 Licheng Sun
Licheng Sun Jinrong Fu
Jinrong Fu Yufeng Zhou
Yufeng Zhou- 1Children’s Hospital and Institute of Biomedical Sciences, Fudan University, Shanghai, China
- 2Key Laboratory of Neonatal Diseases, Ministry of Health, Shanghai, China
Accumulating evidence indicates that metabolism reprogramming is critically important to T cell differentiation, and manipulating metabolic pathways in T cells can shape their fate and function. During T cell differentiation, metabolism provides T cells with energy as well as precursors for various biological processes. Some key metabolic reactions, such as glycolysis, oxidative phosphorylation and fatty acid oxidation, are also considered to play important roles in T cell activation and differentiation. In this review, we will explain why cellular metabolism is important for the Th17/T-regulatory (Treg) cell balance and how metabolism reprogramming impacts this balance. Moreover, we will also discuss some important metabolic sensors, such as mammalian target of rapamycin, AMP-activated protein kinase, and some nuclear receptors. In addition, we will review specific small molecular compounds, which can shift the Th17/Treg cell balance and, therefore, have promising therapeutic roles. Finally, potential methods of manipulating Th17 cell metabolism for treating Th17-associated diseases will be discussed.
Introduction
Naïve T cells are quiescent and largely metabolically inactive. After stimulation, T cells start to proliferate and differentiate (1). During this process, metabolism reprogramming is necessary and crucial and can meet energy requirements and provide various substrates that are indispensable for T cell proliferation and differentiation. Furthermore, increasingly more reports have indicated that, as well as acting as an energy source, metabolic processes can also be regulators of T cell differentiation (2, 3).
Naïve CD4+ T cells can differentiate to various T helper cells (Th), including Th1, Th2, Th9, Th17, and T-regulatory (Treg) cells. These T helper cells have diverse functions and act as a pivotal part in both mediating adaptive immunity and autoimmune diseases (4, 5). According to their special cytokines and function, CD4+ T helper cells were first classified as two subsets, Th1 and Th2. The Th1/Th2 theory was established 20 years ago and reported that dysfunction in the immune system and autoimmune diseases were induced by the unbalance of Th1 and Th2 (6, 7). This hypothesis was not substantially challenged until the discovery of Th17. Th17 cells were first recognized as novel CD4+ T helper cells in 2005; they predominately secrete interleukin-17 (IL-17) and protect the host against pathogens (8, 9). On the other hand, excess Th17 cells can induce autoimmune disease and inflammation (9). Treg cells are also a lineage of CD4+ T cells. The function of Treg cells is to maintain immune homeostasis and limit the excessive immune response. Although the function of Treg and Th17 are completely different, Th17 and Treg share many important elements during differentiation (10, 11).
The unique relationship between Th17 and Treg has been discussed for a long time, and many molecular mechanisms that control the balance of Th17 and Treg have been revealed. Despite the molecular regulating network, accumulating evidence shows that processes involving metabolism are also important in Th17 and Treg differentiation. Therefore, here, we will focus on how metabolism processes influence the balance of Th17 and Treg. Initially, some general ideas about Th17 and cellular metabolism will be discussed, and then we will provide an overview of recent investigations about how metabolic reprogramming regulates the balance of Th17/Treg. Finally, potential therapeutic options will be briefly discussed.
Overview of Th17 And Treg
Th17 cells were identified as a new lineage of CD4+ T helper cells in 2005, although the signature cytokine Th17 was described earlier (12). In fact, the path leading to discovery of Th17 cells began in 2003 with the finding that experimental autoimmune encephalomyelitis (EAE) in the murine model was caused by high levels of IL-23 rather than IL-12 and Th1 cells (13). Consequently, researchers found that the function of IL-23 is to promote differentiation and proliferation of the IL-17-secreting cells, which were eventually identified as Th17. Nevertheless, IL-23 alone is unable to make naïve T cells differentiate into Th17 cells (9, 14, 15). Subsequently, many research teams have found that polarization of Th17 can be efficiently induced by transforming growth factor-β1 (TGF-β1) and IL-6, which activate Smad family proteins and STAT3, respectively (16, 17). STAT3 further promotes and maintains the expression of Th17-specific genes (1, 18). The master transcription factors of Th17 are retinoic acid-related orphan receptors-γt (RORγt), which can directly regulate the expression of Th17-specific genes, such as CCR6, CD161, IL17a, Il17f, and Il23r. On the other hand, RORγt is also a critical component in a larger network of transcription factors like Stat3, IRF4, or BATF (1, 18, 19). In addition to the ability to fight against extracellular microbial organisms and mediate autoimmune disease, Th17 cells are also involved in allograft rejection. Antagonism of IL-17 can promote the survival rate in a rat cardiac allograft model; furthermore, in a lung transplantation model, expression of IL-17 and IL-23 was also demonstrated to be upregulated at the site of allograft rejection (20, 21).
It has been known for a long time that CD4+T cells can either activate or suppress immune responses. Treg is another lineage of CD4+T cells but it has a totally different function compared to Th17 and can modify the immune response, maintain immune tolerance to self, and prevent autoimmune disease (22). Generally, Treg can be classified as nTreg (natural Treg) and iTreg (induced Treg). Treg originating from the thymus is often named as nTreg, otherwise peripherally derived Treg is regarded as iTreg (22). The surface markers of Treg are CD4+ and CD25+, and its master transcription factor is Forkhead box P3 (Foxp3) (23). Foxp3 can provide Treg with suppressive activity and upregulate other Treg-specific genes, such as CD25, CTLA4, and IL-10. In mice, defective Foxp3 causes loss of immune tolerance and autoimmune disease. Moreover, CD4+, CD25+, and Foxp3+ are recognized as good markers for mouse Treg, although recently Foxp3 has also been found to be expressed in CD4+ and CD25− cells (22). Notably, in humans, Foxp3 is not only expressed in Treg but also in some conventional T cells (22, 24). Like other T helper cells, Treg cells are differentiated from their naïve CD4+ T cells, but the main function of Treg is suppressing activation of the immune system (22). A lack of Treg causes autoimmune disease, and a high ratio of Treg/Th17 is associated with cancer incidence (23, 25).
Activation of T cell receptor (TCR) and costimulatory signaling is needed to initialize the differentiation of all CD4+ T helper cells. However, after activation, different lineages of specific cytokines drive the differentiation of distinct cell subsets. TGF-β1 is required for differentiation of both Th17 and peripheral iTreg (26). This common point suggests that differentiation of these two subsets is related. When the TGF-β1 signaling pathway is activated, the expression of RORγt and Foxp3 is upregulated. Whether naïve T cells polarize to a Th17 phenotype or a regulatory phenotype largely depends on the surrounding microenvironments (27, 28). IL-6 has been recognized as a major cytokine that drives the differentiation of Th17 cells. Foxp3 can inhibit Th17 development by directly binding to RORγt. Without IL-6, the TGF-β signaling pathway reinforces this inhibition and favors the formation of Treg from naïve T cells. In the presence of IL-6, STAT3 can be activated and Foxp3 is released from RORγt (29). During differentiation of Th17, the expression of IL-23R is also upregulated, and activated IL-23R can further induce Th17 differentiation. Moreover, activation of IL-23R is also very important for proliferation and maintenance of the phenotype of Th17 cells after differentiation. The IL-23 signaling pathway can also activate STAT3 and inhibit IL-10 production (19, 28, 30). It should also be noted that TGF-β1-induced Th17 differentiation can occur without IL-6, if there is sufficient IL-21. Some studies have shown that human naïve CD4+ T cells can be induced to Th17 by adding exogenous IL-21. Treating human naïve CD4+ T cells with IL-21 and TGF-β can upregulate the expression of both IL-23 and RORγt, while inhibiting the expression of Foxp3 (31). After differentiation, Th17 can secrete IL-21 and form an autocrine loop to further promote the Th17 phenotype. In summary, in the presence of TGF-β1, both IL-6 and IL-21 can induce differentiation of T cells to Th17. Otherwise, T cells will differentiate to Treg (32).
Besides sharing a similar development pathway, in some conditions, Th17 and Treg can also trans-differentiate to each other (33–36). It has been reported that high concentrations of exogenous Th17-generating cytokines can convert Foxp3+ Treg cells into IL-17 secreting cells (35). On the other hand, it has also been reported that Treg can lose Foxp3 expression and go on to acquire an inflammatory function (36). Some investigators also indicate that there are intermediate cells during Th17 to Treg trans-differentiation, which are Foxp3 and IL-17 double-positive T cells (37) (please see Figure 1).
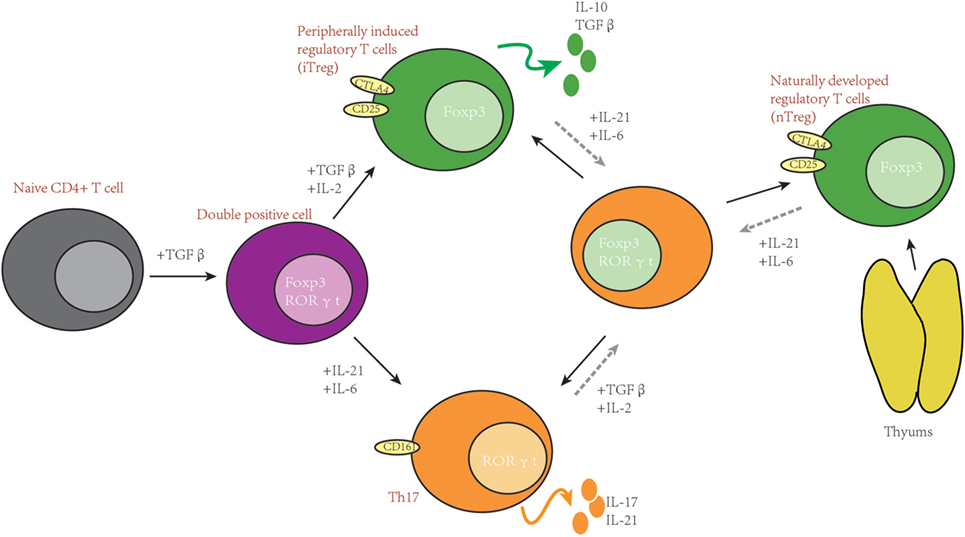
Figure 1. Th17 and T-regulatory (Treg) share important developmental elements. After stimulation, naïve T cells can be induced into RORγt and Forkhead box P3 (Foxp3) double-positive T cells. Furthermore, whether naïve T cells differentiate to Th17 or Treg depends on the surrounding cytokines. In some situations, Th17 and Treg can trans-differentiate.
Overview of Cellular Metabolism
Glycolysis, Tricarboxylic Acid (TCA) Cycle, and Oxidative Phosphorylation (OXPHO)
Glycolysis refers to a sequence of cytosolic enzymatic reactions that convert glucose into pyruvate, generating energy. Generally, after being imported from the extracellular space by glucose transporter (Glut), glucose is initially converted to glucose 6-phosphate (by the rate-limiting enzyme hexokinase) and subsequently to fructose 6-P, spontaneously. A second rate-limiting enzyme, phosphofructokinase-1, enables the formation of fructose1, 6-diphosphate. After several steps, pyruvate is formed, which can then be processed in two ways. It is either imported to the mitochondria, with consequent conversion to acetyl-CoA. Acetyl-CoA can enter the TCA cycle to produce NADH and FADH2, which can generate ATP by OXPHO. One molecule of glucose metabolized by aerobic glycolysis can generate two molecules of ATPs. By contrast, if the products generated by glycolysis can be further metabolized by the TCA cycle and OXPHO, the theoretical maximum number of ATP molecules from one molecule of glucose can be generated: 32 ATPs in total. It should also be noted that glycolysis and the TCA cycle not only provide energy but also generate many intermediates for other metabolic pathways, such as the pentose phosphate pathway (PPP), hexosamine biosynthesis, amino acid biosynthesis, and de novo fatty acid synthesis (FAS). Another fate of pyruvate is conversion to lactate in the cytosol by lactate dehydrogenase (38). Pyruvate converted to lactate often occurs under hypoxic conditions. This process generates NAD+, which is consumed during glycolysis. When cells cannot regenerate NAD+ by OXPHO, the strategy of generating lactate can provide NAD+ and remove the limitation of glycolysis. Interestingly, with sufficient oxygen, sometimes cells still utilize glucose to generate lactate, which is known as aerobic glycolysis or the Warburg effect (3, 38, 39). This unique phenomenon has often been reported in tumor cells and, more recently, in T cells (40, 41). Moreover, it has been shown that aerobic glycolysis is crucial for polarization of T helper cells, especially Th17 (42, 43).
On the other hand, OXPHO is an important metabolic process for generating ATP molecules by means of the electron transport chain (ETC). NADH and FADH2 are catalyzed back into oxidized forms (NAD+ and FAD) by Complex I or II, respectively, in the mitochondria. The reactions can generate electrons and protons. Protons are transferred into the inter-membrane space of the mitochondria. At the same time, electrons are passed along the ETC from Complex III, first to cytochrome C and then to Complex IV, in which protons are finally transferred to molecular oxygen generating water (38, 44). Otherwise, differential electrical charges are created between the mitochondrial membranes during the process, and this electrical potential difference is utilized by ATP synthase, which can pump protons back to the mitochondrial matrix and generate ATP (38).
Fatty Acid Metabolism
The fate of pyruvate generated by glycolysis is participation in de novo FAS. Pyruvate can convert into acetyl-CoA in the mitochondria. However, this acetyl-CoA must be transported back to the cytosol where FAS and cholesterol synthesis occur. Acetyl-CoA cannot be transported into the cytosol directly, it must condensate with oxaloacetate first to generate citrate and is then transported to the cytosol where the reverse reaction occurs, and citrate is re-converted to acetyl-CoA. Furthermore, the rate-limiting step in FAS takes place. Acetyl-CoA is carboxylated into malonyl-CoA by acetyl-CoA Carboxylase 1 (ACC1). Then, acetyl-CoA is converted into malonyl-CoA to generate longer acyl chains, the reaction of which is catalyzed by a multi-enzyme complex, FAS. Acyl chains can continue to elongate by adding acetyl molecules, but this is dependent on the availability of NADPH. The final product of FAS is palmitate, which can be further utilized to generate other fatty acids or lipoproteins (38, 45).
Fatty acid synthesis is an anabolic process that generates fatty acids to reserve energy. Conversely, fatty acid oxidation (FAO) can convert fatty acids to acetyl-CoA in order to generate energy for cell proliferation and activation. Initially, fatty acids can be transported into the mitochondria by carnitine palmitoyl transferase present on the mitochondrial membrane, specifically CPT1 and CPT2. Once transported into the mitochondria, long-chain fatty acids are metabolized by a series of enzymatic reactions (38, 46). At the same time, NADH, FADH2, and acetyl-CoA are generated. Furthermore, acetyl-CoA can feed into the TCA cycle for further oxidization, while NADH and FADH2 can be utilized as electron donors for OXPHO (46). As well as aerobic glycolysis, FAS and FAO are also important regulators for lymphocyte activation and differentiation, especially for Treg (45).
Metabolic Reprogramming and Th17/Treg Balance
Naïve T cells are relatively quiescent cells and their TCR are not stimulated. Compared to activated T cells, naïve T cells are comparatively smaller in size and are arrested at the G0 stage; they rely on IL-7 for survival and maintain their quiescent state (47). Correspondingly, considering metabolic activity, naïve T cells require less energy because they do not divide and secrete significant amounts of cytokines. Without the need to obtain the energy rapidly, naïve T cells usually utilize nutrients in the most effective way to generate maximum ATP through complete oxidization. In this way, OXPHO to yield ATPs is the main energy source for naïve T cells. For glucose, they are usually degraded by glycolysis and then feed into the TCA cycle for further oxidization, which ensures utilizing glucose most efficiently and generating 32 ATPs from one molecule of glucose.
However, after stimulation, CD4+ naïve T cells become highly proliferative and differentiate into T helper cells (Th). Without doubt, T cell activation and differentiation require metabolic reprogramming to support their rapid expansion and bio-function (48), and a series of changes in metabolic pathways occurs. First, the transporters for nutrients are highly expressed, such as glucose transporter (Glut) for transporting glucose and Alanine–Serine–Cysteine Transporter (ASCT2) for transporting glutamine (43, 49). Then glycolysis, OXPHOS, PPP, hexosamine pathway, and fatty acid metabolism all become active. Each aspect of the orchestration is important, and subtle differences in these metabolic programs will lead to differentiation of naïve CD4+ T cells to different T helper cells lineages (48, 50–52). The PPP can generate ribose-5-phosphate (R5P) and NADPH. R5P is used as a substrate to synthesize nucleotides, and NADH is an important mediator in many metabolism reactions. The hexosamine pathway generates UDP-N-acetyl glucosamine, which can be used for posttranslational modifications of proteins (53, 54).
T cell activation needs metabolic reprogramming, and different metabolic profiles can impact the Th17/Treg balance. One research group investigated 400 metabolites by high-resolution non-targeted Q exactive–mass spectrometry, and liquid chromatography/gas chromatography–MS (LC/GC–MS) demonstrated that Th17 and Treg have different metabolic profiles. Compared to Treg, Th17 cells have high levels of pyruvate, lactate, and PPP intermediates, while Treg cells have more TCA-cycle intermediates, which suggests Th17 cells rely more on aerobic glycolysis and glutamine oxidation and Tregs rely more on pyruvate oxidation (43). Examination of the level of amino acids in Th17 and Treg subsets, no clear subtype patterns can be observed. Notably, in both Th17 and Treg, the level of aspartate is much higher than other amino acids (about 10-fold higher), which may indicate that aspartate has an important function in Th17 and Treg (55). Examination of Acyl-carnitines indicates that Treg cells have higher levels of C2 and C4-OH carnitine compared to Th17. These metabolites represent acetyl and β-hydroxybutryl CoA species, respectively. The MS data also show a drop in oleyl-carnitine (C18:1). This fall in a long-chain acyl-carnitine coupled with the rise in C4-OH (hydroxybutyryl carnitine) is consistent with the fact that the level of FAO is high in Treg, and supports the idea that compared to other T helper cells Treg cells use fatty acids as a fuel to generate energy (43). Gene expression data show similar results. Gene arrays demonstrate that glucose metabolism-related genes are highly expressed in Th17. However, the fatty acid transporter CPT1A and ETC component cytochrome c is highly expressed in Treg compared with Th17. Interestingly Th17 and Tregs preferentially expressed a single isoform of each glycolytic enzyme, with utilization of Glut as glucose transporters. In Tregs, only Glut1 is expressed, and in Th17 both Glut1 and Glut3 are expressed, with Glut1 being dominantly expressed. As discussed above, hexokinase is the rate-limiting enzyme in glycolysis. Hk1 is preferentially expressed in Treg, while HK2 and HK3 is preferentially expressed in Th17 (43).
Glycolysis is especially important for the development of Th17. Research has shown that when naïve T cells are stimulated, the PI3K signaling pathway will be activated. Furthermore, while increasing the expression of Glut, PI3K signaling pathway can promote Glut trafficking to the cell membrane (56). Without Glut, the development of T helper cells is impaired, while Tregs are less affected (51). As discussed above, combined with other reports, it can be concluded that glycolysis is much more active in Th17 compared to Treg (43, 57); defective glycolysis can impair Th17 differentiation dramatically. By blocking glycolysis, Th17 cell proliferation and cytokine production are inhibited (58, 59). One reason concerns energy; to some extent, Treg cells are more like naïve T cells and need relatively less energy. Moreover, in order to provide energy for expansion and exerting function, other T helper cells, including Th17, need an abundant amount of ATP in a very short time. Therefore, Th17 cells need to rely on glycolysis for their development. Nevertheless, unlike naïve T cells, pyruvate does not feed into the TCA cycle and it is alternatively converted to lactate. In this process, each molecule of glucose can generate two molecules of ATPs (60). Despite the low usage rate for energy production, glycolysis is still independent of differentiation of T helper cells, especially Th17. This may be due to the fact that glycolysis can generate energy much faster (61).
Another hypothesis is that glycolysis is crucial for initializing metabolic programs and can also generate many metabolic intermediates for other metabolic pathways, such as the synthesis of proteins, nucleic acids, and lipids in the process of development of T helper cells (48, 62, 39). However, this hypothesis is controversial. Some reports have argued that these biosynthetic pathways are only slightly upregulated (less than 10%) by aerobic glycolysis (63). In the past, many investigations were focused on how metabolites impact the synthesis of proteins, nucleic acids, and lipids. Recent reports, however, provide some new ideas. The metabolic pathways may influence Th17 or Tregs in other ways. One report demonstrated that UDP-GlcNAc, a sugar-nucleotide, can affect the balance of Th17 and Treg; UDP-GlcNAc promotes N-acetyl glucosamine branching of Asn (N)-linked glycans. In addition, N-glycan branching can inhibit endocytosis of IL2Rα. Furthermore, this process drives naïve T cell differentiation to Treg. Interestingly, the biosynthesis of UDP-GlcNAc acts via the hexosamine pathway. In Th17 cells, increasing glycolysis and glutaminolysis compete for fructose-6-phosphate and glutamine in the hexosamine pathway, and then lower N-glycan branching. Indeed, GFPT1 is specifically downregulated by Th17 cells, and kifunensine, which blocks GlcNAc branching, can skew the Treg to Th17 (54). Another report has shown that metabolites can impact the epigenetic mechanism, further influencing the Th17 and Treg balance. Compared to Treg, a metabolite named 2-hydroxyglutarate (2-HG) accumulates in Th17. It was also revealed that (aminooxy)acetic acid(AOA) can switch Th17 re-differentiation to Tregs by inhibiting the activity of glutamate-oxaloacetate transaminase (GOT1), which is a transaminase and mediate glutamine metabolism (55). In normal conditions, GOT1 can convert aspartate to oxaloacetic acid, while generating electrons for ETC. However, upon ETC inhibition, it reverses to generate aspartate in the cytosol and rescue cell survival and proliferation (64). However, the reason that AOA can promote Foxp3 expression and inhibit Th17 differentiation is by generating a metabolite, 2-HG, rather than affecting Th17 proliferation (55). It was also reported that 2-HG is an inhibitor of TET1 and TET2; inhibition of TET1 and TET2 leads to hypermethylation of the Foxp3 gene locus and inhibition of Foxp3 transcription (55). Moreover, compared to manipulating glycolysis, AOA and kifunensine are aimed to the branch of the glucose metabolic pathway, which cause less side effects and have the potential to be developed as drugs to treat Th17-associated autoimmune diseases.
In addition to energy requirements and by-products, some reports have given other possible interpretations for the importance of glycolysis in T helper cells. As described above, GAPDH is an important enzyme engaged in glycolysis. When glycolysis is not active, in the absence of substrate, GAPDH will combine with the 3′ UTR of IFN-γ mRNA to disturb its translation. This provides a possible hypothesis that GAPDH enzymes involved in glycolysis (or other metabolic process) may modulate T cell differentiation. Moreover, it has been demonstrated that many metabolic enzymes can indeed transport to the nucleus. Furthermore, these enzymes can interact with mRNA to impact their stability or interfere with their translation (65). Aerobic glycolysis could keep GAPDH or other enzymes fully engaged and prevent their alternative function as inhibitors for T cell development (66). However, this hypothesis cannot explain how aerobic glycolysis can easily impact Th17 cells, apart from the evidence that enzymes involved in glycolysis can influence the expression of IL-17 or RORγt.
Although we have emphasized the importance of aerobic glycolysis, mitochondrial OXPHOS is still indispensable for T cell activation and proliferation. Inhibiting mitochondrial OXPHOS with oligomycin can completely abrogate the proliferation of TCR-activated T cell (66). Conversely, in the glucose-free medium, after TCR stimulation, T cells can still divide and express IL-2, which is regarded as an activation marker of TCR stimulation (44). NAD+ is an important component of ETC and also a co-factor of many biological reactions. A recently published paper shows that, in the presence of oligomycin, Jurkat cells can still proliferate by adding exogenous NAD+ to the medium. In this study, the authors also showed that failure to proliferate was due to the complete inability to synthesize aspartate without NAD+ rather than energy (64, 67). For Th17 and Tregs, NAD+ is reported to have the ability to specifically convert Treg into Th17 through the purinergic receptors, P2RX4 and P2RX7. Treating skin allograft mice in vivo with NAD+ can reduce the frequency of CD4+ CD25+ Foxp3+ Tregs and increase the frequency of Th17 cells. Interestingly, NAD+ can prolong allograft survival, which is paradoxical with the reducing of Tregs and increasing of Th17. The author attributes this to the fact that NAD+ can systematically increase CD4+ IL-10+ producing cells. After treatment with NAD+, around 50% of CD4+ T cells are IL-10-producing cells although Tregs are reduced (68). Sirtuins are a class of NAD-dependent deacetylases, and NAD+ is indispensable for the function of sirtuins. In Th17, SIRT1 can deacetylate RORγt and enhance Th17 cell generation (69). On the other hand, when mitochondrial OXPHOS occurs, mitochondrial complex III generates reactive oxygen species (ROS). Mitochondrial ROS (mtROS) can promote differentiation of Th17 cells. Knockout IEX-1 gene, which suppresses mtROS production can promote Th17 differentiation and aggravate arthritis in a mouse model. However, knockout IEX-1 gene does not impact Treg differentiation. It has also been reported that N-acetylcysteine, which is a specific inhibitor for mtROS production, can dramatically inhibit Th17 differentiation (70).
Lipid metabolism is also an indispensable element for the development of T helper cells. Lipids are the main components of cell membranes and provide energy and participate in cell signaling. Many recent reports have shown that lipid metabolism is an important regulator in T cell development (71). Both FAS and FAO are indispensable for T cell proliferation and differentiation (71–73). As described above, ACC1 is the rate-limiting enzyme for FAS. For Th17 cells, ACC1 is indispensable; Th17 cells depend on ACC1-mediated FAS to produce phospholipids for cellular membranes, while Treg is preferable to take up exogenous fatty acids. Impaired ACC1 function not only decreased FAS, but also decreased the level of glycolysis in Th17 cells, which can further skew Th17 differentiation to Treg (45). Moreover, ACC1 may be regarded as a potential target for clinical trials to treat Th17-related diseases. Soraphen A, which is a specific inhibitor of ACC1 can attenuate EAE in a mouse model (45). Another report also demonstrated that C75, which is an inhibitor of FAS, can reduce EAE disease (74). Madecassic acid has also been reported to ameliorate colitis in mice by downregulating the expression of ACC1 (75). On the other hand, it has also been reported that inhibition of ACC1 can decrease the level of FAS and increase the level of FAO (45, 76). Treg rely more on FAO to supply energy and, therefore, inhibiting ACC1 can also promote the development of Tregs (43, 77).
Important Metabolic Sensors Controlling of Th17/Treg Balance
Mammalian Target of Rapamycin (mTOR)
Mammalian target of rapamycin is a protein kinase of the phosphatidylinositol 3-kinase-related kinase family, which can sense the environment for cells and be activated by hormones, nutrients, and various stress conditions. mTOR contains two complexes, mTORC1 and mTORC2, which each have their own specific functions (78). A lack of mTORC1 signaling, in both human and murine naïve CD4+ T cells resulted in failure to differentiate to the Th17 lineages (79, 80). If both mTORC1 and mTORC2 are lost, naïve CD4+ T cells can only differentiate to Treg cells. Knockout of mTOR in mouse cells causes differentiation of naïve CD4+ T cells only to Treg cells, and expression of Foxp3 will be upregulated significantly (81). Consistent with these results, using rapamycin, an inhibitor of mTOR, can lead to a similar phenotype (81). Moreover, it has been shown that rapamycin can ameliorate EAE in a mouse model by means of the mTOR-STAT3 signaling pathway, which is important to Th17 development (82). In view of a molecular mechanism, some articles have reported that mTORC1 may regulate the development of Th17 cells by decreasing the expression of growth factor independent 1 transcriptional repressor (Gfi1) and increasing the expression of RORγt (79, 80). Inactivation of mTORC1 can cause Th17 cells to become more sensitive to the TGFβ signaling pathway, which can overcome STAT3 signaling further to influence the Th17 and Treg balance (81). In view of glycolysis, mTORC1 has the ability to upregulate the expression of Glut1 gene and promote glycolysis (83). In the early time of development of Th17, glutamine uptake increased significantly (43). Interestingly ASCT2, the transporter of glutamine, has been reported to be associated with the activation of mTORC1. ASCT2 is indispensable for mTORC1 activation in CD4+ T cells, and differentiation of Th17 is impaired in ASCT2-deficient mice. This finding partially explains how glutamine can modulate Th17 and Tregs development (49).
Conversely, the ability of mTOR to promote Th17 differentiation is through promoting the expression of hypoxia-inducible factor 1α (HIF1α). HIF1α is a transcriptional factor. Just like mTOR, it is also a well-known environmental sensor and metabolic regulator. Recently, increasingly more investigations have been focused on the ability of mTOR in T cell differentiation. Notably, both mTOR and HIF1α can promote glucose import and glycolysis at the transcriptional and translational level (84). Th17 cells have been shown to rely more heavily on glycolytic pathways than any other T cell subset (58, 79, 80). In the absence of HIF1α, the ability of murine naïve T cells to differentiate to Th17 cells are dramatically impaired (58). Some researchers have attributed this to the consequence of decreased glycolysis. It has been reported that HIF1α can promote glucose uptake and reinforce glycolysis by upregulating expression of Glut1 and pyruvate dehydrogenase kinase 1 (PDK1), respectively. Glut1 facilitates the transport of glucose across the plasma membranes of mammalian cells. On the other hand, PDK1 can prevent pyruvate feeding into the TCA cycle and drive it to be converted into lactate and then promote glycolysis. Conversely, expression of many glycolysis-related genes is downregulated in HIF1α mutation mice, such as Glut1, HK2, Ldha, and Pkm. All of these genes are rate-limiting enzymes (58, 85). At the transcriptional level, it has been reported that HIF1α can upregulate RORγt expression. Moreover, HIF1α can also form a complex with RORγt and p300 to further promote IL-17 expression. On the contrary, it has also been reported that HIF-1α attenuates Treg development by binding Foxp3 and targeting it for proteasomal degradation (58, 85).
Conversely, after TCR stimulation, naïve CD4+ T cells lacking mTOR tend to differentiate into Tregs, even in the absence of exogenous TGFβ. One research group found that proliferation of Treg is subtly linked to the transient inhibition of mTOR. Constant activation of mTOR can impair the function and proliferation of Treg in vitro. One possible explanation is that high mTOR activity may change the metabolic profile of Treg cells. As a consequence of the activation of mTOR, FA synthesis and aerobic glycolysis is activated, further inhibiting the development of Treg. However, the mechanism for these phenomena remains unclear (79, 86). These results still prove that levels of mTOR activity can influence the balance of Th17 and Treg.
5′ AMP-Activated Protein Kinase (AMPK)
AMP-activated protein kinase is a heterotrimeric kinase complex, which is formed by α, β, and γ subunits. The γ subunit of AMPK contains a special domain, cystathionine-β-synthase that can sense the cellular AMP/ATP ratio and respond to the low energy conditions. In T cells, following TCR stimulation, AMPK is activated by the Ca2+-dependent protein. Once AMPK is activated, the energy-consuming metabolism is downregulated and the catabolic metabolism that can produce energy is upregulated. Unsurprisingly, AMPK can inhibit FAS and promote FAO (61, 87–89). Many important enzymes that are included in FAO and FAS can be regulated by the activation of AMPK, such as ACC1 and ACC2, almitoyl transferase I (CPT I), and sterol regulatory element-binding protein 1c (SREBP-1c). CPT I is a rate-limiting enzyme in FAO; otherwise, phosphorylated ACC1/2 and SREBP-1c can inhibit the synthesis of fatty acids and activate FAO (90). Another phenomenon is the relationship between AMPK and mTOR. AMPK and mTOR are negative regulators for each other, and AMPK can also negatively regulate the level of glycolysis (91). As described above, Th17 cells rely more on glycolysis, and Treg cells rely more on FAO, and the ability of AMPK to modulate metabolism indeed impacts the balance of Th17/Treg; AMPK is highly expressed and active in iTreg (92). According to this report, activated AMPK can drive naïve T cells to differentiate toward Treg cells both in vitro and in vivo. On the contrary, defective function in AMPK leads to an increase in mTOR activity and upregulation of glycolysis (90). Some studies have shown that treating naïve T cells with metformin, which is an activator of AMPK, can impair Th17 differentiation. Treating with metformin can increase lipid oxidation in T cells in vitro. In vivo administration of metformin was shown to be sufficient to decrease Glut1 expression (43, 77). Moreover, treating mice with metformin can also relieve several inflammatory diseases in mouse models (43, 77). In addition, treating mice with metformin can decrease the ratio of Th17:Treg in both colitis and asthma models, which can be attributed to the ability of decreasing the aerobic glycolysis and increasing the FAO (89, 93, 94). Notably, results from genetic and pharmacological approaches are not totally consistent. In vitro results compared to WT, AMPK−/− CD4+ T cells have no more tendency to differentiate to Th17 cells. However, T cells from LKB1 knockout mice can produce more IL-17 than WT mice (95), which is upstream of AMPK and can activate 13 other kinases in the AMPK family (95, 96). The mechanism for this is still unclear and LKB1 may act through other pathways to impact Th17 differentiation (please see Figure 2).
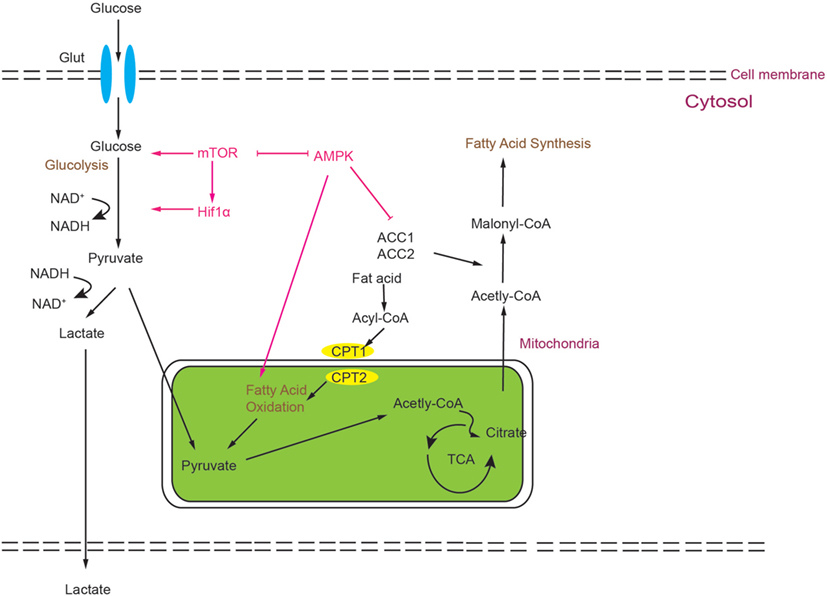
Figure 2. Simplified scheme of the metabolic processes activated during T cell development. Glucose is imported from the extracellular space by Glut1 and degraded by glycolysis to generate two pyruvate molecules, which can be converted to lactate or Acetyl-CoA. Acetyl-CoA can feed into the tricarboxylic acid (TCA) cycle. Alternatively, it can also be utilized as a precursor of FAS. Activation of mammalian target of rapamycin (mTOR) can modulate HIF1α activity, and both can promote glycolysis. mTOR and AMP-activated protein kinase (AMPK) act as negative regulators for each other. AMPK can inhibit glycolysis and promote fatty acid oxidation.
Nuclear Receptors (NRs) in the Th17/Treg Balance
Nuclear receptors are a class of transcriptional factors that can only regulate the expression of target genes when binding with the ligands. Exposure to the ligands leads to a conformational change, and NRs then acquire the ability to start transcription. Ligands of NRs are widely varied from hormones to metabolites (97). Now, many NRs are thought to be important metabolic regulators and they can participate in many metabolic processes during T cell development. These NRs can either promote or inhibit T cell activation and differentiation. Moreover, recent studies have shown that NRs play crucial roles in maintaining the Th17/Treg balances (21).
The aryl hydrocarbon receptor (AHR) is a conserved ligand NR in animals and, as a transcriptional factor, it is classified as being a member of the basic helix-loop-helix family. In the last decade, AHR was established for the ability to sense environmental toxins and protect hosts from being harmed. The external toxin, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), and endogenous ligands, 6-formylindolo[3,2-b]carbazole (FICZ), are well-known ligands of AHR. However, recently the function of AHR in the development of T helper cells has been increasingly more described in research papers (98–100). AHR regulates Treg and Th17 cell differentiation in a ligand-specific manner. AHR activation by TCDD induced Treg cells that suppressed EAE, whereas AHR activation by FICZ interfered with Treg cell differentiation, boosted TH17 cell differentiation, and worsened EAE. But the detail mechanisms remain to be elucidated (99, 100). Some researchers have shown that AHR, together with HIF1α, can regulate the metabolic reprogramming during the development of Treg cells. In the early stage of Treg cells development, expression of metabolism-related genes is controlled by HIF1α, and at the later time points, AHR can promote HIF1α degradation to take over the control of these metabolic genes (101).
Estrogen-related receptor alpha (ERRα) is recognized as a key transcriptional factor in maintaining metabolic homeostasis and can regulate comprehensive genes involved in different metabolic pathways. These genes are related to glycolysis, mitochondrial energy metabolism, OXPHO, and fatty acid metabolism (102, 103). Interestingly, these important NRs are orphan receptors, and the endogenous ligands of ERRα have still not been found (104). It has been shown that in naïve T cells ERRα is weakly expressed. After TCR stimulation, expression of ERRα is dramatically upregulated (102, 103). XCT790, an antagonist of ERRα can decrease the level of energy metabolism in naïve T cells and impair the differentiation of all CD4+T cell lineages. It has been reported that the XCT790-induced reduction in T helper cells function is through upregulation of CPT1 levels, which can activate FAO and reduce glucose metabolism (102). This impairment could be rescued by adding exogenous fat acids. However, exogenous fat acids cannot restore T cell functions completely after differentiation, except for Treg cells (102). Genetic knockout ERRα shows similar results. In the mouse EAE model, the ERRα mutation shows restored symptoms compared to WT mice. Further analysis shows defective ERRα results in a decrease of Th17 polarization, but the differentiation and function of Treg cells are not affected. These results indicate that, compared to Treg, ERRα is more important for Th17 differentiation (102, 103).
The liver x receptors (LXRs) mainly regulate metabolism of cholesterol, fatty acids, and glucose homeostasis. The endogenous ligands of LXRs are recognized as oxysterols. LXRs impact the balance of Th17/Treg by regulating the genes involved in cholesterol metabolism. These genes include Abca1, Abcg1, and SREBP-1c (105). After binding, LXR can negatively regulate the expression of SREBP-1c and inhibit the cholesterol synthesis. These changes are disadvantageous to effector T helper cell development. LXR can also interact with AHR and inhibit its function (106, 107). In vitro, T0901317, which is an agonist of LXRs, can inhibit murine CD4+ naïve T cells differentiation to Th17 (106, 107), and inhibition of Th17 by LXRs can be overcome by adding mevalonate, which is a precursor of cholesterol. Moreover, LXR agonists also suppress expression of IL-17, RORγt, and other Th17-associated genes. In contrast, the antagonist of LXRs, GSK2033, enhances differentiation and proliferation of Th17 (106, 108). In vivo, activation of LXRs can relieve the symptoms of EAE in mouse models by reducing Th17 polarization (106).
Fortunately, most of the NRs involved in the Th17/Treg balance have their own specific inhibitors. Pharmacological modulators make it possible to manipulate the metabolic processes manually, providing an easy way to study the function of metabolic reprogramming in T cell development.
Conclusion
As two important subsets of T helper cells, Th17 and Treg represent two arms of an immune response. Th17 cells can participate in the defense against extracellular bacterial and fungal infections. On the other hand, Treg cells can regulate the immune response and maintain immune homeostasis. Excessive activation of Th17 can lead to inflammation and autoimmune disease. Conversely, enhanced Treg function may cause tumorigenesis and can also be used by pathogens to escape the immune system. The balance of Th17 and Treg cells is critical for the health of the host. An appropriate balance between Th17 and Treg is a critical prerequisite for good health. Considering the close relationship between Th17 and Treg cells, and their plasticity, modulating the immunological balance between Th17 and Treg may represent a promising option for immune therapy.
As mentioned above, the field of immune metabolism has gained increasingly more attention in immunological research. Modulating cellular metabolism may be a feasible solution for adjusting the balance of Th17 and Treg. Both glycolysis and fatty acid can be regulated using small molecule inhibitors of key regulatory enzymes in these processes. Direct manipulation of cellular metabolism can shift the balance between Th17 and Treg cells (109).
Small molecular inhibitors or activators of metabolic enzymes are now becoming available, which makes it possible to manipulate metabolism of T cells much easier than ever before. Moreover, these small molecules can become potential drugs for clinical therapies. However, considering the complexity of metabolism, it is necessary to confirm findings derived from pharmacological inhibitors using animal disease models and clinical trials (please see Figure 3 and Table 1).
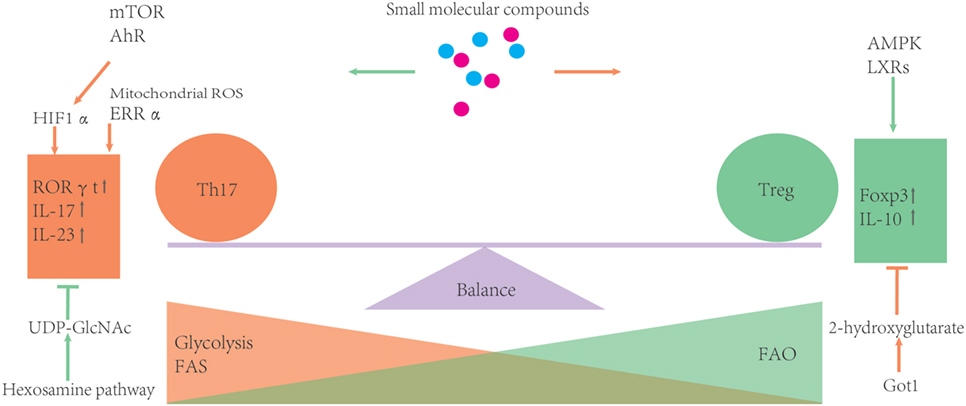
Figure 3. Metabolic processes can influence the balance of Th17/T-regulatory (Treg). During development, Treg rely more on fatty acid oxidation (FAO) as the energy source. Otherwise, glycolysis and FAS can promote Th17 differentiation. Several metabolic regulators or enzymes are involved in the balance of Th17 and Treg. Some small molecular compounds of these regulators or enzymes may provide an easy way to further investigate how certain metabolic routes impact the Th17/Treg balance.
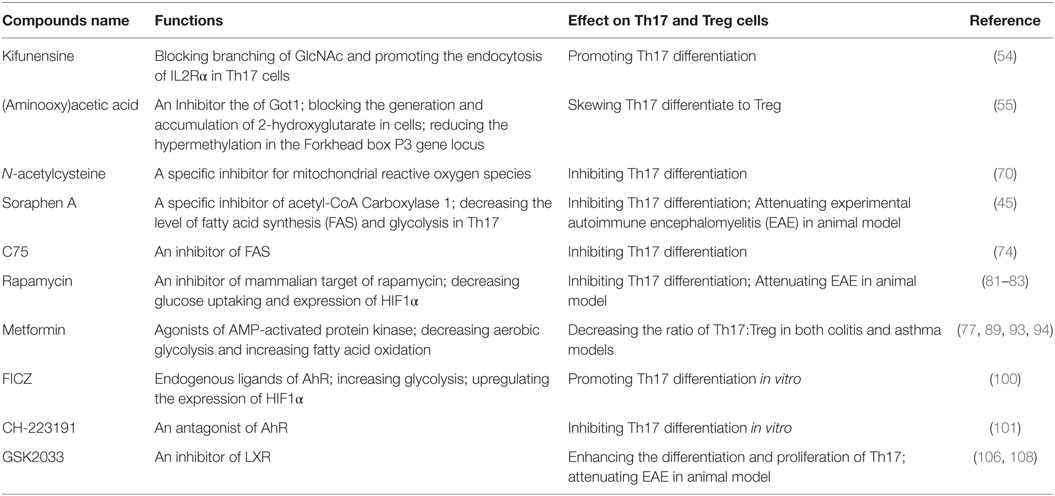
Table 1. Compounds and their effects on Th17 and T-regulatory (Treg) cells.
Metabolic factors driving Th17 development and those shaping the balance between Th17 and Treg have significant biological implications for the design and implementation of novel therapeutic interventions.
Author Contributions
LS, the first author, contributed to collection of references and manuscript preparation. JF and YZ contributed to the editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from National Key R&D Program of China (2016YFC1305102), NSFC of China (81671561), 1000 Young Talents Plan Program of China, Initial Funding for New PI, Fudan Children’s Hospital and Fudan University, and Shanghai Municipal Planning Commission of Science and Research Fund (201740065) (to YZ). Shanghai Pujiang Program 16PJ1401600 (to JF).
References
1. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity (2010) 32(5):605–15. doi:10.1016/j.immuni.2010.05.003
2. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol (2016) 28(5):514. doi:10.1016/j.smim.2016.10.009
3. Park BV, Pan F. Metabolic regulation of T cell differentiation and function. Mol Immunol (2015) 68(2):497–506. doi:10.1016/j.molimm.2015.07.027
4. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30(5):646–55. doi:10.1016/j.immuni.2009.05.001
5. Luckheeram RV, Rui Z, Devi VA, Bing X. CD4+T cells: differentiation and functions. Clin Dev Immunol (2012) 2012(12):925135. doi:10.1155/2012/925135
6. Syrbe U, Siveke J, Hamann A. Th1/Th2 subsets: distinct differences in homing and chemokine receptor expression? Springer Semin Immunopathol (1999) 21(3):263–85. doi:10.1007/BF00812257
7. Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev (2003) 8(3):223–46.
8. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi:10.1146/annurev.immunol.25.022106.141557
9. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi:10.1038/ni1261
10. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
11. Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transpl Int (2016) 30(8):745–53. doi:10.1111/tri.12909
12. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
13. Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J Clin Invest (2003) 112(8):1186–91. doi:10.1172/JCI200319079
14. Lochner M, Wang Z, Sparwasser T. The special relationship in the development and function of T helper 17 and regulatory T cells. Prog Mol Biol Transl Sci (2015) 136:99–129. doi:10.1016/bs.pmbts.2015.07.013
15. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi:10.1038/ni1539
16. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
17. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi:10.1016/j.immuni.2006.01.001
18. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol (2007) 19(6):695–702. doi:10.1093/intimm/dxm045
19. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi:10.1016/j.cell.2006.07.035
20. Yoshida S, Haque A, Mizobuchi T, Iwata T, Chiyo M, Webb TJ, et al. Anti-type V collagen lymphocytes that express IL-17 and IL-23 induce rejection pathology in fresh and well-healed lung transplants. Am J Transplant (2006) 6(4):724–35. doi:10.1111/j.1600-6143.2006.01236.x
21. Burrell BE, Bishop DK. Th17 cells and transplant acceptance. Transplantation (2010) 90(9):945. doi:10.1097/TP.0b013e3181f5c3de
22. Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22(22):531. doi:10.1146/annurev.immunol.21.120601.141122
23. Gaur P, Qadir GA, Upadhyay S, Singh AK, Shukla NK, Das SN. Skewed immunological balance between Th17 (CD4(+)IL17A (+)) and Treg (CD4 (+)CD25 (+)FOXP3 (+)) cells in human oral squamous cell carcinoma. Cell Oncol (Dordr) (2012) 35(5):335–43. doi:10.1007/s13402-012-0093-5
25. Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res (2011) 71(4):1263–71. doi:10.1158/0008-5472.CAN-10-2907
26. Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol (2009) 21(3):274–80. doi:10.1016/j.coi.2009.05.021
27. Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol (2007) 19(6):353–61. doi:10.1016/j.smim.2007.10.008
28. Rizzo A, De Mare V, Rocchi C, Stolfi C, Colantoni A, Neurath MF, et al. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis (2014) 35(7):1536–46. doi:10.1093/carcin/bgu027
29. Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem (2008) 283(25):17003–8. doi:10.1074/jbc.M801286200
30. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem (2007) 282(13):9358–63. doi:10.1074/jbc.C600321200
31. Kyburz D, Corr M. Th17 cells generated in the absence of TGF-beta induce experimental allergic encephalitis upon adoptive transfer. Expert Rev Clin Immunol (2011) 7(3):283–5. doi:10.1586/eci.11.7
32. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448(7152):480–3. doi:10.1038/nature05969
33. Reinerthartwall L, Honkanen J, Salo HM, Nieminen JK, Veijola R, Simell O, et al. Th1/Th17 plasticity is a marker of advanced β cell autoimmunity and impaired glucose tolerance in humans. J Immunol (2015) 194(1):68. doi:10.4049/jimmunol.1401653
34. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, Jager WD, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A (2010) 107(33):14751–6. doi:10.1073/pnas.1003852107
35. Ueno A, Ghosh A, Hung D, Li J, Jijon H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J Gastroenterol (2015) 21(43):12283–95. doi:10.3748/wjg.v21.i43.12283
36. Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature (2015) 523(7559):221. doi:10.1038/nature14452
37. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1β and IL-2 convert human Treg into T H 17 cells. Clin Immunol (2009) 131(2):298–307. doi:10.1016/j.clim.2008.12.008
39. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27(1):441–64. doi:10.1146/annurev-cellbio-092910-154237
40. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi:10.3389/fimmu.2015.00001
41. Bauer DE, Harris MH, Plas DR, Lum JJ, Hammerman PS, Rathmell JC, et al. Cytokine stimulation of aerobic glycolysis in hematopoietic cells exceeds proliferative demand. FASEB J (2004) 18(11):1303–5. doi:10.1096/fj.03-1001fje
42. Kumar P, Natarajan K, Shanmugam N. High glucose driven expression of pro-inflammatory cytokine and chemokine genes in lymphocytes: molecular mechanisms of IL-17 family gene expression. Cell Signal (2014) 26(3):528–39. doi:10.1016/j.cellsig.2013.11.031
43. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi:10.1172/JCI76012
44. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38(2):225–36. doi:10.1016/j.immuni.2012.10.020
45. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. Erratum: de novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi:10.1038/nm.3704
46. Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ, et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep (2015) 12(6):1042–55. doi:10.1016/j.celrep.2015.07.014
47. Vivien L, Benoist C, Mathis D. T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int Immunol (2001) 13(6):763. doi:10.1093/intimm/13.6.763
48. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi:10.1016/j.immuni.2011.09.021
49. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40(5):692–705. doi:10.1016/j.immuni.2014.04.007
50. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16(6):769–77. doi:10.1016/S1074-7613(02)00323-0
51. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20(1):61. doi:10.1016/j.cmet.2014.05.004
52. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi:10.1038/ni.2556
53. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 43(4):2466–85. doi:10.1093/nar/gkv047
54. Araujo L, Khim P, Mkhikian H, Mortales CL, Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife (2017) 6:e21330. doi:10.7554/eLife.21330
55. Xu T, Stewart KM, Wang X, Liu K, Xie M, Kyu RJ, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature (2017) 548(7666):228. doi:10.1038/nature23475
56. Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell (2007) 18(4):1437–46. doi:10.1091/mbc.E06-07-0593
57. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity (2016) 44(6):1337. doi:10.1016/j.immuni.2016.05.009
58. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi:10.1084/jem.20110278
59. Yabu M, Shime H, Hara H, Saito T, Matsumoto M, Seya T, et al. IL-23-dependent and -independent enhancement pathways of IL-17A production by lactic acid. Int Immunol (2011) 23(1):29–41. doi:10.1093/intimm/dxq455
60. Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med (2005) 201(2):259–66. doi:10.1084/jem.20042020
61. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211. doi:10.1016/j.tibs.2015.12.001
62. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
63. Deberardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104(49):19345–50. doi:10.1073/pnas.0709747104
64. Birsoy K, Wang T, Chen WW, Freinkman E, Aburemaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162(3):540–51. doi:10.1016/j.cell.2015.07.016
65. Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem Sci (2016) 41(8):712. doi:10.1016/j.tibs.2016.05.013
66. Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
67. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG, et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162(3):552–63. doi:10.1016/j.cell.2015.07.017
68. Elkhal A, Rodriguez Cetina Biefer H, Heinbokel T, Uehara H, Quante M, Seyda M, et al. NAD(+) regulates Treg cell fate and promotes allograft survival via a systemic IL-10 production that is CD4(+) CD25(+) Foxp3(+) T cells independent. Sci Rep (2016) 6:22325. doi:10.1038/srep22325
69. Lim HW, Kang SG, Ryu JK, Schilling B, Fei M, Lee IS, et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J Exp Med (2015) 212(5):607. doi:10.1084/jem.20132378
70. Zhi L, Ustyugova IV, Chen X, Zhang Q, Wu MX. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Immunol (2012) 189(4):1639–47. doi:10.4049/jimmunol.1200528
71. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi:10.1084/jem.20151159
72. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol (2015) 36(2):81–91. doi:10.1016/j.it.2014.12.005
73. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14(7):435–46. doi:10.1038/nri3701
74. Young KE, Flaherty S, Woodman KM, Sharma-Walia N, Reynolds JM. Fatty acid synthase regulates the pathogenicity of Th17 cells. J Leukoc Biol (2017) 102(5):1229–35. doi:10.1189/jlb.3AB0417-159RR
75. Xu X, Wang Y, Wei Z, Wei W, Peng Z, Bei T, et al. Madecassic acid, the contributor to the anti-colitis effect of madecassoside, enhances the shift of Th17 toward Treg cells via the PPAR|[gamma]||[sol]|AMPK|[sol]|ACC1 pathway. Cell Death Dis (2017) 8(3):e2723. doi:10.1038/cddis.2017.150
76. Jump DB, Torres-Gonzalez M, Olson LK. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem Pharmacol (2011) 81(5):649–60. doi:10.1016/j.bcp.2010.12.014
77. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi:10.4049/jimmunol.1003613
78. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
79. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12(4):295–303. doi:10.1038/ni.2005
80. Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep (2012) 1(4):360–73. doi:10.1016/j.celrep.2012.02.007
81. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi:10.1146/annurev-immunol-020711-075024
82. Hua W, Liu H, Xia LX, Tian BP, Huang HQ, Chen ZY, et al. Rapamycin inhibition of eosinophil differentiation attenuates allergic airway inflammation in mice. Respirology (2015) 20(7):1055–65. doi:10.1111/resp.12554
83. Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, et al. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol (2008) 295(3):C836–43. doi:10.1152/ajpcell.00554.2007
84. Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med (2007) 85(12):1339–46. doi:10.1007/s00109-007-0282-2
85. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi:10.1016/j.cell.2011.07.033
86. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499(7459):485. doi:10.1038/nature12297
87. Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, et al. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol (2010) 80(11):1708. doi:10.1016/j.bcp.2010.08.009
88. Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. Faseb J (2016) 30(11):3800. doi:10.1096/fj.201600522R
89. Park MJ, Lee SY, Moon SJ, Son HJ, Lee SH, Kim EK, et al. Metformin attenuates GVHD via restricting mTOR/STAT3 and promoting AMPK – autophagy for the balance between Th17 and Tregs. Transl Res (2016) 173:115–30. doi:10.1016/j.trsl.2016.03.006
90. Tamás P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med (2006) 203(7):1665–70. doi:10.1084/jem.20052469
91. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30(2):214–26. doi:10.1016/j.molcel.2008.03.003
92. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol (2011) 13(9):1016. doi:10.1038/ncb2329
93. Koh SJ, Kim JM, Kim IK, Ko SH, Kim JS. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J Gastroenterol Hepatol (2014) 29(3):502–10. doi:10.1111/jgh.12435
94. Calixto MC, Lintomen L, Andre DM, Leiria LO, Ferreira D, Lellis-Santos C, et al. Metformin attenuates the exacerbation of the allergic eosinophilic inflammation in high fat-diet-induced obesity in mice. PLoS One (2013) 8(10):e76786. doi:10.1371/journal.pone.0076786
95. Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J (2004) 23(4):833–43. doi:10.1038/sj.emboj.7600110
96. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol (2011) 187(8):4187–98. doi:10.4049/jimmunol.1100367
97. Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol (2003) 65(1):261–311. doi:10.1146/annurev.physiol.65.092101.142528
98. Charlotte E. The aryl hydrocarbon receptor in immunity: tools and potential. Methods Mol Biol (2016) 1371:239–57. doi:10.1007/978-1-4939-3139-2_16
99. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi:10.1038/nature06880
100. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453(7191):106–9. doi:10.1038/nature06881
101. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat Med (2015) 21(6):638–46. doi:10.1038/nm.3868
102. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-{alpha} is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A (2011) 108(45):18348–53. doi:10.1073/pnas.1108856108
103. Mckarns S. Selective deletion of estrogen receptor alpha in FoxP3+ CD4+ T cells influences peripheral T cell homeostasis (BA11P.139). J Immunol (2015) 194(1 Suppl):184.21.
104. Yuk JM, Kim TS, Kim SY, Lee HM, Han J, Dufour CR, et al. Orphan nuclear receptor ERRα controls macrophage metabolic signaling and A20 expression to negatively regulate TLR-induced inflammation. Immunity (2015) 43(1):80–91. doi:10.1016/j.immuni.2015.07.003
105. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med (2003) 9(2):213. doi:10.1038/nm820
106. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest (2011) 121(2):658–70. doi:10.1172/JCI42974
107. Xu J, Wagoner G, Douglas JC, Drew PD. Liver X receptor agonist regulation of Th17 lymphocyte function in autoimmunity. J Leukoc Biol (2009) 86(2):401. doi:10.1189/jlb.1008600
108. Vucic E, Calcagno C, Dickson SD, Rudd JHF, Hayashi K, Bucerius J, et al. Regression of inflammation in atherosclerosis by the LXR agonist R211945: a noninvasive assessment and comparison with atorvastatin. Jacc Cardiovascular Imaging (2012) 5(8):819–28. doi:10.1016/j.jcmg.2011.11.025
Keywords: metabolism, Th17 cell, T-regulatory cell, immunoregulation, cell polarization
Citation: Sun L, Fu J and Zhou Y (2017) Metabolism Controls the Balance of Th17/T-Regulatory Cells. Front. Immunol. 8:1632. doi: 10.3389/fimmu.2017.01632
Received: 29 August 2017; Accepted: 09 November 2017;
Published: 27 November 2017
Edited by:
Wanjun Chen, National Institutes of Health (NIH), United StatesReviewed by:
Hongbo Chi, St. Jude Children’s Research Hospital, United StatesVassiliki A. Boussiotis, Harvard Medical School, United States
Copyright: © 2017 Sun, Fu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yufeng Zhou, yfzhou1@fudan.edu.cn