 Sen Zhang1†
Sen Zhang1† Hongen Xu2,3†
Hongen Xu2,3† Yongan Tian4Danhua Liu3Xinyue Hou1
Yongan Tian4Danhua Liu3Xinyue Hou1 Beiping Zeng4Bei Chen5Huanfei Liu2Ruijun Li2Xiaohua Li5Bin Zuo5
Beiping Zeng4Bei Chen5Huanfei Liu2Ruijun Li2Xiaohua Li5Bin Zuo5 Ryan Tang6
Ryan Tang6 Wenxue Tang2,3,7*
Wenxue Tang2,3,7*- 1School of Basic Medical Sciences, Zhengzhou University, Zhengzhou, China
- 2Precision Medicine Center, Academy of Medical Science, Zhengzhou University, Zhengzhou, China
- 3The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 4BGI College, Zhengzhou University, Zhengzhou, China
- 5Department of Otology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 6Johns Hopkins University, Maryland, MD, United States
- 7Henan Institute of Medical and Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, China
Objective: This study aimed to explore the genetic causes of probands who were diagnosed with Waardenburg syndrome (WS) or congenital sensorineural hearing loss.
Methods: A detailed physical and audiological examinations were carried out to make an accurate diagnosis of 14 patients from seven unrelated families. We performed whole-exome sequencing in probands to detect the potential genetic causes and further validated them by Sanger sequencing in the probands and their family members.
Results: The genetic causes for all 14 patients with WS or congenital sensorineural hearing loss were identified. A total of seven heterozygous variants including c.1459C > T, c.123del, and c.959-409_1173+3402del of PAX3 gene (NM_181459.4), c.198_262del and c.529_556del of SOX10 gene (NM_006941.4), and c.731G > A and c.970dup of MITF gene (NM_000248.3) were found for the first time. Of these mutations, we had confirmed two (c.1459C > T and c.970dup) are de novo by Sanger sequencing of variants in the probands and their parents.
Conclusion: We revealed a total of seven novel mutations in PAX3, SOX10, and MITF, which underlie the pathogenesis of WS. The clinical and genetic characterization of these families with WS elucidated high heterogeneity in Chinese patients with WS. This study expands the database of PAX3, SOX10, and MITF mutations and improves our understanding of the causes of WS.
Introduction
Waardenburg syndrome (WS) is a congenital developmental disorder, which is mainly characterized by congenital sensorineural hearing loss (SNHL) and abnormal pigmentation of the iris, hair, and skin (manifests as heterochromia iridis and brilliant blue eyes, a white forelock, and premature graying, and hypopigmented skin) (Read and Newton, 1997). WS has an incidence rate of approximately 1/42,000 births and is responsible for 2–5% of cases of total congenital deafness (Read and Newton, 1997; Nayak and Isaacson, 2003). Four different types of Waardenburg syndrome (WS I∼IV) have been described based on genotypic and phenotypic variations (Read and Newton, 1997; Pingault et al., 2010). WS I is distinguished from WS II by the presence of dystopia canthorum, which is lateral displacement of the inner canthus in each eye; WS III (Klein–Waardenburg syndrome) is similar to WS I except with additional upper limb abnormalities; WS IV (Waardenburg-Shah syndrome) is characterized by general WS features as well as Hirschsprung’s disease, a disorder that causes severe blockage of the large intestine. Current research suggests that WS I and WS II are more common than WS III and WS IV (Read and Newton, 1997; Pingault et al., 2010).
Waardenburg syndrome shows a high degree of genetic heterogeneity (Hageman and Delleman, 1977; Read and Newton, 1997; Pingault et al., 2010; Song et al., 2016). Six genes have been linked to this syndrome: paired box 3 (PAX3) (Baldwin et al., 1992; Tassabehji et al., 1992; Hoth et al., 1993), melanocyte inducing transcription factor (MITF) (Tassabehji et al., 1994), SRY-box transcription factor 10 (SOX10) (Pingault et al., 1998; Bondurand et al., 2007), endothelin 3 (EDN3) (Edery et al., 1996), endothelin receptor type B (EDNRB) (Puffenberger et al., 1994), and snail family transcriptional repressor 2 (SNAI2) (Sánchez-Martín et al., 2002). PAX3 is responsible for WS I and WS III (Baldwin et al., 1992; Tassabehji et al., 1992; Hoth et al., 1993). SOX10, MITF, and SNAI2 are associated with WS IV (Tassabehji et al., 1994; Pingault et al., 1998; Sánchez-Martín et al., 2002; Bondurand et al., 2007). SOX10, EDNRB, and EDN3 are found to be involved in WS IV (Puffenberger et al., 1994; Edery et al., 1996; Pingault et al., 1998; Bondurand et al., 2007). Although not currently fully understood, all these genes are involved in a complex network in neural crest cells and other derivatives (Read and Newton, 1997; Bondurand et al., 2000; Pingault et al., 2010). The interaction of these genes during the formation and development of melanocytes could be the pathogenesis of WS and other related diseases (Read and Newton, 1997; Bondurand et al., 2000; Pingault et al., 2010).
Diagnosis of WS can be difficult because all features are not present in every patient (Hageman and Delleman, 1977; Newton, 1990; Tamayo et al., 2008; Pingault et al., 2010; Yang et al., 2013). Even within a single family, patients can display different clinical manifestations due to variations in the expressivity of causative genes (Hageman and Delleman, 1977; Newton, 1990; Tamayo et al., 2008; Pingault et al., 2010; Yang et al., 2013). Therefore, genetic testing is an important method for diagnosing this disease and its subtypes (Hageman and Delleman, 1977; Read and Newton, 1997; Pingault et al., 2010; Tang et al., 2015; Song et al., 2016; Wu et al., 2016; Li et al., 2019). To date, ∼400 mutations including missense/nonsense mutations, frameshift mutations, insertions/deletions, and copy number variants (CNVs) have been identified in genes associated with WS (The Human Gene Mutation Database1), with most variants in genes PAX3, SOX10, and MITF (Chen et al., 2010; Pingault et al., 2010; Song et al., 2016). Of these variants, ∼100 mutations were identified in Chinese people. Nevertheless, there are still a number of cases unexplained at the molecular level (Pingault et al., 2010; Song et al., 2016). Discovering novel mutations will lead to a better understanding of the genetic causes of WS pathogenesis.
Recently, next-generation sequencing (NGS) has proven to be a potent tool for the identification of pathogenic mutations related to deafness, which can improve the diagnosis of genetic diseases and the detection of mutations in genes associated with different clinical manifestations (Brownstein et al., 2012; Lin et al., 2012; Tang et al., 2012; Li et al., 2019). In this study, WES was used to identify the possible pathogenic mutations of patients with SNHL or WS. A total of seven novel variants in PAX3, SOX10, and MITF were found, and two of them are de novo confirmed by Sanger sequencing of variants in the probands and their parents. Our results show that WS in China has a high degree of genetic heterogeneity and extend the mutational spectrum of WS-related genes.
Materials and Methods
Patients
From seven Han Chinese families in the Henan province, 14 patients (Table 1) and nine unaffected family members were recruited for our study and asked to perform audiological and general physical examinations (Figure 1). Furthermore, in family WS04, only WS04-II:1 was recruited because he was adopted and had lost contact with his biological family. Among the seven families, WS01 and WS06 were isolated cases, while the remaining families had multiple affected individuals (Figure 1). Photos and blood were collected after informed consent (Figure 2). This study was conducted according to the Declaration of Helsinki and approved by the institutional review board of the Medical Ethics Committee of The Second Affiliated Hospital of Zhengzhou University (Approval No. 2018008).

Table 1. Summary of clinical data for patients.
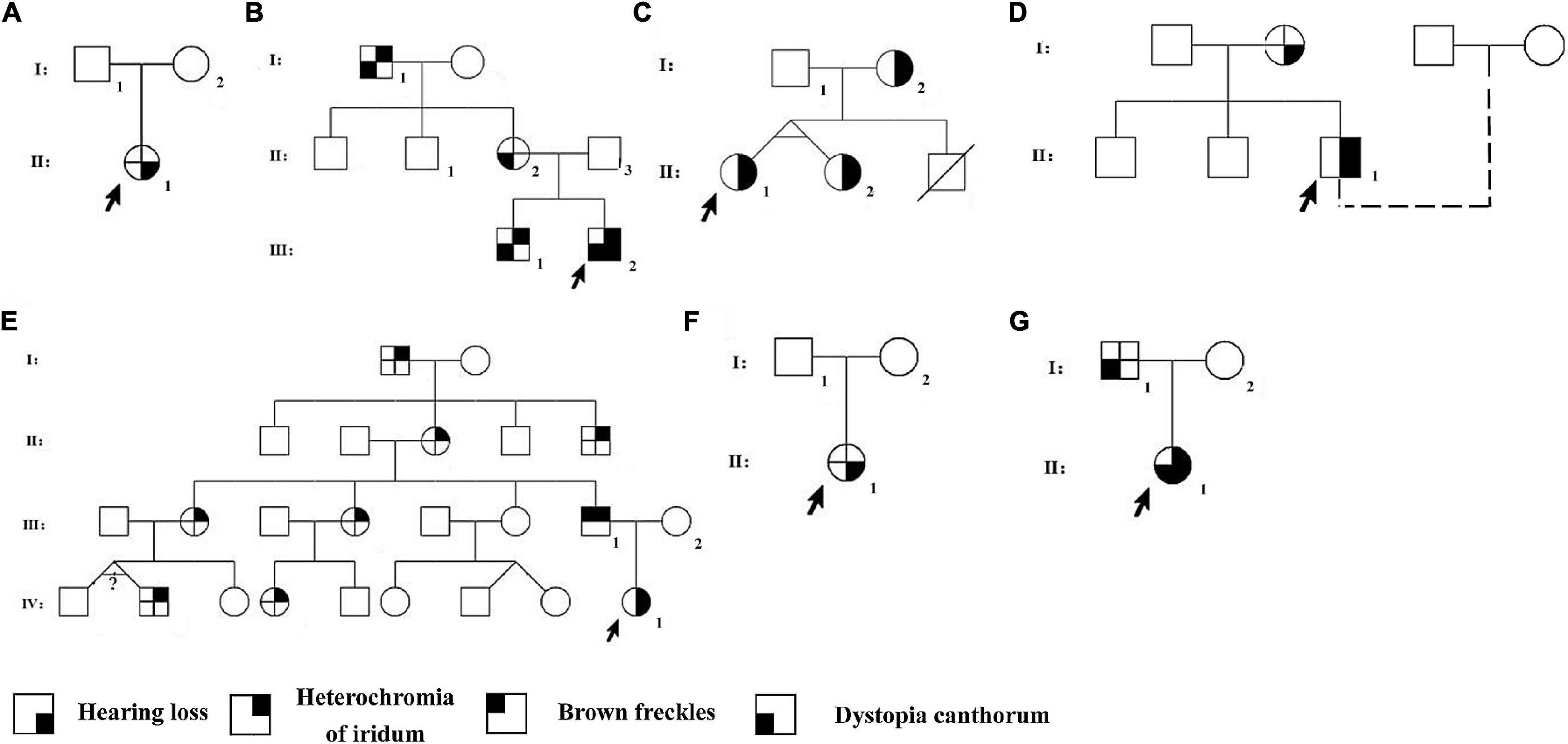
Figure 1. Pedigrees of the Waardenburg syndrome families. Pedigrees of families Individuals with a number assigned participated in the current study. Phenotypes of the rest of the family members were based on the relative’s description. The probands were pointed by arrows. (A) WS01, (B) WS02, (C) WS03, (D) WS04, (E) WS05, (F) WS06, and (G) WS07.
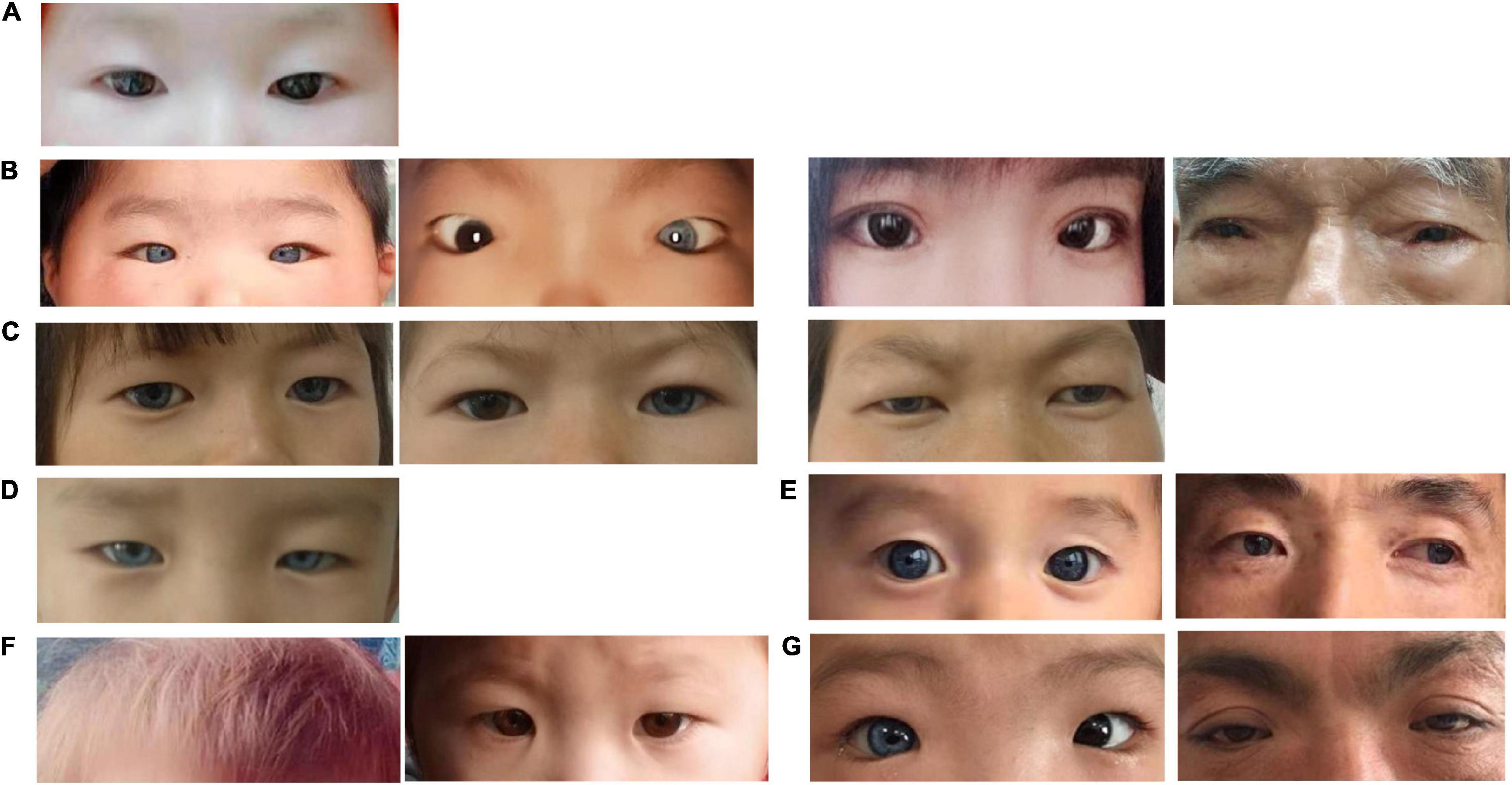
Figure 2. Photographs of affected individuals. (A) WS01-II:1 presented normal pigmentation of the iris, hair, and skin, and without dystopia canthorum. (B) B1, WS02-III:2; B2, WS02-III:1; B3, WS02-II:2; and B4, WS02-I:1. They all presented dystopia canthorum, while WS02-III:2 has bilateral blue iris and WS02-III:1 has unilateral. (C) C1, WS03-II:1; C2, WS03-II:2; and C3, WS03-I:2. They presented bilateral or unilateral blue iris. (D) WS04-II:1 presented complete bilateral blue iris. (E) E1, WS05-IV:1 presented complete bilateral blue iris. E2, WS05-III:1 presented complete bilateral blue iris and special brown freckles on the face. (F) F1, F2, WS06-II:1 presented yellow hair and normal iridis color. (G) G1, WS07-II:1 presented unilateral blue iris and dystopia canthorum. G2, WS07-II:1 presented dystopia canthorum.
Clinical Investigation
All patients (medical history described by parents) received elaborate physical examinations in their hair color and skin pigmentation, joints, skeletomuscular system, digestion, ophthalmology and otology, and intelligence assessment. Patients also underwent audiological examinations, which included auditory steady-state response (ASSR), auditory brainstem response (ABR), and distortion product otoacoustic emission (DPOAE). Additionally, imageological examinations such as computerized tomography (CT) of the temporal bone and magnetic resonance imaging (MRI) were conducted. The characteristics of the patients are summarized in Table 1.
Next-Generation Sequencing-Based Genetic Testing
In this study, WES was applied to identify the potential genetic causes for probands. A standard NGS-based genetic testing, including sample preparation and quantification, library construction, sequencing, and data analyses, was performed as previously described (Pan et al., 2020). Briefly, after library construction, the resulting libraries were hybridized to the Agilent SureSelect Human All Exon V7. Then, sequencing was carried out on an Illumina HiSeq 4000 sequencer (Illumina Inc., San Diego, CA, United States) to generate paired-end reads of 150 bp.
Data analyses were divided into bioinformatics analysis and variant interpretation. Under the framework of bcbio-nextgen2, we used the Burrows–Wheeler Aligner (BWA) (version 0.7.17-r1188) (Li, 2013) to align the sequencing reads to the human reference genome (GRCh37); GATK Haplotype Caller software (version 4.1.2) (McKenna et al., 2010) to identify the single nucleotide variants (SNVs) and short indels; DECoN (Fowler et al., 2016) to identify the CNVs; and Vcfanno software (version 0.3.1) (Pedersen et al., 2016) to annotate the VCF files with external database, including Clinvar (Landrum et al., 2018), ExAC (Lek et al., 2016), dbNSFP (Liu et al., 2016), 1,000 Genomes (Auton et al., 2015), and gnomAD (Karczewski et al., 2019). The filtered variants were interpreted following the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) (Richards et al., 2015) and the ClinGen hearing loss expert group’s recommendation on variant interpretation (Oza et al., 2018). Copy number analysis was performed from NGS data using DECoN with the bam files from the same enrichment panel and sequencing run. Paternity tests were performed on families WS01 and WS06 since the gene tests had shown mutations occurred de novo.
Sanger Sequencing
Sanger sequencing was used to confirm the candidate variants detected by NGS and to conduct co-segregation analyses in family members. The specific primers (Table 2) were designed by NCBI Primer-BLAST and synthesized by Sunya Biotech Co., Ltd. (Zhengzhou, China). Conventional PCR was performed for SNVs and short indels detected in families WS01 to WS06. While long-range PCR (LR-PCR) based on nested-PCR and fragments gel-purified were performed for the CNV of patients WS07-I:1 and WS07-II:1. LR-PCR is a traditional approach to obtain CNV breakpoint junction (Woodward et al., 2005; Zhang et al., 2017), for which several primers were designed from both the proximal and the distal breakpoint regions identified, and used in different combinations until an appropriate size product was generated (Supplementary Figure 1). After PCR amplification, purification, and quality control, Sanger sequencing was run in a SeqStudio Genetic Analyzer (Thermo Scientific, United States) with a mixture of PCR products and BigDyeTM Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, United States). The sequencing results were analyzed by the SnapGene viewer (Figure 3).
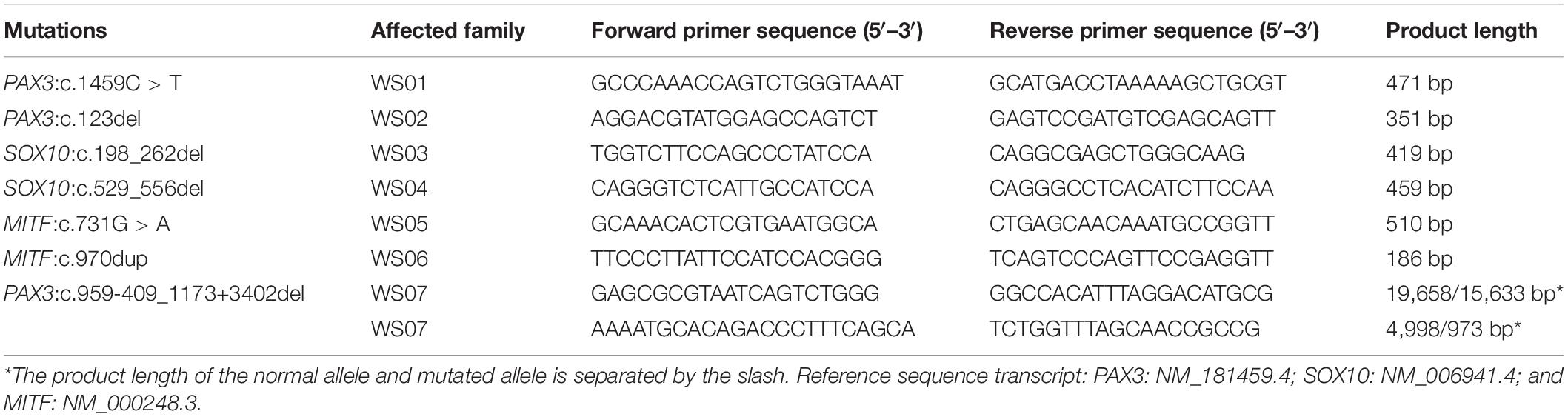
Table 2. Primer pairs of the novel mutations of paired box 3 (PAX3), SRY-box transcription factor 10 (SOX10), and melanocyte inducing transcription factor (MITF).
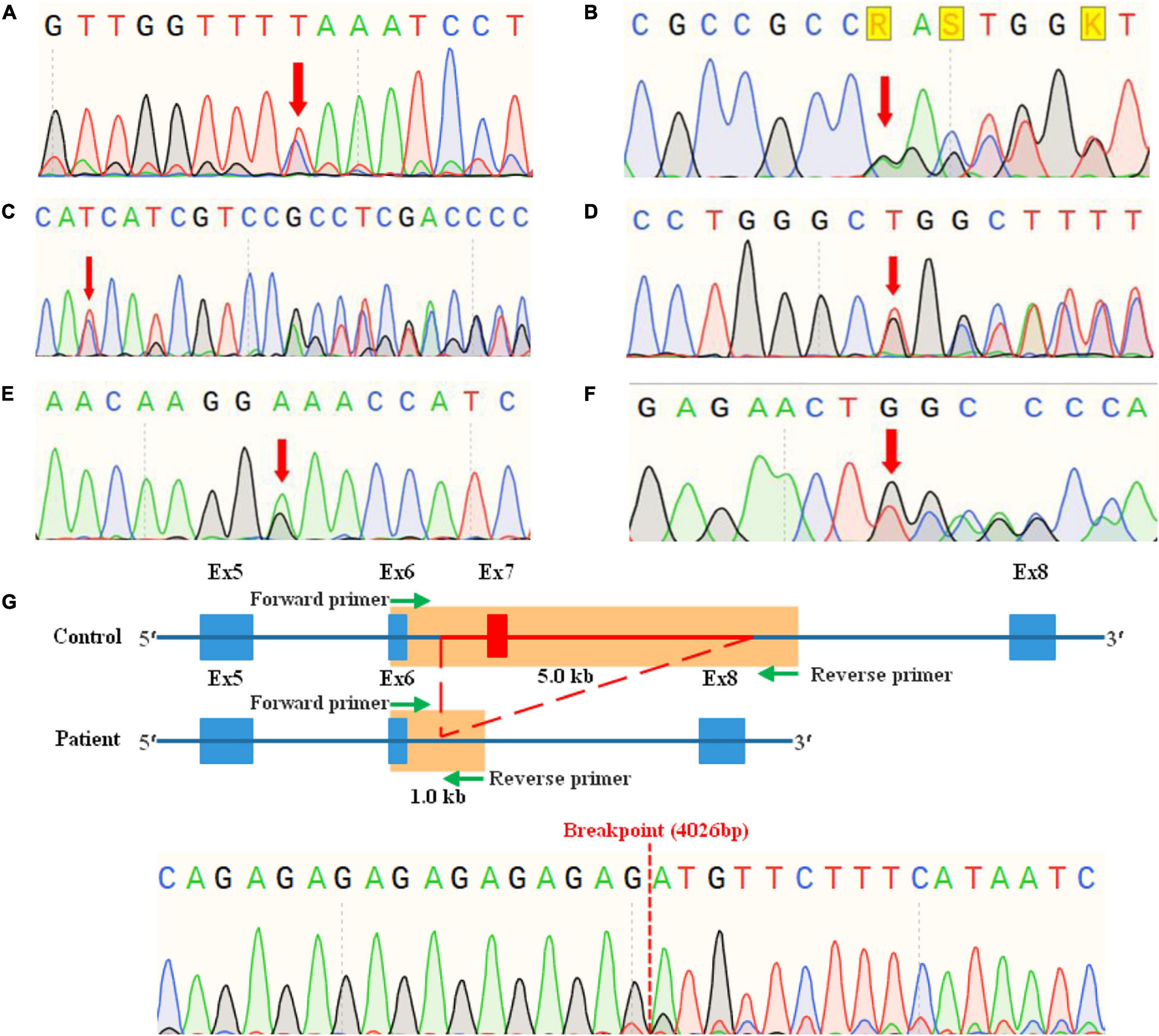
Figure 3. Mutation analyses of Chinese Waardenburg syndrome families WS01 to WS07 by sanger sequencing. (A) Heterozygous mutation c.1459C > T of PAX3 in WS01-II:1. (B) Heterozygous mutation c.123del of PAX3 in WS02-I:1, II:3, III:1, and III:2. (C) Heterozygous mutation c.198_262del of SOX10 in WS03-I:2, II:1, and III:2. (D) Heterozygous mutation c.529_556del of SOX10 in WS04-II:1. (E) Heterozygous mutation c.731G > A of MITF in WS05-III:3, IV:4. (F) Heterozygous mutation c.970dup of MITF in WS06-II:1. (G) Heterozygous mutation c.959-409_1173+3402del of PAX3 in WS07-I:1, II:1.
Results
Clinical Findings
A total of 14 patients from seven unrelated families were involved in this study. Before genetic testing, patients WS01-II:1 and WS06-II:1 were primarily diagnosed with SNHL, while the other 12 patients were diagnosed with WS. These diagnoses were made by otorhinolaryngologists based on the manifestation of the typical symptom of WS, such as SNHL, abnormal pigmentation, and the presence or absence of dystopia canthorum, musculoskeletal anomalies, and intestinal aganglionosis. After a genetic diagnosis, further examinations were performed on the patients WS01-II:1 and WS06-II:1. We found that the hair color of WS06-II:1 is gray, which was previously ignored, but nothing new with WS01-II:1.
Among all the patients, heterochromia iridum and deafness were the most frequent features. Ten affected individuals (10/14, 71.4%) had blue iris, of which three were heterochromia iridum; nine patients (9/14, 64.3%) had a profound sensorineural hearing impairment; six (6/14, 42.9%) had dystopia canthorum; one (1/14, 7.1%) had facial freckles; two (2/14, 14.3%) had abnormal pigmentation of hair (Figures 1, 2). Table 1 lists the clinical data of these WS patients.
Members in the same family can have different symptoms. WS03-II:1 and WS03-II:2 are identical twins; one has bilateral blue iris and the other has unilateral. According to his adoptive father, WS04-II:1 was born with a white forelock that returned to being black (we were unable to collect pictures of the patient with a white forelock). Across four generations, seven members of the family WS05 had unexpectedly brilliant blue eyes without other WS-related symptoms. However, it is impossible to confirm whether they were affected by the same mutation of the proband (WS05-IV:1) since they refused to provide blood samples.
Molecular Etiology
Whole-exome sequencing was performed in probands of these seven families. For each sample, at least 10 Gbp raw data was generated, with more than 82% of bases having a Phred quality score Q ≥ 30 (Q30), 99% of the clean reads can map to the human reference genome (GRCh37/hg19), and the average sequencing depth of target regions was 100×, with 95% of target regions having coverage greater than 20× (Supplementary Table 1).
In this study, the genetic causes for all recruited patients had been confirmed, which contain a total of seven mutations in PAX3, SOX10, and MITF, respectively (Table 3). To our best knowledge, these mutations, including a nonsense mutation, a missense mutation, a CNV, and four frameshift mutations, have not been reported by previous studies or recorded in any public database. Further analysis by Sanger sequencing of patients and their family members shows that all the variants were present in the affected members and absent in the unaffected ones (Figure 3), and variants of probands WS01-II:1 and WS06-II:1 are de novo. Pathogenicity analysis of variants was according to the standards and guidelines for interpreting genetic variants proposed by the ACMG-AMP. The mutations and pathogenicity analysis were summarized in Table 3.
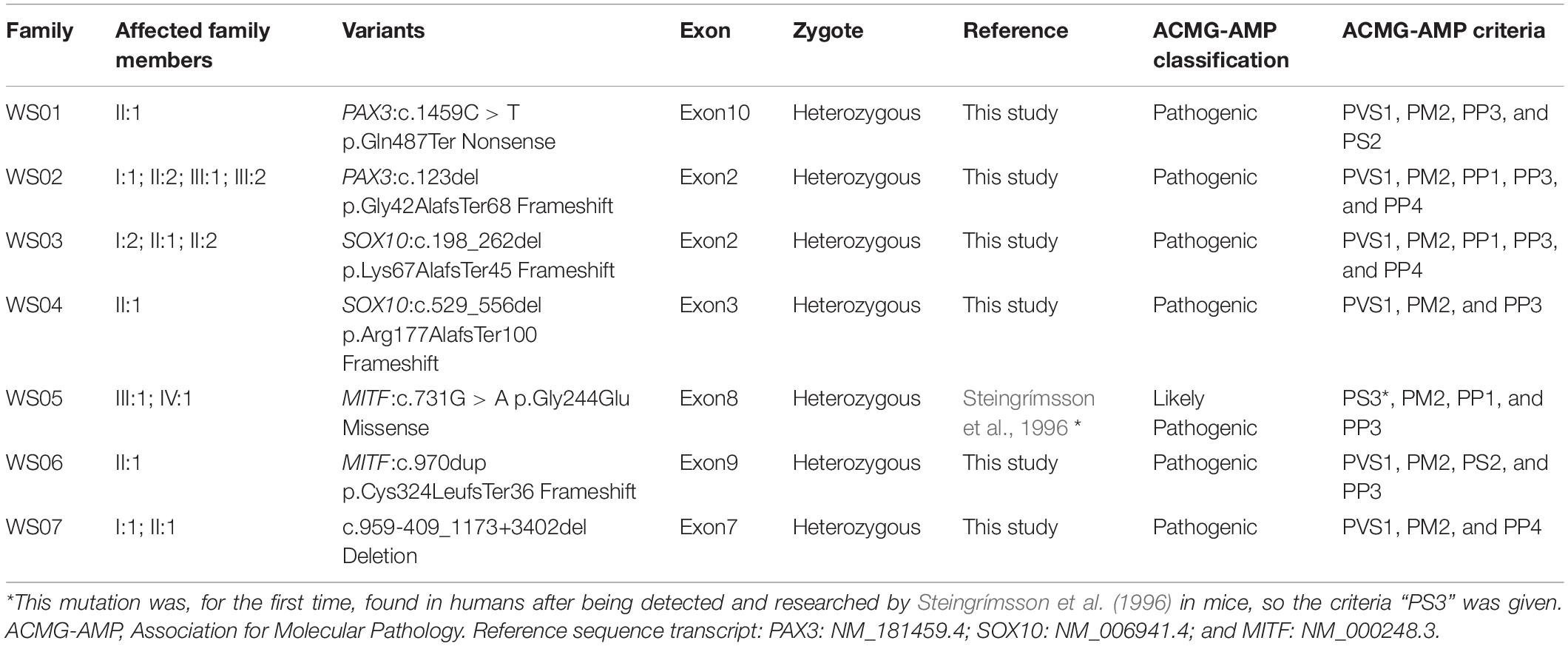
Table 3. Gene variants and pathogenicity analysis of patients.
Genotype–Phenotype Correlation
The phenotypes of WS patients with PAX3 (n = 7), SOX10 (n = 4), and MITF (n = 3) mutations are compared in Supplementary Table 2. Among the WS patients who participated in this study, all patients with SOX10 mutations have hearing loss, while some patients with PAX3 (3/7) or MITF (2/3) have. Similarly, the symptom of the blue iris could be found in all patients with SOX10 mutations, while it was found in some patients with PAX3 (4/7) or MITF (2/3) variants. Abnormal hair pigmentation is rare in patients with PAX3 (0/7), SOX10 (1/4), and MITF (1/3) mutations. Previous reports suggested that freckles could be observed only in Chinese WS II patients with MITF mutations (Chen et al., 2010; Sun et al., 2016). Indeed, in this study, we found one WS II patient with MITF mutation has freckles. Synophridia is only present in WS I patients with PAX3 mutations.
Discussion
In this study, we had confirmed seven novel heterozygous variants which are the genetic causes of 14 WS patients from seven unrelated families, including c.1459C > T (nonsense), c.123del (frameshift), and c.959-409_1173+3402del (deletion) of PAX3 (NM_181459.4), c.198_262del (frameshift) and c.529_556del (frameshift) of SOX10 (NM_006941.4), and c.731G > A (missense) and c.970dup (frameshift) of MITF (NM_000248.3) (Table 3). Among 14 patients, seven each were classified as WS I and WS II, respectively, based on their phenotypes and genotypes, showing that WS I and WS II were two major WS subtypes (Read and Newton, 1997; Pingault et al., 2010). While mutations in PAX3 were the major causes for WS I (7/7), SOX10 (4/7), and MITF (3/7) were two major causative genes attributable to WS II. Our findings had extended the mutational spectrum of WS-related genes and revealed high genetic heterogeneity in Chinese WS patients (Yang et al., 2013; Sun et al., 2016; Liu et al., 2020).
To explore the genotype–phenotype correlation, we compared the phenotypes between WS patients with PAX3, SOX10, and MITF mutations (Supplementary Table 2). Several reports had shown that the clinical features of WS II caused by SOX10 and MITF mutations were indistinguishable, except that freckle was frequent in WS II probands with MITF mutation (Chen et al., 2010; Toriello, 2011; Sun et al., 2016). Indeed, in this study, freckle seems to be unique for patients with MITF mutations (1/3) but was absent in those with PAX3 (0/7) or SOX10 (0/4) mutations. Dystopia canthorum is a rebarbative but crucial clinical feature, because of its value in distinguishing WS I and WS II, but it is not completely applicable for Chinese WS I patients (Sun et al., 2016; Morimoto et al., 2018; Suzuki et al., 2018; Minami et al., 2019). Herein, we had an interesting finding that the synophridia, even though a minor symptom, was only present in WS patients (5/7) but absent in WS II patients (0/7). Our results may have shown the clinical differences between WS II patients with SOX10 and MITF mutations, and between WS II and WS I. However, gene test is as necessary as clinical investigation for the accurate diagnosis and subtype confirmation (Hageman and Delleman, 1977; Read and Newton, 1997; Pingault et al., 2010; Song et al., 2016; Li et al., 2019).
Mutations in PAX3, SOX10, and MITF were the most common genetic causes for WS and responsible for almost all Chinese WS patients (Chen et al., 2010; Wu et al., 2016; Liu et al., 2020). Beyond that, to date, several WS cases associated with mutations in ENDRB, EDN3, and SNAI2 had been reported (Sánchez-Martín et al., 2002; Pingault et al., 2010; Xiong et al., 2015; Somashekar et al., 2019), but the situation is a bit different in Chinese. There were two reported cases of WS type I caused by mutations in the EDNRB gene (Cheng et al., 2019; Li et al., 2019), which is different from the cases in other races (WS II or IV) (Pingault et al., 2010; Issa et al., 2017). Of the few reports about WS type II being caused by mutations in the EDN3 gene, there was no Chinese case reported. Only one research group reported SNAI2 mutations caused WS within two unrelated WS II patients (Sánchez-Martín et al., 2002), which had been questioned recently (Song et al., 2016; Mirhadi et al., 2020). To understand the differences of WS among different races, we need further research on the pathogenesis of WS and more accurate diagnostic means.
The deficiency of melanocytes, the neural crest (NC) derivatives, is common to various WS types (Bondurand et al., 2000; Pingault et al., 2010; Song et al., 2016), which is responsible for the phenotypes of pigmentation defects and hearing loss (Steel and Barkway, 1989). PAX3 encodes a DNA-binding transcription factor, consisting of a paired box (PD) encoded by exons 2, 3, and 4, the homeodomain (HD) by exons 5 and 6, C-terminal transcriptional activation domain by exons 7 and 8 (Read and Newton, 1997; Wildhardt et al., 2013). It is indispensable in the development of somites, skeletal muscle, and the neural crest cells (NCC) and their derivatives like melanocytes. It can cooperate with SOX10 to regulate the expression of the MITF promoter (Pingault et al., 2010). PAX3:c.123del and c.959-409_1173+3402del mutations are predicted to activate the nonsense-mediated mRNA decay (NMD) machinery (Khajavi et al., 2006), thereby resulting in haploinsufficiency, which might be the disease-causing mechanism for WS I. PAX3:c.1459C > T mutation is located in the exon 10 and could only influence the isoform PAX3e (Wang, 2006), which most likely pathogenic mechanism is haploinsufficiency (Barber et al., 1999).
SRY-box transcription factor 10 encodes a transcription factor that contains an HMG (high mobility group) DNA binding domain and a C-terminal transactivation domain (Chan et al., 2003). In the early development of NC, SOX10 plays an important role in promoting cell survival and maintaining the multipotency of NC stem cells (Kapur, 1999; Kelsh, 2006; Pingault et al., 2010; Stolt and Wegner, 2010). Besides synergy with PAX3 to regulate the expression of MITF, it also can directly regulate the expression of genes important for melanin synthesis, suggesting the importance for melanocyte differentiation (Bondurand et al., 2000; Lee et al., 2000; Potterf et al., 2000; Verastegui et al., 2000; Jiao et al., 2004; Wegner, 2005; Pingault et al., 2010). It is also crucial for the peripheral nervous system like sensory, sympathetic, and enteric ganglia and along nerves (Bondurand et al., 2000; Pingault et al., 2010). SOX10: c.198_262del and c.529_556del are located in the HMG domain and predicted to activate the NMD machinery, resulting in haploinsufficiency.
Melanocyte inducing transcription factor, a basic helix–loop–helix leucine zipper (bHLHZip) protein, is the key transcription factor of melanocyte development. The bHLHZip structure binds DNA by basic domain, dimerizes through HLH domain, and is stabilized via the Zip domain (Hodgkinson et al., 1993; Steingrímsson et al., 1994). The C-terminal of MITF contributes to defining the target genes by a serine-rich transcriptional activation domain. Mice with MITF mutations show reduced or absent pigmentation, deafness, and small or absent eyes, etc. (Yasumoto et al., 1995; Bertolotto et al., 1998; Steingrímsson et al., 2004; Pingault et al., 2010). MITF:c.970dup is a frameshift mutation and predicted to activate the NMD machinery, leading to haploinsufficiency. MITF:c.731G > A (p.Gly244Glu) is the genetic cause of III:1 and IV:1 of family WS05 and might be responsible for the other seven affected individuals (Figure 1). We had noticed that only one had hearing loss and blue iris while the other eight only had blue iris in this family, although Song et al. (2016) had suggested that nearly 90% of patients with MITF have hearing loss. The Gly244Glu mutation of MITF was found in humans for the first time, while the mouse model with the same mutation (MITFMi–b) had been found by Steingrímsson et al. (1996). The phenotype of MITFMi–b homozygous animals is mild compared with loss-of-function mi alleles. Gly244 would lie at the very beginning of the second helix, close to the protein-DNA interface. The Gly244Glu alteration is at the junction of the loop and helix 2 of the protein. The MITFMi–b protein largely spares dimerization function, while it is defective in its ability to bind DNA. However, the DNA binding function can be partially compensated by a wild-type partner in the dimer, since MITFMi–b is capable of forming TFE3 (Transcription factor E3) heterodimeric complexes which had a stronger DNA binding than the MITFMi–b homodimers. It may explain why the mutation c.731G > A resulting in a less-severe phenotype in the family WS05.
WS has high genetic heterogeneity and highly variable phenotype expressivity (Hageman and Delleman, 1977; Newton, 1990; Read and Newton, 1997; Tamayo et al., 2008; Pingault et al., 2010; Yang et al., 2013; Song et al., 2016), which makes the diagnosis challenging. NGS of numerous genes is allowed in a single test with lower turnaround time, cost, and higher throughput, which makes it ideal for figuring out the exact genetic mechanism (Brownstein et al., 2012; Tang et al., 2012, 2015). Mutations in PAX3 are responsible for WS I and WS III in most cases; however, using WES, we had detected a heterozygous nonsense mutation of PAX3:c.1459C > T in an SNHL patient (WS01-II:1). To our best knowledge, this is the first mutation found in exon 10 of PAX3 and results in a premature stop codon, which is very close to the normal ending (487/506) (Boudjadi et al., 2018). We suspect that this is why SNHL is the only symptom of WS01-II:1, even though previous studies argued that there was no correlation between genotype and phenotype of WS caused by PAX3 mutations (Tassabehji et al., 1995; Boudjadi et al., 2018). Patient WS06-II:1 was also diagnosed with SNHL initially, before being corrected to WS type II after the genetic testing in which a heterozygous de novo mutation MITF: c.970dup was detected. Besides, the other five families with classic symptoms and clear family histories, these two cases in particular highlight the superiority of NGS in the diagnosis of WS.
In conclusion, the clinical and genetic characteristics of one SNHL patient and six Chinese families of WS had been investigated in this study. Altogether, seven novel pathogenic/likely pathogenic variants in the PAX3, SOX10, and MITF were identified. Our results support that NGS is a useful diagnostic procedure for the diagnosis and subtype differentiation of WS. This report reveals the highly genetic heterogeneity and variable phenotype in Chinese patients with WS and will contribute to a better understanding of the WS by extending the mutational spectrum of WS-related genes.
Data Availability Statement
According to national legislation/guidelines, specifically the Administrative Regulations of the People’s Republic of China on Human Genetic Resources (http://www.gov.cn/zhengce/con tent/2019-06/10/content_5398829.htm, http://english.www.gov. cn/policies/latest_releases/2019/06/10/content_281476708945462.htm), no additional raw data is available at this time. Data of this project can be accessed after an approval application to the Bio-Med Big Data Center, NODE. Please refer to https://www.biosino.org/node/project/detail/OEP001401 for detailed application guidance. The accession code OEP001401 should be included in the application.
Ethics Statement
The studies involving human participants were reviewed and approved by the institutional review board of the Medical Ethics Committee of The Second Affiliated Hospital of Zhengzhou University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
WT and HX: study design. BC, HL, RL, XL, and BZu: patient phenotypic analysis and genetic counseling. SZ, YT, XH, BZe, HL, and RL: next−generation sequencing and Sanger sequencing. HX, DL, SZ, and YT: data analysis and variant interpretation. SZ, HX, DL, WT, and RT: writing and review of original draft of the manuscript. RT: language editing of original draft of the manuscript. All authors have read and approved the final manuscript.
Funding
This study is funded by the Collaborative Innovation Project of Zhengzhou (Zhengzhou University) (Grant No. 18XTZX12004) and the Medical Science and Technology Projects in Henan (Grant No. SBGJ2018043) to WT, and the Joint Project of Medical Science and Technology Research in Henan Province (Grant No. LHGJ20190317) to HX.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank all the family members for their participation in this study. We also thank the Supercomputing Center of Zhengzhou University for providing computational and storage resources.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.643546/full#supplementary-material
Supplementary Figure 1 | LR-PCR for the identification of the large deletion, PAX3:c.959-409_1173+3402del.
Supplementary Table 1 | Detailed information for quality control of WES data.
Supplementary Table 2 | Phenotypes in patients with PAX3, SOX10, and MITF mutations.
Footnotes
References
Auton, A., Abecasis, G. R., Altshuler, D. M., Durbin, R. M., Abecasis, G. R., Bentley, D. R., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Baldwin, C. T., Hoth, C. F., Amos, J. A., da-Silva, E. O., and Milunsky, A. (1992). An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature 355, 637–638. doi: 10.1038/355637a0
Barber, T. D., Barber, M. C., Cloutier, T. E., and Friedman, T. B. (1999). PAX3 gene structure, alternative splicing and evolution. Gene 237, 311–319.
Bertolotto, C., Buscà, R., Abbe, P., Bille, K., Aberdam, E., Ortonne, J. P., et al. (1998). Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol. Cell. Biol. 18, 694–702. doi: 10.1128/MCB.18.2.694
Bondurand, N., Dastot-Le Moal, F., Stanchina, L., Collot, N., Baral, V., Marlin, S., et al. (2007). Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 81, 1169–1185. doi: 10.1086/522090
Bondurand, N., Pingault, V., Goerich, D. E., Lemort, N., Sock, E., Caignec, C. L., et al. (2000). Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum. Mol. Genet. 9, 1907–1917. doi: 10.1093/hmg/9.13.1907
Boudjadi, S., Chatterjee, B., Sun, W., Vemu, P., and Barr, F. G. (2018). The expression and function of PAX3 in development and disease. Gene 666, 145–157.
Brownstein, Z., Bhonker, Y., and Avraham, K. B. (2012). High-throughput sequencing to decipher the genetic heterogeneity of deafness. Genome Biol. 13, 245.
Chan, K. K., Wong, C. K. Y., Lui, V. C. H., Tam, P. K. H., and Sham, M. H. (2003). Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: differential effects on target gene regulation. J. Cell Biochem. 90, 573–585. doi: 10.1002/jcb.10656
Chen, H., Jiang, L., Xie, Z., Mei, L., He, C., Hu, Z., et al. (2010). Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem. Biophys. Res. Communi. 397, 70–74. doi: 10.1016/j.bbrc.2010.05.066
Cheng, H.-H., Ling, S.-Q., Zhao, P.-Z., Li, W.-L., and Deng, J. (2019). The heterozygous EDNRB mutation in a Chinese family with Waardenburg syndrome type I. Int. J. Ophthalmol. 12, 1507–1509. doi: 10.18240/ijo.2019.09.22
Edery, P., Attié, T., Amiel, J., Pelet, A., Eng, C., Hofstra, R. M., et al. (1996). Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat. Genet. 12, 442–444.
Fowler, A., Mahamdallie, S., Ruark, E., Seal, S., Ramsay, E., Clarke, M., et al. (2016). Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 1:20. doi: 10.12688/wellcomeopenres.10069.1
Hageman, M. J., and Delleman, J. W. (1977). Heterogeneity in Waardenburg syndrome. Am. J. Hum. Genet. 29, 468–485.
Hodgkinson, C. A., Moore, K. J., Nakayama, A., Steingrímsson, E., Copeland, N. G., Jenkins, N. A., et al. (1993). Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 74, 395–404. doi: 10.1016/0092-8674(93)90429-t
Hoth, C. F., Milunsky, A., Lipsky, N., Sheffer, R., Clarren, S. K., and Baldwin, C. T. (1993). Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I). Am. J. Hum. Genet. 52, 455–462.
Issa, S., Bondurand, N., Faubert, E., Poisson, S., Lecerf, L., Nitschke, P., et al. (2017). EDNRB mutations cause Waardenburg syndrome type II in the heterozygous state. Hum. Mutat. 38, 581–593. doi: 10.1002/humu.23206
Jiao, Z., Mollaaghababa, R., Pavan, W. J., Antonellis, A., Green, E. D., and Hornyak, T. J. (2004). Direct interaction of Sox10 with the promoter of murine Dopachrome Tautomerase (Dct) and synergistic activation of Dct expression with Mitf. Pigment Cell Res. 17, 352–362. doi: 10.1111/j.1600-0749.2004.00154.x
Kapur, R. P. (1999). Early death of neural crest cells is responsible for total enteric aganglionosis in Sox10(Dom)/Sox10(Dom) mouse embryos. Pediatr. Dev. Pathol. 2, 559–569. doi: 10.1007/s100249900162
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. J. bioRxiv [Preprint] doi: 10.1101/531210bioRxiv:531210
Kelsh, R. N. (2006). Sorting out Sox10 functions in neural crest development. BioEssays 28, 788–798. doi: 10.1002/bies.20445
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 14, 1074–1081. doi: 10.1038/sj.ejhg.5201649
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067.
Lee, M., Goodall, J., Verastegui, C., Ballotti, R., and Goding, C. R. (2000). Direct regulation of the Microphthalmia promoter by Sox10 links Waardenburg-Shah syndrome (WS4)-associated hypopigmentation and deafness to WS2. J. Biol. Chem. 275, 37978–37983. doi: 10.1074/jbc.M003816200
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291.
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv[Preprint] arXiv: 1303.3997,
Li, W., Mei, L., Chen, H., Cai, X., Liu, Y., Men, M., et al. (2019). New genotypes and phenotypes in patients with 3 subtypes of Waardenburg Syndrome identified by diagnostic next-generation sequencing. Neural Plast. 2019, 1–12. doi: 10.1155/2019/7143458
Lin, X., Tang, W., Ahmad, S., Lu, J., Colby, C. C., Zhu, J., et al. (2012). Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hear. Res. 288, 67–76. doi: 10.1016/j.heares.2012.01.004
Liu, Q., Cheng, J., Lu, Y., Zhou, J., Wang, L., Yang, C., et al. (2020). The clinical and genetic research of Waardenburg syndrome type I and II in Chinese families. Int. J. Pediatr. Otorhinolaryngol. 130:109806. doi: 10.1016/j.ijporl.2019.109806
Liu, X., Wu, C., Li, C., and Boerwinkle, E. (2016). dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241. doi: 10.1002/humu.22932
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Minami, S. B., Nara, K., Mutai, H., Morimoto, N., Sakamoto, H., Takiguchi, T., et al. (2019). A clinical and genetic study of 16 Japanese families with Waardenburg syndrome. Gene 704, 86–90.
Mirhadi, S., Spritz, R. A., and Moss, C. (2020). Does SNAI2 mutation cause human piebaldism and Waardenburg syndrome? Am. J. Med. Genet. Part A 182, 3074–3075. doi: 10.1002/ajmg.a.61887
Morimoto, N., Mutai, H., Namba, K., Kaneko, H., Kosaki, R., and Matsunaga, T. (2018). Homozygous EDNRB mutation in a patient with Waardenburg syndrome type 1. Auris Nasus Larynx 45, 222–226. doi: 10.1016/j.anl.2017.03.022
Nayak, C. S., and Isaacson, G. (2003). Worldwide distribution of Waardenburg syndrome. Ann. Otol. Rhinol. Laryngol. 112(9 Pt 1), 817–820. doi: 10.1177/000348940311200913
Newton, V. (1990). Hearing loss and Waardenburg’s syndrome: implications for genetic counselling. J. Laryngol. Otol. 104, 97–103.
Oza, A. M., DiStefano, M. T., Hemphill, S. E., Cushman, B. J., Grant, A. R., Siegert, R. K., et al. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613.
Pan, Z., Xu, H., Tian, Y., Liu, D., Liu, H., Li, R., et al. (2020). Perrault syndrome: clinical report and retrospective analysis. Mol. Genet. Genomic Med. 8:e1445.
Pedersen, B. S., Layer, R. M., and Quinlan, A. R. (2016). Vcfanno: fast, flexible annotation of genetic variants. Genome Biol. 17:118. doi: 10.1186/s13059-016-0973-5
Pingault, V., Bondurand, N., Kuhlbrodt, K., Goerich, D. E., Préhu, M. O., Puliti, A., et al. (1998). SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 18, 171–173.
Pingault, V., Ente, D., Dastot-Le Moal, F., Goossens, M., Marlin, S., and Bondurand, N. (2010). Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 31, 391–406. doi: 10.1002/humu.21211
Potterf, S. B., Furumura, M., Dunn, K. J., Arnheiter, H., and Pavan, W. J. (2000). Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum. Genet. 107, 1–6. doi: 10.1007/s004390000328
Puffenberger, E. G., Hosoda, K., Washington, S. S., Nakao, K., deWit, D., Yanagisawa, M., et al. (1994). A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell 79, 1257–1266.
Read, A. P., and Newton, V. E. (1997). Waardenburg syndrome. J. Med. Genet. 34, 656–665. doi: 10.1136/jmg.34.8.656
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424.
Sánchez-Martín, M., Rodríguez-García, A., Pérez-Losada, J., Sagrera, A., Read, A. P., and Sánchez-García, I. S. L. U. G. (2002). (SNAI2) deletions in patients with Waardenburg disease. Hum. Mol. Genet. 11, 3231–3236.
Somashekar, P. H., Girisha, K. M., Nampoothiri, S., Gowrishankar, K., Devi, R. R., Gupta, N., et al. (2019). Locus and allelic heterogeneity and phenotypic variability in Waardenburg syndrome. Clin. Genet. 95, 398–402. doi: 10.1111/cge.13468
Song, J., Feng, Y., Acke, F. R., Coucke, P., Vleminckx, K., and Dhooge, I. J. (2016). Hearing loss in Waardenburg syndrome: a systematic review. Clin. Genet. 89, 416–425.
Steel, K. P., and Barkway, C. (1989). Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107, 453.
Steingrímsson, E., Copeland, N. G., and Jenkins, N. A. (2004). Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 38, 365–411.
Steingrímsson, E., Moore, K. J., Lamoreux, M. L., Ferré-D’Amaré, A. R., Burley, S. K., Sanders Zimring, D. C., et al. (1994). Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat. Genet. 8, 256–263. doi: 10.1038/ng1194-256
Steingrímsson, E., Nii, A., Fisher, D. E., Ferré-D’Amaré, A. R., McCormick, R. J., Russell, L. B., et al. (1996). The semidominant Mi(b) mutation identifies a role for the HLH domain in DNA binding in addition to its role in protein dimerization. Embo J. 15, 6280–6289.
Stolt, C. C., and Wegner, M. (2010). SoxE function in vertebrate nervous system development. Int. J. Biochem. Cell Biol. 42, 437–440.
Sun, L., Li, X., Shi, J., Pang, X., Hu, Y., Wang, X., et al. (2016). Molecular etiology and genotype-phenotype correlation of Chinese han deaf patients with type I and type II Waardenburg Syndrome. Sci. Rep. 6:35498. doi: 10.1038/srep35498
Suzuki, N., Mutai, H., Miya, F., Tsunoda, T., Terashima, H., Morimoto, N., et al. (2018). A case report of reversible generalized seizures in a patient with Waardenburg syndrome associated with a novel nonsense mutation in the penultimate exon of SOX10. BMC Pediatr. 18:171. doi: 10.1186/s12887-018-1139-2
Tamayo, M. L., Gelvez, N., Rodriguez, M., Florez, S., Varon, C., Medina, D., et al. (2008). Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am. J. Med. Genet. A 146a, 1026–1031. doi: 10.1002/ajmg.a.32189
Tang, H. Y., Fang, P., Lin, J. W., Darilek, S., Osborne, B. T., Haymond, J. A., et al. (2015). DNA sequence analysis and genotype-phenotype assessment in 71 patients with syndromic hearing loss or auditory neuropathy. BMJ Open 5:e007506.
Tang, W., Qian, D., Ahmad, S., Mattox, D., Todd, N. W., Han, H., et al. (2012). A low-cost exon capture method suitable for large-scale screening of genetic deafness by the massively-parallel sequencing approach. Genet. Test. Mol. Biomarkers 16, 536–542. doi: 10.1089/gtmb.2011.0187
Tassabehji, M., Newton, V. E., Liu, X. Z., Brady, A., Donnai, D., Krajewska-Walasek, M., et al. (1995). The mutational spectrum in Waardenburg syndrome. Hum. Mol. Genet. 4, 2131–2137.
Tassabehji, M., Newton, V. E., and Read, A. P. (1994). Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 8, 251–255. doi: 10.1038/ng1194-251
Tassabehji, M., Read, A. P., Newton, V. E., Harris, R., Balling, R., Gruss, P., et al. (1992). Waardenburg’s syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature 355, 635–636.
Verastegui, C., Bille, K., Ortonne, J. P., and Ballotti, R. (2000). Regulation of the microphthalmia-associated transcription factor gene by the Waardenburg syndrome type 4 gene, SOX10. J. Biol. Chem. 275, 30757–30760. doi: 10.1074/jbc.C000445200
Wang, Q. (2006). Functional analysis of alternative isoforms of the transcription factor PAX3 in melanocytes in vitro. Cancer Res. 66, 8574–8580. doi: 10.1158/0008-5472.can-06-0947
Wegner, M. (2005). Secrets to a healthy Sox life: lessons for melanocytes. Pigment Cell Res. 18, 74–85. doi: 10.1111/j.1600-0749.2005.00218.x
Wildhardt, G., Zirn, B., Graul-Neumann, L. M., Wechtenbruch, J., Suckfüll, M., Buske, A., et al. (2013). Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics. BMJ Open 3:e001917.
Woodward, K. J., Cundall, M., Sperle, K., Sistermans, E. A., Ross, M., Howell, G., et al. (2005). Heterogeneous duplications in patients with Pelizaeus-Merzbacher disease suggest a mechanism of coupled homologous and nonhomologous recombination. Am. J. Hum. Genet. 77, 966–987. doi: 10.1086/498048
Wu, H., Feng, Y., Jiang, L., Pan, Q., Liu, Y., Liu, C., et al. (2016). Application of a new genetic deafness microarray for detecting mutations in the deaf in China. PLoS One 11:e0151909. doi: 10.1371/journal.pone.0151909
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K. C., et al. (2015). RNA splicing. the human splicing code reveals new insights into the genetic determinants of disease. Science (New York, NY) 347:1254806. doi: 10.1126/science.1254806
Yang, S., Dai, P., Liu, X., Kang, D., Zhang, X., Yang, W., et al. (2013). Genetic and phenotypic heterogeneity in Chinese patients with Waardenburg syndrome type II. PLoS One 8:e77149. doi: 10.1371/journal.pone.0077149
Yasumoto, K., Yokoyama, K., Shibata, K., Tomita, Y., and Shibahara, S. (1995). Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol. Cell. Biol. 15:1833.
Zhang, L., Wang, J., Zhang, C., Li, D., Carvalho, C. M. B., Ji, H., et al. (2017). Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum. Mol. Genet. 26, 1927–1941. doi: 10.1093/hmg/ddx102
Keywords: PAX3, SOX10, MITF, Waardenburg syndrome, next-generation sequencing, genetic heterogeneity
Citation: Zhang S, Xu H, Tian Y, Liu D, Hou X, Zeng B, Chen B, Liu H, Li R, Li X, Zuo B, Tang R and Tang W (2021) High Genetic Heterogeneity in Chinese Patients With Waardenburg Syndrome Revealed by Next-Generation Sequencing. Front. Genet. 12:643546. doi: 10.3389/fgene.2021.643546
Received: 18 December 2020; Accepted: 23 April 2021;
Published: 04 June 2021.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Duangrurdee Wattanasirichaigoon, Mahidol University, ThailandChufeng He, Central South University, China
Copyright © 2021 Zhang, Xu, Tian, Liu, Hou, Zeng, Chen, Liu, Li, Li, Zuo, Tang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenxue Tang, twx@zzu.edu.cn
†These authors have contributed equally to this work and share the first authorship