 Melita Irving
Melita Irving Romain Vuillefroy de Silly
Romain Vuillefroy de Silly Kirsten Scholten
Kirsten Scholten Nahzli Dilek
Nahzli Dilek George Coukos1,3*
George Coukos1,3*
- 1The Ludwig Institute for Cancer Research, University of Lausanne, Epalinges, Switzerland
- 2Swiss Institute of Bioinformatics, University of Lausanne, Lausanne, Switzerland
- 3Department of Oncology, University Hospital of Lausanne (CHUV), Lausanne, Switzerland
T-cells play a critical role in tumor immunity. Indeed, the presence of tumor-infiltrating lymphocytes is a predictor of favorable patient prognosis for many indications and is a requirement for responsiveness to immune checkpoint blockade therapy targeting programmed cell death 1. For tumors lacking immune infiltrate, or for which antigen processing and/or presentation has been downregulated, a promising immunotherapeutic approach is chimeric antigen receptor (CAR) T-cell therapy. CARs are hybrid receptors that link the tumor antigen specificity and affinity of an antibody-derived single-chain variable fragment with signaling endodomains associated with T-cell activation. CAR therapy targeting CD19 has yielded extraordinary clinical responses against some hematological tumors. Solid tumors, however, remain an important challenge to CAR T-cells due to issues of homing, tumor vasculature and stromal barriers, and a range of obstacles in the tumor bed. Protumoral immune infiltrate including T regulatory cells and myeloid-derived suppressor cells have been well characterized for their ability to upregulate inhibitory receptors and molecules that hinder effector T-cells. A critical role for metabolic barriers in the tumor microenvironment (TME) is emerging. High glucose consumption and competition for key amino acids by tumor cells can leave T-cells with insufficient energy and biosynthetic precursors to support activities such as cytokine secretion and lead to a phenotypic state of anergy or exhaustion. CAR T-cell expansion protocols that promote a less differentiated phenotype, combined with optimal receptor design and coengineering strategies, along with immunomodulatory therapies that also promote endogenous immunity, offer great promise in surmounting immunometabolic barriers in the TME and curing solid tumors.
Natural Tumor Immunity and Response to Immunotherapy
Immune Checkpoint Blockade
T lymphocytes play a critical role in tumor immunity through the recognition of tumor-associated antigens processed and presented as peptides at the cell surface by major histocompatibility complex (MHC) molecules (1). For various tumor types, the presence of tumor-infiltrating T lymphocytes (TILs) predicts longer disease-free survival and overall patient survival (2–6). Cancers employ numerous mechanisms of immune evasion (7) that dampen T-cell activity, which in some patients can be successfully reversed by monoclonal antibodies (mAbs) targeting immune checkpoints, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4), and the programmed cell death 1 (PD-1)/PD ligand 1 (PD-L1) axis. These immunomodulatory mAbs have enabled regression of a range of malignancies including melanoma (8, 9), lung (10), bladder (11), Hodgkin’s lymphoma (12), renal-cell carcinoma (13), ovarian (14), as well as gastrointestinal and endometrial cancers with DNA mismatch-repair defects (15), thus providing proof that the majority of solid tumor types can be spontaneously recognized by the host’s T-cells. PD-1 inhibition alone is active in about 30% of cancer patients, but in combination with CTLA-4 the fraction of responding metastatic melanoma patients increases to 57% (16). Notably, clinical responses to immunotherapy are usually associated with durability (17).
A number of studies have attempted to elucidate mechanisms underlying resistance to checkpoint blockade. The presence of CD8+ T-cells within the tumor or its invasive margin is a key requirement for responses to PD-1 inhibition (18). Many studies indicate that the neoantigen load may also be an influencing factor (19–22). However, as the case of Merkel-cell carcinoma (MCC) has shown, the quality of tumor epitopes and the corresponding TILs, and not only the mutation rate, can confer sensitivity to checkpoint blockade; MCC that is virally induced and having a low mutation rate responds similar to checkpoint blockade as MCC caused by ultraviolet radiation (23, 24). The presence of CD8+ TILs has been linked to the expression of a type I interferon (IFN) signature in tumors (25, 26) and the recruitment of a subset of CD103+/CD8α+ DCs driven by the transcription factor Batf3 (27, 28). WNT/β-catenin signaling upregulation may be one of the tumor cell-intrinsic pathways driving a non-T-cell inflamed phenotype, at least in metastatic melanoma (29) for which a genetically engineered mouse model revealed reduced expression of the chemokine CCL4, impaired recruitment of Batf3+ DCs and T-cells, and resistance to checkpoint blockade (29). Interestingly, commensal bacteria in the gut can also shape innate immunity and responses to immune checkpoint therapy (30–32). Finally, metabolic circuitries play an important role in regulating immune function in tumors. It has recently been demonstrated that CTLA-4, PD-1, and PD-L1 blockade can restore glucose levels in the tumor microenvironment (TME), thereby improving T-cell fitness. Anti-PD-L1 mAb was specifically shown to block the mechanistic target of rapamycin [mTOR, a central regulator of metabolism and physiology (33)] and decrease the expression of glycolytic enzymes in tumor cells (34). Further, for PD-L1+ renal-cell carcinoma patients, non-responsiveness to PD-1 blockade has been associated with metabolic gene upregulation as well as of solute transport functions such as UGT1A family members, whereas responders present an immune response profile including upregulation of CCL3, a chemokine involved in leukocyte migration (35).
TIL and Chimeric Antigen Receptor (CAR) T-Cell Therapy
Another powerful form of immunotherapy is adoptive T-cell therapy (ACT), which entails the ex vivo expansion of tumor-specific T-cells and their infusion into a patient. For TIL therapy, in which T lymphocytes are enriched from tumor biopsies, patients are typically lymphodepleted and receive high-dose interleukin-2 (IL-2) (36–38). TIL therapy has proven successful in advanced metastatic melanoma, mediating objective responses in about 50% of patients, and durable complete responses in up to 20% of patients receiving a single TIL infusion (36). It is now evident that in the case of metastatic melanoma an important target of TILs are mutated gene products (39). TIL therapy has also been anecdotally successful in common carcinomas (40), suggesting that this approach could be applied to other solid tumor indications. For various reasons, however, ranging from tumor vasculature barriers to a lack of type I IFN signaling, not all tumors are infiltrated by T-cells at baseline (27, 41–43).
In the absence of endogenous T-cell infiltrate due to aberrant antigen processing and presentation, for example, which precludes the use of TIL therapy and immune checkpoint blockade, a promising solution for treating cold tumors is the transfer of mAb-modified T-cells, so-called CAR T-cells (39). In recent years, CD19-targeted CAR T-cell therapy has yielded spectacular clinical responses against hematologic liquid tumors (44), including up to 90% complete response in relapsed or treatment-refractory acute lymphoblastic leukemia (ALL) patients (45). In the solid TME, however, T-cells face a battery of physical and immunometabolic barriers (46, 47), to which CAR T-cells, like endogenous T-cells, are vulnerable (48, 49). CAR T-cells may thus similarly require combinatorial regimens of immunomodulation such as kinase inhibitors (50), chemotherapy (51), radiotherapy (RT) (52), or checkpoint blockade (53), to unleash their full therapeutic potential (54–56). CAR T-cells can also be armored through additional gene modification (57). For example, they have been coengineered to express stimulatory ligands, such as CD40 ligand (CD40L) (58), or to secrete stimulatory cytokines, such as IL-12 (57), for improved antitumor responses. With an emerging awareness of the role played by metabolism in both cancer progression and T-cell activity in the TME (59), it is apparent that further development of CAR T-cell therapy for maximizing functionality in harsh, nutrient-depleted conditions is critical. Here, we review the design and function of CAR T-cells, immunometabolic barriers in the solid TME, and different ex vivo expansion, coengineering and combinatorial therapy approaches for overcoming them.
CAR T-Cell Engineering
Basic CAR Design
Chimeric antigen receptors, first conceived in the late 1980s (60), are hybrid receptors comprising (i) an extracellular tumor-binding moiety, typically an Ab-derived single-chain variable fragment (scFv), (ii) a hinge/spacer, (iii) a transmembrane (TM) region, and (iv) various combinations of intracellular signaling domains associated with T-cell activation (61). First-generation CARs include the endodomain of CD3ζ only (for signal 1 of T-cell activation), while second- and third-generation CARs also have one or more costimulatory endodomains (for signal 2), respectively (Figure 1) (62). Finally, armored CAR T-cells are further gene modified to express or block molecules and/or receptors to enhance immune activity. Patient responses to first-generation CAR T-cells were disappointing, probably due to poor expansion and persistence (63–65) as a result of an anergic phenotype (66–68), and most ongoing trials involve second-generation CARs incorporating either CD28 or 4-1BB (CD137) (39, 69). CARs can be transiently expressed in primary T-cells by RNA electroporation, typically for about 1 week with current technology, or they can be stably incorporated into the genome by lentiviral or gamma-retroviral transduction (70), as well as by transposon/transposase-mediated integration using the sleeping beauty system (71). RNA electroporation along with dosing escalation is often used in the testing of new CARs in the clinic. To minimize toxicity, molecules secreted by armored CAR T-cells can be placed under an inducible promoter (72). To date, there have been no safety issues related to viral or transposon-mediated genomic integration of CARs (38, 73).
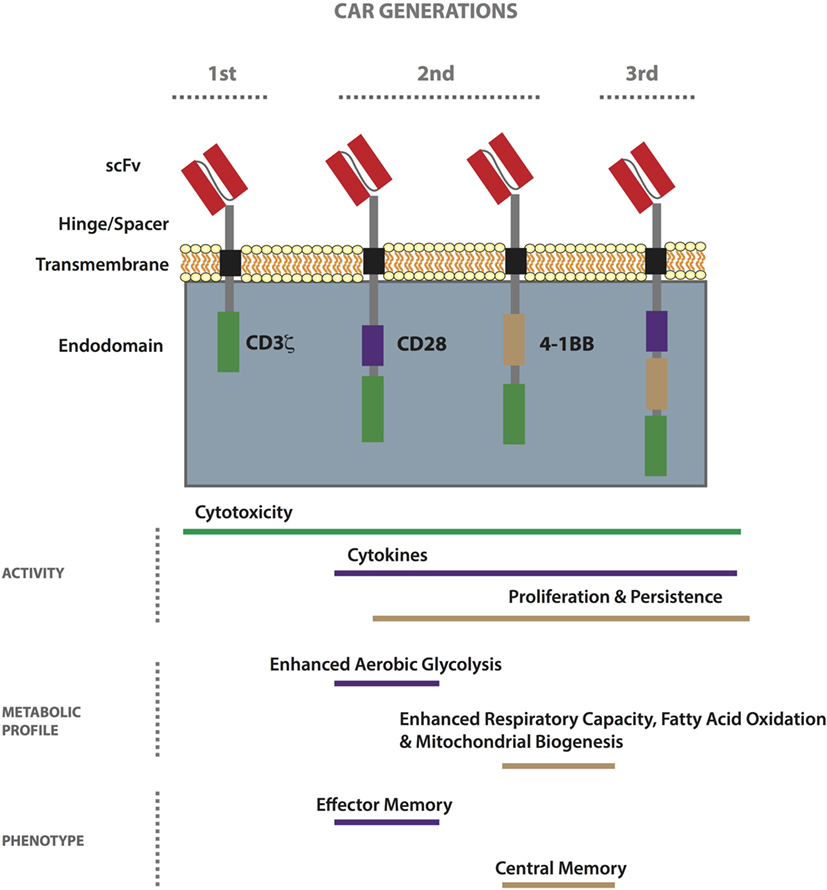
Figure 1. Properties of first-, second-, and third-generation chimeric antigen receptor (CAR) T-cells. CARs comprise an extracellular tumor-binding moiety, typically a single-chain variable fragment (scFv), followed by a hinge/spacer of varying length and flexibility, a transmembrane (TM) region, and various combinations of endodomains associated with T-cell signaling. While first-generation CARs include the endodomain of CD3ζ, second- and third-generation CARs also have one or more costimulatory endodomain, respectively. The most commonly used costimulatory endodomains are from CD28 and 4-1BB. CAR T-cell functionality can vary based on design (scFv affinity, hinge/spacer length, TM domain, etc.) and generation. In general, the 4-1BB endodomain confers the highest level of in vivo persistence and resistance to exhaustion. Metabolically, second-generation CD28-based CAR T-cells exhibit enhanced aerobic glycolysis as compared to 4-1BB-based ones which demonstrate enhanced respiratory capacity, fatty acid oxidation, and mitochondrial biogenesis. Stimulated CD28-based CAR T-cells acquire an effector memory-like phenotype, whereas 4-1BB-based ones take on a central memory-like phenotype.
Unlike T-cell receptors (TCRs) that are MHC restricted, CARs can potentially bind any cell surface-expressed antigen and can thus be more universally used to treat patients. CARs have been developed against not only proteins, including the pMHC complex (74), but also against carbohydrates and glycolipids, as well as targets upregulated in the tumor stroma (75) and the tumor vasculature (42). Notably, not all scFvs are suitable for CAR development. A recent study demonstrated antigen-independent clustering of an anti-GD2 CAR, caused by the framework region sequences, thereby exhausting the cells and limiting antitumor responses. Interestingly, it was further shown that 4-1BB was superior to CD28 in alleviating exhaustion from this tonic signaling (76). In addition, caution should be taken in the use of scFvs of murine origin, as human-anti-mouse Ab responses can deplete the transferred CAR T-cells, and even result in patient death by anaphylactic shock following multiple infusions (77, 78). Interestingly, similar to TCRs, increasing receptor-binding strength can augment T-cell function, but there is an affinity threshold beyond which there is no further gain in activity, and target density impacts CAR T-cell activation (79–81). Along with scFv, other tumor-binding moieties including an anti-integrin peptide α5β6 (82), heregulin (83), interleukin 13-zetakine (84), NKG2D (85), vasculature endothelial growth factor (VEGF) (86), and TCRs (87) have been incorporated into functional CARs. Finally, universal CARs, including an avidin ectodomain (88) and an anti-FITC scFv (89) for recognizing targets bound by biotinylated and FITC-labeled mAbs, respectively, have been developed.
CAR T-Cell Safety and Next-Generation Receptor Design
Chimeric antigen receptor T-cells are a potent living drug and a primary consideration in their development is the choice of target antigen. Ideally, it is highly expressed on the tumor and not at all on normal cells. Currently, there are about 30 solid tumor antigens being evaluated for CAR therapy including mesothelin, carcinoembryonic antigen (CEA), the diganglioside GD2, interleukin 13 receptor alpha (IL13Ra), human epidermal growth factor 2 (HER-2), fibroblast-activating protein (FAP), and L1 cell adhesion molecule (L1CAM) (90, 91). Unfortunately, there have been instances of severe on-target/off-site toxicity (92) such as a HER-2 CAR that may have caused patient death via reactivity with low levels of cognate antigen expressed on lung epithelium (93). One approach to circumvent this is to use a lower affinity scFv such that the CAR T-cells are only activated in the presence of high cell-surface expression of antigen (i.e., on the tumor cells only) (94–96). Or one can target an antigen that is tumor restricted, such as epidermal growth factor receptor variant III (EGFRvIII) (97), but there are few such examples.
In recent years various novel, next-generation engineering strategies have been devised to improve CAR T-cell safety. For example, the signaling can be split by coexpressing two CARs, one incorporating CD3ζ and the other the costimulatory endodomain, such that the T-cell will only be fully activated when both receptors are engaged (98, 99). Bispecific tandem CARs (TanCARs; the extracellular domain of one receptor can engage two distinct antigens) (100) have also been developed that synergistically enhance T-cell activity levels when coengaged. More recently, novel synthetic Notch-based receptors have been designed that enable combinatorial activation of T-cells—binding by the Notch-based CAR upregulates expression of the second CAR (101, 102). In addition, innovative ON-Switch/Remote Control CARs have been developed that restrict T-cell activation to tumor cell antigen encounter in the presence of a remotely provided heterodimerizing small molecule that links the antigen-binding receptor with intracellular components that initiate signaling (103). Finally, it is also possible to gene-modify CAR T-cells with various suicide genes or safety switches such as inducible caspase 9 to cause rapid T-cell destruction in the case of an adverse reaction in a treated patient (104, 105). In the event of CAR T-cell toxicity, such as cytokine release syndrome (CRS), early and aggressive supportive patient care is critical. The current mainstay treatment for CRS is IL-6 receptor blockade with the monoclonal Ab tocilizumab, but in the case of neurologic toxicities corticosteroids are employed (106).
CAR T-Cell Activity As a Function of Receptor Design
The distance between a T-cell and its target antigen-presenting cell (APC) of approximately 15 nm is dictated by the TCR–pMHC interaction and is critical for the exclusion of large phosphatases such as CD45 and CD148 to the periphery of the immune synapse (107), thereby enabling TCR clustering and triggering (1). The precise mechanism by which CARs activate T-cells has not been fully elucidated, but the hinge/spacer, which governs the spatial distance between a CAR T-cell and its target, can significantly impact function (108, 109). Various hinge/spacers have been incorporated into CARs including regions from CD8α and CD4, as well as CH2–CH3 from the Fc domains of IgG1 and IgG4. In the case of CH2 spacers derived from IgG4, sequence modification is required to prevent Fc receptor binding by myeloid cells that can cause activation-induced T-cell death (AICD) (110, 111). Most CARs comprise a TM domain from a type I membrane protein such as CD4, CD8, CD3ζ, or CD28. Interestingly, it was demonstrated that CARs comprising a CD3ζ TM domain engage endogenous TCR–CD3 complexes for optimal activity (112). CARs can function, however, in the absence of endogenous TCR (113), and important efforts are being undertaken to develop universal allogeneic T-cell donors for immunotherapy (114).
In general, distal membrane epitopes require a shorter hinge/spacer, whereas an epitope that is closer to the tumor cell membrane may necessitate a longer and more flexible one to facilitate scFv binding and optimal T-cell activity (115). Recently, a Strep-tag II has been used to modify spacer length and at the same time provide a means of identifying and rapidly purifying CAR-engineered T-cells, during both the manufacturing process and patient monitoring (116). A notable study comparing three L1CAM CARs varying only in hinge/spacer length (short, medium, and long) revealed that the CAR conferring the highest in vitro function (long; IgG4 hinge–CH2–CH3 spacer) fared poorly in vivo, and, conversely, the weakest CAR in vitro (short; IgG4 hinge) performed the best in tumor-bearing mice. The authors hypothesized that the limited duration of CAR signaling required for in vitro assays does not reflect the recursive rounds of activation needed to eradicate a tumor in vivo. Thus, they set up a coculture stress test for which CAR T-cells were repeatedly harvested and transferred to culture dishes seeded with fresh tumor cells. Interestingly, after three rounds, the CAR T-cells bearing the long linker (i.e., conferring the best activity in round 1) underwent the highest level of AICD as a result of upregulated FasL–Fas interactions (110). This work highlights a disaccord between T-cell responses observed in standard in vitro testing versus in vivo challenges. Establishing in vitro T-cell parameters such as receptor affinity/kinetics and two-dimensional interactions (117), functionality upon stress testing (110), gene expression profiles upon activation, etc., that correlate to maximum antitumor responses in vivo is an important area of research for the efficient screening of new leads for preclinical and clinical testing.
Optimal T-Cell Subsets for CAR Activity
Another important factor governing CAR T-cell activity in vivo is the subset of input T-cells used. Based on their differentiation and level of maturity, T-cells are presently classified into naïve (TN), and four main activated subtypes: stem cell memory (TSCM), central memory (TCM), effector memory (TEM), and terminally differentiated effector cells (TEFF) (118). For both mouse and humans, it has been demonstrated that the less differentiated subsets (TSCM and TCM) display better expansion, persistence, and antitumor activity in vivo (119–122). The phenotypic and functional properties of human CD8+ TN versus the different memory subsets are illustrated in Figure 2. Retrospective studies from ACT trials have correlated objective clinical responses in patients with the transfer of less differentiated T-cells (123, 124). A recent analysis of CD19 CAR-treated patients revealed a correlation between in vivo expansion and the level of infused TSCM phenotype (125). There are also studies demonstrating better antitumor responses when both CD4+ and CD8+ CAR T-cells are transferred (126), and it has proven beneficial to gene-engineer virus-specific memory T-cells that can persist long term, presumably via TCR-mediated survival signals and the quality of the original TCR-mediated priming of the cell (127). Although T-cell metabolism has long been an active field of study due to the remarkable cellular rewiring required to accommodate changes in energetic requirements when naïve, quiescent T-cells encounter and respond to cognate pMHC, it is only recently that it has come to the forefront of cancer research.
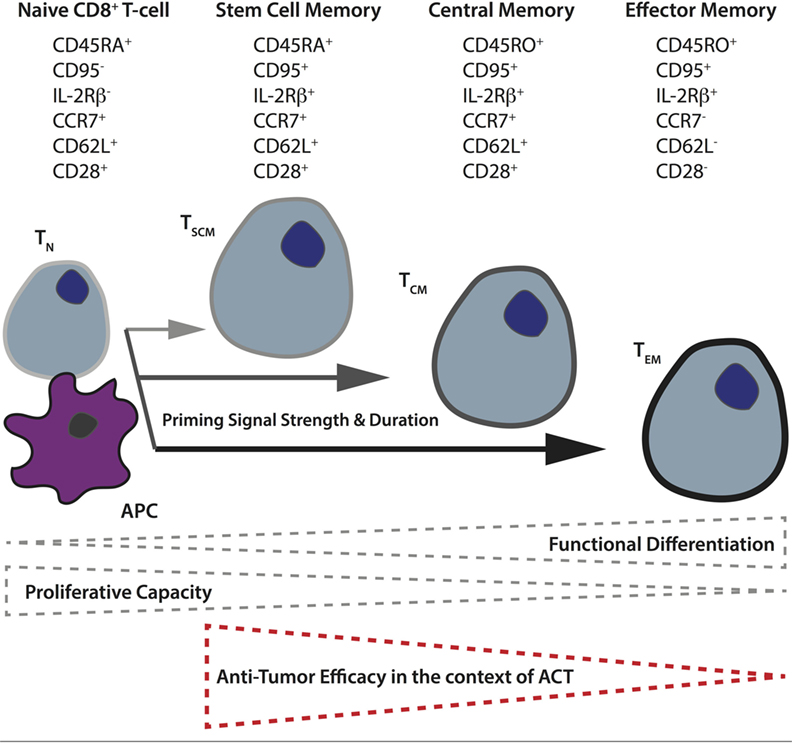
Figure 2. Differentiation of naïve T-cells to memory subsets upon priming in secondary lymphoid organs. The differentiation state of T-cells depends upon the strength and duration of priming naïve T-cells receive from antigen-presenting cells in the secondary lymphoid organs. In general, the more differentiated the T-cell, as a result of more robust priming, the lower its proliferative and self-renewal capacity, and the poorer its antitumor control in the context of adoptive T-cell therapy. The different memory T-cell subsets can be distinguished based on their expression of various cell-surface markers including the tyrosine phosphatase CD45 (full length = CD45RA, truncated form = CD45RO), the lymph node homing receptors, chemokine receptor 7, and CD62 ligand, as well as CD95 (Fas receptor), interleukin-2 receptor β-chain, and the costimulatory receptor CD28.
Interplay Between the Metabolic Activity of T-Cells and Function
Metabolic Activity of Naïve T-Cells
All T-cells use glucose as their primary source of fuel for the generation of adenosine triphosphate (ATP), but there are important differences in the metabolic requirements and pathways used by naïve, activated, and memory T-cells, which correspond to their specific functional state (128). TN cells are quiescent and have limited biosynthetic needs because their major role is to circulate the host with the aim of being primed by an APC. Quiescent T-cells, comprising both naïve and memory T-cells, rely on catabolic metabolism, whereby nutrients including glucose, fatty acids, and amino acids are broken down for fuel (128). Indeed, quiescent T-cells mainly undertake oxidative phosphorylation (OXPHOS), a process that takes place in the mitochondria involving the oxidation of substrates in the tricarboxylic acid (TCA) cycle to generate ATP (129) (summarized in Figure 3A). The TCA cycle itself is a series of chemical reactions that use metabolic substrates to produce reducing agents such as the coenzyme reduced nicotinamide adenine dinucleotide (NADH), which donate electrons to the electron transport chain. Up to 36 molecules of ATP can be generated by OXPHOS per glucose molecule.
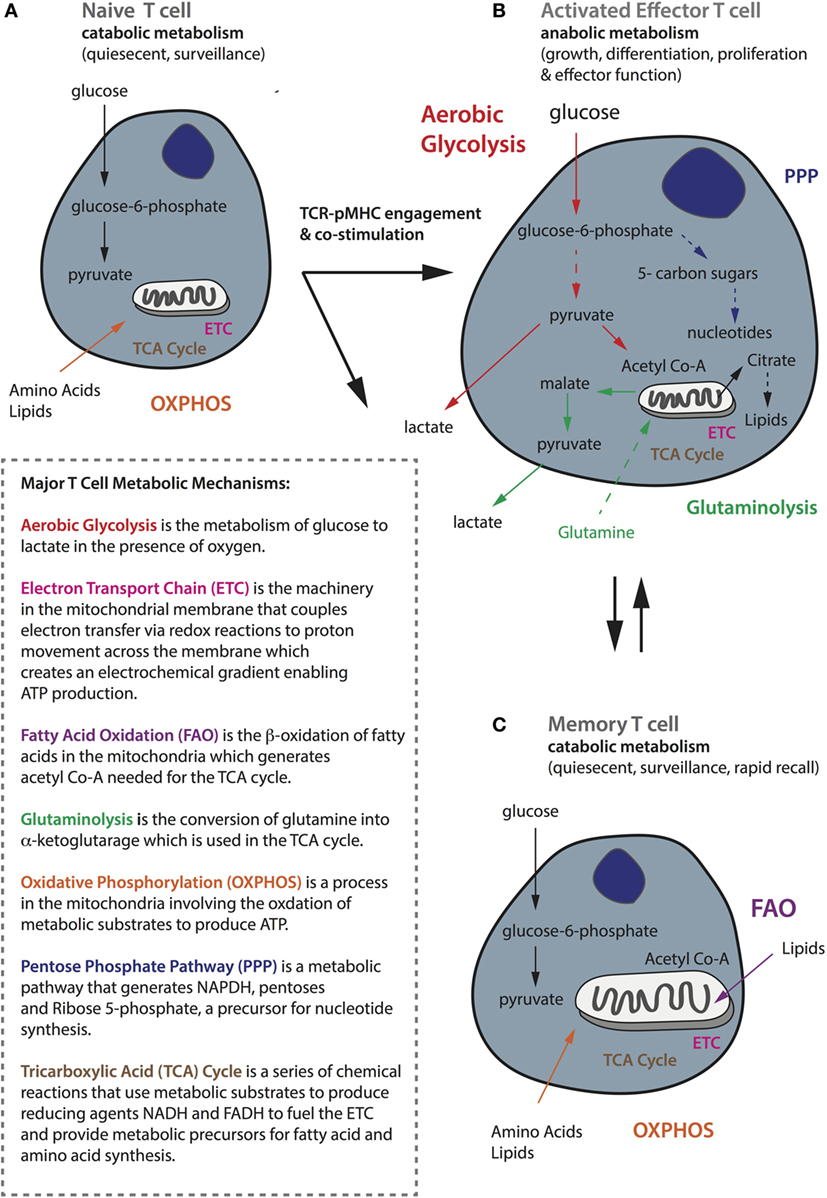
Figure 3. General metabolic profiles of naïve, activated, and memory T-cells. (A) Naïve T-cells are quiescent and perform catabolic metabolism to accommodate basic energy requirements. (B) Activated effector T-cells, whereas switch to anabolic metabolism to meet increasing energy demands as well as for biosynthesis to support functionality including proliferation, growth, and cytokine secretion. (C) Memory cells, like naïve cells, are quiescent and perform metabolic catabolism. While naïve and memory T-cells mostly rely on oxidative phosphorylation (shown in orange), activated ones are metabolically rewired to upregulate aerobic glycolysis (shown in red). Activated T-cells also require glutaminolysis (in green) and the pentose phosphate pathway (in navy blue) to support their activities. Memory T-cells rely on fatty acid oxidation (in purple) and they have increased mitochondrial mass and maintain substantial spare respiratory capacity to enable survival and rapid recall to antigen challenge. A brief summary of the major metabolic mechanisms is described in the inlet.
Metabolic Activity of Effector T-Cells
Upon productive TCR–pMHC engagement and costimulation, naïve T-cells become activated and undergo extensive metabolic rewiring causing them to enlarge in size, and enabling extensive proliferation and the acquisition of effector functions such as cytokine secretion (130). Activated T-cells switch to anabolic metabolism meaning that nutrients are used to construct molecular building blocks. This transition is associated with mTOR induction, and the expression of the transcription factors Myc and hypoxia-inducible factor-1α (HIF-1α) (131, 132). Although activated T-cells increase mitochondrial OXPHOS and reactive oxygen species production, they also upregulate and rely heavily upon aerobic glycolysis (133) to meet their metabolic needs (134, 135) (Figure 3B). Upon T-cell activation, there is an increase in the expression of the glucose receptor Glut1, as well as glycolytic enzymes to enable increased import of glucose that is converted to glucose-6-phosphate and eventually pyruvate that is finally secreted in the form of lactate. It has recently been demonstrated that T-cells activated under hypoxic conditions upregulate significantly higher levels of Glut1 than under atmospheric oxygen (136), but the reactivation of CD8+ T-cells under hypoxia has also been shown to switch them toward a poorly proliferative and IL-10 secreting phenotype (137). This phenomenon of aerobically fermenting glucose to lactate, despite sufficient oxygen to support OXPHOS, a process also utilized by tumor cells, which themselves are heavy consumers of glucose, is known as the Warburg effect (138, 139).
Aerobic glycolysis is energetically less efficient than OXPHOS, generating only two molecules of ATP per glucose molecule, but the process is rapid and in addition supplies a critical source of metabolic intermediates needed for the synthesis of nucleic acids, proteins, carbohydrates, and lipids, and it provides a means of maintaining the NAD+-NADH redox balance (138). Glycolytic metabolites also help to sustain effector functions through transcriptional and translation regulation (59). For example, glyceraldehyde-3-phosphate promotes IFNγ production by relieving its restraint by glyceraldehyde-3-phosphate dehydrogenase (140). In addition, Ho et al. demonstrated that phosphophenolpyruvate (141) accumulation can help sustain the Ca2+-NFAT pathway, which controls the production of effector molecules, by inhibiting ER Ca2+ reuptake. It has been shown that activated T-cells can switch between OXPHOS and aerobic glycolysis depending on their environment, but glycolysis is required for full effector function—if activated T-cells revert to OXPHOS it will be at the expense of IL-2 and IFNγ production (140). In addition to glucose, activated T-cells rely upon an extracellular supply of glutamine to replenish intermediates for the TCA cycle through the process of glutaminolysis, and this can also contribute to the citrate pool used for lipid synthesis by reductive carboxylation (142, 143). Finally, activated T-cells decrease fatty acid oxidation (FAO), decrease pyruvate flux into the TCA cycle, and increase glucose flux into the pentose phosphate pathway to produce nucleotides for DNA synthesis (Figure 3B).
Metabolic Activity of Memory T-Cells
By contrast, memory T-cells transition from aerobic glycolysis to OXPHOS, fueled in part by the catabolism of intracellular fatty acids in the mitochondria (144–146). Furthermore, memory T-cells have increased mitochondrial mass and activity (147, 148) and upregulate mitochondrial biogenesis to build substantial spare respiratory capacity (SRC), thereby enabling both survival and rapid recall to antigen challenge (130) (Figure 3C). Interestingly, memory T-cells activated in vitro increase aerobic glycolysis, and consequently IFNγ production, more rapidly than their naive counterparts. How this relates to their capacity to compete for glucose in the TME remains to be established (149). Moreover, memory T-cells can potentially better adapt to a nutrient crisis because they have healthier mitochondria (147). AMP-activated protein kinase (AMPK) is another important metabolic regulator in T-cells that senses a high AMP to ATP ratio and can promote catabolic pathways and conservation of energy during metabolic stress. AMPK is important for memory T-cell development (150); treating activated CD8+ T-cells with either rapamycin (inhibits mTOR) or metformin (activates AMPK) to augment catabolic pathways enhances CD8+ memory formation (151, 152).
Immunometabolism and T-Cell Therapy
It has been observed that several genes involved in metabolism are downregulated in exhausted T-cells (153). Moreover, blocking leucine or glucose metabolism during T-cell activation leads to an anergic phenotype (154). Mechanistically, leucine can stimulate mTOR via leucyl-tRNA synthetase and hence low leucine levels may impair mTOR activation (155, 156). It has also been demonstrated that both PD-1 and CTLA-4 ligation inhibits glycolysis in activated T-cells but through distinct mechanisms; CTLA-4 inhibits Akt via the serine–threonine phosphatase PP2A, while PD-1 inhibits Akt phosphorylation by blocking CD28-mediated activation of PI3K (157). PD-1 signaling also blocks amino acid metabolism but promotes fatty acid β-oxidation (158). With an increased appreciation for the high competition for nutrients including glucose in the TME which can promote tumor progression, as well as the dynamic interplay between T-cell phenotype, metabolism, and immune checkpoint, it is clear that an important consideration in the development of CAR T-cells is how their culture prior to transfer, as well as their design, affects their metabolic profile.
Traditionally, T-cells for ACT are cultured with high doses of IL-2, followed by a rapid expansion protocol comprising agonistic anti-CD3 Ab and allogeneic feeder cells (159). However, it is now known that TCR signaling coupled with high-dose IL-2 drives TEFF differentiation (160) which is not an ideal phenotype for ACT. Alternative common gamma chain (γc) signaling cytokines have also been assessed for T-cell culture. Naïve mouse T-cells cultured in the presence of IL-15 (145) or IL-21 (161), for example, acquire phenotypic, functional, and metabolic properties of naturally occurring TCM cells. Moreover, IL-15 cultured murine T-cells confer superior in vivo antitumor activity than ones cultured in IL-2 (162). Similarly, human T-cells cultured with artificial APCs and IL-15 exhibit a TCM phenotype, demonstrate clonotypic persistence, and can mediate objective clinical responses upon transfer (163, 164). Human T-cells cultured in IL-21 also maintain a minimally differentiated profile (161, 165). Interestingly, it has been demonstrated that IL-15 regulates SRC and oxidative metabolism by mitochondrial biogenesis as well as the expression of carnitine palmitoyl transferase, a metabolic enzyme involved in the rate-limiting step in FAO (145). IL-7 also enhances T-cell survival in a metabolically driven manner by Glut1 trafficking via STAT5 and Akt (166), as well as by inducing glycerol transport (via AQP9) and triglyceride synthesis (167). Finally, metabolically robust T-cells isolated with the lipophilic cationic dye tetramethylrhodamine methyl ester (TMRM; staining can be used to distinguish mitochondrial membrane potential) conferred enhanced persistence and tumor eradication upon ACT (168).
A T-cell’s commitment between a memory versus effector phenotype is governed by its metabolic state (145, 152, 169), which in turn is controlled by stimuli from TCR, costimulatory and cytokine receptors that converge at common development, and differentiation signal transduction pathways including PI3K/Akt/mTOR and Wnt/β-catenin (130). Thus, small molecule modulators of these pathways have been assessed for the in vitro culture of T-cells for ACT. Promotion of the canonical Wnt/β-catenin pathway with the GSK3β inhibitor TWS119, for example, favors the formation of TCM and TSCM cells with improved in vivo antitumor responses as compared to untreated cells (170). Similarly, Akt inhibition during ex vivo priming and expansion of T-cells favors TSCM-like cells having higher rates of OXPHOS and FAO and enabling enhanced in vivo tumor control (171). Another consideration in T-cell culture for ACT is glucose concentration in the media. Although media often comprise about 5.5 mM glucose (similar to that of blood), many used for ACT are in the range of 10–25 mM—this may program high dependency on glucose and thereby further impair T-cell responses in the nutrient-deprived TME (169, 172). Interestingly, it has been shown that the activation of CD8+ T-cells in the presence of 2-deoxyglucose, an inhibitor of glycolysis, enhances memory generation and antitumor responses (169). Several amino acids have been implicated in immunomodulation including cysteine, glutamine, phenylalanine, tryptophan, and arginine (173). It has been observed that low arginine, for example, induces the loss of CD3ζ and inhibits proliferation and cytokine production by T-cells, but that this state can be reversed by exposure to excess arginine (174). Recently, it has been further demonstrated that the culture of murine T-cells with elevated l-arginine promotes the generation of TCM-like cells, shifts the metabolic profile from glycolysis to OXPHOS, and endows the T-cells with enhanced antitumor activity in vivo (175).
CAR Endodomains and Metabolism
As previously mentioned, in recent years, there have been important clinical responses to CD19 CAR T-cell therapy against hematological tumors including ALL, chronic lymphocytic leukemia (CLL), and diffuse large B cell lymphoma, using second-generation receptors comprising either CD28 or 4-1BB [reviewed in Ref. (44)]. It is oftentimes difficult to compare the success of trials because of differences in the study design, including the scFv used, the gene transfer protocols, and interventions undertaken post-transfer. Initial clinical response rates against ALL have been the same for CD19 CAR trials incorporating either CD28 or 4-1BB (45, 176, 177). In the case of CLL, however, 4-1BB-based CAR T-cells appear superior (178), demonstrating persistence of greater than 4 years in some patients versus 30 days in the case of CD28 (179). Signaling pathways induced by CD28 versus 4-1BB, a member of the tumor-necrosis receptor family, are distinct. While CD28 activates the PI3K/Akt pathway that can enhance glycolysis (180), 4-1BB has been linked to long-term survival of T-cells (181). Recently, the metabolic pathways induced by the CD28 versus the 4-1BB costimulatory endodomain of second-generation CD19 CAR T-cells were described for the first time (182). T-cells engineered with a 4-1BB-bearing CAR had an increased frequency of TCM phenotype, mitochondrial biogenesis, and oxidative metabolism upon activation, and had greater survival, as compared to the CD28-based CAR T-cells that had enhanced aerobic glycolysis and a predominantly TEM phenotype (Figure 1). This work has important implications in the choice of CAR costimulatory endodomains for targeting different solid TMEs. For example, in the case of a tumor such as BRAF V600E melanoma, which has a high glycolytic rate (183), it may be prudent to develop a CAR incorporating 4-1BB. Pretreatment with a BRAF inhibitor may also help in restoring glucose levels prior to ACT for improved T-cell activity in the TME. Alternatively, it may be beneficial to transfer both CD28 and 4-1BB-based CAR T-cells for both immediate effector and persistent antitumor activity.
Armored CAR T-Cells and Combination Therapies to Overcome Immunometabolic Obstacles in Solid Tumors
Immunometabolic Obstacles in Solid Tumors
The clinical efficacy of CAR T-cells against solid tumors remains to be proven (184). The two most successful trials to date involve HER-2 CAR T-cells against sarcoma, for which 4/17 patients showed stable disease (185), and GD2 CAR T-cells to treat neuroblastoma, for which 3/11 patients underwent a complete response (186). There are various physical and physiological hurdles faced by T-cells in the context of solid tumors (46). To begin with, T-cells must successfully, (i) home to the tumor bed, often in the face of mismatches between T-cell chemokine receptors and chemokines present in the TME. Furthermore, T-cells must migrate along, (ii) an aberrant vasculature that is not conducive to transendothelial migration of T-cells due to downregulation of adhesion molecules (ICAM-1) (42) and the upregulation of FasL (187), etc., and they can encounter, (iii) additional barriers in the stroma, including a dense collagen matrix and suppressive cancer-associated fibroblasts (47). If T-cells are successful in penetrating the tumor bed, there they can face a battery of obstacles, including (iv) suppressive immune infiltrate comprising T regulatory cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, tumor-associated neutrophils (188), and immature DCs (189), (v) a range of suppressive molecules such as transforming growth factor beta (TGFβ) VEGF and adenosine, (vi) suppressive ligands including PD-L1/L2, VISTA, and FasL (190), (vii) competition for, and downregulation of, costimulatory ligands such as CD80/86, and (viii) T-cell-intrinsic regulatory mechanisms including PD-1 and CTLA-4 upregulation, and ultimately exhaustion (191) or anergy (192). Finally, (ix) the T-cells must function in an environment that is acidic, hypoxic (193), nutritionally depleted (194) and comprising toxic metabolic by-products such as lactic acid, glutamate, and ketone bodies (195) (summarized in Figure 4).
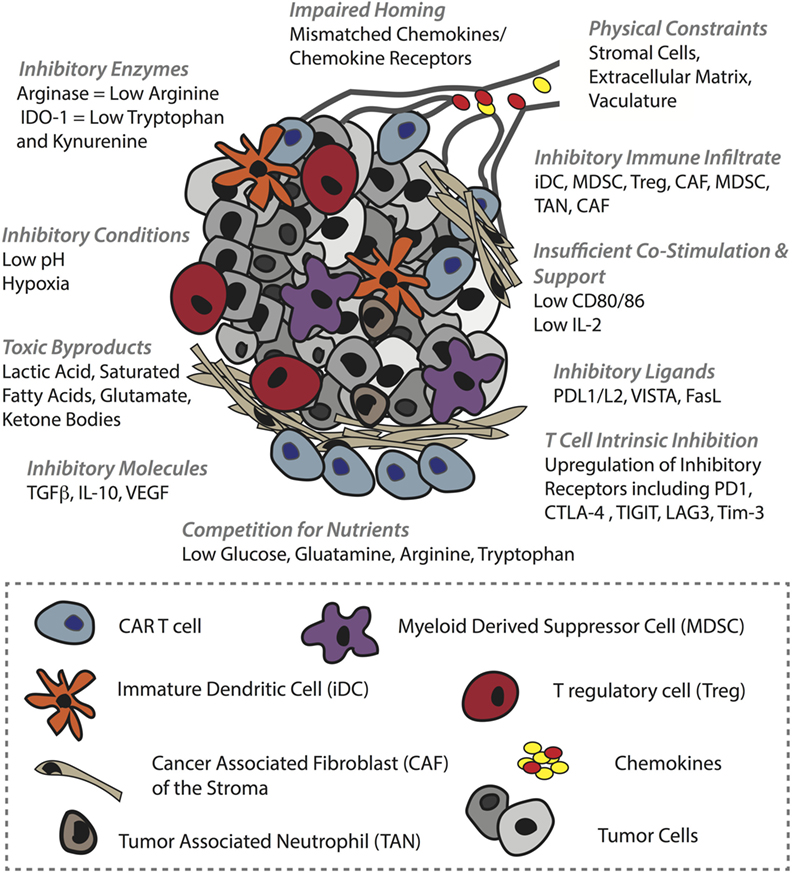
Figure 4. Barriers in the solid tumor microenvironment (TME) that can hinder chimeric antigen receptor (CAR) T-cell activity. In order to mediate an antitumor response, CAR T-cells must first successfully home to and penetrate the tumor bed. Obstacles to these events include tumor chemokine and T-cell chemokine receptor mismatches, and various inhibitory mechanisms in the vasculature such as ICAM-1 downregulation. In the stroma, T-cells can be physically blocked by dense extracellular matrix and by inhibitory cancer-associated fibroblasts. The TME itself is also hostile, comprising a range of inhibitory immune cells, inhibitory receptors such as PD-L1 on tumor cells and immune infiltrate alike, and soluble molecules such as TGFβ that impair T-cell activity (some examples are shown). In addition, the TME is typically acidic, hypoxic, full of toxic metabolites, and nutrient depleted which can inhibit not only T-cell activity but also other immune infiltrate such as DCs that lose their ability to mature and provide support to T-cells. Moreover, some of these conditions promote protumoral immune cells.
CAR T-Cell Coengineering and Combinatorial Therapy for Tumor Homing and Migration into the Tumor Bed
The predominant chemokine receptor mediating effector T-cell recruitment to tumors is CXCR3 via chemokines CXCL9 and CXCL10 (196). Chemokines commonly secreted in TMEs, however, rather than attracting cytotoxic T-cells often recruit inhibitory immune cells such as Tregs and MDSCs, whose presence is associated with poor patient prognosis (197). The chemokine CCL22, for example, present in breast and prostate cancer, mediates CCR4-dependent Treg trafficking (197, 198), while hypoxia-dependent expression of the chemokine ligand CCL28 in ovarian cancer recruits Tregs via CCR10 (199), and in pancreatic cancer the upregulation of CCL5 induces the migration of CCR5-expressing Tregs (200). Tregs are potent inhibitors of CD8+ T-cells as they compete for IL-2 (201), generate adenosine by CD39/CD73 (202), and via CTLA-4 downregulate CD80/CD86 on DCs that are needed by T-cells for costimulation (203) while upregulating indoleamine 2,3-dioxygenase-1 (IDO-1) in DCs (204, 205). MDSCs can be recruited to different TMEs by CCL2, CXCL5, CXCL12, and stem cell factor (206). They also inhibit tumor immunity by various mechanisms including the expression of arginase, TGFβ, cyclooxygenase 2 that controls prostaglandin E2 (PGE2) production (207) (a powerful repressor of TCR signaling), and IL-10. In addition, MDSCs can sequester cysteine and induce Tregs (188). One approach to direct CAR T-cells (Figure 5A) toward a tumor that does not express CXCL9 or CXCL10 is to coengineer them with a borrowed chemokine receptor (Figure 5B). For example, CXCR2-engineered T-cells (208, 209) demonstrated improved localization and control of melanoma tumors expressing CXCL1 and CXCL8, chemokines that enable the migration of CXCR2+ monocytes.
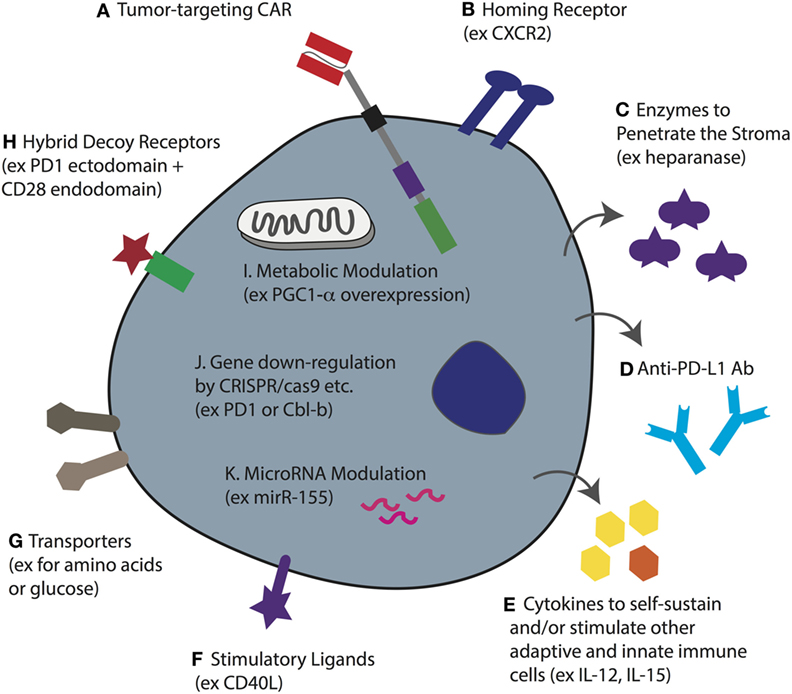
Figure 5. T-cell coengineering strategies to target and attack solid tumors, and overcome immunometabolic barriers. Human T-cells, ideally minimally differentiated, can be gene-engineered ex vivo in various ways including transient RNA electroporation and stable genome integration by lentiviral transduction and sleeping beauty. (A) To enable tumor specificity, the T-cell can be engineered with a chimeric antigen receptor (CAR) directed against a surface-expressed tumor antigen (or a TCR against a target pMHC). (B) To enable tumor homing the T-cell can be coengineered to express a specific chemokine receptor that matches the chemokine profile of the tumor being targeted. (C) To better penetrate dense stroma CAR T-cells can be coengineered to secrete heparanase. (D) Instead of injecting the patient with checkpoint blockade monoclonal antibodies (mAbs), the T-cells can be gene engineered to secrete anti-PD ligand 1 (PD-L1) mAb. (E) The tumor microenvironment (TME) often lacks T-cell promoting cytokines, either because they are not produced, or because they are competitively consumed, such as IL-2 by T regulatory cells. Thus, CAR T-cells can be coengineered to forcibly secrete various cytokines that can serve not only to self-sustain the CAR T-cell but also to stimulate other adaptive and innate immune cells in the TME. (F) Forced expression of the stimulatory ligand CD40L will help activate antigen-presenting cells as well as upregulate adhesion molecules on endothelial cells. (G) The upregulation of nutrient transporters for glucose or amino acids may help to increase T-cell competition for limited resources in the TME. (H) Hybrid receptors such as ones comprising the extracellular domain of PD-1 fused to the endodomain of CD28 can turn a negative signal (i.e., from PD-L1) into one that costimulates the cell. This may be further improved by designing a higher affinity variant of PD-1. (I) The overexpression of intracellular proteins such as the transcription coactivator PGC1-α, can help to reverse metabolic exhaustion. (J) Using various techniques including CRISPR/cas9, it is possible to knockout gene expression such as of PD-1 and the master regulator Cbl-b to enhance T-cell activity in the TME. (K) microRNAs play an important role in T-cell activity and the overexpression of miR-155, for example, can be used to enhance sensitivity to homeostatic γc cytokines. Note that some of these approaches have yet to be demonstrated for engineered CAR T-cells.
In order to tackle the stroma, FAP-directed CAR T-cells have been developed, and in murine tumor models have been shown to slow tumor growth (49). The observation that in vitro cultured T-cells downregulate heparanase, an enzyme that is required for the degradation of heparin sulfate proteoglycans, the primary component of the extracellular matrix of tumor stroma, led to the development of CAR T-cells coengineered to secrete it (210) (Figure 5C). Such CAR T-cells may be potent against stroma-rich solid tumors. Various approaches can be taken to normalize the tumor vasculature (42). For example, blocking endothelin B receptor (211), and the pharmacologic inhibition of VEGF and PGE2 to attenuate FasL expression, enables enhanced CD8+ T-cell influx and tumor control (187). In addition, CAR T-cells that disrupt the tumor vasculature and mediate tumor regression have been developed (212).
CAR T-Cell Coengineering and Combinatorial Therapy for Overcoming Immunometabolic Challenges in the Tumor Bed
The solid TME is hostile for effector T-cells. As a result of high aerobic glycolysis by tumor cells, as well as the fact that nutrients and oxygen must be supplied, and waste removed, by an aberrant vasculature, tumors are typically nutrient depleted, hypoxic, acidic, and toxic. As previously described, low glucose levels and a lack of critical amino acids such as leucine and arginine will alter T-cell metabolism and directly impair their function (156, 213, 214). Gene-engineering approaches to render CAR T-cells more competitive in nutrient acquisition, such as by overexpressing transporters (Figure 5G), or to rewire their metabolism, may improve their activity in solid tumors. For example, overexpression of PPAR-gamma coactivator 1-α (PGC1-α), a transcriptional coactivator involved in mitochondrial biogenesis, could in part reverse metabolic exhaustion (decreased mitochondrial mass and function induced by chronic Akt signaling) of T-cells in the TME (215) (Figure 5I). Interestingly, modulating cholesterol metabolism can enhance antitumor response of CD8+ T-cells; both pharmacological inhibition and gene knockdown of the cholesterol esterification enzyme ACAT1 increased plasma membrane concentration of cholesterol, enabling more efficient immune synapse formation, TCR clustering, and enhanced signaling (216). Whether such strategies would also augment CAR T-cell activity remains to be determined.
Various other immunometabolic gene-engineering strategies have been proposed for increasing the activity of TCR- or CAR-engineered T-cells in the TME. With respect to the PD-1/PD-L1 checkpoint blockade axis, at least three different approaches have been taken. PD-1 has been knocked down in T-cells (217, 218), hybrid receptors comprising the ectodomain of PD-1 and the endodomain of CD28 have been expressed to divert PD-L1 binding toward costimulatory intracellular signaling (219), and CAR T-cells have been engineered to secrete anti-PD-L1 Abs (220), all of which have been reported to increase antitumor responses (Figures 5D,H,J). Others have knocked-down master regulators of T-cell activity such as the E3 ubiquitin ligase Cbl-b (Figure 5J) and have shown enhanced antitumor T-cell responses (221, 222). microRNAs, such as miR155, have also been manipulated for enhanced tumor control (223, 224) (Figure 5K), and T-cells have been gene engineered to overexpress cytokines including IL-12 (225) and IL-15 (226) (Figure 5E) for improved activity. The advantage of secreted molecules is that they can support not only the T-cell that produces them but also endogenous immune cells in the TME. As a final example, coengineering CAR T-cells to constitutively express CD40L demonstrated enhanced T-cell proliferation and secretion of pro-inflammatory cytokines (Figure 5F). The CD40L+ CAR T-cells also increased the immunogenicity of CD40+ tumor cells through the upregulation of costimulatory, adhesion, and human leukocyte antigen molecules, as well as the Fas death receptor, and they induced the maturation and secretion of IL-12 by monocyte-derived DCs (58).
The TME can also reprogram other immune infiltrate to the detriment of T-cell activity. For example, the maturation, function, and phenotype of DCs can be impaired by VEGF (227), IL-6, macrophage colony-stimulating factor (228), and TGFβ (229). In addition, PD-L1 expression by DCs can be induced by IL-10 and VEGF (230). It was also recently demonstrated that HIFα expression [the main transcriptional factors responding to limited oxygen supply (231)] elevates miR-210 in MDSCs, which in turn increases both arginase activity and the production of nitric oxide (232). Along with hypoxia (233), lactate, a major byproduct of aerobic glycolysis in tumor cells, can also directly and indirectly inhibit cytotoxic T lymphocyte activity. Indeed, high lactate in the TME can block the export of endogenous lactate produced by aerobic glycolysis in T-cells via the gradient-dependent transporter monocarboxylate transporter-1, and thereby disturb T-cell metabolism (234). Lactic acid can also promote M2-polarization and expression of arginase-1 by HIF-1α stabilization (235). Moreover, hypoxia, via HIF-1, can induce glycolysis as well as a switch from glucose to glutamine as the major substrate for FA synthesis in tumor cells (236), further depleting the TME of vital nutrients needed for T-cell function. Thus, pretreatment of tumors with inhibitors of either HIF-1 or metabolic enzymes could potentially impair the metabolic flexibility of cancer cells and inhibitory immune infiltrate, thus rendering tumors more sensitive to CAR T-cell transfer—the CAR T-cells will benefit from entering a more nutrient replete and less aggressive/suppressive TME. Alternatively, metabolic drugs could be targeted to tumors with Abs, or the pharmacologic inhibitors could be designed in such a way that they are preferentially taken up by tumor cells.
A range of other strategies can be used to pretreat or cotreat tumors for enhanced responses to CAR T-cell therapy. Localized RT, for example, can reprogram the TME by various mechanisms including by inducing immunogenic cell death (52, 237), supporting T-cell trafficking to the tumor (238), and by promoting the polarization of macrophages (239) from a suppressive M2 (240) to M1 phenotype (241). An inhibitor of DNA methyltransferase 1 (catalyzes the methylation of genetic loci), 5-aza-2′-deoxycytidine, has been used to enhance expression of the epigenetically silenced chemokines CXCL9 and CXCL10 in the TME and thereby promote T-cell infiltration and responses to checkpoint blockade (242). CAR T-cells expressing CXCR3 could benefit from such treatment. Cyclophosphamide, such as RT, can be used not only to direct tumor cell destruction but also to deplete Tregs from the TME and thereby enhance responses to immunotherapy (51). As a final example, IDO-1 inhibition is a powerful approach for promoting tumor immunity. IDO-1, the rate-limiting enzyme involved in the conversion of the essential amino acid tryptophan to its catabolic product kynurenine (Kyn) (204, 243), can be upregulated in DCs, by Tregs as mentioned above, as well as by tumor cells and myeloid cells in response to IFNγ (244, 245), and has been associated with poor prognosis for several types of cancer including ovarian (246), endometrial (247), colorectal (248), and lung (249). The depletion of tryptophan in the TME activates stress response kinases in T-cells, including general control non-depressing 2 (GCN2), which detects uncharged tRNAs, and ultimately blocks T-cell proliferation and triggers the caspase pathway (250, 251). In addition, Kyn binds the arylhydrocarbon receptor, a ligand-activated transcription factor that promotes the polarization of naïve T-cells toward a Treg phenotype (251). Thus, IDO-1 inhibition promotes tryptophan availability for effector T-cells and limits Tregs in the TME.
Concluding Remarks
Metabolism is an important driver of cancer progression that must be addressed in the context of CAR T-cell immunotherapy to improve clinical responses against solid tumors. High levels of aerobic glycolysis by tumor cells leads to an accumulation of metabolic by-products that, along with oxygen deprivation and low pH, can drive protumoral activity of various immune cells, including Tregs and M2 macrophages, as well as directly inhibit effector T-cell function. In addition, competition for critical nutrients, such as the amino acids tryptophan, glutamine, and arginine, as well as glucose, all contribute to the suppression of T-cell activity. Checkpoint pathways are intimately linked with the metabolic status of both tumor cells and T-cells; non-responsiveness to anti-PD-1 mAb has been linked to the upregulation of a metabolic gene-signature in tumors (35), whereas successful PD-L1 blockade has been demonstrated to block tumor cell glycolysis, thereby enhancing T-cell fitness (34).
Immunometabolic barriers can be targeted therapeutically prior to and/or during ACT to enhance responses to CAR T-cell therapy and to support endogenous immunity. In addition, CAR T-cells can be optimally designed based on the metabolic properties of the tumor being targeted and cultured to promote a less differentiated, long-lived phenotype that can efficiently self-renew and differentiate in vivo into potent effector cells (252, 253). Further, CAR T-cells can be coengineered to enhance both their own activity and that of other immune cells in the TME. Emerging knowledge on the immunometabolic pathways regulating T-cell function in tumors offers new opportunities for gene-engineering to drive favorable T-cell energetics and optimize their activity. Next-generation CAR T-cell immunotherapy based on combinatorial engineering and treatments to reprogram T-cell properties and the TME offer unprecedented hope for curing solid tumors.
Author Contributions
MI and GC conceived the manuscript; revised and approved the final manuscript. MI, RS, KS, and ND drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This project was supported by an ERC Advanced Grant to GC (1400206AdG-322875) and the Leenaards Foundation.
References
1. van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol (2011) 11(1):47–55. doi:10.1038/nri2887
2. Jass JR. Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol (1986) 39(6):585–9. doi:10.1136/jcp.39.6.585
3. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi:10.1056/NEJMoa020177
4. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
5. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12(4):298–306. doi:10.1038/nrc3245
6. Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol (2012) 124(2):192–8. doi:10.1016/j.ygyno.2011.09.039
7. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi:10.1016/j.coi.2014.01.004
8. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
9. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
10. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi:10.1056/NEJMoa1504627
11. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515(7528):558–62. doi:10.1038/nature13904
12. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi:10.1056/NEJMoa1411087
13. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi:10.1056/NEJMoa1510665
14. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol (2015) 33(34):4015–22. doi:10.1200/JCO.2015.62.3397
15. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi:10.1056/NEJMoa1500596
16. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi:10.1056/NEJMoa1504030
17. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol (2014) 32(10):1020–30. doi:10.1200/JCO.2013.53.0105
18. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi:10.1038/nature13954
19. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi:10.1056/NEJMoa1406498
20. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
21. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi:10.1038/nature13988
22. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. doi:10.1200/JCO.2012.47.7521
23. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med (2016) 374(26):2542–52. doi:10.1056/NEJMoa1603702
24. Lipson EJ, Vincent JG, Loyo M, Kagohara LT, Luber BS, Wang H, et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res (2013) 1(1):54–63. doi:10.1158/2326-6066.CIR-13-0034
25. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi:10.1158/0008-5472.CAN-08-2281
26. Salerno EP, Olson WC, McSkimming C, Shea S, Slingluff CL Jr. T cells in the human metastatic melanoma microenvironment express site-specific homing receptors and retention integrins. Int J Cancer (2014) 134(3):563–74. doi:10.1002/ijc.28391
27. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi:10.1038/ni.2703
28. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208(10):2005–16. doi:10.1084/jem.20101159
29. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi:10.1038/nature14404
30. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342(6161):967–70. doi:10.1126/science.1240527
31. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350(6264):1084–9. doi:10.1126/science.aac4255
32. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84. doi:10.1126/science.aad1329
33. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249(1):43–58. doi:10.1111/j.1600-065X.2012.01152.x
34. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi:10.1016/j.cell.2015.08.016
35. Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, et al. The intratumoral balance between metabolic and immunologic gene expression is associated with anti-PD-1 response in patients with renal cell carcinoma. Cancer Immunol Res (2016) 4(9):726–33. doi:10.1158/2326-6066.CIR-16-0072
36. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
37. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12(4):269–81. doi:10.1038/nri3191
38. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348(6230):62–8. doi:10.1126/science.aaa4967
39. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev (2014) 257(1):56–71. doi:10.1111/imr.12132
40. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344(6184):641–5. doi:10.1126/science.1251102
41. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi:10.1016/j.immuni.2013.07.012
42. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi:10.1016/j.coi.2015.01.011
43. Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol (2016) 28(8):383–91. doi:10.1093/intimm/dxw014
44. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood (2014) 123(17):2625–35. doi:10.1182/blood-2013-11-492231
45. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi:10.1056/NEJMoa1407222
46. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
47. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi:10.1126/science.aaa6204
48. Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology (2014) 3(11):e970027. doi:10.4161/21624011.2014.970027
49. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res (2014) 20(16):4262–73. doi:10.1158/1078-0432.CCR-13-2627
50. Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest (2015) 125(5):1780–9. doi:10.1172/JCI76094
51. Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res (2012) 72(14):3439–44. doi:10.1158/0008-5472.CAN-11-3912
52. Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer (2016) 2(6):286–94. doi:10.1016/j.trecan.2016.05.002
53. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
54. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity (2013) 39(1):61–73. doi:10.1016/j.immuni.2013.07.005
55. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov (2015) 14(8):561–84. doi:10.1038/nrd4591
56. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med (2016) 22(1):26–36. doi:10.1038/nm.4015
57. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol (2016) 13(5):273–90. doi:10.1038/nrclinonc.2016.25
58. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther (2015) 23(4):769–78. doi:10.1038/mt.2015.4
59. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13(10):907–15. doi:10.1038/ni.2386
60. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A (1989) 86(24):10024–8. doi:10.1073/pnas.86.24.10024
61. Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol (2015) 36(8):494–502. doi:10.1016/j.it.2015.06.004
62. Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol (2001) 167(11):6123–31. doi:10.4049/jimmunol.167.11.6123
63. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121(5):1822–6. doi:10.1172/JCI46110
64. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol (2004) 172(1):104–13. doi:10.4049/jimmunol.172.1.104
65. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A (2009) 106(9):3360–5. doi:10.1073/pnas.0813101106
66. Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood (2000) 96(5):1999–2001.
67. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20 Pt 1):6106–15. doi:10.1158/1078-0432.CCR-06-1183
68. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24(13):e20–2. doi:10.1200/JCO.2006.05.9964
69. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16(9):566–81. doi:10.1038/nrc.2016.97
70. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics (2016) 3:16015. doi:10.1038/mto.2016.15
71. Singh H, Huls H, Kebriaei P, Cooper LJ. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev (2014) 257(1):181–90. doi:10.1111/imr.12137
72. Pluta K, Luce MJ, Bao L, Agha-Mohammadi S, Reiser J. Tight control of transgene expression by lentivirus vectors containing second-generation tetracycline-responsive promoters. J Gene Med (2005) 7(6):803–17. doi:10.1002/jgm.712
73. June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol (2009) 9(10):704–16. doi:10.1038/nri2635
74. Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther (2001) 8(21):1601–8. doi:10.1038/sj.gt.3301570
75. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi:10.1158/2326-6066.CIR-13-0027
76. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi:10.1038/nm.3838
77. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant (2010) 16(9):1245–56. doi:10.1016/j.bbmt.2010.03.014
78. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res (2013) 1:26–31. doi:10.1158/2326-6066.CIR-13-0006
79. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol (2004) 173(12):7647–53. doi:10.4049/jimmunol.173.12.7647
80. Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, et al. Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J Biol Chem (2012) 287(27):23068–78. doi:10.1074/jbc.M112.357673
81. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol (2010) 184(9):4936–46. doi:10.4049/jimmunol.1000173
82. Pameijer CR, Navanjo A, Meechoovet B, Wagner JR, Aguilar B, Wright CL, et al. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther (2007) 14(1):91–7. doi:10.1038/sj.cgt.7700993
83. Muniappan A, Banapour B, Lebkowski J, Talib S. Ligand-mediated cytolysis of tumor cells: use of heregulin-zeta chimeras to redirect cytotoxic T lymphocytes. Cancer Gene Ther (2000) 7(1):128–34. doi:10.1038/sj.cgt.7700100
84. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res (2004) 64(24):9160–6. doi:10.1158/0008-5472.CAN-04-0454
85. Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res (2006) 66(11):5927–33. doi:10.1158/0008-5472.CAN-06-0130
86. Niederman TM, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci U S A (2002) 99(10):7009–14. doi:10.1073/pnas.092562399
87. Govers C, Sebestyén Z, Roszik J, van Brakel M, Berrevoets C, Szöőr Á, et al. TCRs genetically linked to CD28 and CD3epsilon do not mispair with endogenous TCR chains and mediate enhanced T cell persistence and anti-melanoma activity. J Immunol (2014) 193(10):5315–26. doi:10.4049/jimmunol.1302074
88. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res (2012) 72(7):1844–52. doi:10.1158/0008-5472.CAN-11-3890
89. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res (2012) 18(23):6436–45. doi:10.1158/1078-0432.CCR-12-1449
90. Fousek K, Ahmed N. The evolution of T-cell therapies for solid malignancies. Clin Cancer Res (2015) 21(15):3384–92. doi:10.1158/1078-0432.CCR-14-2675
91. Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev (2016) 30(3):157–67. doi:10.1016/j.blre.2015.10.003
92. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech (2015) 8(4):337–50. doi:10.1242/dmm.018036
93. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
94. Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother (2007) 30(7):684–93. doi:10.1097/CJI.0b013e3180de5d90
95. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res (2015) 75(17):3596–607. doi:10.1158/0008-5472.CAN-15-0159
96. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res (2015) 75(17):3505–18. doi:10.1158/0008-5472.CAN-15-0139
97. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi:10.1089/hum.2012.041
98. Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol (2015) 33:9–15. doi:10.1016/j.coi.2015.01.002
99. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res (2013) 1(1):43–53. doi:10.1158/2326-6066.CIR-13-0008
100. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi:10.1038/mtna.2013.32
101. Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell (2016) 164(4):780–91. doi:10.1016/j.cell.2016.01.012
102. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164(4):770–9. doi:10.1016/j.cell.2016.01.011
103. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science (2015) 350(6258):aab4077. doi:10.1126/science.aab4077
104. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol (2014) 5:235. doi:10.3389/fphar.2014.00235
105. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol (2014) 5:254. doi:10.3389/fphar.2014.00254
106. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi:10.1182/blood-2016-04-703751
107. Cordoba SP, Choudhuri K, Zhang H, Bridge M, Basat AB, Dustin ML, et al. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood (2013) 121(21):4295–302. doi:10.1182/blood-2012-07-442251
108. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol (2008) 180(10):7028–38. doi:10.4049/jimmunol.180.10.7028
109. Hombach AA, Schildgen V, Heuser C, Finnern R, Gilham DE, Abken H. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J Immunol (2007) 178(7):4650–7. doi:10.4049/jimmunol.178.7.4650
110. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res (2015) 3(2):125–35. doi:10.1158/2326-6066.CIR-14-0127
111. Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘ff-target’ activation and unintended initiation of an innate immune response. Gene Ther (2010) 17(10):1206–13. doi:10.1038/gt.2010.91
112. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol (2010) 184(12):6938–49. doi:10.4049/jimmunol.0901766
113. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119(24):5697–705. doi:10.1182/blood-2012-01-405365
114. Yang Y, Jacoby E, Fry TJ. Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol (2015) 22(6):509–15. doi:10.1097/MOH.0000000000000181
115. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother (2005) 28(3):203–11. doi:10.1097/01.cji.0000161397.96582.59
116. Liu L, Sommermeyer D, Cabanov A, Kosasih P, Hill T, Riddell SR. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat Biotechnol (2016) 34(4):430–4. doi:10.1038/nbt.3461
117. Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature (2010) 464(7290):932–6. doi:10.1038/nature08944
118. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi:10.1146/annurev.immunol.22.012703.104702
119. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115(6):1616–26. doi:10.1172/JCI24480
120. Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev (2014) 257(1):264–76. doi:10.1111/imr.12135
121. Gattinoni L, Restifo NP. Moving T memory stem cells to the clinic. Blood (2013) 121(4):567–8. doi:10.1182/blood-2012-11-468660
122. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc (2013) 8(1):33–42. doi:10.1038/nprot.2012.143
123. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi:10.1038/leu.2015.247
124. Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother (2012) 35(9):651–60. doi:10.1097/CJI.0b013e31827806e6
125. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123(24):3750–9. doi:10.1182/blood-2014-01-552174
126. Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol (2014) 44(1):69–79. doi:10.1002/eji.201343718
127. Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood (2012) 119(1):72–82. doi:10.1182/blood-2011-07-366419
128. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5(11):844–52. doi:10.1038/nri1710
129. Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem (1993) 212(1):95–9. doi:10.1111/j.1432-1033.1993.tb17637.x
130. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249(1):27–42. doi:10.1111/j.1600-065X.2012.01150.x
131. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi:10.1016/j.immuni.2011.09.021
132. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol (2013) 14(11):1173–82. doi:10.1038/ni.2714
133. Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature (1976) 261(5562):702–5. doi:10.1038/261702a0
134. Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol (2004) 172(8):4661–5. doi:10.4049/jimmunol.172.8.4661
135. Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell (2000) 6(3):683–92. doi:10.1016/S1097-2765(00)00066-6
136. Cretenet G, Clerc I, Matias M, Loisel S, Craveiro M, Oburoglu L, et al. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci Rep (2016) 6:24129. doi:10.1038/srep24129
137. Vuillefroy de Silly R, Ducimetière L, Yacoub Maroun C, Dietrich P-Y, Derouazi M, Walker PR. Phenotypic switch of CD8+ T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol (2016) 45:2263–75. doi:10.1002/eji.201445284
138. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
140. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
141. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi:10.1016/j.cell.2015.08.012
142. Yaqoob P, Calder PC. Glutamine requirement of proliferating T lymphocytes. Nutrition (1997) 13(7–8):646–51. doi:10.1016/S0899-9007(97)83008-0
143. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
144. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol (2015) 36(2):81–91. doi:10.1016/j.it.2014.12.005
145. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36(1):68–78. doi:10.1016/j.immuni.2011.12.007
146. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41(1):75–88. doi:10.1016/j.immuni.2014.06.005
147. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110(35):14336–41. doi:10.1073/pnas.1221740110
148. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. doi:10.1016/j.cell.2016.05.035
149. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14(10):1064–72. doi:10.1038/ni.2687
150. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42(1):41–54. doi:10.1016/j.immuni.2014.12.030
151. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460(7251):108–12. doi:10.1038/nature08155
152. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460(7251):103–7. doi:10.1038/nature08097
153. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
154. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183(10):6095–101. doi:10.4049/jimmunol.0803510
155. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi:10.1038/ni.2556
156. Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell (2012) 149(2):410–24. doi:10.1016/j.cell.2012.02.044
157. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
158. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
159. Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods (1987) 102(1):127–41. doi:10.1016/S0022-1759(87)80018-2
160. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32(1):79–90. doi:10.1016/j.immuni.2009.11.012
161. Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood (2008) 111(11):5326–33. doi:10.1182/blood-2007-09-113050
162. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A (2004) 101(7):1969–74. doi:10.1073/pnas.0307298101
163. Butler MO, Lee JS, Ansén S, Neuberg D, Hodi FS, Murray AP, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res (2007) 13(6):1857–67. doi:10.1158/1078-0432.CCR-06-1905
164. Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP, et al. Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med (2011) 3(80):80ra34. doi:10.1126/scitranslmed.3002207
165. Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol (2005) 175(4):2261–9. doi:10.4049/jimmunol.175.4.2261
166. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111(4):2101–11. doi:10.1182/blood-2007-06-096297
167. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell (2015) 161(4):750–61. doi:10.1016/j.cell.2015.03.021
168. Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab (2016) 23(1):63–76. doi:10.1016/j.cmet.2015.11.002
169. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi:10.1172/JCI69589
170. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15(7):808–13. doi:10.1038/nm.1982
171. van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, Falkenburg JH, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood (2014) 124(23):3490–500. doi:10.1182/blood-2014-05-578583
172. O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol (2015) 36(2):71–80. doi:10.1016/j.it.2014.12.004
173. Sikalidis AK. Amino acids and immune response: a role for cysteine, glutamine, phenylalanine, tryptophan and arginine in T-cell function and cancer? Pathol Oncol Res (2015) 21(1):9–17. doi:10.1007/s12253-014-9860-0
174. Rodriguez PC, Quiceno DG, Ochoa AC. l-Arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi:10.1182/blood-2006-06-031856
175. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42.e13. doi:10.1016/j.cell.2016.09.031
176. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi:10.1126/scitranslmed.3005930
177. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
178. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7(303):303ra139. doi:10.1126/scitranslmed.aac5415
179. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118(18):4817–28. doi:10.1182/blood-2011-04-348540
180. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi:10.1016/S1074-7613(02)00323-0
181. Martinez-Forero I, Azpilikueta A, Bolaños-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63-polyubiquitin-dependent signals from endosomes. J Immunol (2013) 190(12):6694–706. doi:10.4049/jimmunol.1203010
182. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44(2):380–90. doi:10.1016/j.immuni.2016.01.021
183. Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget (2013) 4(4):584–99. doi:10.18632/oncotarget.965
184. Beatty GL, O’Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Ther (2016) 166:30–9. doi:10.1016/j.pharmthera.2016.06.010
185. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi:10.1200/JCO.2014.58.0225
186. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118(23):6050–6. doi:10.1182/blood-2011-05-354449
187. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20(6):607–15. doi:10.1038/nm.3541
188. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125(9):3356–64. doi:10.1172/JCI80005
189. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25(2):268–76. doi:10.1016/j.coi.2013.02.009
190. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi:10.1038/nri3405
192. Schwartz RH. T cell anergy. Annu Rev Immunol (2003) 21:305–34. doi:10.1146/annurev.immunol.21.120601.141110
193. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol (2014) 24(8):472–8. doi:10.1016/j.tcb.2014.06.001
194. Zhang Y, Ertl HC. Starved and asphyxiated: how can CD8(+) T cells within a tumor microenvironment prevent tumor progression. Front Immunol (2016) 7:32. doi:10.3389/fimmu.2016.00032
195. Justus CR, Sanderlin EJ, Yang LV. Molecular connections between cancer cell metabolism and the tumor microenvironment. Int J Mol Sci (2015) 16(5):11055–86. doi:10.3390/ijms160511055
196. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun (2015) 6:7458. doi:10.1038/ncomms8458
197. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res (2009) 69(5):2000–9. doi:10.1158/0008-5472.CAN-08-2360
198. Miller AM, Lundberg K, Ozenci V, Banham AH, Hellström M, Egevad L, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol (2006) 177(10):7398–405. doi:10.4049/jimmunol.177.10.7398
199. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi:10.1038/nature10169
200. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol (2009) 182(3):1746–55. doi:10.4049/jimmunol.182.3.1746
201. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi:10.1038/ni1536
202. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi:10.1084/jem.20062512
203. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
204. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi:10.1038/nm934
205. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117(5):1147–54. doi:10.1172/JCI31178
206. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer (2008) 8(8):618–31. doi:10.1038/nrc2444
207. Chen EP, Smyth EM. COX-2 and PGE2-dependent immunomodulation in breast cancer. Prostaglandins Other Lipid Mediat (2011) 96(1–4):14–20. doi:10.1016/j.prostaglandins.2011.08.005
208. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res (2010) 16(22):5458–68. doi:10.1158/1078-0432.CCR-10-0712
209. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther (2002) 13(16):1971–80. doi:10.1089/10430340260355374
210. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21(5):524–9. doi:10.1038/nm.3833
211. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi:10.1038/nm1699
212. Santoro SP, Kim S, Motz GT, Alatzoglou D, Li C, Irving M, et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res (2015) 3(1):68–84. doi:10.1158/2326-6066.CIR-14-0192
213. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi:10.3389/fimmu.2015.00001
214. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi:10.1158/0008-5472.CAN-04-0465
215. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity (2016) 45(2):374–88. doi:10.1016/j.immuni.2016.08.009
216. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531(7596):651–5. doi:10.1038/nature17412
217. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. doi:10.1172/JCI83092
218. Borkner L, Kaiser A, van de Kasteele W, Andreesen R, Mackensen A, Haanen JB, et al. RNA interference targeting programmed death receptor-1 improves immune functions of tumor-specific T cells. Cancer Immunol Immunother (2010) 59(8):1173–83. doi:10.1007/s00262-010-0842-0
219. Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol Immunol (2012) 51(3–4):263–72. doi:10.1016/j.molimm.2012.03.023
220. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7(23):34341–55. doi:10.18632/oncotarget.9114
221. Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest (2010) 120(10):3722–34. doi:10.1172/JCI41991
222. Paolino M, Thien CB, Gruber T, Hinterleitner R, Baier G, Langdon WY, et al. Essential role of E3 ubiquitin ligase activity in Cbl-b-regulated T cell functions. J Immunol (2011) 186(4):2138–47. doi:10.4049/jimmunol.1003390
223. Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol (2013) 14(6):593–602. doi:10.1038/ni.2576
224. Ji Y, Wrzesinski C, Yu Z, Hu J, Gautam S, Hawk NV, et al. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic gammac cytokines. Proc Natl Acad Sci U S A (2015) 112(2):476–81. doi:10.1073/pnas.1422916112
225. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119(18):4133–41. doi:10.1182/blood-2011-12-400044
226. Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon-optimized IL-15 gene resist cytokine withdrawal-induced apoptosis and persist long-term in the absence of exogenous cytokine. J Immunol (2005) 175(11):7226–34. doi:10.4049/jimmunol.175.11.7226
227. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2(10):1096–103. doi:10.1038/nm1096-1096
228. Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, et al. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood (1998) 92(12):4778–91.
229. Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, et al. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol (1999) 162(8):4567–75.
230. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi:10.1038/nm863
231. Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res (2012) 18(5):1207–13. doi:10.1158/1078-0432.CCR-11-1591
232. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res (2015) 75(18):3771–87. doi:10.1158/0008-5472.CAN-15-0405
233. Vuillefroy de Silly R, Dietrich PY, Walker PR. Hypoxia and antitumor CD8+ T cells: an incompatible alliance? Oncoimmunology (2016) 5(12):e1232236. doi:10.1080/2162402X.2016.1232236
234. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
235. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi:10.1038/nature13490
236. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123(9):3664–71. doi:10.1172/JCI67230
237. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin (2016) 67(1):65–85. doi:10.3322/caac.21358
238. Burnette B, Fu YX, Weichselbaum RR. The confluence of radiotherapy and immunotherapy. Front Oncol (2012) 2:143. doi:10.3389/fonc.2012.00143
239. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol (2014) 5:514. doi:10.3389/fimmu.2014.00514
240. Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci U S A (1996) 93(23):13119–24. doi:10.1073/pnas.93.23.13119
241. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24(5):589–602. doi:10.1016/j.ccr.2013.09.014
242. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi:10.1038/nature15520
243. Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol (2002) 168(8):3771–6. doi:10.4049/jimmunol.168.8.3771
244. Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J (1991) 5(11):2516–22.
245. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med (2013) 5(200):200ra116. doi:10.1126/scitranslmed.3006504
246. Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res (2005) 11(16):6030–9. doi:10.1158/1078-0432.CCR-04-2671
247. Ino K, Yoshida N, Kajiyama H, Shibata K, Yamamoto E, Kidokoro K, et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer (2006) 95(11):1555–61. doi:10.1038/sj.bjc.6603477
248. Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res (2006) 12(4):1144–51. doi:10.1158/1078-0432.CCR-05-1966
249. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov (2012) 2(8):722–35. doi:10.1158/2159-8290.CD-12-0014
250. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22(5):633–42. doi:10.1016/j.immuni.2005.03.013
251. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi:10.1016/j.it.2012.10.001
252. Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res (2012) 72(13):3125–30. doi:10.1158/0008-5472.CAN-11-4094
Keywords: immunotherapy, chimeric antigen receptor, T-cells, gene-engineering, immunometabolism, solid tumors, tumor microenvironment
Citation: Irving M, Vuillefroy de Silly R, Scholten K, Dilek N and Coukos G (2017) Engineering Chimeric Antigen Receptor T-Cells for Racing in Solid Tumors: Don’t Forget the Fuel. Front. Immunol. 8:267. doi: 10.3389/fimmu.2017.00267
Received: 10 January 2017; Accepted: 23 February 2017;
Published: 03 April 2017
Edited by:
Christoph Hess, University of Basel, SwitzerlandCopyright: © 2017 Irving, Vuillefroy de Silly, Scholten, Dilek and Coukos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melita Irving, melita.irving@unil.ch;
George Coukos, george.coukos@chuv.ch