 Helen Chapel1*
Helen Chapel1* Johan Prevot2
Johan Prevot2 Hubert Bobby Gaspar3
Hubert Bobby Gaspar3 Teresa Español4
Teresa Español4 Francisco A. Bonilla5
Francisco A. Bonilla5 Leire Solis2
Leire Solis2 Josina Drabwell2
Josina Drabwell2 The Editorial Board for Working Party on Principles of Care at IPOPI†
The Editorial Board for Working Party on Principles of Care at IPOPI†- 1University of Oxford, Oxford, UK
- 2International Patient Organisation for Primary Immunodeficiencies (IPOPI), Downderry, UK
- 3University College London Institute of Child Health, London, UK
- 4Hospital General Vall d’Hebron, Barcelona, Spain
- 5Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA
Primary immune deficiencies (PIDs) are a growing group of over 230 different disorders caused by ineffective, absent or an increasing number of gain of function mutations in immune components, mainly cells and proteins. Once recognized, these rare disorders are treatable and in some cases curable. Otherwise untreated PIDs are often chronic, serious, or even fatal. The diagnosis of PIDs can be difficult due to lack of awareness or facilities for diagnosis, and management of PIDs is complex. This document was prepared by a worldwide multi-disciplinary team of specialists; it aims to set out comprehensive principles of care for PIDs. These include the role of specialized centers, the importance of registries, the need for multinational research, the role of patient organizations, management and treatment options, the requirement for sustained access to all treatments including immunoglobulin therapies and hematopoietic stem cell transplantation, important considerations for developing countries and suggestions for implementation. A range of healthcare policies and services have to be put into place by government agencies and healthcare providers, to ensure that PID patients worldwide have access to appropriate and sustainable medical and support services.
Introduction
Why a Principles of Care Document/Call to Action
Primary immune deficiencies (PIDs) are a large and growing group of over 230 different disorders, caused when some components of the immune system (mainly cells and proteins) are defective. While PIDs are generally recognized as rare disorders, some are more common than others. Taken as a whole, they represent an important group of conditions that, if not treated, can be chronic, life-long, serious, and even fatal. The lives of patients with PIDs are profoundly impacted by their condition. The immune system normally helps the body to fight infections caused by germs (or “micro-organisms”) such as bacteria, viruses, fungi, and protozoa. Owing to defective immune systems, people with PIDs are more prone to infections. In addition, a poorly regulated immune system may start to attack tissues, leading to inflammation, and autoimmunity (1, 2). When PIDs are left undiagnosed or are misdiagnosed, chronic illness and disability take a heavy toll on healthcare resources (3, 4).
The immune system is divided into two parts, each of which contains two components: on the one hand, soluble proteins may be particular for one germ (antibodies) or non-specific (complement). The other components are cellular – those that are specific for one germ only (lymphocytes) and innate cells that are involved in clearing all types of infections (such as phagocytes including macrophages and neutrophils).
Primary immune deficiencies are currently classified into groups, depending on the part(s) of the immune system affected. Over half the affected patients have antibody deficiencies and their treatment consists of replacing the missing antibodies (5). Cellular defects of lymphocytes are more severe and require replacement of stem cells that can mature to effective immune blood cells [hematopoietic stem cell transplantation (HSCT)] (6, 7) or replacement of the faulty gene.
While it is considered that many PIDs can be diagnosed easily with two simple blood tests (8), unfortunately many PIDs remain undiagnosed due to failure to consider this diagnosis. In addition, access to treatment varies widely between different regions of the world (9) and even from country to country within the same continent.
In order to tackle these disparities and ensure early diagnosis and appropriate access to treatments for all PID patients worldwide, a range of healthcare policies and services need to be put into place. While some countries have managed to do this, most have not.
A worldwide multi-disciplinary team of specialists prepared this document, which sets out comprehensive principles of care for PIDs. These include the role of specialized centers, the importance of registries, the need for multinational research, the role of patient organizations, the need for sustained access to all treatments, including immunoglobulin (Ig) therapies and HSCT, worldwide.
It is hoped that these principles of care will guide stakeholders and decision-makers in a common goal to ensure that all PID patients can access the care they are entitled to in order to live normal and productive lives. There are examples of national and international networks in other rare diseases that provide opportunities for collaborations to improve patient care, an example being hemophilia. Such networks are fruitful and although already underway in some areas, provision of sustainable infra-structures is important to overcome major obstacles in the care of PID patients throughout the world.
Brief PIDs Overview: Rare and Chronic
Primary immune deficiencies are rare diseases of relatively recent description. Infections were a common cause of death in the general population up to the first half of the nineteenth century, so these diseases were not suspected. With the advent of improved hygiene and the development of vaccines and antibiotic therapies, physicians began to realize that not all people with infections died, and with increased life span, those with recurrent infections were recognized.
In the second half of the twentieth century, the occurrence of unusual (opportunistic) infections led to the understanding of individual susceptibility to infection. Furthermore, increased susceptibility seemed to occur in some members of a given family, but not in others. Hence, the realization that in some instances, susceptibility was inherited and this led to the discovery that defects in immune mechanisms resulted in pathologies known as PIDs. However, the effective action of antibiotics can also “mask” the recognition of PIDs. When an infection occurs and resolves, not all doctors will suspect a PID. So many patients have a long history of infections before a PID diagnosis is made and chronic sequelae have already developed (bronchiectasis, malabsorption, etc.). Since PIDs are chronic diseases, once recognised, patients require specific care for the rest of their lives.
Need for Awareness: Role of Key Stakeholders
Greater awareness is key to ensure PID patients can be diagnosed, treated, and lead productive lives all over the world. Target groups include the public, patient, and healthcare professionals, especially primary care physicians and specialists to whom PID patients may present – see Section “Support for PID Diagnosis and Complications in Other Medical Specialties.” Whilst awareness of PIDs has been increasing dramatically in certain regions of the world, the fact is that today a large majority of PID patients remain undiagnosed and do not have access to appropriate treatment.
Increasing awareness is of utmost importance to ensure that the principles of care set out in this paper can be implemented as widely as possible. The nature of awareness campaigns will vary depending on the country. In many developing countries, greater medical awareness is needed; political and financial factors play a key role in educational efforts aimed at the medical and nursing communities, which are crucial to ensure the first steps are taken to improve the situation (10).
Awareness of the public is also important. In 2010, a worldwide awareness campaign was created with the launch of World Primary Immunodeficiencies Week (WPIW), which seeks to drive such awareness. Patient organizations have been working tirelessly on awareness campaigns to inform and educate the general public, health policymakers and decision makers, schools and families about PIDs in order to improve diagnosis rates and optimize access to treatment.
Importance of Access to Early Diagnosis and Specialist Care
Access to specialist care is a problem in many countries. Diagnosis of PIDs is often delayed, resulting in unnecessary complications, and appropriate management is suboptimal or even unavailable, especially in less developed regions. Access to early diagnosis and specialist care ensures the best health outcomes not only for the individual but also for society.
Early diagnosis shortens diagnostic delay that is distressing to the family, damaging to the patient and wasteful of health-care resources (11). Before the diagnosis is made, an individual suffering from recurrent bouts of infections, autoimmune, or inflammatory disease due to PIDs is often investigated by many different specialists but without appropriate treatment or management. The end result is deterioration of the patient’s condition, inappropriate use of health resources, and a feeling of helplessness among all parties. The data resulting from a new service to over 1,000 suspected PID patients in Asia showed that families often lost one or more children from an undiagnosed PID before the current child was diagnosed (Pamela Lee, personal communication). Precise diagnosis can lead not only to specialist care for the patient but also to genetic counseling/prenatal diagnosis for future children.
Unfortunately, PID specialist care is often absent in less developed regions, even once an accurate diagnosis of PID is made. Many governments, even in fast-growing economies, do not fund life-long Ig replacement or HSCT, though both therapies are effective and lifesaving. HSCT is a one-off curative treatment for patients with several PIDs. Hence, access to early diagnosis must be coupled with access to specialist care, to ensure appropriate therapy. This is a financial, and initially also a technical, challenge for many countries, which require at least temporary international collaborations to provide the necessary treatment, especially for HSCT. Therefore governments should recognize established reference centers and work in collaboration with them.
Principle 1: The Role for Specialized Centers
Diagnostic Facilities
Patient diagnoses
Although it is estimated that about 70% of PID patients are undiagnosed, even in countries with existing PID facilities, it is impossible to know (12). Prompt PID diagnosis results in better use of health facilities (13) and has been demonstrated to result in lower healthcare costs overall (14).
Criteria for fast and reliable PID diagnoses are given in Table 1. Several multi-stage diagnostic protocols are available, according to the clinical presentation that will optimize speed of diagnosis and referral for therapy (2, 15).

Table 1. Criteria for fast and reliable PID diagnoses.
Newborn screening
Severe combined immune deficiency (SCID) is a rare group of disorders and is characterized by inadequate T lymphocyte production or severe abnormalities of function (16). SCID is fatal due to overwhelming infection(s) in the first year of life unless definitive treatment with stem cells from a healthy donor’s bone marrow or blood or gene therapy can be used to correct the underlying immune defect. In most cases, infants already have serious infections at diagnosis; if these do not respond to standard treatments, the infants die before immune reconstitution can even be established. The key to improving the outcome for SCID is early diagnosis and treatment, so that severe infections can be prevented (17, 18). This significantly improves HSCT results, with an overall survival rate above 90% in those infants diagnosed at birth due to a positive family history of SCID, compared to 40% in cases diagnosed later because of serious infection or significant complications (7). Diagnosis at birth means that babies can be both protected from infection and transplanted earlier in a better clinical state, all of which improves the chances of survival.
Newborn screening for SCID and other conditions with very low T-cell numbers can now be performed by detecting markers of healthy T-cell development in DNA extracted from the dried blood spots already routinely obtained from infants by a simple heel prick in the first few days of life (19). Results from recently established programs demonstrate that newborn screening dramatically improves the outcomes of infants with SCID (20–22) and should be implemented widely, not least as it is cost-effective.
Specialist Centers and Networks for Patient Management
Patients with PIDs should be managed in regional specialist PID centers (see Table 2) to enable equitable geographical access to medical and nursing expertise in these diseases. Formal links should exist between these regional immunology centers, with recognized referral pathways for treatments. National centers in different countries will vary depending on geography, available resources and expertise but all should reach internationally agreed standards of care, as in other rare diseases (23).

Table 2. Criteria for regional specialist PID centers for adults/children.
A national professional network raises standards of care through dissemination of guidelines, registration, and peer review of PID centers, patient registries, and professional leadership in PID (24). Established professional networks, for which there are different models in different countries, could be adopted as the basis for provision of PID management by national healthcare providers, whether governmental or insurance companies.
Support for PID Diagnosis and Complications in Other Medical Specialties
The immune system is distributed throughout the entire body. Accordingly, disturbances of immune function will have repercussions on all other organ systems and patients may present to a variety of medical and pediatric specialists. In addition, clinical immunologists (adult and pediatric) cannot have the necessary expertise to manage all potential medical and social complications and must therefore have access to a broad range of supporting specialist services, with which they have good working relationships and mutual understanding of the problems that are particular to PID patients. The importance of access to biopsy services and microbiological investigations cannot be over emphasized in all specialties. These services (listed in Table 3) should ideally be in the same institution, to facilitate consultation and be accredited to appropriate national/international standards.

Table 3. Key disciplines related to PID services.
Adolescent Care
Transition from adolescence to adulthood is characterized by changes of many kinds: physical, social, psychological, educational, and domestic (25). Coping with a chronic condition as well can make adolescence more complicated.
Pediatric patients are usually treated from an early age by the same staff, making it more difficult for the parent or patient to trust new faces. Without a defined and coordinated pathway guiding them toward adult services, the adolescent patient can become lost to the system, leading to poor compliance with treatment, potential irreversible organ damage, lower life expectancy, and reduced quality of life, all of which have health and cost implications.
As in other chronic diseases, planned transitional care for PID patients is often overlooked but is a cost-effective way of preventing poor compliance resulting in further damage. It is vital to ensure easy access to essential healthcare and support during this period.
Role of New Genetics
There are currently over 230 genes that, when mutated are known to disrupt immune function, and so cause a PID. This list is rapidly expanding with the detection of more genes that are proving to be important in explaining disorders of immune regulation and autoimmunity as well as defects of infection protection.
Recent advances in genetic technology have helped immensely in the diagnosis of PIDs. These include traditional sequencing of specific genes (particularly in families for genetic counseling), whole exome (and soon whole genome) sequencing (both with appropriate bioinformatics for interpretation) and chromosomal microarrays (detailing which parts of the genome are missing or duplicated). Another important area of investigation is the “mutational load” of genetic lesions causing PIDs, which is the combined impact of mutated genes, and copy number variations, which cause missing or duplicated parts of genes or entire genes (26). The latter has been especially relevant in common variable immunodeficiency disorders (CVID) (27), the commonest form of symptomatic PID. Future studies will likely show that regulatory regions of DNA are also important for expression of immune system genes and changes involving genes during life – epigenetics – may also be involved. Activating mutations can cause PIDs and somatic mutations may require analysis of DNA from particular cell populations to establish a diagnosis.
These technologies are already available in some centers and will be increasingly used in patient care in all medical specialties. As costs tumble and access to these technologies becomes more widespread, a combination of techniques will be used to diagnose previously unknown PIDs as well as provide earlier detection, with prognostic, therapeutic, and genetic counseling benefits.
Principle 2: The Importance of Registries
National Registries
Primary immune deficiencies are recognized as rare conditions and data on epidemiology of PIDs is scarce, although many countries across the EU and worldwide have implemented registries for PIDs. An example is that established in France in 2005; the Reference Center for PIDs (CEREDIH) (28) runs the largest national PID registry worldwide, with dedicated and highly trained staff. It is based on a tight network of all university teaching hospitals, with 130 clinicians and at least 30 diagnostic immunology laboratories (29). National registries are important tools for assessing the proportion of affected individuals among the general population (prevalence) as well as measuring the number of new cases diagnosed each year (incidence), detection of areas of low-diagnostic rates and provision of insights on diagnostic delay associated with increased morbidity and mortality. A registry also provides information that is helpful to governments regarding estimates of those not diagnosed, to aid planning of educational programs and provision of treatments and their costs. Presentation of this data to pharmaceutical industries helps to ensure that the supply of relevant medical products meets demand.
Thus, a national registry is an important tool for health policy makers, stakeholders, and health insurers, enabling plans for allocation of therapies and the development of innovative treatments, as also demonstrated in the UK Demand Management Plan (30).
International Experience of Registries
Studies on PIDs require international collaboration. Even in large individual European countries, no more than six to eight new patients with particularly rare genetic variants of PIDs are diagnosed per year. To collect information on all PIDs including the very uncommon cases, the European Society for Immune Deficiencies (ESID) set up an initial registry in 1994 in Sweden, supplemented by an online database in 2004. In the last 2 years, the ESID registry has been completely revised in order to provide information for national registries, healthcare providers and the European Commission. Evaluation of large registry data also provides information for individual physicians, with limited personal experience of PIDs, to answer pressing questions relating to patients or families (31).
Other continents have followed with the formation of continental registries by the Latin American Society for Immune Deficiencies (LASID) and more recently the African Society for Immune Deficiencies (ASID), and others are planned. These registries also have a role in raising awareness in countries in which the only immunodeficiency receiving attention from healthcare providers and the general public is HIV – a totally unrelated condition.
The United States Immune Deficiency Network (USIDNET), a program of the Immune Deficiency Foundation (IDF), was created in 2003 for patients in Canada and the US. The aim was to solicit, develop, evaluate, and implement clinical research strategies to advance detection, understanding, diagnosis, and treatment of PIDs. The USIDNET registry records patient data to provide both support for retrospective research studies and a source of help to physicians in making clinical decisions. USIDNET is also utilized by patients seeking information on PIDs and engages them holistically (32, 33).
International registries also provide information to centers in developing countries in which there are no national networks as yet.
Principle 3: The Need for International Collaborations for Scientific Research
International collaboration is not only important for diagnosis (genetics) and treatment (HSCT) of patients but also for research. Clinical research on PIDs faces a range of difficulties, because of scarcity of these diseases. International collaboration is necessary in order to collect sufficient patient numbers for adequate clinical research, as well as to develop new diagnostic and therapeutic strategies, for epidemiologic studies and the identification of novel disease-associated genes (34). PID disorders are caused by a large variety of genetic defects, and not all monogenetic defects have been discovered, to date, let alone those that are oligogenic or disease modifying genes. Worldwide networks, such as the JMF Network (35), need to be encouraged and developed in all countries and continents in which these support facilities do not yet exist. It is important to note that the EU, among others, has created special programs for rare diseases (23), which help to fund the necessary financial coverage for this type of research. The International Rare Diseases Research Consortium (36) provide general guidelines for diagnostic investigations, encouragement for drug/treatment development, visibility for funding calls, and publication of new results.
Principle 4: The Role of Patient Groups
Patient organizations have a significant role to play in healthcare systems and are increasingly key stakeholders in the political decision-making processes in healthcare. Today, it is well recognized that patient representatives have become true experts in their conditions and relevant treatments, and can bring unique and personal perspectives on the impact of diagnosis and treatment to their communities. All countries should aim to have an efficient national patient organization, representing all PID patients – children and adults – in order to give them a voice and represent their interest in policymaking. Effective national patient organizations provide advice, education, and support to patients and families and their healthcare providers too. They have a pivotal role to play in ensuring that their community is kept informed and updated about latest developments in a wide range of areas including medical and scientific advances, political and regulatory decisions, supply and safety of life-saving treatments.
Patient groups are increasingly active in collecting clinical data and participating in the management of registries, which yield important useful information and help to guide decisions affecting their health. They are also the driving force behind awareness campaigns around the world. To do this successfully, collaboration with doctors and other key stakeholders such as nurses, regulators, civil servants, decision makers, and industry is of vital importance, as many voices resonate louder than one. Patient-centered policies improve compliance, and therefore outcomes, once the interest of the patient is at the center of the decision making process.
At the international level, organizations such as the International Patient Organisation for Primary Immunodeficiencies (IPOPI) (37) have 51 national patient member organizations so far (38) and are actively involved in assisting new countries to start such an organization. They are involved on many different fronts, providing the patients’ expertise in international committees and institutions, including making representations to the European institutions and the World Health Organization.
Principle 5: Management and Treatment Options for PIDs
Nature of Ig Replacement Therapies
There are several types or “classes” of human Ig in blood; these include IgG, IgA, and IgM. Among these, IgG has the highest concentration in blood and body fluids and is critical for protection against infection. IgG is purified safely from human plasma and administered to people who are unable to produce a sufficient amount of good quality of IgG themselves, as a result of PID. Such IgG replacement treatment is literally life saving for such individuals and usually needs to be continued for life.
The IgG products used for replacement therapy are protective against a broad range of infections. This explains why therapeutic Ig cannot be made by recombinant technology, as for other drugs. Ig therapy is also distinct from “monoclonal antibodies” that have recently been developed to treat specific diseases rather than to protect from all types of infections.
Immunoglobulin replacement therapy is absolutely essential for the treatment of the majority of patients with a PID. There are a variety of IgG products currently marketed worldwide (39). Each new product undergoes clinical trials to establish efficacy (protection from infection), safety (no disease transmission or seriously harmful adverse events), and tolerability (minimal side effects) before it can be sold.
Although Ig therapies have been in clinical practice for over 60 years, there are still challenges since Ig products are not yet available to all PID patients (Table 4).

Table 4. Current challenges to Ig therapy.
Safety of immunoglobulin therapies
Immunoglobulin products are prepared from plasma collected from tens of thousands of screened blood/plasma donors under carefully controlled conditions and thus contain a vast repertoire of neutralizing antibodies to defend the recipient against the range of pathogens. Each company uses slightly different proprietary methods to purify IgG molecules to a high degree (typically >98%) and to reduce the possibility of disease transmission. All available products have been shown to protect against infections.
Over the last 50 years, purification and manufacturing methods have improved tremendously. There have been no known cases of disease transmission by therapeutic Ig in the past 20 years. Since 1994, viral safety has been robust, due to specific genetic testing of individual donations and implementation of a number of viral inactivation/removal steps. Authorized products contain at least two viral inactivation/removal steps. To date, there has been no known transmission of pathological prions (the agent causing variant Creutzfeldt–Jakob-disease) with Ig.
Most PID patients are able to receive Ig replacement therapy without side effects at all. A few patients may need some medicines (non-steroidal anti-inflammatory drugs and antihistamines) to treat mild side effects. A very few (1%) may have more troublesome side effects, but serious reactions are very rare indeed. With the variety of products and infusion options available, almost all patients can be treated successfully with Ig. However, it is important to realize that there is no single Ig product or method of administration that is suitable for all PID patients.
All blood products carry an infection risk, and like others, Ig products are regulated by national or international regulatory bodies to be sure that they are safe and of high quality. These bodies share their information worldwide. Excipients, preservatives, pH, IgA content, and protein concentration vary between Ig products and thus alter the side-effect profiles; respective warnings are stated in the product specific labeling. Constant vigilance is necessary to detect any new clusters of side effects of Ig therapy that may result from changes in manufacturing, concentration of the product, infusion rates, or expansion to new indications. Recent examples of investigations following unexpected side effects include the risk of thrombotic events and hemolytic episodes.
Optimal treatment levels of Ig replacement therapy
The key principle goal of Ig replacement therapy is to prevent bacterial infections and avoid organ damage that leads to chronic disease and poor quality of life. Infection prevention rather than a targeted serum IgG level is the goal of Ig replacement therapy as the protective serum IgG level varies with individual patients (40). In a meta-analysis of 17 clinical trials, there was a 27% decrease in the incidence of pneumonia for every 100 mg/kg/28 days increase in the dose of Ig therapy (41). Patients with chronic gut disease or bronchiectasis require higher doses of Ig replacement therapy (29). Other studies of patients with antibody deficiency receiving subcutaneous Ig replacement therapy show similar findings. However, cost implications may prevent optimal doses being used and it is important for patient groups and healthcare professionals to unite to explain to healthcare providers the importance, both medically and financially, of optimal dosing.
Replacement Ig therapy has been shown to be cost-effective in preventing hospitalizations and emergency departments visits, unscheduled physician visits, expensive antibiotic treatments and missed days of school or work. These studies emphasize that the appropriate dose of Ig replacement therapy enhances quality of life measures too (42).
Access to a wide choice of Ig therapies
Human Ig preparations reflect the infectious or immunization exposures of the plasma donors. However, due to patent law, they cannot be considered as generic drugs (i.e., connected together by the same chemical composition, pharmacological effects, treatment use, and same adverse effects) as Ig production methods vary. Therapeutic Ig is administered by three routes: subcutaneously (SC), intravenously (IV), or intramuscularly, though the latter is no longer recommended due to high rates of infusion related reactions. SC and IV have specific advantages and disadvantages, which may depend on the patient’s medical background or on personal preferences. Frequency of treatment, the availability of good venous access and other factors all play a role and it is impossible to say that one brand or route of administration is generally better than another. Therefore, all countries and immunodeficiency centers should have access to a wide spectrum of Ig products, to provide optimal treatment for all immunodeficient patients.
Recognition of PIDs as priority indications for Ig therapy
Immunoglobulin replacement therapy for PIDs was primarily developed for patients with antibody deficiencies in the last half of the twentieth century. Since then many other indications in autoimmunity or immune dysregulation have been recognized, resulting in much larger amounts of intravenous Ig being used in these conditions, often in off label usage not recognized by the regulatory authorities. For some autoimmune diseases, alternative therapies are available and monoclonal antibodies, such as rituximab, may supersede the use of high dose Ig therapies in autoimmunity in time. However, the available amount of Ig is limited, depending as it does on blood donors. Therefore, for now, Ig therapy should be prioritized for PIDs since there are clear indications, proven efficacy, and no alternative treatments available. This was the reason why Ig products were accepted to the WHO Lists of Essential Medicines both for adults (43) and children (44).
Site of Ig Treatment (Specialist Center, Local Hospital, Home)
To replace missing antibodies, the antibody deficiency patients need Ig on a regular basis for the duration of their lives. Although initial treatments are started under supervision in a day care facility with experienced staff, once stable, patients can either self-treat or be treated at the local hospital or health-care center. There still needs to be regular contact with the specialist immunology nurses and both easy access to and regular follow-up by the PID doctor, since complications can occur later and significant damage from infection or chronic inflammation, particularly for children or adults who are diagnosed late, needs to be monitored. Research has shown that self-infusions at home after education are effective, safe (45), and appreciated by patients and families, as this results in less disruption to work and school as well as social lives (Table 5) (31, 46). This is also appropriate for some children or infants awaiting HSCT if a donor is difficult to find.

Table 5. Advantages of programs for self-infusion at home.
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (from blood or bone marrow) is the only cure for severe, otherwise fatal PIDs that present in infancy or early childhood and all SCID children should have access to this life-saving therapy, regardless of where they live.
Bone marrow resides in the cavities of the long bones of the body and contains stem cells for all the cellular components of the blood. Stem cells are capable of repopulating a new immune system throughout life, so some are present in blood as well and can be purified following adult or cord blood donation.
The first bone marrow transplants were performed over 30 years ago but until recently were too risky to do in any but the sickest patients. Today, progress has made HSCT feasible for an increasing number of patients, though outcomes are determined by a variety of factors (Table 6).

Table 6. Current challenges to hematopoietic stem cell transplantation.
Tissue types are diverse around the world and statistically the chances of finding a match are higher within one’s own ethnicity. Donor recruitment strategies vary from country to country but patients who come from under-represented ethnic backgrounds can have a difficult time finding a “match.” Knowledge regarding transplantation, cultural, and technical limitations can hamper the broad recruitment needed to ensure that every patient who requires it has access to this life-saving treatment.
Raising awareness of the success of HSCT and additional research into outcomes is needed to continue the impressive successes in this field. Optimal treatment to prepare the body for the transplant has been studied for a decade, with tremendously improved outcomes (6), yet children and adults still die due to the risks of the process and patients require great stamina for the rigors of maintaining ideal conditions. Gentler strategies are under development and additional research may pave the way for our goal of curing 100% of patients who need HSCT.
Additional Antimicrobial Measures
Infection is the most common presentation, the nature of which depends on the underlying immune defect. More than one infection may be present and more than one organ affected. Occult infections require careful imaging, tissue sampling, and molecular techniques needed to identify the pathogen. Treatment of infection in PID patients is complex, often requiring one or more broad-spectrum antimicrobials and for prolonged time-courses, and physiotherapy is essential for those with lung complications.
If an innate PID is diagnosed for which there is no specific effective therapy, prophylactic antimicrobials may be given. Physical barriers range from sterile positive pressure ventilation for extremely immunodeficient infants to specific measures such as boiling water or avoidance of exposure to fungi. Anti-bacterial prophylaxis is used to prevent pneumococcal or meningococcal disease in patients with complement deficiency or hyposplenism. Treating existing fungal infections in patients with innate deficiencies is a huge challenge, with larger doses of increasingly toxic antifungal agents and even neutrophil transfusions as well as surgical drainage.
Immunological and Other Treatments
Other replacement therapies or even immunosuppression to counter the aberrant immune responses may be necessary. Examples include granulocyte-colony stimulating factor (G-CSF) to boost the production of neutrophils or gamma interferon in patients with defective neutrophils.
Long acting adenosine deaminase (PEG-ADA), an enzyme replacement therapy, is administered to patients who lack ADA prior to HSCT.
Anti-inflammatory agents, such as corticosteroids, and immunosuppressive agents are useful for particular complications (including respiratory, gastro-intestinal, and dermatological). Nutritional supplements and other types of therapies (physiotherapy, psychotherapy) are also used to treat PID patients with specific complications.
This list of other treatments is not exhaustive but provides examples of the complexity of therapies that should be available to ensure appropriate care for all PID patients.
Gene Therapy
A new approach to replacement of faulty genes has been developed in recent years, though currently remains a research tool. Gene therapy is defined as the addition of a normal copy of a gene to a patient’s purified stem cells to supplement the one that is defective or absent. This is currently reserved for children for whom HSCT is not available (generally because they do not have a suitable matched donor for HSCT). Successful clinical trials started in Paris in 1999 for patients with X-linked SCID. More recently, these have been extended at several centers to include patients with ADA SCID, CGD, and Wiskott–Aldrich syndrome. Most of the patients have been successfully treated and are at home, off all therapy, thus, proving that such therapy is possible and effective (8).
However, there have been a small number of patients who developed leukemia-like disorders as a result of unwelcome mutations in the cells as a result of using the original retroviral vectors – a process known as insertional mutagenesis. New improved self-inactivating retroviral and lentiviral vectors have been developed incorporating additional safety features and these are now being used in clinical trials in the same patient groups.
Vaccines
Vaccination – or more commonly – immunization is the administration of material derived from infectious material in order to produce a protective immune response to a specific pathogen (bacteria or virus) without developing the infection. The material in the vaccine can be modified (attenuated) or killed (inactivated) whole micro-organisms, defined components of the microorganism or specific proteins such as modified toxins (known as toxoids). Those that consist of live-attenuated bacteria or virus can undergo reversion to a more virulent form and cause disease (unlike the killed micro-organisms). In general, attenuated vaccines including BCG and rotavirus, must be avoided in all the severe forms of PID (SCID, CGD, T-cell defects, etc.), as in such patients there will be no protective immune response and therefore a clear risk of developing the disease itself (47). If BCG is given in the first month of life, infants with severe PIDs or MSMD develop generalized BCG-osis that compromises HSCT and is often fatal. To postpone the age of BCG vaccine would prevent this (48). If BCG administration cannot be delayed, newborn screening is important to identify at-risk babies.
Inactivated vaccines are safe for use in most PID patients but are ineffective if there is no or only a limited immune response, depending on the type of immune failure. SCID patients are only immunized after HSCT has restored a healthy immune system. If the patient cannot produce antibodies, vaccines are ineffective though patients may produce a T-cell response that will prevent or lessen any subsequent, particularly viral, infection.
In those PIDs due to neutrophil defects, complement deficiencies and some mild forms of antibody failure, vaccination is beneficial (41) and forms part of the treatment protocol.
It is necessary to discuss the risks and benefits to the specific individual with the patient’s pediatrician or specialist immunologist as well as the patient (49). Immunization of household relatives to prevent close-contact infection, such in an influenza outbreak or to provide on-going protection in a meningitis or chickenpox outbreak, is helpful.
Killed vaccines are also used in diagnostic procedures if antibody failure is suspected; failure of specific antibody production indicates an immune defect.
Comprehensive and Holistic Approach to the Patient
Primary immune deficiency patients encounter a diversity of clinical manifestations during their lifetime (infections, autoimmunity, granuloma, allergy, lymphoproliferative disorders, and malignancies). Thus, expert management according to the best international standard of care is warranted and acknowledgment of the personal context of the patients. Treated patients now have an increased life expectancy but there are new and unexpected complications seen with longer life, as well as difficulties in transition to adolescence and adulthood. Thought should be given to long-term care plans and access to genetic counseling when patients consider having children of their own. As their life progresses, modifications to treatment will be needed to fit in with changing circumstances such as work, studying, travel, or pregnancy. Patients need to have assistance and choice in treatment modalities and to be able to change the route, dose and location of Ig replacement (IVIgs versus SCIgs and home versus clinic) especially as children grow.
Impacts on a patient’s life include stress, anxiety, chronic fatigue, pain, disability, fertility, and psychosocial aspects. Advice about sports to relieve complications such as bronchiectasis, safety measures for traveling (including places where safe drinking water is not easily available), avoidance of inappropriate hobbies, how to obtain health insurance, etc., should be given by trained healthcare professionals. Further studies are needed.
Emergency Medicine
Primary immune deficiency patients may need emergency treatment, so every patient should have an individually tailored plan (medical diagnosis, specific therapy, expert center contact), outlining management of emergencies common to their immunodeficiency, and ensuring access to specialist medication. If direct access to the specialist center is impractical (for example, because of distance), shared care should be sought with local direct-access staff who are informed in the care required. Emergency staff are unlikely to be familiar with management of immunodeficiency and the patient may be at-risk of delayed or inappropriate care; a 24-h contact number for specialist advice must be included in the patient’s plan. This is particularly important for patients with complement deficiencies in whom the correct emergency therapy undoubtedly saves lives.
Principle 6: Managing PID Diagnosis and Care in all Countries
Access to PID Care World-Wide
The broad scope of PID clinical presentations presents special challenges (Table 7), especially in developing countries lacking even basic health care; infectious diseases are common and so PIDs are missed. Yet, diseases that are rarer than PIDs still achieve good diagnostic and treatment levels in low-income countries through organized efforts.

Table 7. Integrated approaches for PIDs.
In the context of high prevalence of infectious diseases in developing countries (in particular, infections resulting from HIV) PIDs, which are due to intrinsic failure of the immune system, are now more likely to be exposed.
Access to PID Diagnosis
Diagnosis of PID is a challenge under any circumstances, but even more so in a resource-constrained setting of developing countries. Clinicians must be sure to select scarce (often expensive) laboratory resources judiciously to avoid an unnecessary work-up in a suspected PID. Understanding the clinical clues (Table 8) and using a small set of basic tests enable diagnosis of most PIDs enables appropriate treatment to be sought. As there are few centers and few immunologists in developing countries, it is important to set up a network, using the Internet, to discuss clinical cases and support of physicians who live far from specialized centers. International links are helpful too.
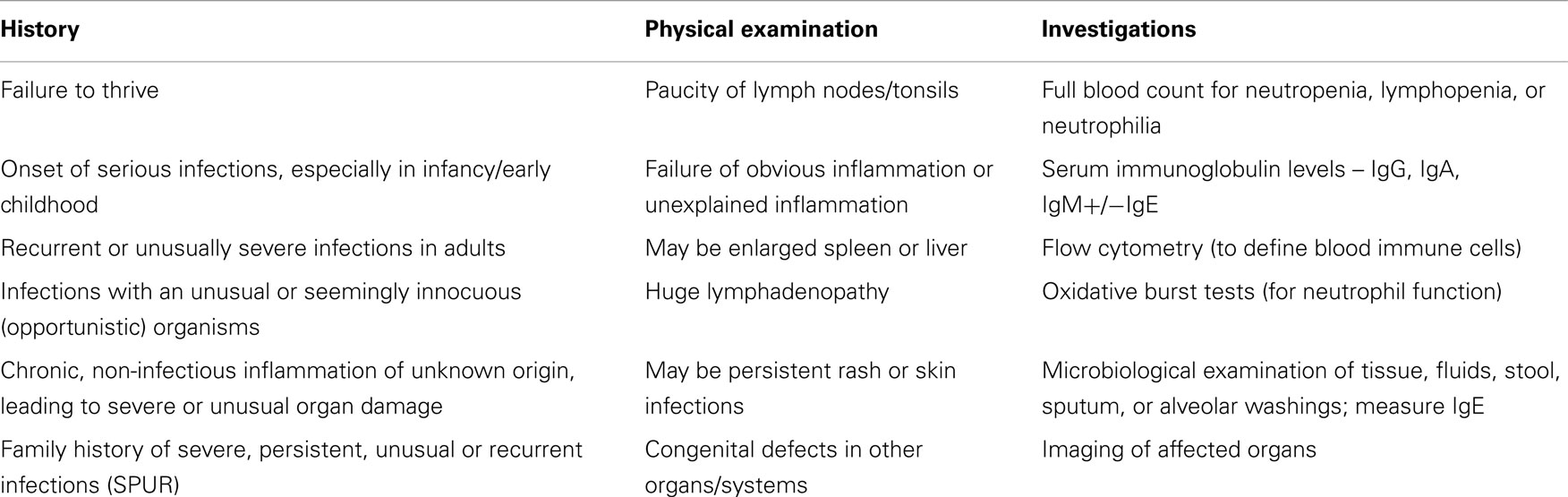
Table 8. Clues toward a diagnosis of a PID.
With this simplified approach, it is possible to diagnose the vast majority of common PIDs (50).
Access to PID Treatments
Best level of available PID care must be pursued, even if the full range of treatment options is not available. Knowledge must be promoted on treatment and prevention of infectious diseases with hygiene measures, nutritional support, vaccines, and antibiotics. Very often developing countries are specifically faced with important challenges with regards to relatively expensive therapies used in the treatment of PIDs, mostly because of cost constraints or a lack of expertise.
Immunoglobulin therapies are listed as Essential Medicines by the WHO (43, 44) and should be made available for PID patients in all developing countries. While HSCT has been the standard of care in developed countries for most forms of severe PIDs, it is frequently not available in developing countries though the situation is slowly improving. The cost of building appropriate facilities and of providing expert training locally may seem significant but actually allows for more efficient management of healthcare resources, as many SCID patients in developing regions currently have to travel to developed countries where the cost is higher. International collaboration and the creation of regional societies such as ESID in Europe, LASID in Latin America, and ASID in Africa have been and will continue to be pivotal in increasing PID expertise in developing countries.
Implementation
Vast gaps in knowledge of PIDs and translation of diagnosis and management into clinical practice exist between regions, countries, and even areas. This causes unnecessary suffering and deaths, particularly in developing countries. PID facilities should be at least as good as those for other, even rarer, diseases. Services for diagnosis and treatment can be developed initially alongside facilities for diagnosis and monitoring of HIV patients or located with immunization centers. In order for the best level of available PID care to be pursued, even if the full range of treatment options may not be available, knowledge of PIDs must be promoted, and infra-structure for diagnosis and care implemented.
This involves, by stages:
• Raising awareness and understanding of the clinical clues among local healthcare workers, general physicians, and pediatricians so that patients are diagnosed quickly;
• Provision of inexpensive tests (often already available in district hospitals/healthcare centers) plus guidance on their interpretation and indications for further complex testing;
• Defining practical pathways to obtain appropriate therapies for the patient.
With this simplified approach, it is possible to diagnose and treat the majority of common PIDs.
To achieve these aims, professional stakeholders in PIDs have to come together in a network to:
• Share information informally regarding individual patients and access to all relevant treatments including HSCT, and to make this knowledge available to others;
• Agree to share relevant specialist investigations so that all useful assays are available to patients throughout every country;
• Construct and publish a consensus document on the need for and provision of PID services nationally (51) and use this to persuade healthcare providers to establish regional and national centers for complex investigations and therapies;
• Seek advice from established centers in other countries or continents and to establish links for treatment, training, and education with these centers;
• Meet at least annually and to correspond regularly or post information and questions on a website;
• Include patient organizations, who are key stakeholders in healthcare decisions and who can also assist with clinical data collection.
Established professional networks should be recognized, formalized, and adopted as the basis for provision of PID management by national healthcare providers, whether governmental or insurance companies. Network responsibilities also include registration of patients (33), setting of national standards, and auditing PID services to these standards (52).
As in the provision of other rare diseases, services for PID patients usually involve recognized expert centers for complex diagnostic facilities as well as experience in treatments, whether Ig replacement or HSCT. In well-established countries, there may be several expert centers, each concentrating on a particular type of PID, with a broad range of supporting specialist services; this has the advantage of concentrating resources for research. Good links with pediatric services are essential for planned transitional care and the importance of a national registry has been emphasized above (53).
Conclusion
In conclusion, we call upon international and national healthcare policy makers to join us in taking strong and decisive action to ensure that people with PIDs are diagnosed as early as possible and have appropriate access to safe, efficient life-saving treatments, and optimum care throughout the world.
We endorse the above principles, as elements of PID care provision that should be available and implemented in each country. These include specialized centers, PID registries, provision for transnational research, patient organizations, access to services for diagnosis and treatment, the need for sustained access to all treatments including Ig therapies and HSCT, and important considerations for developing countries.
Authorship
The Principles of Primary Immunodeficiency Care were led by the International Patient Organisation for Primary Immunodeficiencies (IPOPI) and developed by an interdisciplinary working group of 30 leading experts in PID care from 12 countries in 6 continents. Over a 2-year period, this group reviewed current best practice care and guidelines, as well as assessed the needs of those both providing and receiving PID care and treatment worldwide. Representatives from patient organizations and specialist immunology nurses were included.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Review Editor Klaus Warnatz declares that, despite having collaborated on a publication in the last 2 years with the authors Helen Chapel and Hubert Bobby Gaspar, the review process was handled objectively.
References
1. Al-Herz W, Bousfiha A, Casanova J-L, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:54. doi: 10.3389/fimmu.2011.00054
2. Maggadottir SM, Sullivan KE. The intersection of immune deficiency and autoimmunity. Curr Opin Rheumatol (2014) 26(5):570–8. doi:10.1097/BOR.0000000000000091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Blore J, Haeney M. Primary antibody deficiency and diagnostic delay. Br Med J (1989) 298:516–7. doi:10.1136/bmj.298.6672.516
4. Cunningham-Rundles C. Clinical and immunologic analyses of 103 patients with common variable immunodeficiency. J Clin Immunol (1989) 9:22–33. doi:10.1007/BF00917124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. De Vries E, Driessen G. Primary immunodeficiencies in children: a diagnostic challenge. Eur J Pediatr (2011) 170(2):169–77. doi:10.1007/s00431-010-1358-5
6. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med (2014) 371:434–46. doi:10.1056/NEJMoa1401177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther (2014) 14(6):789–98. doi:10.1517/14712598.2014.895811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. De Vries E; European Society for Immunodeficiencies (ESID) members. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol (2012) 167:108–19. doi:10.1111/j.1365-2249.2011.04461.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Hernandez-Trujillo HS, Chapel H, Lo Re V III, Notarangelo LD, Gathmann B, Grimbacher B, et al. Comparison of American and European practices in the management of patients with primary immunodeficiencies. Clin Exp Immunol (2012) 169:57–69. doi:10.1111/j.1365-2249.2012.04588.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Modell F, Puente D, Modell V. From genotype to phenotype. Further studies measuring the impact of a Physician Education and Public Awareness Campaign on early diagnosis and management of primary immunodeficiencies. Immunol Res (2009) 44(1–3):132–49. doi:10.1007/s12026-008-8092-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Gardulf A, Björvell H, Gustafson R, Hammarström L, Smith CI. The life situations of patients with primary antibody deficiency untreated or treated with subcutaneous gammaglobulin infusions. Clin Exp Immunol (1993) 92:200–4. doi:10.1111/j.1365-2249.1993.tb03380.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Gardulf A, Nicolay U, Math D, Asensio O, Bernatowska E, Böck A, et al. Children and adults with primary antibody deficiencies gain quality of life by subcutaneous IgG self-infusions at home. J Allergy Clin Immunol (2004) 114:936–42. doi:10.1016/j.jaci.2004.06.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Gardulf A, Andersen V, Björkander J, Ericson D, Frøland SS, Gustafson R, et al. Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: safety and costs. Lancet (1995) 345:365–9. doi:10.1016/S0140-6736(95)90346-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Bousfiha AA, Jeddane L, Ailal F, Al-Herz W, Conley ME, Cunningham-Rundles C, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol (2013) 33:1078–87. doi:10.1007/s10875-013-9901-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Buckley R. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol (2004) 22:625–55. doi:10.1146/annurev.immunol.22.012703.104614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency (SCID) in the neonatal period leads to superior thymic output and improved survival. Blood (2002) 99:872–8. doi:10.1182/blood.V99.3.872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood (2011) 117:3243–6. doi:10.1182/blood-2010-08-300384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol (2005) 115:391–8. doi:10.1097/MOP.0b013e32834cb9b0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T cell lymphopenia in California: results of the first two years. J Allergy Clin Immunol (2013) 132:140–50. doi:10.1016/j.jaci.2013.04.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gaspar HB, Hammarström L, Mahlaoui N, Borte M, Borte S. The case for mandatory newborn screening for severe combined immunodeficiency (SCID). J Clin Immunol (2014) 34:393–7. doi:10.1007/s10875-014-0029-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. doi:10.1001/jama.2014.9132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Available from: http://www.rare-diseases.eu
24. UKPIN: UK Primary Immunodeficiency Network [Internet]. UKPIN (2014) [cited 2014 Aug 07]. Available from: www.ukpin.org.uk
25. DH Partnerships for Children, Families and Maternity/CNO Directorate (UK). Transition: Moving on Well. A Good Practice Guide for Health Professionals and Their Partners on Transition Planning for Young People with Complex Health Needs or a Disability [Internet]. NIH Publication. (2008) [cited 2014 Aug 07]. 81 p. Available from: http://www.bacdis.org.uk/policy/documents/transition_moving-on-well.pdf
26. Keller M, Glessner J, Resnick E, Perez E, Chapel H, Lucas M, et al. Burden of copy number variation in common variable immunodeficiency. Clin Exp Immunol (2014) 177:269–71. doi:10.1111/cei.12255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol (2011) 127(6):1360–7. doi:10.1016/j.jaci.2011.02.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. CEREDIH: Le Centre de Référence Déficits Immunitaires Héréditaires [Internet]. Paris: CEREDIH (2014) [cited 2014 Sep 04]. Available from: https://www.ceredih.fr/
29. CEREDIH: The French PID Study Group. The French national registry of primary immunodeficiency diseases. Clin Immunol (2010) 135:264–72. doi:10.1016/j.clim.2010.02.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. UK Department of Health. Demand Management Plan for Immunoglobulin Use [Internet]. London: Department of Health (2008) [cited 2014 Aug 07]. 11 p. Available from: http://www.ivig.nhs.uk/documents/Demand_Management_Plan_SECOND_EDITION.pdf
31. ESID: European Society for Immunodefiencies [Internet]. ESID [cited 2014 Aug 07]. Available from: http://esid.org/
32. USIDNET. A Program of IDF. (2014) [cited 2014 Aug 07]. Available from: http://usidnet.org/usidnet-registry/publications
33. Sullivan KE, Puck JM, Notarangelo LD, Fuleihan R, Caulder T, Wang C, et al. USIDNET: a strategy to build a community of clinical immunologists. J Clin Immunol (2014) 34:428–35. doi:10.1007/s10875-014-0028-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Parvaneh N, Casanova JL, Notarangelo LD, Conley ME. Primary immunodeficiencies: a rapidly evolving story. J Allergy Clin Immunol (2013) 131:314–23. doi:10.1016/j.jaci.2012.11.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jeffrey Modell Foundation. Global PI Village [Internet]. Jeffrey Modell Foundation [cited 2014 Aug 07]. Available from: http://www.info4pi.org/jmf/
36. Available from: http://www.irdirc.org
37. International Patient Organisation for Primary Immunodeficiences (IPOPI) [Internet]. [cited 2014 Aug 7]. Available from: http://www.ipopi.org/
38. National Member Organisations of IPOPI [Internet]. [cited 2014 Aug 07]. Available from: http://www.ipopi.org/index.php?page=member-organisations
39. IPOPI Immunoglobulin list [Internet]. IPOPI [cited 2014 Aug 07]. Available from: http://www.ipopi.org/index.php?page=immunoglobulin-countries
40. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol (2010) 125:1354–60. doi:10.1016/j.jaci.2010.02.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Orange JS, Ballow M, Stiehm ER, Ballas ZK, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American academy of allergy, asthma and immunology. J Allergy Clin Immunol (2012) 130:1–24. doi:10.1016/j.jaci.2012.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Gardulf A, Nicolay U. Replacement IgG therapy and self-therapy at home improve the health-related quality of life in patients with primary antibody deficiencies. Curr Opin Allergy Clin Immunol (2006) 6:434–42. doi:10.1097/01.all.0000246619.49494.41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. World Health Organization. 18th WHO Model List of Essential Medicines [Internet]. (2013) [cited 2014 Aug 7]. Available from: http://apps.who.int/iris/bitstream/10665/93142/1/EML_18_eng.pdf?ua=1
44. World Health Organization. 4th WHO Model List of Essential Medicines for Children [Internet]. (2013) [cited 2014 Aug 7]. Available from: http://apps.who.int/iris/bitstream/10665/93142/1/EML_18_eng.pdf?ua=1
45. Brennan VM, Salomé-Bentley NJ, Chapel H. Prospective audit of adverse reactions occurring in 459 primary antibody-deficient patients receiving intravenous immunoglobulin. Clin Exp Immunol (2003) 133:247–51. doi:10.1046/j.1365-2249.2003.02199.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Espanol T, Prevot J, Drabwell J, Sondhi S, Olding L. Improving current Improving current immunoglobulin therapy for patients with primary immunodeficiency: quality of life and treatment views [Internet]. Patient Prefer Adherence (2014) 8:621–9. doi:10.2147/PPA.S60771
47. Obenga G, Mahlaoui N, Launay O. Recommandations vaccinales chez les patients atteints de déficit immunitaire héréditaire [Internet]. Centre de Référence Déficits Immunitaires Héréditaires (2009) [cited 2014 Aug 07]. 7 p. Available from: http://www.ceredih.fr/documents/Recos_Vaccins_20091113.pdf
48. Marciano BE, Huang CY, Joshi G, Rezaei N, Carvalho BC, Allwood Z, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol (2014) 133(4):1134–41. doi:10.1016/j.jaci.2014.02.028
49. Shearer WT, Fleischer TA; For the Medical Advisory Committee of the Immune Deficiency Foundation. Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol (2014) 133:961–6. doi:10.1016/j.jaci.2013.11.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Costa-Carvalho BT, Grumach AS, Franco JL, Espinosa-Rosales FJ, Leiva LE, King A, et al. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J Clin Immunol (2014) 34:10–22. doi:10.1007/s10875-013-9954-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Chapel HM. Consensus on diagnosis and management of primary antibody deficiencies. Consensus panel for the diagnosis and management of primary antibody deficiencies. BMJ (1994) 308:581–5. doi:10.1136/bmj.308.6928.581
52. Spickett GP, Askew T, Chapel HM. Management of primary antibody deficiency by consultant immunologists in the United Kingdom: a paradigm for other rare diseases. Qual Health Care (1995) 4:263–8. doi:10.1136/qshc.4.4.263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Gathmann B, Binder N, Ehl S, Kindle G; ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: update 2011. Clin Exp Immunol (2012) 167:479–91. doi:10.1111/j.1365-2249.2011.04542.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: primary immunodeficiencies, awareness, diagnosis, management, treatments, worldwide
Citation: Chapel H, Prevot J, Gaspar HB, Español T, Bonilla FA, Solis L, Drabwell J and The Editorial Board for Working Party on Principles of Care at IPOPI (2014) Primary immune deficiencies – principles of care. Front. Immunol. 5:627. doi: 10.3389/fimmu.2014.00627
Received: 08 September 2014; Accepted: 24 November 2014;
Published online: 15 December 2014.
Edited by:
Guzide Aksu, Ege University School of Medicine, TurkeyReviewed by:
Klaus Warnatz, University of Freiburg, GermanyLaurence E. Cheng, University of California San Francisco, USA
Copyright: © 2014 Chapel, Prevot, Gaspar, Español, Bonilla, Solis, Drabwell and The Editorial Board for Working Party on Principles of Care at IPOPI. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helen Chapel, Nuffield Department of Medicine, Oxford University Hospitals, Level 7, Headley Way, Oxford OX3 9DU, UK e-mail: helen.chapel@ndm.ox.ac.uk
†Contributors: M. Ballow (Children’s Hospital of Buffalo, USA); L. Brown (UCL Institute of Child Health, London, UK); B. T. Costa-Carvalho (Federal University of São Paulo, Brazil); J. D. Edgar (United Kingdom Primary Immunodeficiency Network, UK); W. Egner (British Society for Immunology, UK); S. Ehl (Center for Chronic Immunodeficiency, Freiburg, Germany); M. Esser (University of Stellenbosch, South Africa); A. Gardulf (Karolinska Institute at Huddinge University Hospital, Sweden); A. Gennery (University of Newcastle upon Tyne, Newcastle, UK); J. Kerr (Paul-Ehrlich-Institut, Langen, Germany); C. Kinnon (UCL Institute of Child Health, London, UK), Y. L. Lau (The University of Hong Kong, Hong Kong, China); J. Litzman (St. Anne’s University Hospital Brno, Czech Republic); H. J. Longhurst (Barts Health NHS Trust, London, UK); N. Mahlaoui (Centre de Référence Déficits Immunitaires Héréditaires – CEREDIH, Hôpital Universitaire Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, France); J. S. Orange (Texas Children’s Hospital and Baylor College of Medicine, USA); C. Picard (Hôpital Universitaire Necker-Enfants Malades, Assistance Publique-Hôpitaux de Paris, France); J. Puck (University of California San Francisco, UCSF Benioff Children’s Hospital, San Francisco, USA); S. Sánchez Ramon (Hospital Clínico San Carlos, Madrid, Spain); R. E. Schmidt (Medical University Hanover, Hanover, Germany); S. Singh (Advanced Pediatrics Center, PGIMER, Chandigarh, India); G. Spickett (University of Newcastle upon Tyne, Newcastle, UK); K. Sullivan (Children’s Hospital of Philadelphia, USA); A. Symes (Royal Free Hospital NHS Trust, London, UK)