J Am Soc Bone Miner Res (1989) 4:493–500. doi: 10.1002/jbmr.5650040407
4. Miyauchi A, Fukase M, Tsutsumi M, Fujita T. Hemangiopericytoma-induced osteomalacia: tumor transplantation in nude mice causes hypophosphatemia and tumor extracts inhibit renal 25-hydroxyvitamin D 1-hydroxylase activity. J Clin Endocrinol Metab (1988) 67:46–53. doi: 10.1210/jcem-67-1-46
5. ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet (2000) 26:345–8. doi: 10.1038/81664
6. Levi M, Gratton E, Forster IC, Hernando N, Wagner CA, Biber J, et al. Mechanisms of phosphate transport. Nat Rev Nephrol (2019) 15:482–500. doi: 10.1038/s41581-019-0159-y
7. Bär L, Stournaras C, Lang F, Föller M. Regulation of fibroblast growth factor 23 (FGF23) in health and disease. FEBS Lett (2019) 593:1879–900. doi: 10.1002/1873-3468.13494
8. Erben RG, Andrukhova O. FGF23-Klotho signaling axis in the kidney. Bone (2017) 100:62–8. doi: 10.1016/j.bone.2016.09.010
9. Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun (2004) 314:409–14. doi: 10.1016/j.bbrc.2003.12.102
10. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res Off J Am Soc Bone Miner Res (2004) 19:429–35. doi: 10.1359/JBMR.0301264
11. Hattenhauer O, Traebert M, Murer H, Biber J. Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol (1999) 277:G756–762. doi: 10.1152/ajpgi.1999.277.4.G756
12. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol - Ren Physiol (2009) 297:F282–91. doi: 10.1152/ajprenal.90742.2008
13. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest (2007) 117:4003–8. doi: 10.1172/JCI32409
14. Krajisnik T, Björklund P, Marsell R, Ljunggren O, Akerström G, Jonsson KB, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol (2007) 195:125–31. doi: 10.1677/JOE-07-0267
15. Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med (2010) 61:91–104. doi: 10.1146/annurev.med.051308.111339
16. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet TIG (2004) 20:563–9. doi: 10.1016/j.tig.2004.08.007
17. Richter B, Faul C. FGF23 Actions on Target Tissues-With and Without Klotho. Front Endocrinol (2018) 9:189:189. doi: 10.3389/fendo.2018.00189
18. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol (2013) 75:503–33. doi: 10.1146/annurev-physiol-030212-183727
19. Yamashita T. Structural and Biochemical Properties of Fibroblast Growth Factor 23. Ther Apher Dial (2005) 9:313–8. doi: 10.1111/j.1744-9987.2005.00288.x
20. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol (2007) 27:3417–28. doi: 10.1128/MCB.02249-06
21. Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U.S.A. (2010) 107:407–12. doi: 10.1073/pnas.0902006107
22. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U.S.A. (2001) 98:6500–5. doi: 10.1073/pnas.101545198
23. Yamamoto H, Ramos-Molina B, Lick AN, Prideaux M, Albornoz V, Bonewald L, et al. Posttranslational processing of FGF23 in osteocytes during the osteoblast to osteocyte transition. Bone (2016) 84:120–30. doi: 10.1016/j.bone.2015.12.055
24. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci USA (2014) 111:5520–5. doi: 10.1073/pnas.1402218111
25. Lindberg I, Pang HW, Stains JP, Clark D, Yang AJ, Bonewald L, et al. FGF23 is Endogenously Phosphorylated in Bone Cells. J Bone Miner Res Off J Am Soc Bone Miner Res (2015) 30:449–54. doi: 10.1002/jbmr.2354
26. Wang X, Wang S, Li C, Gao T, Liu Y, Rangiani A, et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PloS Genet (2012) 8:e1002708. doi: 10.1371/journal.pgen.1002708
27. Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3 , encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet (2004) 36:579–81. doi: 10.1038/ng1358
28. Qin C, D’Souza R, Feng JQ. Dentin matrix protein 1 (DMP1): new and important roles for biomineralization and phosphate homeostasis. J Dent Res (2007) 86:1134–41. doi: 10.1177/154405910708601202
29. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet (2006) 38:1310–5. doi: 10.1038/ng1905
30. Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem (2015) 290:10447–59. doi: 10.1074/jbc.M114.609230
31. Martin A, David V, Li H, Dai B, Feng JQ, Quarles LD. Overexpression of the DMP1 C-terminal fragment stimulates FGF23 and exacerbates the hypophosphatemic rickets phenotype in Hyp mice. Mol Endocrinol Baltim Md (2012) 26:1883–95. doi: 10.1210/me.2012-1062
32. Dussold C, Gerber C, White S, Wang X, Qi L, Francis C, et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res (2019) 7. doi: 10.1038/s41413-019-0051-1
33. Lecoq A-L, Brandi ML, Linglart A, Kamenický P. Management of X-linked hypophosphatemia in adults. Metabolism (2020) 103S:154049. doi: 10.1016/j.metabol.2019.154049
34. Abe K, Masatomi Y, Nakajima Y, Shintani S, Moriwaki Y, Sobue S, et al. The occurrence of interglobular dentin in incisors of hypophosphatemic mice fed a high-calcium and high-phosphate diet. J Dent Res (1992) 71:478–83. doi: 10.1177/00220345920710031101
35. Barratt KR, Sawyer RK, Atkins GJ, St-Arnaud R, Anderson PH. Vitamin D supplementation improves bone mineralisation independent of dietary phosphate in male X-linked hypophosphatemic (Hyp) mice. Bone (2020), 115767. doi: 10.1016/j.bone.2020.115767
36. Addison WN, Masica DL, Gray JJ, McKee MD. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J Bone Miner Res (2010) 25:695–705. doi: 10.1359/jbmr.090832
37. Liu S, Guo R, Simpson LG, Xiao Z-S, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem (2003) 278:37419–26. doi: 10.1074/jbc.M304544200
38. Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive Osteocytic Fgf23 Secretion Contributes to Pyrophosphate Accumulation and Mineralization Defect in Hyp Mice. PloS Biol (2016) 14:e1002427. doi: 10.1371/journal.pbio.1002427
39. Rowe PSN. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr (2012) 22:61–86. doi: 10.1615/critreveukargeneexpr.v22.i1.50
40. Yuan B, Feng JQ, Bowman S, Liu Y, Blank RD, Lindberg I, et al. Hexa-D-arginine treatment increases 7B2•PC2 activity in hyp-mouse osteoblasts and rescues the HYP phenotype. J Bone Miner Res Off J Am Soc Bone Miner Res (2013) 28:56–72. doi: 10.1002/jbmr.1738
41. Robinson M-E, AlQuorain H, Murshed M, Rauch F. Mineralized tissues in hypophosphatemic rickets. Pediatr Nephrol (2019) 35:1843–54. doi: 10.1007/s00467-019-04290-y
42. Orstavik KH, Orstavik RE, Halse J, Knudtzon J. X chromosome inactivation pattern in female carriers of X linked hypophosphataemic rickets. J Med Genet (1996) 33:700–3. doi: 10.1136/jmg.33.8.700
43. Owen CJ, Habeb A, Pearce SHS, Wright M, Ichikawa S, Sorenson AH, et al. Discordance for X-linked hypophosphataemic rickets in identical twin girls. Horm Res (2009) 71:237–44. doi: 10.1159/000201113
44. Jørgensen AL, Philip J, Raskind WH, Matsushita M, Christensen B, Dreyer V, et al. Different patterns of X inactivation in MZ twins discordant for red-green color-vision deficiency. Am J Hum Genet (1992) 51:291–8.
45. Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol (2005) 289:G1036–1042. doi: 10.1152/ajpgi.00243.2005
46. Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, et al. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab (2005) 288:E1101–1109. doi: 10.1152/ajpendo.00502.2004
47. Kinoshita Y, Ito N, Makita N, Nangaku M, Fukumoto S. Changes in bone metabolic parameters following oral calcium supplementation in an adult patient with vitamin D-dependent rickets type 2A. Endocr J (2017) 64:589–96. doi: 10.1507/endocrj.EJ16-0583
48. Sirajudeen S, Shah I, Al Menhali A. A Narrative Role of Vitamin D and Its Receptor: With Current Evidence on the Gastric Tissues. Int J Mol Sci (2019) 20:3832. doi: 10.3390/ijms20153832
49. Collins MT, Lindsay JR, Jain A, Kelly MH, Cutler CM, Weinstein LS, et al. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J Bone Miner Res Off J Am Soc Bone Miner Res (2005) 20:1944–50. doi: 10.1359/JBMR.050718
50. Saito H, Maeda A, Ohtomo S-I, Hirata M, Kusano K, Kato S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem (2005) 280:2543–9. doi: 10.1074/jbc.M408903200
51. Saji F, Shigematsu T, Sakaguchi T, Ohya M, Orita H, Maeda Y, et al. Fibroblast growth factor 23 production in bone is directly regulated by 1{alpha},25-dihydroxyvitamin D, but not PTH. Am J Physiol Renal Physiol (2010) 299:F1212–1217. doi: 10.1152/ajprenal.00169.2010
52. Yashiro M, Ohya M, Mima T, Nakashima Y, Kawakami K, Yamamoto S, et al. Active vitamin D and vitamin D analogs stimulate fibroblast growth factor 23 production in osteocyte-like cells via the vitamin D receptor. J Pharm BioMed Anal (2020) 182:113139. doi: 10.1016/j.jpba.2020.113139
53. Marcinkowska E. The Vitamin D System in Humans and Mice: Similar but Not the Same. Y Rep (2020) 3:1. doi: 10.3390/reports3010001
54. Panda DK, Miao D, Tremblay ML, Sirois J, Farookhi R, Hendy GN, et al. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U.S.A. (2001) 98:7498–503. doi: 10.1073/pnas.131029498
55. Kim CJ, Kaplan LE, Perwad F, Huang N, Sharma A, Choi Y, et al. Vitamin D 1alpha-hydroxylase gene mutations in patients with 1alpha-hydroxylase deficiency. J Clin Endocrinol Metab (2007) 92:3177–82. doi: 10.1210/jc.2006-2664
56. Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet (1997) 16:391–6. doi: 10.1038/ng0897-391
57. Feldman D, J Malloy P. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. BoneKEy Rep (2014) 3:510. doi: 10.1038/bonekey.2014.5
58. Blau JE, Collins MT. The PTH-Vitamin D-FGF23 axis. Rev Endocr Metab Disord (2015) 16:165–74. doi: 10.1007/s11154-015-9318-z
59. Burnett-Bowie S-AM, Henao MP, Dere ME, Lee H, Leder BZ. Effects of hPTH(1-34) infusion on circulating serum phosphate, 1,25-dihydroxyvitamin D, and FGF23 levels in healthy men. J Bone Miner Res Off J Am Soc Bone Miner Res (2009) 24:1681–5. doi: 10.1359/jbmr.090406
60. Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJS, et al. Effects of parathyroid hormone on Wnt signaling pathway in bone. J Cell Biochem (2005) 95:1178–90. doi: 10.1002/jcb.20506
61. Maeda A, Okazaki M, Baron DM, Dean T, Khatri A, Mahon M, et al. Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH. Proc Natl Acad Sci U.S.A. (2013) 110:5864–9. doi: 10.1073/pnas.1301674110
62. Datta NS. Muscle-bone and fat-bone interactions in regulating bone mass: do PTH and PTHrP play any role? Endocrine (2014) 47:389–400. doi: 10.1007/s12020-014-0273-3
63. Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol (2010) 299:F882–889. doi: 10.1152/ajprenal.00360.2010
64. Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int (2014) 86:1106–15. doi: 10.1038/ki.2014.215
65. An Y, Zhao J, Nie F, Wu Y, Xia Y, Li D. Parathyroid hormone (PTH) promotes ADSC osteogenesis by regulating SIK2 and Wnt4. Biochem Biophys Res Commun (2019) 516:551–7. doi: 10.1016/j.bbrc.2019.06.084
66. Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun (2016) 7:13176. doi: 10.1038/ncomms13176
67. Olauson H, Lindberg K, Amin R, Jia T, Wernerson A, Andersson G, et al. Targeted deletion of Klotho in kidney distal tubule disrupts mineral metabolism. J Am Soc Nephrol JASN (2012) 23:1641–51. doi: 10.1681/ASN.2012010048
68. Hum JM, O’Bryan L, Smith RC, White KE. Novel functions of circulating Klotho. Bone (2017) 100:36–40. doi: 10.1016/j.bone.2016.11.025
69. Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, et al. A translocation causing increased α-Klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci (2008) 105:3455–60. doi: 10.1073/pnas.0712361105
70. Smith RC, O’Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, et al. Circulating αKlotho influences phosphate handling by controlling FGF23 production. J Clin Invest (2012) 122:4710–5. doi: 10.1172/JCI64986
71. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci U.S.A. (2019) 116:11418–27. doi: 10.1073/pnas.1815166116
72. Hum JM, O’Bryan LM, Tatiparthi AK, Cass TA, Clinkenbeard EL, Cramer MS, et al. Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho. J Am Soc Nephrol JASN (2017) 28:1162–74. doi: 10.1681/ASN.2015111266
73. Ferrari SL, Bonjour J-P, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab (2005) 90:1519–24. doi: 10.1210/jc.2004-1039
74. Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab (2006) 91:3144–9. doi: 10.1210/jc.2006-0021
75. Gutiérrez OM, Wolf M, Taylor EN. Fibroblast growth factor 23, cardiovascular disease risk factors, and phosphorus intake in the health professionals follow-up study. Clin J Am Soc Nephrol CJASN (2011) 6:2871–8. doi: 10.2215/CJN.02740311
76. Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int (2006) 70:2141–7. doi: 10.1038/sj.ki.5002000
77. Ito N, Fukumoto S, Takeuchi Y, Takeda S, Suzuki H, Yamashita T, et al. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J Bone Miner Metab (2007) 25:419–22. doi: 10.1007/s00774-007-0779-3
78. Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol Baltim Md (2009) 23:1505–18. doi: 10.1210/me.2009-0085
79. Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab (2018) 11:197–204. doi: 10.1016/j.molmet.2018.02.007
80. Camalier CE, Yi M, Yu L-R, Hood BL, Conrads KA, Lee YJ, et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J Cell Physiol (2013) 228:1536–50. doi: 10.1002/jcp.24312
81. Hori M, Kinoshita Y, Taguchi M, Fukumoto S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J Bone Miner Metab (2016) 34:132–9. doi: 10.1007/s00774-015-0651-9
82. Ito N, Findlay DM, Anderson PH, Bonewald LF, Atkins GJ. Extracellular phosphate modulates the effect of 1α,25-dihydroxy vitamin D3 (1,25D) on osteocyte like cells. J Steroid Biochem Mol Biol (2013) 136:183–6. doi: 10.1016/j.jsbmb.2012.09.029
83. Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int (2013) 83:1029–41. doi: 10.1038/ki.2012.439
84. Rodriguez-Ortiz ME, Lopez I, Muñoz-Castañeda JR, Martinez-Moreno JM, Ramírez AP, Pineda C, et al. Calcium Deficiency Reduces Circulating Levels of FGF23. J Am Soc Nephrol (2012) 23:1190–7. doi: 10.1681/ASN.2011101006
85. Akiyama K, Miura Y, Hayashi H, Sakata A, Matsumura Y, Kojima M, et al. Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int (2020) 97:702–12. doi: 10.1016/j.kint.2019.10.019
86. Soriano S, Ojeda R, Rodríguez M, Almadén Y, Rodríguez M, Martín-Malo A, et al. The effect of phosphate binders, calcium and lanthanum carbonate on FGF23 levels in chronic kidney disease patients. Clin Nephrol (2013) 80:17–22. doi: 10.5414/CN107764
87. David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium Regulates FGF-23 Expression in Bone. Endocrinology (2013) 154:4469–82. doi: 10.1210/en.2013-1627
88. Okada M, Imamura K, Fuchigami T, Omae T, Iida M, Nanishi F, et al. [2 cases of nonspecific multiple ulcers of the small intestine associated with osteomalacia caused by long-term intravenous administration of saccharated ferric oxide]. Nihon Naika Gakkai Zasshi J Jpn Soc Intern Med (1982) 71:1566–72.
89. Wolf M, White KE. Coupling FGF23 Production and Cleavage: Iron Deficiency, Rickets and Kidney Disease. Curr Opin Nephrol Hypertens (2014) 23:411–9. doi: 10.1097/01.mnh.0000447020.74593.6f
90. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron Modifies Plasma FGF23 Differently in Autosomal Dominant Hypophosphatemic Rickets and Healthy Humans. J Clin Endocrinol Metab (2011) 96:3541–9. doi: 10.1210/jc.2011-1239
91. Noonan ML, Clinkenbeard EL, Ni P, Swallow EA, Tippen SP, Agoro R, et al. Erythropoietin and a hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHDi) lowers FGF23 in a model of chronic kidney disease (CKD). Physiol Rep (2020) 8:e14434. doi: 10.14814/phy2.14434
92. David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int (2016) 89:135–46. doi: 10.1038/ki.2015.290
93. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res (2013) 28:1793–803. doi: 10.1002/jbmr.1923
94. Dounousi E, Torino C, Pizzini P, Cutrupi S, Panuccio V, D’Arrigo G, et al. Intact FGF23 and α-Klotho during acute inflammation/sepsis in CKD patients. Eur J Clin Invest (2016) 46:234–41. doi: 10.1111/eci.12588
95. Holecki M, Chudek J, Owczarek A, Olszanecka-Glinianowicz M, Bożentowicz-Wikarek M, Duława J, et al. Inflammation but not obesity or insulin resistance is associated with increased plasma fibroblast growth factor 23 concentration in the elderly. Clin Endocrinol (Oxf) (2015) 82:900–9. doi: 10.1111/cen.12759
96. McKnight Q, Jenkins S, Li X, Nelson T, Marlier A, Cantley LG, et al. IL-1β drives production of FGF-23 at the onset of chronic kidney disease in mice. J Bone Miner Res Off J Am Soc Bone Miner Res (2020) 35:1352–62. doi: 10.1002/jbmr.4003
97. Ito N, Wijenayaka AR, Prideaux M, Kogawa M, Ormsby RT, Evdokiou A, et al. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol Cell Endocrinol (2015) 399:208–18. doi: 10.1016/j.mce.2014.10.007
98. Onal M, Carlson AH, Thostenson JD, Benkusky NA, Meyer MB, Lee SM. Pike JW. A Novel Distal Enhancer Mediates Inflammation-, PTH-, and Early Onset Murine Kidney Disease-Induced Expression of the Mouse Fgf23 Gene. JBMR Plus (2017) 2:31–46. doi: 10.1002/jbm4.10023
99. Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol (2006) 290:L622–45. doi: 10.1152/ajplung.00477.2005
100. Fajol A, Honisch S, Zhang B, Schmidt S, Alkahtani S, Alarifi S, et al. Fibroblast growth factor (Fgf) 23 gene transcription depends on actin cytoskeleton reorganization. FEBS Lett (2016) 590:705–15. doi: 10.1002/1873-3468.12096
101. Zhang B, Yan J, Umbach AT, Fakhri H, Fajol A, Schmidt S, et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J Mol Med Berl Ger (2016) 94:557–66. doi: 10.1007/s00109-015-1370-3
102. Clinkenbeard EL, Hanudel MR, Stayrook KR, Appaiah HN, Farrow EG, Cass TA, et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica (2017) 102:e427–30. doi: 10.3324/haematol.2017.167882
103. Hanudel MR, Eisenga MF, Rappaport M, Chua K, Qiao B, Jung G, et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol Dial Transplant (2019) 34:2057–65. doi: 10.1093/ndt/gfy189
104. Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat Rev Nephrol (2020) 16:7–19. doi: 10.1038/s41581-019-0189-5
105. Rabadi S, Udo I, Leaf DE, Waikar SS, Christov M. Acute blood loss stimulates fibroblast growth factor 23 production. Am J Physiol Renal Physiol (2018) 314:F132–9. doi: 10.1152/ajprenal.00081.2017
106. Ruschitzka FT, Wenger RH, Stallmach T, Quaschning T, de Wit C, Wagner K, et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc Natl Acad Sci U.S.A. (2000) 97:11609–13.
107. Daryadel A, Natale L, Seebeck P, Bettoni C, Schnitzbauer U, Gassmann M, et al. Elevated FGF23 and disordered renal mineral handling with reduced bone mineralization in chronically erythropoietin over-expressing transgenic mice. Sci Rep (2019) 9. doi: 10.1038/s41598-019-51577-z
108. Flamme I, Ellinghaus P, Urrego D, Krüger T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PloS One (2017) 12:e0186979. doi: 10.1371/journal.pone.0186979
109. Zhang Q, Doucet M, Tomlinson RE, Han X, Quarles LD, Collins MT, et al. The hypoxia-inducible factor-1α activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res (2016) 4:1–6. doi: 10.1038/boneres.2016.11
110. Hanks LJ, Casazza K, Judd SE, Jenny NS, Gutiérrez OM. Associations of fibroblast growth factor-23 with markers of inflammation, insulin resistance and obesity in adults. PloS One (2015) 10:e0122885. doi: 10.1371/journal.pone.0122885
111. Garland JS, Holden RM, Ross R, Adams MA, Nolan RL, Hopman WM, et al. Insulin resistance is associated with Fibroblast Growth Factor-23 in stage 3-5 chronic kidney disease patients. J Diabetes Complications (2014) 28:61–5. doi: 10.1016/j.jdiacomp.2013.09.004
112. Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. Diabetes mellitus and inflammation. Curr Diabetes Rep (2013) 13:435–44. doi: 10.1007/s11892-013-0375-y
113. Cabrera SM, Henschel AM, Hessner MJ. Innate inflammation in type 1 diabetes. Transl Res J Lab Clin Med (2016) 167:214–27. doi: 10.1016/j.trsl.2015.04.011
114. Bär L, Feger M, Fajol A, Klotz L-O, Zeng S, Lang F, et al. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc Natl Acad Sci U.S.A. (2018) 115:5804–9. doi: 10.1073/pnas.1800160115
115. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell (2017) 169:381–405. doi: 10.1016/j.cell.2017.04.001
116. Zhang Y, Chua S. Leptin Function and Regulation. Compr Physiol (2017) 8:351–69. doi: 10.1002/cphy.c160041
117. Matsunuma A, Kawane T, Maeda T, Hamada S, Horiuchi N. Leptin Corrects Increased Gene Expression of Renal 25-Hydroxyvitamin D3-1α-Hydroxylase and -24-Hydroxylase in Leptin-Deficient, ob/ob Mice. Endocrinology (2004) 145:1367–75. doi: 10.1210/en.2003-1010
118. Tsuji K, Maeda T, Kawane T, Matsunuma A, Horiuchi N. Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1α,25-dihydroxyvitamin D3 synthesis in leptin-deficient ob/ob Mice. J Bone Miner Res (2010) 25:1711–23. doi: 10.1002/jbmr.65
119. Mirza MAI, Alsiö J, Hammarstedt A, Erben RG, Michaëlsson K, Tivesten A, et al. Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler Thromb Vasc Biol (2011) 31:219–27. doi: 10.1161/ATVBAHA.110.214619
120. Bonewald LF, Wacker MJ. FGF23 Production by Osteocytes. Pediatr Nephrol Berl Ger (2013) 28:563–8. doi: 10.1007/s00467-012-2309-3
121. Zhang C, Bakker AD, Klein-Nulend J, Bravenboer N. Studies on Osteocytes in Their 3D Native Matrix Versus 2D In Vitro Models. Curr Osteoporos Rep (2019) 17:207–16. doi: 10.1007/s11914-019-00521-1
122. Kato Y, Boskey A, Spevak L, Dallas M, Hori M, Bonewald LF. Establishment of an Osteoid Preosteocyte-like Cell MLO-A5 That Spontaneously Mineralizes in Culture. J Bone Miner Res (2001) 16:1622–33. doi: 10.1359/jbmr.2001.16.9.1622
123. Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res (2011) 26:2634–46. doi: 10.1002/jbmr.465
124. Spatz JM, Wein MN, Gooi JH, Qu Y, Garr JL, Liu S, et al. The Wnt Inhibitor Sclerostin Is Up-regulated by Mechanical Unloading in Osteocytes in Vitro. J Biol Chem (2015) 290:16744–58. doi: 10.1074/jbc.M114.628313
125. Wang K, Le L, Chun BM, Tiede-Lewis LM, Shiflett LA, Prideaux M, et al. A Novel Osteogenic Cell Line That Differentiates Into GFP-Tagged Osteocytes and Forms Mineral With a Bone-Like Lacunocanalicular Structure. J Bone Miner Res Off J Am Soc Bone Miner Res (2019) 34:979–95. doi: 10.1002/jbmr.3720
126. Bellido T. Osteocyte-Driven Bone Remodeling. Calcif Tissue Int (2014) 94:25–34. doi: 10.1007/s00223-013-9774-y
127. Martin TJ, Ingleton PM, Underwood JCE, Michelangeli VP, Hunt NH, Melick RA. Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature (1976) 260:436–8. doi: 10.1038/260436a0
128. Sawa N, Fujimoto H, Sawa Y, Yamashita J. Alternating Differentiation and Dedifferentiation between Mature Osteoblasts and Osteocytes. Sci Rep (2019) 9. doi: 10.1038/s41598-019-50236-7
129. Choudhary S, Sun Q, Mannion C, Kissin Y, Zilberberg J, Lee WY. Hypoxic Three-Dimensional Cellular Network Construction Replicates Ex Vivo the Phenotype of Primary Human Osteocytes. Tissue Eng Part A (2018) 24:458–68. doi: 10.1089/ten.tea.2017.0103
130. Brolese E, Buser D, Kuchler U, Schaller B, Gruber R. Human bone chips release of sclerostin and FGF-23 into the culture medium: an in vitro pilot study. Clin Oral Implants Res (2015) 26:1211–4. doi: 10.1111/clr.12432
131. Hanudel MR, Chua K, Rappaport M, Gabayan V, Valore E, Goltzman D, et al. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am J Physiol-Ren Physiol (2016) 311:F1369–77. doi: 10.1152/ajprenal.00281.2016
132. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci USA (2011) 108:E1146–55. doi: 10.1073/pnas.1110905108
133. Chefetz I, Kohno K, Izumi H, Uitto J, Richard G, Sprecher E. GALNT3, a gene associated with hyperphosphatemic familial tumoral calcinosis, is transcriptionally regulated by extracellular phosphate and modulates matrix metalloproteinase activity. Biochim Biophys Acta (2009) 1792:61–7. doi: 10.1016/j.bbadis.2008.09.016
134. Knutson MD. Iron-sensing proteins that regulate hepcidin and enteric iron absorption. Annu Rev Nutr (2010) 30:149–71. doi: 10.1146/annurev.nutr.012809.104801
135. Simic P, Kim W, Zhou W, Pierce KA, Chang W, Sykes DB, et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J Clin Invest (2020) 130:1513–26. doi: 10.1172/JCI131190
136. Zhang B, Umbach AT, Chen H, Yan J, Fakhri H, Fajol A, et al. Up-regulation of FGF23 release by aldosterone. Biochem Biophys Res Commun (2016) 470:384–90. doi: 10.1016/j.bbrc.2016.01.034
137. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet (2019) 20:675–91. doi: 10.1038/s41576-019-0158-7
138. Qian D-Y, Yan G-B, Bai B, Chen Y, Zhang S-J, Yao Y-C, et al. Differential circRNA expression profiles during the BMP2-induced osteogenic differentiation of MC3T3-E1 cells. BioMed Pharmacother Biomed Pharmacother (2017) 90:492–9. doi: 10.1016/j.biopha.2017.03.051
139. Pathak JL, Bravenboer N, Klein-Nulend J. The Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases. Front Endocrinol (2020) 11:405. doi: 10.3389/fendo.2020.00405
140. Imel EA, Biggin A, Schindeler A, Munns CF. FGF23, Hypophosphatemia, and Emerging Treatments. JBMR Plus (2019) 3. doi: 10.1002/jbm4.10190
141. Athonvarangkul D, Insogna KL. New Therapies for Hypophosphatemia-Related to FGF23 Excess. Calcif Tissue Int (2020) 108:143–57. doi: 10.1007/s00223-020-00705-3
142. Meyerhoff N, Haffner D, Staude H, Wühl E, Marx M, Beetz R, et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol Berl Ger (2018) 33:447–56. doi: 10.1007/s00467-017-3820-3
Keywords: FGF23, osteocytes, phosphate, vitamin D, iron deficiency, hypoxia, inflammation, erythropoiesis
Citation: Ratsma DMA, Zillikens MC and van der Eerden BCJ (2021) Upstream Regulators of Fibroblast Growth Factor 23. Front. Endocrinol. 12:588096. doi: 10.3389/fendo.2021.588096
Received: 28 July 2020; Accepted: 11 January 2021;
Published: 26 February 2021.
Edited by:
Gudrun Stenbeck, Brunel University London, United KingdomReviewed by:
Monzur Murshed, McGill University, CanadaMaren Leifheit-Nestler, Hannover Medical School, Germany
Copyright © 2021 Ratsma, Zillikens and van der Eerden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bram C. J. van der Eerden, b.vandereerden@erasmusmc.nl